

Abstract
Background & Aims:
Intraductal papillary mucinous neoplasms (IPMNs) are lesions that can progress to invasive pancreatic cancer and an important system for studies of pancreatic tumorigenesis. We performed comprehensive genomic analyses of entire IPMNs to determine the diversity of somatic mutations in genes that promote tumorigenesis.
Methods:
We microdissected neoplastic tissues from 6–24 regions each of 20 resected IPMNs, resulting in 227 neoplastic samples that were analyzed by capture-based targeted sequencing. Somatic mutations in genes associated with pancreatic tumorigenesis were assessed across entire IPMN lesions, and the resulting data were supported by evolutionary modeling, whole-exome sequencing, and in situ detection of mutations.
Results:
We found a high prevalence of heterogeneity among mutations in IPMNs. Heterogeneity in mutations in KRAS and GNAS was significantly more prevalent in IPMNs with low-grade dysplasia than in IPMNs with high-grade dysplasia (P<.02). Whole-exome sequencing confirmed that IPMNs contained multiple independent clones, each with distinct mutations, as originally indicated by targeted sequencing and evolutionary modeling. We also found evidence for convergent evolution of mutations in RNF43 and TP53, which are acquired during later stages of tumorigenesis.
Conclusions:
In an analysis of the heterogeneity of mutations throughout IPMNs, we found that early-stage IPMNs contain multiple independent clones, each with distinct mutations, indicating their polyclonal origin. These findings challenge the model in which pancreatic neoplasms arise from a single clone. Increasing our understanding of the mechanisms of IPMN polyclonality could lead to strategies to identify patients at increased risk for pancreatic cancer.
Keywords: PDAC, Carcinogenesis, driver gene, oncogene
Graphical Abstract
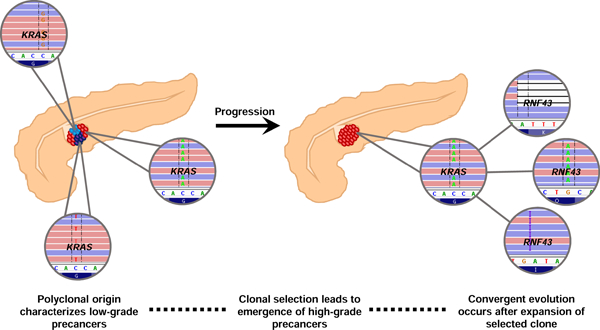
Lay Summary
Genomic analyses reveal that low-grade precancerous neoplasms in the pancreas arise from multiple independent clones, while selection of a dominant clone leads to neoplastic progression.
INTRODUCTION
Invasive pancreatic ductal adenocarcinoma (PDAC) is a deadly disease with a dismal prognosis and is expected to be the second leading cause of cancer-related deaths in the United States by 20301. PDAC arises from non-invasive precancerous lesions, including pancreatic intraepithelial neoplasia (PanIN) and intraductal papillary mucinous neoplasm (IPMN), which are curable if detected and treated before they progress to invasive carcinoma. In addition to being a key target for early detection and intervention, direct analysis of these precancerous lesions represents a critical strategy to better understand the earliest steps in pancreatic tumorigenesis. Precancerous lesions in the pancreas are intraepithelial neoplasms which are classified by grade of dysplasia based on the architectural and cytological atypia2. Current consensus guidelines recommend a two-tiered grading system, with low-grade lesions showing minimal to moderate dysplasia and high-grade showing severe dysplasia2. Another key feature of the classification of these precancers is their size – PanINs are microscopic lesions (by definition <0.5cm), and IPMNs are larger (by definition ≥1cm) and thus can be identified using imaging modalities2. While the majority of PDACs likely arise through PanINs, IPMNs present unique clinical challenges as they are common in the population and are often incidentally identified on routine abdominal imaging3. Moreover, their size and resultant early clinical intervention provide critical human tissue samples, which can be used to interrogate early pancreatic neoplasia.
Molecular studies of IPMN suggest that the progression from low-grade to high-grade dysplasia is associated with an accumulation of genetic alterations in oncogenes and tumor suppressor genes, eventually leading to the development of invasive PDAC. Previous studies have characterized several key driver gene alterations in IPMNs: mutations in the oncogenes KRAS and GNAS are thought to be the earliest driver gene alterations, while mutations in tumor suppressor genes such as RNF43, CDKN2A, and TP53 occur later4. With the exception of GNAS, these driver genes are also commonly altered in invasive PDACs occurring in the absence of IPMN5. Most studies of malignant progression in IPMNs analyzed a single tissue section from each lesion across many patients, allowing comparison of the genetic alterations among different IPMNs based on grade of dysplasia or other clinical and pathological features6–10. Although such studies allow estimation of mutation prevalence in specific IPMN subgroups, they do not provide comprehensive data about the diversity of genetic alterations within a single lesion. Recent studies that analyzed more than one section from each IPMN suggest the potential for substantial genetic heterogeneity within these neoplasms. For instance, targeted sequencing on two regions per IPMN demonstrated that 23% had multiple KRAS and/or GNAS mutations11; however, analyses systematically interrogating this genetic heterogeneity are currently lacking. Such studies can provide important insights into the evolutionary history of early pancreatic tumorigenesis. Importantly, recent data have shown a lack of genetic heterogeneity with respect to driver gene alterations in invasive PDAC, raising the possibility that unique evolutionary processes govern the earlier stages of tumorigenesis in the pancreas12,13. As our current understanding of pancreatic tumor evolution has mostly been inferred from sequencing advanced cancers, there is a critical need for analyses of bona fide precancers to directly describe initiation and progression in pancreatic neoplasia. The importance of studying precancers has been exemplified in other tumor types, such as esophageal carcinogenesis14,15. These studies reshaped our understanding of the origin and dynamics of tumorigenesis in these organs and have significant biologic and therapeutic implications.
To explore the evolutionary history of early pancreatic tumorigenesis, we performed multi-region targeted next generation sequencing on surgically resected IPMNs. These data provide novel insights into IPMN progression, which will advance our understanding of the origins of invasive pancreatic cancer.
METHODS
Specimen acquisition
This study was approved by the Institutional Review Board of the Johns Hopkins Hospital. We retrospectively collected multi-region samples from 20 patients diagnosed with IPMN who had undergone surgical resection at the Johns Hopkins Hospital between 2008 and 2015. Slides from every available formalin-fixed paraffin embedded (FFPE) block from each IPMN were stained with hematoxylin-and-eosin (H&E). An H&E slide from each block was reviewed by an expert pancreatic pathologist to confirm diagnosis and determine regions for microdissection. One H&E slide from each matched normal (duodenum or spleen) was also reviewed by an expert pathologist prior to DNA extraction.
Laser capture microdissection
Five 10µm serial tissue sections from FFPE blocks were cut onto membrane slides (Zeiss MembranSlide 1.0 PEN). Deparaffinization was performed in fresh xylenes for 2 min, followed by 100% ethanol for 2 min, 95% ethanol for 2 min, and 70% ethanol for 2 min. Subsequently, the slides were stained by crystal-violet (Sigma Aldrich; diluted 1:4 in 70% ethanol) for 30 sec and dehydrated by ascending ethanol solutions. The stained slides were microdissected within 1 h. Regions of IPMN epithelium were identified under the microscope and microdissected for enrichment of neoplastic cellularity on a Leica LMD7000 instrument. Microdissected tissues were collected into 0.5ml Lo-bind tubes (Eppendorf) and immediately processed for subsequent DNA extraction. For matched-normal samples, five 5µm sections were cut onto regular slides and tissue was scraped off using a sterile razor blade (Personna).
DNA extraction and quantification
DNA was extracted from each sample using the QIAamp DNA FFPE Tissue kit (Qiagen) and the MagMAX FFPE Isolation kit (Applied Biosystems) (See Supplementary Methods). DNA concentration was measured using the Qubit dsDNA High Sensitivity Assay kit (Invitrogen) according to the manufacturer’s protocol on a Qubit 2.0 fluorometer (Invitrogen). DNA was stored at −20C until library preparation.
Targeted sequencing and analysis
A targeted sequencing approach analyzed the entire coding regions of 15 known driver genes in IPMN tumorigenesis (KRAS, GNAS, BRAF, RNF43, CDKN2A, PIK3CA, PTEN, APC, CTNNB1, MAP2K4, STK11, ATM, TP53, TGFBR2, SMAD4). Library preparation was performed using the Agilent SureSelectXT Target Enrichment System (Agilent) following the manufacturer’s instructions with several modifications (see Supplementary Methods). The barcoded libraries were sequenced using an Illumina MiSeq system (Illumina), generating 300 base pairs (2×150bp reads) per fragment. The average distinct sequencing depth was 570x per sample. We identified candidate mutations that were altered in ≥5% of distinct reads with coverage >100X. All candidate mutations were validated by visual inspection in Integrated Genome Viewer (IGV)16.
Somatic mutation evolutionary reconstruction
We reconstructed somatic mutation evolutionary trees based on the neoplastic cell fractions (NCFs) of the mutations we identified, using SCHISM software17. Mutation NCFs were calculated using VAF, estimated tumor purity (p), tumor copy number (CNT) at the mutation site, germline copy number (CNN) at mutation site, and multiplicity (m), i.e. estimated number of mutant alleles.
Tumor purity and copy number are difficult to estimate from a small targeted panel. Rather than assuming a single value for purity, on which both CNT and NCF depend, we tried purity values in the range 0.5 to 0.9, in increments of 0.1. CNT and m were estimated at each purity level. Tumor integer copy number was estimated by scaling the tumor-to-normal read ratios to consider purity. In the absence of allele-specific copy number information, m was set to 1 unless VAF>0.8. For mutations with VAF>0.8, m was set to the CNT if CNT ≤ 2, and CNT-1 if CNT >2.
For each IPMN, SCHISM was used to estimate an optimal evolutionary tree at each purity level. We chose the optimal tree based on the highest fitness value calculated by SCHISM. When SCHISM identified multiple trees with the same highest fitness value, we present all trees in Supplementary Figure 1. If SCHISM could not infer a tree with a fitness >0.1, we excluded the case from the evolutionary analysis. SCHISM was originally designed to model only tumors of monoclonal origin, and we extended it to handle topologies with multiple originating clones.
Whole-exome sequencing and analysis
Human exome capture was performed using Agilent’s SureSelect Human All Exon 50Mb Kit 5.0 (Agilent). The captured libraries were sequenced with an Illumina XTEN system, generating 300 base pairs (2×150bp reads) per fragment. The average distinct sequencing depth was 104x per sample. We identified candidate mutations that were altered in ≥10% of distinct reads. All candidate mutations were validated by visual inspection in Integrated Genome Viewer (IGV)16 (see Supplementary Methods).
Single-molecule RNA in situ hybridization
The BaseScope assays (Advanced Cell Diagnostics, Inc., Newark, CA) were developed to achieve point mutation-specific detection of the KRAS transcripts (see Supplementary Methods). The 1-ZZ probe BA-Hs-KRAS-G12V was designed to target KRAS G12V (nt. G35T) mRNA, the 1-ZZ probe BA-Hs-KRAS-G12D was designed to target KRAS G12D (nt. G35A) mRNA, and the 1-ZZ probe BA-Hs-KRAS-G12R was designed to target KRAS G12R (nt. G34C) mRNA. The BaseScope™ Reagent Kit – RED (Advanced Cell Diagnostics, Newark, CA) was used for sample pretreatment, hybridization and signal development according to the manufacturer’s instructions. FFPE tissue section samples were prepared according to manufacturer’s recommendations.
Estimating genetic heterogeneity
Genetic heterogeneity was quantitatively measured using Jaccard similarity coefficients. The Jaccard similarity coefficient is defined as the ratio of shared variants to all variants (shared + discordant) between two samples. We calculated Jaccard similarity coefficients (based on KRAS and GNAS mutations only) between all samples for each IPMN. Next, we calculated the average Jaccard similarity coefficient for each IPMN. Additionally, the average coefficient of high-grade IPMNs was recalculated after excluding low-grade samples. The average Jaccard similarity coefficient was plotted for all three groups (low-grade IPMNs, high-grade IPMNs (whole lesion analysis), and high-grade IPMNs (high-grade only analysis)) using ggplot2 function in R18 and significant differences between groups were determined by Mann-Whitney U test.
Software
OncoPrinter software available through Memorial Sloan Kettering Cancer Center’s cBioPortal was used to generate Figure 1. SCHISM software is available for non-profit use at https://karchinlab.org/apps/appSchism.html.
Figure 1. Landscape of somatic driver gene mutations in IPMNs.

Integrated genomic data for 227 samples (bars) from 20 IPMNs (columns). The histologic subtype and grade of dysplasia data for each sample are shown as tracks at the top. IPMNs are grouped by grade of dysplasia with low-grade on the left and high-grade on the right. A. Bar plot representing the total number of KRAS and GNAS mutations per sample. B. Oncoprint heatmap depicting mutations occurring in KRAS, GNAS, or others (see color legend) or absence (gray bar). Single-nucleotide variants are listed for KRAS and GNAS, all other mutations are grouped by gene (rows). The percentage of samples with a given mutation is noted at the left. C. Proportion of heterogeneous mutations (green) and homogeneous mutations (blue) in each IPMN. *For mixed-grade IPMNs, the proportion of heterogeneous and homogenous mutations was recalculated after excluding low-grade samples (high-grade only).
RESULTS
Clinical and pathological features
This study included 227 neoplastic samples from 20 surgically resected IPMNs (Table 1). Of the 20 IPMNs analyzed, ten had low-grade dysplasia and ten had high-grade dysplasia. Of the ten high-grade IPMNs, three had co-occurring pancreatic ductal adenocarcinoma (PDAC), one of which occurred in the same blocks as the IPMN. The size of IPMNs in our cohort ranged from 1.6 cm to 6.5 cm, resulting in 6 to 24 FFPE blocks per case. For each case, IPMN epithelium was microdissected from every FFPE block. Importantly, because IPMNs are classified based on the highest grade of dysplasia present in the lesion, high-grade IPMNs will often also contain areas with low-grade dysplasia, even in the same FFPE block. Therefore, areas of low-grade dysplasia were separately microdissected from the areas of high-grade dysplasia. We refer to each FFPE block as an analyzed “region” from which we isolated one or two DNA “samples” based on the grade(s) of dysplasia present within the FFPE block. Of the ten high-grade IPMNs, seven also contained low-grade dysplasia – resulting in more total samples than regions in these cases. We microdissected a total of 117 low-grade samples, 105 high-grade samples, and five invasive PDAC samples. Of the 117 low-grade samples, 42 were from IPMNs that were overall classified as high-grade. Matched normal DNA for each case from duodenum or spleen was also analyzed using the same sequencing pipeline and used as a germline comparator (Supplementary Table S1).
Table 1.
Clinicopathological and molecular data from analyzed IPMNs
| Case | Sex | Age (at surgery)* | Location | Size (cm) | Predominant Histological Subtype | Final Diagnosis | Tissue Analyzed | # of regions | # of samples (LGD,HGD,PDAC) | Mutations Identified | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IPC01 | F | 80 | Head | 1.6 | Gastric | IPMN with LGD | IPMN | 6 | 6 | (6,0,0) | KRAS(p.G12D, p.G12R, p.G12V, p.GQ60GK), RNF43(p.A169T) |
| IPC02 | M | 50 | Tail | 4.5 | Gastric | IPMN with LGD | IPMN | 8 | 8 | (8,0,0) | KRAS(p.G12D, p.G12R, p.G12V), GNAS(p.R201C, p.R201H), RNF43(p.R286W), TP53(p.P72R) |
| IPC03 | F | 60 | Tail | 2.3 | Gastric | IPMN with LGD | IPMN | 6 | 6 | (6,0,0) | KRAS(p.G12D, p.G12R, p.G12V), GNAS(p.R201H), CDKN2A(p.R80P) |
| IPC04 | M | 70 | Head | 4.2 | Gastric | IPMN with LGD | IPMN | 6 | 6 | (6,0,0) | KRAS(p.G12R), GNAS(p.R201H) |
| IPC05 | F | 80 | Tail | 2.8 | Gastric | IPMN with LGD | IPMN | 8 | 8 | (8,0,0) | KRAS(p.G12V), GNAS(p.R201C) |
| IPC06 | M | 60 | Body | 3 | Gastric | IPMN with LGD | IPMN | 9 | 9 | (9,0,0) | KRAS(p.G12V) GNAS(p.R201C), BRAF(p.N486_P490d el), CTNNB1(p.S45F), TP53(p.L369M) |
| IPC07 | M | 60 | Head | 5 | Gastric | IPMN with LGD | IPMN | 11 | 11 | (11,0,0) | KRAS(p.G12D p.G12V), GNAS(p.R201C), RNF43(p.W15*, p.A115Pfs*43), PIK3CA(p.N1044K) |
| IPC08 | F | 70 | Head | 3 | Gastric | IPMN with LGD | IPMN | 8 | 8 | (8,0,0) | KRAS(p.G12D, p.G12R, p.G12V, p.A146V), GNAS(p.R201C, p.Q227H), RNF43(p.P231L), BRAF(p.N486_P490del), ATM(p.P1526P) |
| IPC09 | M | 80 | Tail | 3 | Intestinal | IPMN with LGD | IPMN | 7 | 7 | (7,0,0) | KRAS(p.G12D, p.G12V), PIK3CA(p.H1047R) |
| IPC10 | M | 70 | Head | 3 | Gastric | IPMN with LGD | IPMN | 6 | 6 | (6,0,0) | KRAS(p.G12D, p.G12V), GNAS(p.R201C, p.R201H), RNF43(p.L418M), STK11(p.P203S) |
| IPC11 | M | 70 | Head | 5 | Pancreatobiliary | IPMN with HGD | IPMN | 10 | 13 | (6,7,0) | KRAS(p.G12V, p.G12S), GNAS(p.R201C, p.R201H, p.Q227E) |
| IPC12 | F | 90 | Head | 5.5 | Intestinal | IPMN with HGD | IPMN | 23 | 25 | (3,22,0) | KRAS(p.A146V, p.Q61H), GNAS(p.R201C, p.Q227H), RNF43(p.Q6Rfs*9), ATM(p.L2475Yfs*2), TP53(p.P72R) |
| IPC13 | M | 70 | Head | 6.5 | Intestinal | IPMN with invasive carcinoma (ductal) | IPMN and carcinoma | 22 | 25 | (0,20,5) | KRAS(p.G12D), GNAS(p.R201H) |
| IPC14 | M | 70 | Head | 2.9 | Intestinal | IPMN with invasive carcinoma (ductal) | IPMN | 11 | 12 | (5,7,0) | KRAS(p.G12R), GNAS(p.R201H), BRAF(p.E26D), ATM(p.L1517F) |
| IPC15 | M | 70 | Head | 3 | Pancreatobiliary | IPMN with HGD | IPMN | 8 | 9 | (5,4,0) | KRAS(p.G12D, p.G12V), GNAS(p.R201C, p.R201S), RNF43(p.E37*) |
| IPC16 | F | 70 | Tail | 2.4 | Intestinal | IPMN with HGD | IPMN | 6 | 10 | (7,3,0) | KRAS(p.G12D), GNAS(p.R201H), RNF43(p.R49Sfs*3, p.R132*, p.Q152*, p.T76Nfs*13) |
| IPC17 | M | 80 | Head | 5 | Intestinal | IPMN with HGD | IPMN | 18 | 19 | (15,4,0) | KRAS(p.A146T, p.Q61H), GNAS(p.R201C), RNF43(p.C119Lfs*6), BRAF(p.T241M), CDKN2A(p.H83N) |
| IPC18 | M | 60 | Head | 4 | Pancreatobiliary | IPMN with HGD | IPMN | 8 | 9 | (1,8,0) | KRAS(p.G12V), RNF43(p.A115Pfs*43, p.S216L) |
| IPC19 | M | 70 | Head | 4 | Intestinal | IPMN with HGD | IPMN | 6 | 6 | (0,6,0) | KRAS(p.G12D), GNAS(p.R201H), RNF43(p.A169T, p.R337*), APC(p.R99W) |
| IPC20 | M | 70 | Head | 2.7 | Intestinal | IPMN with invasive carcinoma (ductal) | IPMN | 24 | 24 | (0,24,0) | KRAS(p.G13D), GNAS(p.R201C), RNF43(p.I141N, p.I201N), TP53(p.V172D, p.P152L, p.D208A, p.R248Q) |
To ensure patient privacy, age (at surgery) was rounded in the nearest decade
LGD: low-grade dysplasia; HGD: high-grade dysplasia
Multi-region sequencing reveals striking driver gene heterogeneity in IPMN
Targeted next generation sequencing (mean distinct coverage >500x) was used to interrogate the entire coding regions of 15 well characterized driver genes in pancreatic tumorigenesis (Supplementary Table S2–S4). Based on previous literature that characterized these driver gene mutations, we consider mutations in KRAS, GNAS, and BRAF to be “early drivers,” while we consider mutations in the other genes interrogated by our panel to be “later-occurring drivers”4,6,10. Mutations (including single nucleotide variants and small insertions/deletions) were identified in KRAS, GNAS, BRAF, RNF43, CDKN2A, PIK3CA, CTNNB1, STK11, ATM, APC, and TP53 (Supplementary Table S5). The total number of somatic driver gene mutations called in each analyzed IPMN ranged from 2 to 9 (Table 1). The most commonly mutated genes were KRAS and GNAS – all IPMNs had at least one KRAS mutation, and 17 had at least one GNAS mutation (Figures 1A and 1B). Of the 20 IPMNs, 15 had multiple mutations called in the same driver gene.
This sequencing approach identified mutations that were present in all regions of an IPMN (hereafter referred to as “homogeneous mutations”), as well as mutations that were only present in a subset of the analyzed regions (hereafter referred to as “heterogeneous mutations”). On average, a greater proportion of mutations were heterogeneous in low-grade IPMNs, relative to high-grade (Figure 1C). Of the ten low-grade IPMNs, three did not contain any homogeneous mutations, and nine contained at least one heterogeneous mutation (Figure 1C and Supplementary Table S6). In order to more precisely characterize the molecular features of IPMN epithelium with different grades of dysplasia, we analyzed IPMNs classified as high-grade in two different ways: (1) we considered both the low-grade and high-grade samples for each IPMN, which we call “whole lesion analysis”, and (2) we considered only the high-grade samples, which we call “high-grade only analysis.” Using whole lesion analysis, 57% of mutations were heterogeneous in high-grade IPMNs. However, this proportion decreased to 36% in the high-grade only analysis, suggesting that high-grade samples were less heterogeneous than low-grade samples within the same IPMN. (Figure 1C). For example, IPC15 appeared to contain only heterogeneous mutations via whole lesion analysis; yet, less than half of the mutations were heterogeneous after excluding low-grade samples. These data demonstrate a striking degree of driver gene heterogeneity in IPMNs, especially those classified as low-grade, indicating that the identification of a driver mutation in one region of an IPMN does not necessarily imply its occurrence throughout the entire lesion.
Early driver genetic heterogeneity is pervasive in IPMNs with low-grade dysplasia
All of the IPMNs in our cohort harbored mutations in GNAS and/or KRAS, and we identified 11 with multiple mutations in these early driver genes. These 11 IPMNs all had multiple mutations in KRAS, including seven with two different KRAS mutations, two with three different KRAS mutations, and two with four different KRAS mutations. Of these 11 IPMNs with multiple KRAS mutations, six also had multiple GNAS mutations. Strikingly, IPC02 and IPC08 had five and six different total mutations in KRAS and GNAS, respectively (Figures 1A and 1B). Of the ten low-grade IPMNs, seven harbored multiple mutations in GNAS and/or KRAS, while this was true for just one high-grade IPMN when performing high-grade only analysis (Fisher’s exact test; p=0.02). We quantitatively measured genetic heterogeneity with respect to KRAS and GNAS by calculating Jaccard similarity coefficients12,13,19 (Supplementary Table S7 and S8). When using whole lesion analysis, we found that low-grade IPMNs were more genetically heterogeneous than high-grade IPMNs with respect to KRAS and GNAS (0.63 v. 0.87; Mann-Whitney U test, p=0.070). Moreover, when using high-grade only analysis, high-grade IPMNs were significantly less genetically heterogeneous that low-grade IPMNs with respect to these early driver gene alterations (0.63 v. 0.92; Mann-Whitney U test, p=0.019) (Figure 2A). Altogether, these data reveal genetic heterogeneity with respect to early drivers is significantly more prevalent in low-grade IPMNs.
Figure 2. Early driver gene heterogeneity and polyclonal origin of IPMNs.

A. Dot plot of the average Jaccard similarity coefficient for each case based on KRAS and GNAS mutations only. Low-grade IPMNs are significantly more heterogeneous than high-grade IPMNs via whole lesion analysis (p value = 0.070; Mann-Whitney U test) and high-grade IPMNs via high-grade only analysis (p value = 0.019; Mann-Whitney U test). B-D. Neoplastic cancer cell fractions (NCFs) are presented in heat maps with each row representing a mutation and each column representing a region or sample. Each NCF heat map corresponds to a schematic of tumor evolution – SCHISM was used to reconstruct somatic mutation hierarchy trees. B. Low-grade IPMN, IPC03. C. Whole lesion analysis (left) or high-grade only analysis (right) of IPC15. D. Whole lesion analysis (left) or high-grade only analysis (right) of IPC14.
Evolutionary modeling suggests polyclonal origin of early pancreatic tumorigenesis
In every sample that contained multiple KRAS mutations, each mutation was found on distinct sequencing reads and therefore did not occur on the same allele. However, sequencing data cannot directly resolve two KRAS mutations in the same cell (bi-allelic KRAS mutation) from single KRAS mutations in two different cells. Thus, we used evolutionary modeling to more accurately estimate the clonal architecture of each neoplasm. Evolutionary trees built with SCHISM17 (see Methods) suggested that several IPMNs were polyclonal in origin – having initiated from more than one separate and independent clone (Figure 2 and Supplementary Figure S1). For example, we identified three different KRAS mutations (KRAS p.G12D, p.G12R, p.G12V) in IPC03, each marking a distinct clone (Figure 2B). In another example, whole lesion analysis of IPC15 inferred it originated from three clones, each marked by a different early driver (KRAS p.G12V, KRAS p.G12D, GNAS p.R201C) (Figure 2C). Interestingly, the clone containing KRAS p.G12D and GNAS p.R201S was absent in the high-grade samples, demonstrating this clone was limited to the low-grade dysplasia. Strikingly, whole lesion analysis of IPC14 suggested this IPMN initiated from two clones with either a GNAS p.R201H or KRAS p.G12R mutation – however, high-grade only analysis revealed that the high-grade neoplasia in IPC14 initiated from a single clone (GNAS p.R201H) (Figure 2D). Altogether, some high-grade IPMNs appear polyclonal in origin, but IPC15 is the only high-grade IPMN that retains polyclonality after excluding low-grade samples from the analysis.
Whole exome sequencing confirms independent origin of clones with discordant mutations in early driver genes
While our targeted sequencing data coupled with evolutionary modeling suggests polyclonal origin for several IPMNs in our cohort, it is also possible that these multiple KRAS mutations occurred as divergent later events in a monoclonal IPMN initiated by an alteration not examined by our targeted panel. In order to more definitively characterize the clonal origin of the analyzed IPMNs, we performed whole exome sequencing (WES) on two regions each from four IPMNs (IPC01, IPC08, IPC12, and IPC14) (Supplementary Table S9). For each IPMN analyzed by WES, we chose two regions with mutually exclusive early driver gene mutations based on our targeted sequencing. For example, samples T5 HG and T8 LG from IPC14 were chosen for WES because T5 HG had a GNAS p.R201H mutation, while T8 LG had a KRAS p.G12R and BRAF p.E26D (Figure 2D). In each of the four IPMNs, we did not identify any shared alterations between the two regions (Supplementary Table S10). Additionally, all mutations called using our targeted sequencing approach at >10% VAF were also found via WES at similar frequencies, further validating the accuracy of our multi-region targeted sequencing. Although our WES was not designed to find novel driver genes, we identified two genes with mutations in multiple IPMN samples: distinct ATR mutations in two regions of IPC08 and two different SCAPER mutations in IPC08 and IPC14. Mutations in SCAPER have been reported at low frequency in a variety of tumor types, including <0.5% of PDACs20. Overall, this comprehensive approach has corroborated our evolutionary modeling and confirms these IPMNs originated from more than one distinct clone – providing strong evidence for polyclonal origin of these neoplasms. If these results are extrapolated to all IPMNs in our cohort harboring multiple early driver gene mutations, this suggests 60% of the analyzed IPMNs are polyclonal in origin.
RNA in-situ hybridization confirms spatial separation of mutant KRAS subclones
Our group previously used single-cell sequencing to demonstrate that, in IPMNs with multiple KRAS mutations, the discordant mutations occur in different cells21. The targeted sequencing and evolutionary modeling in the current study also suggest that multiple KRAS mutations observed within a single IPMN occurred in different clones; however, these data cannot resolve whether the cells containing discordant KRAS mutations are intermixed or spatially separated. To determine the spatial location of cells with discordant KRAS mutations we employed BaseScope, which uses RNA in situ hybridization to specifically detect hotspot alterations in KRAS (see Methods). We analyzed one region each from five IPMNs by probing for KRAS p.G12V, p.G12D, and p.G12R, which were validated using KRAS mutant cell lines (Supplementary Figure S2). We chose regions to analyze based on several factors, including age of tissue block, amount of IPMN epithelium within block, and identification of KRAS mutations targeted by BaseScope probes. Interestingly, in each region that contained multiple KRAS mutations, we identified clones that were spatially separated (Figure 3, Supplementary Figure S3–S7). For example, IPC09 B was a low-grade region with three distinct areas of IPMN epithelium, which were pooled for sequencing (Figure 3A). Targeted sequencing indicated the neoplastic cells in this region had KRAS p.G12D and p.G12V mutations. The IPMN cells in areas 1 and 2 expressed KRAS p.G12D, but not p.G12V (Figures 3B–3E). Conversely, the IPMN cells in area 3 expressed KRAS p.G12V, but not p.G12D (Figure 3F and 3G). Overall, while areas 1 and 2 were spatially separated, they expressed the same KRAS mutation and therefore are likely of the same originating clone. Area 3 was spatially separated from areas 1 and 2 and expressed a different KRAS mutation, suggesting this region likely originated from a distinct clone (Figure 3H). Importantly, the results of the BaseScope assay for each analyzed IPMN region were concordant with our targeted sequencing with respect to the KRAS mutations identified and the relative proportions of cells containing each mutation.
Figure 3. BaseScope in-situ RNA detection of mutant KRAS in FFPE IPMN tissues.

A. Hematoxylin stain of low-grade IPMN sample IPC09 B. B-C. Representative image of Area 1 probed for either KRAS p.G12D (B) or KRAS p.G12V (C). D-E. Representative image of Area 2 probed for either KRAS p.G12D (D) or KRAS p.G12V (E). F-G. Representative image of Area 3 probed for either KRAS p.G12D (F) or KRAS p.G12V (G). H. Hematoxylin stain of IPC09 B overlaid with green, indicating areas of positive staining for KRAS p.G12D or red, indicating areas of positive staining for KRAS p.G12V. Dark red dots designate positive signal (black arrows point to representative cells with positive staining). Nuclei were counterstained with hematoxylin. A and H = Magnification 0.5X. B-G = Magnification 15X.
Convergent evolution and heterogeneity of later-occurring driver genes in IPMNs with high-grade dysplasia
In addition to heterogeneity with respect to early driver gene mutations, we also identified genetic heterogeneity with respect to later-occurring driver gene mutations in several IPMNs in our cohort. Multiple RNF43 mutations were identified in five IPMNs from our cohort. Four IPMNs (IPC07, IPC18, IPC19, and IPC20) had two different RNF43 mutations, while IPC16 had four different RNF43 mutations (Figure 4A). The four RNF43 mutations identified in IPC16 were either nonsense or frameshift, and all four were identified in distinct regions. One RNF43 mutation (p.R49Sfs*3) was present in both the low-grade and high-grade dysplasia from a single region. A second RNF43 mutation (p.R132*) was only found in a different region with low-grade dysplasia. The remaining two RNF43 mutations (p.Q152*and T76Nfs*13) were found in separate regions with only high-grade dysplasia. These data demonstrate multiple, distinct mutations occurring in the RNF43 gene, suggesting there is a specific selection for these mutations in a subset of IPMNs. We also observed multiple mutations in the TP53 gene. One high-grade IPMN (IPC20) had four different TP53 mutations (Figure 4B). One TP53 missense mutation (p.V172D) was present in every region, while the other TP53 missense mutations (p.P152L, p.D208A, and p.R248Q) were present in only one to three regions each.
Figure 4. Convergent evolution and heterogeneity in later-occurring driver genes in IPMNs with high-grade dysplasia.

A-B. Mutations are presented in tables with each row representing a mutation and each column representing a sample. Blue color indicates a mutation call, with variant allele frequencies in each cell of the table. Both depicted IPMNs have multiple mutations called in a later-driver gene. A. IPC16, a high-grade IPMN, has four distinct RNF43 mutations. B. IPC20, a high-grade IPMN, has two different RNF43 mutations and four distinct TP53 mutations. C-D. The 20 IPMNs are represented around the perimeter of the circle in clockwise numerical order – grade of dysplasia/analysis-type for each IPMN is indicated by the colored key. Only heterogeneous mutations are displayed (not all mutations). Each colored rectangle represents a heterogeneous mutation in a given gene, which is indicated by colored key. The heterogeneous mutations in each IPMN are presented by gene in low-grade and whole lesion analysis of high-grade IPMNs (C) and low-grade and high-grade only analysis of high-grade IPMNs (D). Note: Only genes with mutations in at least two IPMNs were included in C-D.
While our data suggest low-grade IPMNs are significantly more heterogeneous with respect to early driver mutations, heterogeneity amongst later-occurring driver mutations is prominent in high-grade IPMNs. Of the five IPMNs harboring multiple RNF43 mutations, four were high-grade IPMNs. Interestingly, while early driver heterogeneity was less prevalent in high-grade IPMNs relative to low-grade, this was not true for later-occurring drivers (Figure 4C and D). Heterogeneity with respect to these later-occurring driver mutations seems to be a feature of high-grade IPMNs.
Mutations in less commonly mutated driver genes also occurred in our cohort. Four IPMNs (IPC06, IPC08, IPC14, and IPC17) had mutations in BRAF. Interestingly, IPC06 and IPC08 had the same in-frame deletion of BRAF (p.N486_P490del), which has previously been reported in IPMNs and numerous other cancers22,23. In IPC08, we identified two regions which lacked a KRAS mutation, yet harbored this BRAF deletion (Figure 1 and Supplemental Table S4). Two IPMNs (IPC03 and IPC17) had missense mutations in CDKN2A. Two IPMNs (IPC07 and IPC09) had mutations in oncogenic hotspots of PIK3CA (p.N1044K and p.H1047R). In IPC09, every region harbored PIK3CA p.H1047R and KRAS p.G12V at similar NCF; therefore, we could not determine which mutation occurred first, or if these mutations represent distinct clones (Supplementary Figure S1). One IPMN (IPC06) had a mutation in an oncogenic hotspot of CTNNB1, and one IPMN (IPC19) had a missense APC mutation. One IPMN (IPC10) had a missense mutation in STK11. Three IPMNs (IPC08, IPC12, and IPC14) had mutations in ATM, one of which (IPC12) was an inactivating frameshift insertion. None of the IPMNs in our cohort had mutations in SMAD4.
Clinical implications of genetic heterogeneity in IPMN
An independent study analyzed somatic mutations in cyst fluid from patients diagnosed with IPMN prior to surgical resection (manuscript in press). Seven cases from this study overlapped with our cohort (IPC03, IPC04, IPC06, IPC08, IPC12, IPC18, and IPC20). We compared driver gene mutations identified in DNA collected from cyst fluid to our multi-region targeted sequencing. For all cases, mutations found in the cyst fluid were also found via our multi-region sequencing approach except for those at very low VAF in the cyst fluid analysis (range 0.00076–0.034 VAF). In all cases, there were mutations identified by our multi-region sequencing approach that were not identified in the cyst fluid (Supplementary Table S11). For example, both approaches found KRAS p.G12V in IPC03; yet, two additional KRAS mutations (p.G12D and p.G12R), and a GNAS mutation (p.R201H) were not identified in the cyst fluid analysis. In general, the mutations not identified in the cyst fluid were present in only a subset of regions within a given IPMN.
DISCUSSION
Because IPMNs are often diagnosed on imaging studies, they can be resected prior to the development of invasive PDAC – as such, IPMNs represent an ideal system in which to perform analyses of precancerous pancreatic neoplasia. Our study represents one of the most comprehensive genetic analyses of driver gene mutations in IPMNs to date, and to our knowledge, the only study to interrogate such mutations throughout entire IPMNs. The resulting data strongly suggest that these precancers are derived from multiple independent clones, transforming our understanding of the origins of pancreatic tumorigenesis.
Here, we demonstrate that the vast majority of analyzed IPMNs harbor multiple mutations within the same driver gene. In addition, we report that many IPMNs do not have any homogeneous mutations across the entire lesion, demonstrating that single-region sequencing approaches are not adequate to accurately identify the complete repertoire of driver gene mutations within an IPMN. Furthermore, the genes that harbor multiple mutations in IPMNs differ depending on the grade of dysplasia and thus the stage of neoplastic progression. We found IPMNs with low-grade dysplasia have significantly more heterogeneity with respect to the early drivers KRAS and GNAS, compared to IPMNs with high-grade dysplasia. These data demonstrate that early pancreatic tumorigenesis is characterized by heterogeneity in initiating driver genes, while progression to high-grade dysplasia leads to decreased heterogeneity in these early driver genes. This likely represents selection and expansion of a single clone after acquisition of additional driver gene alterations, thus eliminating prior clonal diversity. This finding is consistent with previous studies which have demonstrated a lack of driver gene heterogeneity in invasive PDACs and their metastases12,13, suggesting that this heterogeneity and clonal selection are limited to precancerous pancreatic neoplasms.
Using our multi-region sequencing approach, coupled with evolutionary modeling, we show that a substantial proportion of IPMNs in our study evolved from multiple distinct clones. This polyclonal origin is typically marked by multiple KRAS mutations, yet clones can also originate from GNAS mutations, albeit less frequently. This suggests that the presence of multiple mutations in a single early driver gene probably underestimates polyclonality. Previously, we have shown using single cell sequencing21 that these distinct KRAS mutations occur in different cells, and in the current study, we use in situ mutation detection to demonstrate that they are even spatially separate. Interestingly, a previous study by The Cancer Genome Atlas used computational analyses to suggest that multiple KRAS mutations occurred in the same cells in invasive PDACs20. The discordant results between these two studies reveal a potential difference between pancreatic precancerous lesions and advanced cancers and highlight the importance of directly measuring individual single-cell genotypes.
Taken together, these data support a revised model of early pancreatic tumorigenesis in which some IPMNs originate from multiple clones that evolve independently, one of which may acquire additional driver gene mutations that lead to clonal expansion and progression (Figure 5). Importantly, in the IPMNs originating from multiple independent clones, our WES data did not identify a single shared mutation preceding KRAS, suggesting that there is not an earlier coding genetic event initiating these lesions. Moreover, there were no shared passenger alterations in these distinct sections. Because a subset of such passenger alterations accumulate prior to tumor development, the lack of shared passenger mutations suggests that the different clones arose from completely separate cells24. The mechanisms that drive the emergence of these independent clones are not known. Polyclonality in precancerous lesions in other organ systems has previously been demonstrated, including esophagus and skin14,15,25. Intriguingly, many of these organs sites have known strong carcinogen exposures which may serve as a potential mechanism underlying polyclonal tumor origin, raising the possibility of “field carcinogenesis” (or a so-called “field defect”) in the pancreas. It is possible that an as-yet unidentified environmental exposure leads to increased DNA mutations and emergence of clones with KRAS and GNAS mutations. For example, recent work raises the possibility that elevated glucose levels predispose to the acquisition of KRAS hotspot mutations in pancreatic cells26. Alternatively, the emergence of a second clone may be specifically selected after the initial KRAS-mutant clone arises – while the mechanism for such selection is unknown, there is some evidence that clonal evolution may be shaped by paracrine signaling and cell-cell interactions27. Future work should focus on the mechanism underlying the polyclonal origin of IPMNs, as it may reveal new risk factors for the development or progression of pancreatic neoplasms.
Figure 5. Revised clonal evolution model for early pancreatic tumorigenesis.
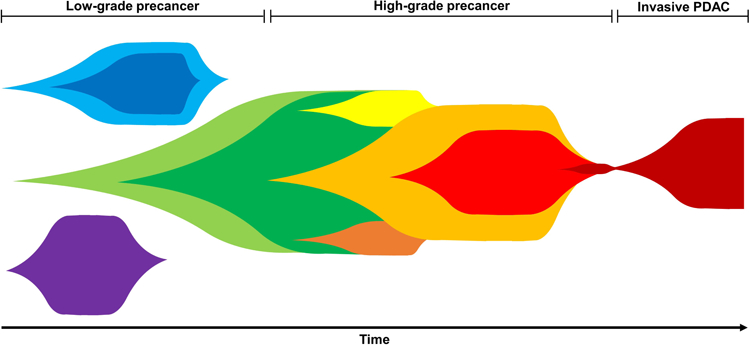
Multiple, independent clones arise with distinct mutations in early driver genes (i.e. GNAS and/or KRAS). Early in tumorigenesis, some selective pressures eventually lead to expansion of a dominant clone. Often during later-stages of tumorigenesis, convergent evolution leads to multiple mutations in the same later-occurring driver gene (i.e. RNF43, TP53). Eventually, a dominant clone invades giving rise to pancreatic ductal adenocarcinoma.
In addition to multiple mutations in early driver genes, we also identified multiple mutations in later-occurring driver genes in the same IPMN, raising the possibility of convergent evolution. We identified five IPMNs harboring multiple RNF43 mutations, which were often located in different regions, as well as one IPMN containing four different TP53 mutations. Nearly all of the IPMNs with multiple mutations in TP53 and/or RNF43 had high-grade dysplasia, suggesting heterogeneity with respect to later-occurring driver gene mutations may be a feature of IPMNs with greater malignant potential. These data add to our revised tumorigenesis model – following the selective sweep and fixation of a dominant clone, additional mutations arise in later-occurring drivers, often multiple mutations in the same gene (Figure 5). Similar evidence for convergent evolution has been reported in other tumor types, including colorectal and clear cell renal carcinoma28,29. The selective forces that lead to convergence of multiple mutations in the same gene are not known. Further investigation into the conditions that select for multiple mutations within specific genes may provide novel insights into later stages of IPMN progression, as well as the functional outcomes of specific driver gene alterations.
Many studies have demonstrated the value of analyzing cyst fluid collected by endoscopic ultrasound fine needle aspiration (EUS-FNA) from patients diagnosed with pancreatic cysts30. Several reports highlight the diagnostic value of molecular markers in cyst fluid for differentiating likely benign cysts from those which have greater risk of malignant progression31,32. Integration of our multi-region sequencing data with cyst fluid molecular analysis on a subset of patients in our cohort reveals important clinical implications for interpreting such molecular biomarkers. Not all driver gene mutations within an IPMN may be captured by cyst fluid analysis, even with high-depth sequencing strategies. Because most of the mutations not identified in cyst fluid were present focally in one or a few regions of the IPMN, it is likely that the genetic material containing these mutations was not present in the cyst fluid. These findings highlight the potential challenges with IPMN risk stratification.
There are some limitations to our study. First, while our study encompassed a large cohort of 227 neoplastic tissue samples, these represent only 20 IPMNs. We prioritized comprehensive analysis of the entire IPMN but could only perform such in-depth analysis on a limited number of IPMN lesions. As such, our findings will need to be confirmed in larger IPMN cohorts. Nevertheless, this case size was sufficient to detect statistically significant differences in genetic heterogeneity between IPMNs with different grades of dysplasia. Second, the majority of samples were sequenced using a small targeted panel of 15 pancreatic driver genes. The use of such a focused sequencing approach allowed us to analyze a large number of samples at high sequencing depth, and thus confidently report the most biologically important driver gene mutations even when they were subclonal. Our sequencing strategy was not designed to provide sufficient data to call copy number alterations or allelic imbalance, and such limitations prevented us from definitively resolving the evolutionary history of every IPMN. Third, our cohort of low-grade and high-grade IPMNs does not have a balanced distribution of histological subtypes. However, histologic subtype is not independent of grade of dysplasia, and the distribution of histological subtypes in our cohort is representative of low-grade and high-grade IPMNs33. Finally, our experimental design required the analysis of surgically resected IPMNs. Because only a small fraction of IPMNs identified radiologically are eventually resected, and the decision to resect is based on specific clinical and radiological criteria34, our cohort is not representative of the entire spectrum of IPMNs in the population. It is possible that enrichment for larger IPMNs in our resected cohort may also enrich for polyclonality. Analysis of low-grade IPMNs that were not targeted for surgical resection will be required to investigate this.
In conclusion, using IPMNs as a system in which to analyze early pancreatic neoplasia, our data provide several insights into the acquisition of somatic mutations during pancreatic tumorigenesis. We show that genetic heterogeneity with respect to driver gene alterations is pervasive in IPMNs. Heterogeneity of the early drivers KRAS and GNAS is more prevalent during early stages of tumorigenesis, with discordant KRAS mutations marking multiple independent clones arising from separate cells. In addition, convergent evolution with respect to later-occurring driver gene mutations is present in high-grade IPMNs, demonstrating unique selective pressures at different stages of tumor progression. The results of this study challenge the traditional monoclonal origin of pancreatic tumors, highlighting distinct evolutionary features of precancerous lesions and transforming our understanding of the clonal evolution of pancreatic neoplasia.
Supplementary Material
What You Need to Know.
Background and Context:
Intraductal papillary mucinous neoplasms (IPMNs) are precancerous pancreatic lesions. A deeper understanding of the origin and progression of these lesions can provide a rational foundation for surveillance approaches.
New Findings:
Low-grade IPMNs are characterized by multiple mutations in early driver genes (KRAS and GNAS) which represent independent clones that are spatially separate. High-grade IPMNs lack this heterogeneity in early drivers but frequently have multiple mutations in later driver genes, suggestive of convergent evolution.
Limitations:
This study comprehensively examined 20 surgically resected IPMNs. Confirmation of the findings will require studies in larger cohorts, ideally also including IPMNs that did not meet criteria for surgical resection, in order to ensure broad applicability of the findings.
Impact:
The results of this study challenge the traditional monoclonal origin of pancreatic neoplasms, highlighting distinct evolutionary features of precancerous lesions and transforming our understanding of the clonal evolution of pancreatic neoplasia.
ACKNOWLEDGEMENTS
The authors thank Joseph W. Fischer for helpful discussions and assistance with data presentation.
The authors acknowledge the following sources of funding: NIH/NCI P50 CA62924; NIH/NIDDK K08 DK107781; Sol Goldman Pancreatic Cancer Research Center; Buffone Family Gastrointestinal Cancer Research Fund; Kaya Tuncer Career Development Award in Gastrointestinal Cancer Prevention; AGA-Bernard Lee Schwartz Foundation Research Scholar Award in Pancreatic Cancer; Sidney Kimmel Foundation for Cancer Research Kimmel Scholar Award; AACR-Incyte Corporation Career Development Award for Pancreatic Cancer Research; American Cancer Society Research Scholar Grant; Emerson Collective Cancer Research Fund; Rolfe Pancreatic Cancer Foundation; Joseph C Monastra Foundation; The Gerald O Mann Charitable Foundation (Harriet and Allan Wulfstat, Trustees); Susan Wojcicki and Denis Troper; Lustgarten Foundation for Pancreatic Cancer Research; CAMS Innovation Fund for Medical Sciences 2016-I2M-1–001
EDT receives salary support from Bristol-Meyers Squibb.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The other authors report no conflict of interest.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: A Cancer Journal for Clinicians 2018; 68: 7–30 [DOI] [PubMed] [Google Scholar]
- 2.Basturk O, Hong S-M, Wood LD, et al. A REVISED CLASSIFICATION SYSTEM AND RECOMMENDATIONS FROM THE BALTIMORE CONSENSUS MEETING FOR NEOPLASTIC PRECURSOR LESIONS IN THE PANCREAS. Am J Surg Pathol 2015; 39: 1730–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laffan TA, Horton KM, Klein AP, et al. Prevalence of Unsuspected Pancreatic Cysts on MDCT. AJR Am J Roentgenol 2008; 191: 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fischer CG, Wood LD. From somatic mutation to early detection: insights from molecular characterization of pancreatic cancer precursor lesions. The Journal of Pathology 2018; 246: 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Felsenstein M, Hruban RH, Wood LD. New Developments in the Molecular Mechanisms of Pancreatic Tumorigenesis. Advances in Anatomic Pathology 2018; 25: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amato E, Molin M dal, Mafficini A, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol 2014; 233: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furukawa T, Kuboki Y, Tanji E, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep 2011; 1 [DOI] [PMC free article] [PubMed]
- 8.Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS Mutations Define an Unexpected Pathway for Pancreatic Cyst Development. Sci Transl Med 2011; 3: 92ra66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan MC, Basturk O, Brannon AR, et al. GNAS and KRAS Mutations Define Separate Progression Pathways in Intraductal Papillary Mucinous Neoplasm-Associated Carcinoma. J Am Coll Surg 2015; 220: 845–854.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Omori Y, Ono Y, Tanino M, et al. Pathways of Progression From Intraductal Papillary Mucinous Neoplasm to Pancreatic Ductal Adenocarcinoma Based on Molecular Features. Gastroenterology 2019; 156: 647–661.e2 [DOI] [PubMed] [Google Scholar]
- 11.Felsenstein M, Noë M, Masica DL, et al. IPMNs with co-occurring invasive cancers: neighbours but not always relatives. Gut March 2018: gutjnl - 2017–315062 [DOI] [PMC free article] [PubMed]
- 12.Connor AA, Denroche RE, Jang GH, et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019; 35: 267–282.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makohon-Moore AP, Zhang M, Reiter JG, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet 2017; 49: 358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross-Innes CS, Becq J, Warren A, et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nature Genetics 2015; 47: 1038–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stachler MD, Taylor-Weiner A, Peng S, et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nature Genetics 2015; 47: 1047–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative Genomics Viewer. Nat Biotechnol 2011; 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niknafs N, Beleva-Guthrie V, Naiman DQ, et al. SubClonal Hierarchy Inference from Somatic Mutations: Automatic Reconstruction of Cancer Evolutionary Trees from Multi-region Next Generation Sequencing. PLoS Comput Biol 2015; 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.R Core Team. R: A Language and Environment for Statistical Computing Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 19.Yates LR, Gerstung M, Knappskog S, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med 2015; 21: 751–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017; 32: 185–203.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuboki Y, Fischer CG, Guthrie VB, et al. Single-cell sequencing defines genetic heterogeneity in pancreatic cancer precursor lesions. The Journal of Pathology 2019; 247: 347–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang MT, Asthana S, Gao SP, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016; 34: 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zehir A, Benayed R, Shah RH, et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat Med 2017; 23: 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci U S A 2013; 110: 1999–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martincorena I, Roshan A, Gerstung M, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015; 348: 880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu C-M, Tien S-C, Hsieh P-K, et al. High Glucose Triggers Nucleotide Imbalance through O-GlcNAcylation of Key Enzymes and Induces KRAS Mutation in Pancreatic Cells. Cell Metabolism March 2019 [DOI] [PubMed]
- 27.Gupta RG, Somer RA. Intratumor Heterogeneity: Novel Approaches for Resolving Genomic Architecture and Clonal Evolution. Mol Cancer Res 2017; 15: 1127–1137 [DOI] [PubMed] [Google Scholar]
- 28.Gerlinger M, Horswell S, Larkin J, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46: 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misale S, Nicolantonio FD, Sartore-Bianchi A, et al. Resistance to Anti-EGFR Therapy in Colorectal Cancer: From Heterogeneity to Convergent Evolution. Cancer Discov 2014; 4: 1269–1280 [DOI] [PubMed] [Google Scholar]
- 30.Genevay M, Mino-Kenudson M, Yaeger K, et al. Cytology Adds Value to Imaging Studies for Risk Assessment of Malignancy in Pancreatic Mucinous Cysts. Ann Surg 2011; 254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Springer S, Wang Y, Molin MD, et al. A Combination of Molecular Markers and Clinical Features Improve the Classification of Pancreatic Cysts. Gastroenterology 2015; 149: 1501–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singhi AD, McGrath K, Brand RE, et al. Preoperative next-generation sequencing of pancreatic cyst fluid is highly accurate in cyst classification and detection of advanced neoplasia. Gut September 2017: gutjnl - 2016–313586 [DOI] [PMC free article] [PubMed]
- 33.Pittman ME, Rao R, Hruban RH. Classification, Morphology, Molecular Pathogenesis, and Outcome of Premalignant Lesions of the Pancreas. Archives of Pathology & Laboratory Medicine 2017; 141: 1606–1614 [DOI] [PubMed] [Google Scholar]
- 34.Tanaka M, Fernández-del Castillo C, Adsay V, et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology 2012; 12: 183–197 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.