

Abstract
Ion mobility spectrometry (IMS) is of significant interest as a platform for glycoanalysis. While much attention has been focused on the resolution of isomeric carbohydrates and glycoconjugates, another appealing aspect of IMS is the ability to sort different classes of biomolecules into distinct regions of mass vs. mobility space. This capability has potential to greatly simplify glycoproteomic analyses, as glycosylated and non-glycosylated peptides can be rapidly partitioned in the gas phase. Nevertheless, the physical and chemical characteristics of glycopeptides that dictate their mass vs. mobility loci have yet to be systematically investigated. This report presents an IMS study of model protonated glycopeptide ions with systematically varied oligosaccharide topologies, polypeptide sequences, and charge states. In all, over 110 ion-neutral collision cross sections (CCSs) were measured and analyzed in the context of the physicochemical characteristics of the analytes. Glycan size and composition emerged as a decisive factor in dictating the CCS space occupied by the glycopeptides and exerted this influence in a charge state dependent fashion. Furthermore, elongation of the glycan group was found to either increase or decrease glycopeptide CCSs depending on the ion charge state and the size of the glycan. Molecular dynamics (MD) simulations of the gas phase structures and CCSs of selected glycopeptides revealed that the experimental observations were consistent with a glycan size and charge state dependent interplay between destabilizing Coulombic repulsion effects (tending to result in more extended structures) and stabilizing charge solvation effects in which the glycan plays a major role (tending to result in more compact structures). Taken together, these IMS and MD findings suggest the possibility of predicting and delineating glycopeptide-enriched regions of mass vs. mobility space for applications in glycoproteomics.
Keywords: Glycosylation, glycoproteins, glycopeptides, glycoproteomics, ion mobility spectrometry, ion-neutral collision cross section
Graphical Abstract
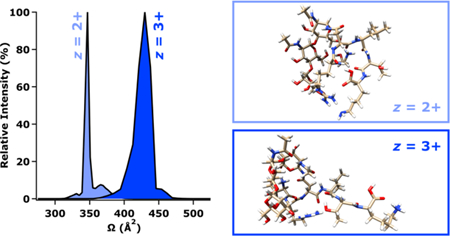
Textual Abstract
This study suggests the possibility of predicting and delineating glycopeptide-enriched regions of mass vs. mobility space for applications in glycoproteomics.
Introduction
The combination of ion mobility spectrometry (IMS) with mass spectrometry (MS) to address needs in the field of glycoanalysis has gained considerable momentum in recent years.1–4 Particularly vigorous study has been focused on pursuing the potential of IMS methods to rapidly resolve or distinguish isomeric carbohydrates in the gas phase.5–12 Indeed, these efforts have resulted in significant progress towards alleviating the general isomer problem in oligosaccharide analysis. In a complementary fashion, IMS-MS methods also provide for the millisecond-order separation of different types of biomolecules into characteristic mass vs. mobility domains depending upon the intrinsic conformational ordering of various analyte classes.13–16 In the context of glycoproteomic analyses, accurate mass measurement17, 18 and tandem mass spectrometry (MS/MS) methods19, 20 are often used to infer site specific protein glycosylation profiles based on deduced compositions and structures of proteolytic glycopeptides.21–26 In such experiments, the glycosylated complement of peptides is typically accompanied by a stoichiometrically more abundant ensemble of non-glycosylated peptides that tend to have more favorable ionization efficiencies than their glycan-bearing counterparts. The potential of IMS to achieve rapid partitioning of these ion populations presents an opportunity to enhance the selectivity and depth of glycoproteomic analyses. Encouragingly, the sorting of glycopeptides and peptides into unique mass vs. mobility coordinates was previously observed by Hill and coworkers when directly infusing an unfractionated tryptic digests of human-α1-acid glycoprotein and antithrombin III for IMS-MS analysis.27 Aside from that study, surprisingly little else has been reported on glycopeptide analysis via IMS.28–33 In particular, the underlying compositional, structural, and physical determinants of glycopeptide mass vs. mobility sorting have not yet been scrutinized in detail. In this study, a collection of glycopeptides with systematically varied polypeptide sequences and oligosaccharide connectivities were prepared by tryptic proteolysis and exoglycosidase digestion. The resultant glycopeptides were analyzed by traveling wave ion mobility spectrometry (TWIMS) as protonated molecular ions in multiple charge states. TWIMS arrival time distributions (ATDs) and ion-neutral collision cross sections (CCSs) were evaluated in relation to the physicochemical features of the glycopeptides under study. Computational chemistry methods were also applied to predict gas-phase structures for selected glycopeptide ions and their corresponding CCSs. The combination of experimental and theoretical results suggested some interesting glycan size dependent and charge state dependent relationships between structure and CCS. These trends appeared to arise largely from transitions between Coulombic repulsion controlled and charge solvation-controlled conformations, where in the latter of these the glycan participated in stabilizing the sites of protonation. These results imply that the domains of mass vs. mobility space that are occupied by glycopeptides can be predicted from first principles and rationalized in terms of analyte characteristics. Collectively, our findings also highlight the potential of an expanded role for IMS in glycoproteomic inquiry.
Experimental
N-Glycopeptide Preparation.
RNase B from Bos taurus (UniProtKB P61823) was acquired from Sigma-Aldrich (St. Louis, MO, USA). Model glycopeptides were generated by tryptic digestion of the target glycoprotein as previously described elsewhere.34–38 Briefly, a 50 μL aliquot of RNase B stock solution (2 μg/μL in 8 M urea and 50 mM ammonium bicarbonate, pH 7.5) was mixed with 10 μL of 450 mM dithiothreitol and incubated for 1 h at 55°C to reduce disulfide linkages. The resulting free thiols were acetamidated by addition of 10 μL of 500 mM iodoacetamide, followed by a 1 h incubation in the dark and at room temperature. The mixture was next combined with 175 μL of 50 mM ammonium bicarbonate (pH 7.5) and 5 μL of 0.5 μg/μL proteomics grade trypsin (Sigma-Aldrich), then incubated for 18 h at 37°C. Tryptic digests prepared in this manner were either subjected to N-glycan truncation or directly purified and analyzed with the glycans intact.
N-Glycan Truncation.
Glycopeptides harboring smaller N-glycan structures were prepared via sequential exoglycosidase digestion of the tryptic peptides using α(1→2,3) and α(1→6) mannosidases (New England Biolabs; Ipswich, MA, USA). Initially, 2 μL of 0.1 nmol/μL α(1→2,3) mannosidase was added to the glycopeptide preparation, followed by incubation at 37°C. The digestion was allowed to proceed for a total of 18 h, with a subsample of the digest being taken after 2 h incubation. The 18 h digest was then subsampled prior to treatment with 2 μL of 0.1 nmol/μL α(1→6) mannosidase. Incubation was again carried out at 37°C, with samples again being taken following 2 h and 18 h digestion. The resulting preparations were subsequently purified and analyzed as detailed below.
N-Glycopeptide Purification.
N-glycopeptides were purified according to previously described protocols.34–38 In short, each digest of interest was vacuum centrifuged using a Speed Vac SC110 (Thermo Savant, Holbrook, NY, USA) to reduce the volume from 150–250 μL to approximately 10 μL. The digests were reconstituted to approximately 100 μL by addition of 0.1% formic acid. Glycopeptide desalting and enrichment was then performed using solid phase extraction (SPE) micropipette tips loaded with zwitterionic hydrophilic interaction liquid chromatography (ZIC-HILIC) stationary phase (Protea Biosciences; Somerset, NJ, USA). The ZIC-HILIC SPE tip was first wet with water, equilibrated with 80% acetonitrile / 0.1% formic acid, then loaded with a 20 μL portion of reconstituted digest. The tip was next washed with 80% acetonitrile / 0.1% formic acid and eluted into 20 μL of 0.1% formic acid.
Ion Mobility Spectrometry and Mass Spectrometry.
All IMS and MS measurements were carried out using a Synapt G2-S HDMS TWIMS-MS instrument (Waters, Manchester, UK) equipped with a modified static-mode nanoflow electrospray ionization (nESI) source. Glass nESI emitters were fashioned from Pyrex melting point capillaries (100 mm x 1.5–1.8 mm; Corning, NY, USA) using a vertical micropipette puller (David Kopf Instruments, Tujunga, CA, USA). The nESI emitters were loaded with 10 μL of analyte solution and fitted onto the ion source stage using an electrode holder (Warner Instruments, Holliston, MA, USA) such that a platinum wire (Alfa Aesar, Haverhill, MA, USA) applied the nESI capillary potential directly to the analyte solution. The nESI capillary potential was optimized for each emitter and was typically in the range 1.0–1.5 kV. The sampling cone voltage was adjusted between 10–20 V, the source offset voltage was set to 10 V, and the source temperature was held at 80°C. During TWIMS analysis, the helium cell gas flow was set to 180 mL/min, with the flow of nitrogen to the mobility cell set to 60 mL/min. The trap DC bias was held constant at 44 V, the TWIMS traveling DC wave height was held constant at 40 V, and the TWIMS traveling DC wave velocity was held constant at 650 m/s. The RF amplitudes applied to the stacked ring ion guides in the pre-TWIMS, TWIMS, and post-TWIMS regions of the instrument were 350 V (trap cell), 250 V (TWIMS cell), and 380 V (transfer cell). Initial data analysis and processing was carried out in MassLynx 4.1 and DriftScope 2.7 (Waters). Further data analysis and visualization was accomplished using SigmaPlot 13 (Systat, Chicago, IL, USA) and with custom routines written and implemented in IGOR Pro 7 (WaveMetrics, Lake Oswego, OR, USA).
Ion-Neutral Collision Cross Section Calibration.
Drift times measured by TWIMS were converted to CCS values using previously described calibration procedures.8, 9, 39–42 To concisely summarize, protonated polyalanine ions were used as CCS calibrants since these are among the most well-characterized and broadly adopted calibrants for CCS measurement by TWIMS.43–50 Advantageously, both He and N2 CCSs have been established for these ion series, allowing their use as standards for the measurement of analyte CCSs on either a He or N2 basis.44 Although CCSs are more commonly reported for He drift gas, N2 CCS values are likely to be more relevant to TWIMS experiments carried out using N2 drift gas. Therefore, both values are reported in this work. The polyalanine mixture (Sigma-Aldrich) was prepared at 12.5 µg/µL in 50% acetonitrile / 0.1% formic acid and analyzed as described above. Calibration curves were then generated that related the known He and N2 CCSs of the polyalanine peptides to their corresponding TWIMS drift times. Separate calibration curves were constructed for each drift gas (He and N2), and for each ion charge state considered (z = +2 and z = +3). The CCSs of doubly charged and triply charged analytes were measured using the corresponding charge state matched calibration as previously recommended.42 While every effort was made to ensure the reliability of this widely accepted calibration method, we acknowledge that some non-idealities may arise from calibrating TWIMS drift times to CCSs in this manner. However, prior work suggests that this procedure results in CCS errors that are comparable to the inherent reproducibility of these measurements.42
Computational Chemistry.
Gas phase structures and CCSs were computed using a previously reported method.51 Briefly, global minimum structures of glycopeptide ions were identified by using the Merck Molecular Force Field force field (MMFF94)52–55 and a simple molecular dynamics (MD) optimization method implemented in the Quantum Chemistry Polarizable Force Field program (QuanPol)56 which is integrated in the General Atomic and Molecular Electronic Structure System (GAMESS).57, 58 In this optimization approach (QuanPol keyword MDOPT=1000), an MD simulation is conducted at 600 K. Every 1000 MD steps, the MD is paused, but not interrupted, for a steepest descent geometry optimization to locate a minimum-energy structure. The globally optimized ion structures were then used for MD simulations (100 ns each) of the ion mobility in He or N2 drift gases. Force field parameters for He and N2 drift gases are described in Tables S1–S2 of the Electronic Supplementary Information. In all the simulations, 512 He atoms or N2 molecules were included in a cubic periodic boundary condition (PBC) box with fixed side length of 77.04 Å for He and 131.74 Å for N2. The volumes of the ions, estimated by using a density of 1.0 g/cm3, were deducted from the total volume. The MD simulations were performed with drift gases at 290 K. The average pressure was ~50 bar for He, and ~10 bar for N2. The electric fields were selected so that they resulted in Townsend numbers (~30–40 Td) and drift velocities (~200 m/s and ~80 m/s in He and N2, respectively) that were comparable to experimental conditions. As a result, the effective thermodynamic ion temperatures (considering vibrations and rotations) were all controlled at 300 ± 2 K. The average temperature of the ions (without considering ion drift) were also around 300 K.
A mixed force field was used for the ion and gas system. The partial atomic charges, bond stretching terms, bond angle bending terms, stretching-bending coupling terms, dihedral rotation terms and wagging terms for the ion atoms were from the original MMFF94 force field. Conventional 12–6 Lennard-Jones (LJ) terms were used for interactions between the ion atoms (note the original MMFF94 uses a drifted 14–7 LJ), between the gas atoms, and between the ion atoms and gas atoms. The 12–6 LJ potential parameters for the ion atoms are shown in Tables S1–S2 of the Electronic Supplementary Information. Within the ion, the LJ potentials were excluded for the 1–2 and 1–3 atom pairs that were described with bond stretching and bending terms and were fully included for the 1–4 atoms. Due to the use of a PBC, a switching function was used for the LJ potential (QuanPol keywords ISWITCH=2, SWRA=10.0 Å, SWRB=12.0 Å).
The dipole polarization of the He and N2 molecules were included by using the dipole polarizability from experiments (0.2051 Å3 for He; 1.740 Å3 for N2; see Tables S1–S2 of the Electronic Supplementary Information).59, 60 No polarizability points were used for the ion atoms. The QuanPol keyword IDOPOL=1 was used so the mutual polarization between the gas molecules was not considered. This is identical to the R−4 charge-polarizability potential commonly used in trajectory methods. Due to the use of a PBC, a switching function was used for the charge-polarizability potential (QuanPol keywords IPOLSHF=0, ISWITCH=2, SWRA=10.0 Å, SWRB=12.0 Å).
Results
Overview.
The model glycoprotein RNase B was selected to provide tryptic N-glycopeptide ions with multiple amino acid sequences (NLTK, SRNLTK, NLTKDR, SRNLTKDR), glycan compositions (GlcNAc2 + Mann, or simply “Man n,” with n = 1–8), and charge states (doubly and triply protonated) appropriate for a study of physical and chemical factors affecting the glycopeptide CCSs. Over the course of this study, some interesting trends in measured CCS were noted that prompted the consideration of N-glycopeptides with truncated glycans in addition to the intact, native N-glycans (Man5 through Man8). Thus, a sequential exoglycosidase digestion strategy was used to produce the Man1 through Man4 glycopeptides, as illustrated in Figure 1. In all, over 110 CCS values were obtained for this pool of analytes. Computational modeling and prediction of theoretical CCSs were also performed to rationalize several key experimental findings in structural and conformational terms.
Figure 1.
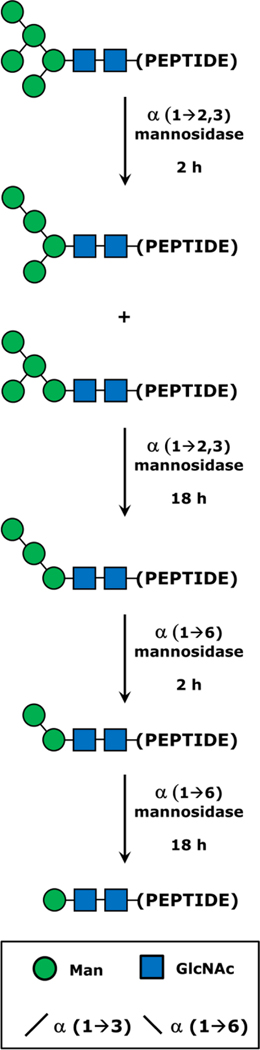
Schematic summary of the enzymatic deconstruction of N-glycan moieties to yield truncated N-glycopeptides. For simplicity, this process is illustrated starting with the smallest and most abundant RNase B glycoform, Man5. The larger N-glycans present in the mixture would be decomposed in a similar fashion. Glycopeptide preparations were first treated with α(1→2,3) mannosidase, which after a 2 h yielded a mixture of Man4 structures via hydrolysis of one of the two susceptible glycosidic linkages of Man5. With 18 h of incubation, both α(1→3) linked residues were cleaved from Man5, producing the Man3 structure. The digest was next treated with α(1→6) mannosidase, which sequentially degraded the remaining branch of Man3 to yield Man2 and Man1 after 2 h and 18 h of incubation, respectively. A key to the monosaccharide and glycosidic bond symbology is provided in the inset.
Glycopeptide Arrival Time Distributions.
The TWIMS ATDs for all doubly and triply protonated glycoforms of SRNLTK, NLTKDR, and SRNLTKDR are presented in Figure 2. As expected, the doubly and triply charged glycopeptides sort into distinct drift time windows (2.5–5.0 ms for z = 2+; 1.5–3.0 ms for z = 3+). Interestingly, very different shifts in ATD position were observed with the addition of each mannose residue. In general, the doubly protonated ions exhibited more uniform shifts between successive glycoforms, with the inclusion of additional mannose residues shifting the ATDs to longer drift times in comparable increments. Some exceptions to this trend can be found, perhaps most notably in the very small shift in drift time observed between the doubly protonated Man4 and Man5 glycoforms of SRNLTK (Figure 2a). Nevertheless, the addition of monosaccharide units tended to shift the mobilities of the doubly charged glycopeptides in a monotonic and incremental fashion. By contrast, the triply charged glycopeptides demonstrated markedly different behavior characterized by highly variable shifts in mobility with successive addition of monosaccharides. Indeed, in several cases involving the triply protonated glycoforms of SRNLTK and NLTKDR, the order of arrival time did not follow the size of the glycan (Figures 2b, 2d). In both of these instances, the ATDs for Man3-Man6 glycoforms were heavily overlapped, with larger gaps in drift time separating the lower and higher glycoforms. The triply protonated SRNLTKDR glycopeptides exhibited some compression of the ATDs for the intermediate glycoforms; however, the order of arrival times still followed the mannose number of the glycan involved. Overall, these findings were interpreted as potentially revealing of an interplay between Coulombic repulsion effects (which tend to bring about more extended gas phase structures to minimize charge-charge interactions) and charge solvation effects (which could result in more compact structures owing to increased intramolecular interaction contributing to stabilization of charge sites). The dependence on glycan size also suggested some involvement of the glycan moiety in charge solvation, at least for sufficiently large glycans.
Figure 2.

Representative TWIMS ATDs for the Man1 through Man8 glycoforms of SRNLTK with z = 2+ (a) and z = 3+ (b); NLTKDR with z = 2+ (c) and z = 3+ (d); and SRNLTKDR with z = 2+ (e) and z = 3+ (f). Each ATD is labelled with the number of mannose residues comprising the corresponding glycoform.
Glycopeptide Collision Cross Sections.
In Figure 3, all measured He and N2 CCSs values are visualized as a function of m/z, which resulted in distinct groupings of the doubly and triply charged glycopeptide ions. The CCS values and their uncertainties are provided in Table S3 of the Electronic Supplementary Information. Regardless of whether the CCS values were calibrated to a He or N2 axis, the doubly protonated glycopeptide ions under study generally exhibited a rather linear increase in CCS as a function of m/z. The only contribution to increasing m/z for a given peptide group was the addition of mannose residues, resulting in the Man1–Man8 glycoforms. Thus, increases in CCS increased in direct proportion to the number of mannose residues for the doubly charged ions under investigation. This result is consistent with a previous finding of Costello and coworkers, which demonstrated linear increases in the CCSs of various glycopeptides as monosaccharide residues were added.32 Contrastingly, the triply charged glycopeptide ions examined here exhibited significantly greater scatter in the CCS dimension, particularly in the region about 600 m/z. Interestingly, CCSs of the Man1–Man8 glycoforms of the triply charged SRNLTKDR peptide increased in an approximately linear fashion, similar to the behavior noted for the doubly charged glycopeptides. This may be related to the larger size of the peptide group and the presence of an additional stably protonated amino acid side chain. Overall, these results are consistent with the observations noted from the TWIMS ATDs and demonstrate that glycopeptide ion mobilities can scale with glycan size in charge state dependent manners that may seem counterintuitive upon initial examination.
Figure 3.

CCS (Ω) vs. mass-to-charge ratio (m/z) for all glycopeptides measured, with TWIMS arrival times calibrated to provide He (a) and N2 (b) CCS values. Where visible, error bars represent the standard deviation of four replicate measurements.
Trends in Charge State and Glycan Size Dependence.
CCS values for the z = 2+ and z = 3+ ions from each glycoform of SRNLTK, NLTKDR, and SRNLTKDR are directly compared in Figure 4. Overall, the z = 3+ glycopeptide ions were consistently found to have significantly larger CCS values than their z = 2+ counterparts for the smaller glycoforms studied (Man1–Man4); however, the larger glycoforms (Man5–Man8) exhibited more varied behavior with respect to the relative CCSs of the doubly and triply charged ions. For instance, He CCSs for the SRNLTK glycoforms (Figure 4a) were significantly larger for z = 3+ ions between Man1 and Man4, but for Man5–Man8 the z = 2+ CCSs were found to be larger. Comparable trends can be seen for the He CCSs of NLTKDR and SRNLTKDR glycoforms, as well (Figures 4c,e). When viewed as N2 CCSs, this reversal in relative size was not apparent for any of the peptide compositions involved; however, in some cases the gap in N2 CCS between the z = 2+ and z = 3+ forms of the glycopeptides was found depend heavily on the glycoform at hand. For SRNLTK and NLTKDR, this gap was initially quite large for the Man1 species but had diminished significantly by the Man8 glycoform (Figure 4b,d). A similar, though less pronounced manifestation of this behavior was also noted for the N2 CCSs of the SRNLTKDR glycoforms (Figure 4f). In aggregate, these comparisons suggest that Coulombic repulsion is a major driving force dictating the conformations of the smaller glycoforms, forcing these triply charged ions into extended structures that are markedly larger than those of their doubly charged counterparts. Meanwhile, for the larger glycoforms, the CCS differences between z = 2+ and z = 3+ ions are far less pronounced, suggesting that the addition of mannose residues allows the doubly and triply charged ions to take on similar CCSs. This is intriguing, since the addition of neutral monosaccharide residues would not seem to provide favorable new sites of protonation to a glycopeptide ion already containing multiple sites of high gas phase basicity. Thus, the means by which the triply protonated structures achieve CCSs similar to the corresponding doubly protonated form may not be through the relief of charge-charge interaction by accessing alternative protonation motifs during the nESI process. Instead, there is also the possibility that intramolecular interactions, perhaps driven by charge solvation and involving the glycan itself, lead to the generation of more compact structures. Furthermore, the ability to sample these condensed conformers may depend on the glycan being of sufficient size to facilitate the decisive interactions.
Figure 4.

CCS (Ω) for the Man1 through Man8 glycoforms of SRNLTK as doubly protonated and triply protonated ions calibrated to He (a) and N2 (b) drift gases; the Man1 through Man8 glycoforms of NLTKDR as doubly protonated and triply protonated ions calibrated to He (c) and N2 (d) drift gases; and the Man1 through Man8 glycoforms of SRNLTKDR as doubly protonated and triply protonated ions calibrated to He (e) and N2 (f) drift gases. For each glycoform, the CCS values measured for the z = 2+ charge state (solid bars) are compared to those for the z = 3+ charge state (hatched bars). Where visible, error bars represent the standard deviation of four replicate measurements.
Structural Corollaries from Computational Modeling.
Global minimization of doubly charged glycopeptides SRNLTK with N-glycan compositions and structures of GlcNAc2Man1–6 indicated that, when the peptides are protonated at the arginine residue and the N-terminal serine (the C-terminal lysine is not protonated), the ions have lower energies, as seen in Figure 5 and all models shown in Figure S1 of the Electronic Supplementary Information. The simulated He and N2 CCS values for these doubly and triply charged glycopeptides are shown in Table S4 of the Electronic Supplementary Information. In general, the triply charged glycopeptides have larger simulated CCS values than the corresponding doubly charged ions. This is mainly caused by the higher charge state, but conformational differences also contribute significantly. As shown in Figure 4, the doubly charged ions are more compact than their triply charged counterparts. With the addition of a third proton on the C-terminal lysine residue and thus stronger internal Coulomb repulsion, triply charged glycopeptides tend to be less compact and exhibit larger CCS values. However, as the glycan becomes larger (e.g., Man5 and Man6), the internal Coulomb repulsion becomes less severe, so the compactness of triply charged glycopeptides is comparable to the doubly charged counterparts. Another contributing factor may be the size dependence of charge-gas interaction: larger sized ions tend to exhibit weaker electric fields due to greater charge delocalization, so their charge induced dipole interactions are lower. The glycopeptides with larger oligosaccharide structures also have more atoms that can provide stronger internal self-solvation of the charges via intramolecular hydrogen bonding and charge-dipole interaction. The net effect of these contributions was that, doubly and triply charged glycopeptide ions with larger glycans (e.g., Man5 and Man6) tend to have more similar CCSs than those with smaller glycans (e.g., Man1–3). For example, in N2 gas, the difference between simulated CCS values of doubly and triply charged SRNLTK + GlcNAc2Man5 is 26 Å2, and the CCS difference between doubly and triply charged SRNLTK + GlcNAc2Man6 is 23 Å2. These differences are significantly smaller than those for SRNLTK + GlcNAc2Man1 (48 Å2 difference between z = +2 and z = +3), SRNLTK + GlcNAc2Man2 (57 Å2 difference between z = +2 and z = +3), and SRNLTK + GlcNAc2Man3 (67 Å2 difference between z = +2 and z = +3). The simulated results are in good agreement with the experimental data, as the average percent difference between experimental and theoretical CCSs was 3.8% for He CCSs and 2.3% for N2 CCSs (see Table S4 of the Electronic Supplementary Information). Full Cartesian coordinates for all modeled glycopeptide ions are provided in Table S5 of the Electronic Supplementary Information.
Figure 5.

Energy-minimized gas phase structures of the SRNLTK + GlcNAc2Man1 glycopeptide with z = 2+ (a) and z = 3+ (b); and the SRNLTK + GlcNAc2Man5 glycopeptide with z = 2+ (c) and z = 3+ (d). The corresponding experimentally measured (Ωexpt) and theoretically calculated (Ωcalc) N2 CCS values are indicated for each ion.
Conclusions
For the glycopeptides studied here, some general conclusions can be drawn regarding the influence of charge state and glycan size upon the mobility of their doubly and triply protonated ions. As size of the glycan is increased from Man1 to Man8, the TWIMS arrival times and CCSs of both doubly and triply charged ions generally tend to increase. Nevertheless, there also appear to be pivotal transition regions for some triply charged ions in which the addition of monosaccharide units causes the ion mobilities, as monitored through ATDs and CCSs, to plateau or even decrease before eventually increasing again. We note here that similar qualitative trends have been well-documented in IMS analyses of synthetic polymers.61–65 Distinct grouping of z = 2+ and z = 3+ ions was observed in CCS vs. m/z space, though the manner in which the various glycoforms of each peptide were distributed depended to a large extent on the charge state. Direct comparisons of CCSs for the doubly and triply protonated series of peptide glycoforms suggests an interplay between two competing modes of structural stabilization: minimizing Coulombic repulsion and maximizing charge solvation. The CCS dependencies observed appear to suggest that the glycan moiety itself participates in charge stabilizing intermolecular interactions, though this is only possible for glycans of sufficient size to participate in these interactions. When the glycan has been truncated such that these interactions are no longer accessible, Coulombic repulsion is the major driver of the glycopeptide ion conformation. These structural arguments were supported by MD simulations of the gas phase ion structures and MD based CCS prediction. The latter of these produced N2 CCS values that agreed with experimental results to within 2.3%, on average. On the whole, these results provide useful fundamental insights into some physical and structural determinants of glycopeptide ion sorting in IMS. Such insights have potential to facilitate the further development of IMS as an analytical tool for glycoproteomic analysis.
Supplementary Material
Acknowledgements
Funding from the National Institutes of Health, National Institute of General Medical Sciences, supported this work through a Maximizing Investigators’ Research Award to E.D.D. (grant number R35GM128926), a fellowship to A.S.G. from the Molecular Mechanisms of Disease Predoctoral Training Program (grant number T32GM107001), and a seed grant to H.L. and E.D.D from the Nebraska Center for Integrated Biomolecular Communication (grant number P20GM113126). Funding to E.D.D. from the National Science Foundation, Division of Chemistry, through the Chemical Measurement and Imaging Program (grant number 1507989) is also acknowledged. This work was carried out using core facilities supported in part by the National Institutes of Health, National Institute of General Medical Sciences through the Nebraska Center for Integrated Biomolecular Communication (grant number P20GM113126). Computational chemistry calculations were performed using resources at the University of Nebraska Holland Computing Center. Finally, A.S.G. would like to thank Dr. Yuting Huang for valued guidance and mentorship in the early stages of this project.
Footnotes
Electronic Supplementary Information
Electronic supplementary information (ESI) is available: Tabulated atomic charges, Lennard-Jones potentials, and dipole polarizabilities used in the molecular dynamics simulations of ion mobility; tabulated experimental ion-neutral collision cross section values; tabulated comparisons of experimental and theoretical ion-neutral collision cross section values; gas-phase structures for additional selected glycopeptide ions; and Cartesian coordinates of optimized gas-phase glycopeptide structures.
References
- 1.Huang Y, Gelb AS and Dodds ED, Curr. Metabolom, 2013, 1, 291–305. [Google Scholar]
- 2.Gray CJ, Thomas B, Upton R, Migas LG, Eyers CE, Barran PE and Flitsch SL, Biochim. Biophys. Acta, 2016, 1860, 1688–1709. [DOI] [PubMed] [Google Scholar]
- 3.Hofmann J and Pagel K, Angew. Chem, 2017, 56, 8342–8349. [DOI] [PubMed] [Google Scholar]
- 4.Morrison KA and Clowers BH, Curr. Opin. Chem. Biol, 2018, 42, 119–129. [DOI] [PubMed] [Google Scholar]
- 5.Clowers BH, Dwivedi P, Steiner WE, Hill HH Jr and Bendiak B, J. Am. Soc. Mass Spectrom, 2005, 16, 660–669. [DOI] [PubMed] [Google Scholar]
- 6.Williams JP, Grabenauer M, Holland RJ, Carpenter CJ, Wormald MR, Giles K, Harvey DJ, Bateman RH, Scrivens JH and Bowers MT, Int. J. Mass Spectrom, 2010, 298, 119–127. [Google Scholar]
- 7.Fenn LS and McLean JA, Phys. Chem. Chem. Phys, 2011, 13, 2196–2205. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y and Dodds ED, Anal. Chem, 2013, 85, 9728–9735. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y and Dodds ED, Analyst, 2015, 140, 6912–6921. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y and Dodds ED, Anal. Chem, 2015, 87, 5664–5668. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann J, Hahm HS, Seeberger PH and Pagel K, Nature, 2015, 526, 241–244. [DOI] [PubMed] [Google Scholar]
- 12.Zheng X, Zhang X, Schocker NS, Renslow RS, Orton DJ, Khamsi J, Ashmus RA, Almeida IC, Tang K, Costello CE, Smith RD, Michael K and Baker ES, Anal. Bioanal. Chem, 2017, 409, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenn LS and McLean JA, Anal. Bioanal. Chem, 2008, 391, 905–909. [DOI] [PubMed] [Google Scholar]
- 14.Fenn LS, Kliman M, Mahsut A, Zhao SR and McLean JA, Anal. Bioanal. Chem, 2009, 394, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLean JA, J. Am. Soc. Mass Spectrom, 2009, 20, 1775–1781. [DOI] [PubMed] [Google Scholar]
- 16.May JC, Goodwin CR, Lareau NM, Leaptrot KL, Morris CB, Kurulugama RT, Mordehai A, Klein C, Barry W and Darland E, Anal. Chem, 2014, 86, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desaire H and Hua D, Int. J. Mass Spectrom, 2009, 287, 21–26. [Google Scholar]
- 18.Froehlich JW, Dodds ED, Wilhelm M, Serang O, Steen JA and Lee RS, Mol. Cell. Proteomics, 2013, 12, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wuhrer M, Catalina MI, Deelder AM and Hokke CH, J. Chromatogr. B, 2007, 849, 115–128. [DOI] [PubMed] [Google Scholar]
- 20.Dodds ED, Mass Spectrom. Rev, 2012, 31, 666–682. [DOI] [PubMed] [Google Scholar]
- 21.Dalpathado DS and Desaire H, Analyst, 2008, 133, 731–738. [DOI] [PubMed] [Google Scholar]
- 22.An HJ, Froehlich JW and Lebrilla CB, Curr. Opin. Chem. Biol, 2009, 13, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alley WR Jr., Mann BF and Novotny MV, Chem. Rev, 2013, 113, 2668–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desaire H, Mol. Cell. Proteomics, 2013, 12, 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolli V, Schumacher KN and Dodds ED, Bioanalysis, 2015, 7, 113–131. [DOI] [PubMed] [Google Scholar]
- 26.Gaunitz S, Nagy G, Pohl NLB and Novotny MV, Anal. Chem, 2017, 89, 389–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Bendiak B, Siems WF, Gang DR and Hill HH, Int. J. Ion Mobil. Spectrom, 2013, 16, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Creese AJ and Cooper HJ, Anal. Chem, 2012, 84, 2597–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Both P, Green AP, Gray CJ, Sardzik R, Voglmeir J, Fontana C, Austeri M, Rejzek M, Richardson D and Field RA, Nat. Chem, 2014, 6, 65. [DOI] [PubMed] [Google Scholar]
- 30.Hinneburg H, Hofmann J, Struwe WB, Thader A, Altmann F, Varón Silva D, Seeberger PH, Pagel K and Kolarich D, Chem. Commun, 2016, 52, 4381–4384. [DOI] [PubMed] [Google Scholar]
- 31.Campbell JL, Baba T, Liu C, Lane CS, Le Blanc JCY and Hager JW, J. Am. Soc. Mass Spectrom, 2017, 28, 1374–1381. [DOI] [PubMed] [Google Scholar]
- 32.Glaskin RS, Khatri K, Wang Q, Zaia J and Costello CE, Anal. Chem, 2017, 89, 4452–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barroso A, Giménez E, Konijnenberg A, Sancho J, Sanz-Nebot V and Sobott F, J. Proteomics, 2018, 173, 22–31. [DOI] [PubMed] [Google Scholar]
- 34.Kolli V and Dodds ED, Analyst, 2014, 139, 2144–2153. [DOI] [PubMed] [Google Scholar]
- 35.Aboufazeli F, Kolli V and Dodds ED, J. Am. Soc. Mass Spectrom, 2015, 26, 587–595. [DOI] [PubMed] [Google Scholar]
- 36.Kolli V, Roth HA, De La Cruz G, Fernando GS and Dodds ED, Anal. Chim. Acta, 2015, 896, 85–92. [DOI] [PubMed] [Google Scholar]
- 37.Kolli V, Schumacher KN and Dodds ED, Analyst, 2017, 142, 4691–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aboufazeli F and Dodds ED, Analyst, 2018, 143, 4459–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruotolo BT, Benesch JLP, Sandercock AM, Hyung SJ and Robinson CV, Nat. Protoc, 2008, 3, 1139–1152. [DOI] [PubMed] [Google Scholar]
- 40.Thalassinos K, Grabenauer M, Slade SE, Hilton GR, Bowers MT and Scrivens JH, Anal. Chem, 2008, 81, 248–254. [DOI] [PubMed] [Google Scholar]
- 41.Smith D, Knapman T, Campuzano I, Malham R, Berryman J, Radford S and Ashcroft A, Eur. J. Mass Spectrom, 2009, 15, 113–130. [DOI] [PubMed] [Google Scholar]
- 42.Gelb AS, Jarratt RE, Huang Y and Dodds ED, Anal. Chem, 2014, 86, 11396–11402. [DOI] [PubMed] [Google Scholar]
- 43.Henderson SC, Li J, Countermann AE and Clemmer DE, J. Phys. Chem. B, 1999, 103, 8780–8785. [Google Scholar]
- 44.Bush MF, Campuzano IDG and Robinson CV, Anal. Chem, 2012, 84, 7124–7130. [DOI] [PubMed] [Google Scholar]
- 45.Campuzano I, Bush MF, Robinson CV, Beaumont C, Richardson K, Kim H and Kim HI, Anal. Chem, 2012, 84, 1026–1033. [DOI] [PubMed] [Google Scholar]
- 46.Pagel K and Harvey DJ, Anal. Chem, 2013, 85, 5138–5145. [DOI] [PubMed] [Google Scholar]
- 47.Hofmann J, Struwe WB, Scarff CA, Scrivens JH, Harvey DJ and Pagel K, Anal. Chem, 2014, 86, 10789–10795. [DOI] [PubMed] [Google Scholar]
- 48.Paglia G, Williams JP, Menikarachchi L, Thompson JW, Tyldesley-Worster R, Halldorsson S, Rolfsson O, Moseley A, Grant D, Langridge J, Palsson BO and Astarita G, Anal. Chem, 2014, 86, 3985–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forsythe JG, Petrov AS, Walker CA, Allen SJ, Pellissier JS, Bush MF, Hud NV and Fernandez FM, Analyst, 2015, 140, 6853–6861. [DOI] [PubMed] [Google Scholar]
- 50.Hines KM, May JC, McLean JA and Xu L, Anal. Chem, 2016, 88, 7329–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lai R, Dodds ED and Li H, J. Chem. Phys, 2018, 148, 064109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halgren TA, J. Comput. Chem, 1996, 17, 520–552. [Google Scholar]
- 53.Halgren TA, J. Comput. Chem, 1996, 17, 553–586. [Google Scholar]
- 54.Halgren TA and Nachbar RB, J. Comput. Chem, 1996, 17, 587–615. [Google Scholar]
- 55.Halgren TA, J. Comput. Chem, 1996, 17, 616–641. [Google Scholar]
- 56.Thellamurege NM, Si DJ, Cui FC, Zhu HB, Lai R and Li H, J. Comput. Chem, 2013, 34, 2816–2833. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M and Montgomery JA, J. Comput. Chem, 1993, 14, 1347–1363. [Google Scholar]
- 58.Gordon MS and Schmidt MW, in Theory and Applications of Computational Chemistry, eds. Dykstra CE, Frenking G, Kim KS and Scuseria GE, Elsevier, Amsterdam, 2005, 10.1016/B978-044451719-7/50084-6, pp. 1167–1189. [DOI] [Google Scholar]
- 59.Newell AC and Baird RC, J. Appl. Phys, 1965, 36, 3751–3759. [Google Scholar]
- 60.Alms GR, Burnham AK and Flygare WH, J. Chem. Phys, 1975, 63, 3321–3326. [Google Scholar]
- 61.Trimpin S and Clemmer DE, Anal. Chem, 2008, 80, 9073–9083. [DOI] [PubMed] [Google Scholar]
- 62.Weidner SM and Trimpin S, Anal. Chem, 2010, 82, 4811–4829. [DOI] [PubMed] [Google Scholar]
- 63.Morsa D, Defize T, Dehareng D, Jerome C and De Pauw E, Anal. Chem, 2014, 86, 9693–9700. [DOI] [PubMed] [Google Scholar]
- 64.Haler JRN, Far J, Aqil A, Claereboudt J, Tomczyk N, Giles K, Jerome C and De Pauw E, J. Am. Soc. Mass Spectrom, 2017, 28, 2492–2499. [DOI] [PubMed] [Google Scholar]
- 65.Wesdemiotis C, Angew. Chem. Int. Ed, 2017, 56, 1452–1464. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.