

Abstract
The human plasma contact system is an immune surveillance system activated by the negatively charged surfaces of bacteria and fungi and includes the kallikrein-kinin, the coagulation, and the fibrinolytic systems. Previous work shows that the contact system also activates complement, and that plasma enzymes like kallikrein, plasmin, thrombin, and FXII are involved in the activation process. Here, we show for the first time that kallikrein cleaves the central complement component C3 directly to yield active components C3b and C3a. The cleavage site within C3 is identical to that recognized by the C3 convertase. Also, kallikrein-generated C3b forms C3 convertases, which trigger the C3 amplification loop. Since kallikrein also cleaves factor B to yield Bb and Ba, kallikrein alone can trigger complement activation. Kallikrein-generated C3 convertases are inhibited by factor H; thus, the kallikrein activation pathway merges with the amplification loop of the alternative pathway. Taken together, these data suggest that activation of the contact system locally enhances complement activation on cell surfaces. The human pathogenic microbe Candida albicans activates the contact system in normal human serum. However, C. albicans immediately recruits factor H to the surface, thereby evading the alternative and likely kallikrein-mediated complement pathways.
Keywords: Candida albicans, Complement system, Contact activation, Factor H, Host defense, Kallikrein
Introduction
The plasma contact activation system is part of the innate immune system [1] and is activated spontaneously by negatively charged surfaces (e.g., bacterial or fungal surfaces) and by polyphosphates or orthophosphates released by platelets. During the first step, FXII binds to these negatively charged surfaces and undergoes a conformational change that induces limited proteolytic activity. Activated FXII (FXIIa) then cleaves plasma prekallikrein to yield kallikrein, which enters an amplification loop and further activates FXII zymogens [2, 3]. Generated kallikrein subsequently cleaves high molecular weight kininogen (HMWK) to release the proinflammatory peptide bradykinin, which in turn causes vascular leakage and the sensation of pain. Kallikrein also activates the blood pressure-regulating renin-angiotensin system by converting the substrate prorenin to renin [4]. Furthermore, kallikrein activates the fibrinolytic system, and plasminogen is converted to plasmin. The simultaneous activation of all 3 pathways (contact, coagulation, and complement) is a phylogenetic and ancient host response to infection and tissue injury, leading to thromboinflammation [5]. Thus, it is not surprising that the plasma contact and complement systems interact.
The complement system is an immune surveillance system that detects invading microbes and modified self surfaces and opsonizes them with C3b/iC3b. This results in 3 outcomes: recognition and clearance by phagocytic cells, induction of terminal lytic pathways, and induction of inflammation and adaptive immune responses [6]. Three complement activation pathways have been described to date: the alternative pathway (AP) (induced by deposition and amplification of C3b on foreign surfaces), the classical pathway (CP) (induced by deposition of immunoglobulins on target surfaces), and the lectin pathway (LP) (induced by carbohydrate structures present on foreign, mainly microbial, surfaces). Each pathway generates C3 convertases (the alternative pathway generates convertase C3bBb and the classical and lectin pathways generate convertase C4bC2a), which cleave C3 into C3b and the anaphylatoxin C3a. Newly generated C3b also forms C3 convertases, thereby amplifying the production of C3b. The C3 convertase can act in conjunction with a second C3b molecule, the C5 convertase (which cleaves C5 into C5b and C5a), to initiate the terminal pathway, culminating in generation of the terminal complement complex (TCC). The TCC inserts into the cell membrane to induce lysis and death. Since activation of the complement system is accompanied by the generation of toxic molecules such as C3a and C5a, complement is tightly regulated by complement inhibitors. The main regulator of the alternative pathway is complement factor H, whereas that of the classical/lectin pathway is C4b binding protein. Both regulators dissociate C3 convertases and mediate cofactor activity, thereby enabling protease factor I to degrade C3b [5, 7, 8, 9, 10]. Previous studies show that the complement, coagulation, and contact systems interact: thrombin FIXa, FXa, FXIa, and plasmin cleave C3 and C5 [11, 12], FXIIf activates the protease C1s of the classical pathway [13], and kallikrein cleaves complement factor B [14]. In addition, a recent study shows that MASP2 cleaves C3 in the absence of C2 and C4 [15, 16], and cleaves prothrombin to yield active thrombin [17]. However, it is not known how effective these cross-reactions are or whether they contribute to local complement activation.
The pathogenic fungus Candida albicans is a common opportunistic organism that plays a major role in human pathology. C. albicans is commensal on mucous membranes and the skin, but can invade immunocompromized patients (e.g., those with neutropenia, HIV, diabetes, or those receiving immunosuppressive therapy) [18, 19]. Once microbes such as C. albicans come into “contact” with human plasma, the contact (kallikrein-kinin) and complement systems are activated. C3b is deposited on the cell surface and complement is amplified to opsonize and target the microbe for phagocytosis and to prime the adaptive immune response. Similarly, factor XII is activated by the foreign negatively charged surface, leading to cleavage of zymogen plasma prekallikrein and subsequent activation of the protease kallikrein. This protease then cleaves HMWK to release the potent inflammatory mediator bradykinin [20]. Thus, the contact and complement activation cascades interact and act synergistically to mount an immune response.
Here, we show that kallikrein activates the complement system and demonstrate that factor H restricts kallikrein-induced complement activation. Our group previously showed that C. albicans recruits factor H to the cell surface [21, 22, 23]. In this study, we further investigate kallikrein-mediated complement activation and whether factor H is capable of inhibiting complement activation via the contact activation system and alternative pathway.
Materials and Methods
Cell Growth
C. albicans wild-type (SC5314) cells were grown overnight in YPD medium (2% D-glucose, 1% peptone, 5% yeast extract in water) at 30°C, reseeded in YPD medium and grown for 4 h at 30°C into the midlog-phase. Escherichia coli (XL1-Blue) was grown overnight in tryptic soy broth and Luria-Bertani broth at 37°C, reseeded in the same broth, and grown to OD 1 at 37°C, resembling the midlog phase. Renal cortex/proximal tubular cells (HK2, ATCC® CRL-2190TM) were grown in complete DMEM with epidermal growth factor (10 ng/mL; Sigma-Aldrich) at 37°C and CO2 (5%). Cells were passaged every 2 days and used for experiments until the 30th passage. Complement-active normal human serum (NHS) was prepared from fresh whole blood obtained from healthy volunteers, which was immediately centrifuged (10 min, 2,000 g, 4°C), mixed in a pooled stock, and stored in aliquots at −80°C.
Cleavage
C3 (100 nM; Comptech) was incubated with kallikrein (Athens Research and Technology), plasmin, thrombin, or neutrophil elastase (each 125 nM; all from Technoclone GmbH) in PBS for 60 min at 37°C. Proteins were separated by SDS-PAGE (10 or 12%), transferred to a membrane, and immunoblotted using polyclonal C3a (1:3,000; Comptech) or C3 antibodies (1:3,000; Calbiochem). For dose-dependent cleavage assays, C3 (50 nM) or C3b (50 nM; Comptech) was incubated with kallikrein or plasmin (10, 25, and 100 nM) for 60 min at 37°C and processed as above. Similarly, C3 (100 nM) was incubated with kallikrein (100 nM) for 5, 60, and 180 min at 37°C and immunoblotted as described above. To follow the cleavage of C3, NHS (1%) was incubated with kallikrein (200 nM), plasmin (200 nM), or properdin (350 nM; Comptech) for 30 min at 37°C. Probes were separated by SDS-PAGE (12%) and immunoblotted using C3 antiserum as described above. For the detection of C3a, probes were boiled for 10 min at 95°C in probe buffer (Roti®-Load, Carl Roth, Karlsruhe, Germany), separated by SDS-PAGE (15%), and immunoblotted using polyclonal C3a antibodies (1:3,000; Comptech). To measure the efficiency of kallikrein compared to thrombin in cleaving C3, kallikrein (0.14 units/mL; Athens Research & Technology) and thrombin (0.14 units/mL; Sigma-Aldrich) were incubated with C3 (100 nM; Comptech) over a time period of 60 min and separated by SDS-PAGE (15%). C3a generation was determined by Western blot analysis and quantified using Image J software. Factor B (110 nM; Comptech) was incubated in the presence of NiCl2 (2 mM) with C3b (5, 25, 50, or 100 nM) and kallikrein (80 nM) or with plasmin (80 nM) for 60 min at 37°C. Factor D (80 nM; Comptech) was used in parallel. Factor B cleavage was determined by Western blot analysis using factor B antiserum. Cleavage of factor B was assayed in NHS (2.5%) with kallikrein (300 nM), plasmin (300 nM), or properdin (100 nM). Cleavage was performed upon addition of EDTA or EGTA (each 10 mM) to the plasma, separated by SDS-PAGE (10%) and immunoblotted with polyclonal factor B antibodies (1:4,000; Comptech).
To analyze C3b generation by kallikrein and the deposition of C3b on C. albicans, fungal cells (1 × 106) were incubated with C3 (1 µM) and kallikrein or thrombin (each 0.3 units/mL) in DPBS for 60 min at 37°C. Candida cells were then washed and incubated in reducing buffer (Roti®-Load 1, Carl Roth) for 10 min at 95°C. C. albicans proteins were separated by SDS-PAGE (10%) and immunoblotted using polyclonal C3 antibodies (1:1,000; Comptech). Factor H binding to C. albicans cells was elucidated by incubating the fungal cells (1 × 106) for 60 min with purified factor H (320 nM; Comptech) and the binding was visualized with fluorescent microscopy (LSM 780, Zeiss; equipped with a ×63/1.25–0.75 numerical aperture plan apochromat oil objective) using polyclonal factor H antibodies (1:2,000; Comptech) and donkey anti-goat Alexa 647 (1:400; Life Technologies).
Complement Activation
To determine convertase activity, kallikrein or plasmin (each 160 nM) were added together with factor B (215 nM) to immobilized C3b (55 nM) for 120 min at 37 °C. After washing, the substrate C3 (155 nM) was added and C3 cleavage was determined by Western blot analysis as described above. In parallel, a convertase was formed by adding factor B (215 nM), factor D (20 nM), and factor P (190 nM) to the immobilized C3b.
Contact Activation
Binding of purified FXII to C. albicans was determined by a whole cell ELISA. C. albicans (1 × 104) diluted in DPBS were coated on an ELISA plate for 1 h at 30°C. Cells were blocked with 100 µL of blocking buffer 1 (AppliChem) for 60 min at room temperature (RT). Plasma-purified human factor XII (10, 20, 40, 100 µg/mL, ab62423; Abcam) was incubated with the coated C. albicans cells for 60 min at RT. FXII binding was detected with polyclonal factor XII antibodies (1:150; BP2296, F12; Acris) and HRP-labeled polyclonal sheep IgGs (1:500, P0163; Dako). Between each step, cells were washed with PBS Tween (0.05% Tween 20).
To determine C3b deposition on the C. albicans surface, C. albicans (4 × 107) was incubated in NHS (4% with 20 mM EDTA in Tris buffer, pH 7.8). 50 µL of substrate Chromogenix S-2302 (2 mM; Haemochrom Diagnostica GmbH) was added and incubated for 20 min at 37°C. The reaction was stopped with 20% acetic acid. The cells were pelleted and 100 µL of the supernatant was measured at 405 nm using a microplate reader (TECAN, Safire2). C3b deposition on C. albicans was evaluated by incubating C. albicans in NHS (5% or supplemented with 10 mM EDTA in DPBS with or without 0.1 mM of AEBSF [4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride]; A8456; Sigma-Aldrich) or 25 μM of PMSF (phenylmethanesulfonyl fluoride; P7626; Sigma-Aldrich) for 30 min at 37°C. The cells were blocked with blocking buffer 1 (100 µL; AppliChem) for 1 h at RT. C3 cleavage products were detected with anti C3d antiserum (A0063; Dako) combined with anti-rabbit Alexa Fluor 647 (A21246; Invitrogen; both 1:500 in blocking buffer) for 30 min at RT. Between each step the cells were washed twice with DPBS. The cells were analyzed by flow cytometry (BD AccuriTM C6).
N-Terminal Amino Acid Sequencing
To localize the cleavage site of kallikrein in the C3 protein, kallikrein (300 nM) was added to C3 (1800 nM) in DPBS (30 µL). Following incubation for 1 h at 37°C, the reaction was stopped by the addition of Roti-Load 1 (10 µL), then the mixture was boiled for 10 min at 95°C and subjected to SDS-PAGE (7%). Upon transfer to a PVDF membrane, the membrane was stained with Coomassie blue R250 (1% freshly prepared in 40% methanol/1% acidic acid) for 2 h at RT to identify the position of the bands. The membrane was destained with methanol (50%) and washed 3 times with degased water. Then, the 110-kDa band was cut out and subjected to N-terminal protein sequence analysis (Alphalyse, Denmark).
Decay Acceleration
Dissociation of the kallikrein-generated convertase C3bBb was measured by ELISA. Immobilized C3b (28 nM, overnight at 4 °C) was incubated with factor B (5 nM), factor P (20 nM), and kallikrein (25 nM), or with plasmin (25 nM) or factor D (1 nM) and incubated for 1 h at 37°C. Subsequently factor H (0.5, 1.5, 3, or 6 nM) was added and dissociation of the convertase was followed by detecting the remaining factor B/Bb bound to C3b using ELISA with antipolyclonal factor B antibodies (1:4,000; Comptech).
Cofactor Activity
Cofactor activity of factor H was assayed as previously described [23]. Factor H (60 nM) was added to C3 (100 nM), kallikrein (100 nM), and factor I (8 nM), and incubated for 1 h at 37°C in buffer A (140 mM NaCl, 10 mM Tris, 2 mM CaCl2, 1 mM MgCl2, pH 7.4). In parallel, factor H (60 nM) was added to C3 (100 nM), factor B (100 nM), factor P (190 nM), factor D (20 nM), and NiCl2 (2 mM), and also incubated for 1 h at 37°C in buffer A. Then, samples were separated by SDS-PAGE (8%), transferred to a membrane, and C3b as well as C3 degradation fragments were identified using polyclonal C3 antibodies (1:3,000; Comptech).
Mouse Model of Disseminated Candidiasis
All animal experiments were conducted in compliance with European and German regulations. Protocols were approved by the responsible Federal State authority and ethics committee (Thüringer Landesamt für Verbraucherschutz, permit No. 03-007/13). Female BALB/c mice (Charles River, Germany) weighing 18–20 g were housed in groups of 5 in individually ventilated cages with free access to water and food. For infection, C. albicans was grown for 12 h at 30°C in YPD medium, washed 3 times with sterile PBS, and diluted to the desired concentrations in pyrogen-free sterile PBS (pH 7.2). The concentration was confirmed by plating serial dilutions on YPD. Mice were infected via the lateral tail vein with 2.5 × 104C. albicans CFU/g body weight and sacrificed after 6 h. Upon necropsy, the liver was removed, fixed in 10% neutral buffered formalin (Histofix, Carl Roth), embedded in paraffin, and sectioned at a thickness of 4 µm.
Immunohistochemistry
Paraffin-embedded mice liver sections were deparaffinized by treating them consecutively in Roticlear (Carl Roth), 100% ethanol, and 95% ethanol. Sectioned tissues were boiled in 10 mM Na citrate buffer (pH 6.5) for antigen retrieval and blocked in 1% BSA supplemented PBS for 30 min. For detection of factor H or FXIIa on erythrocytes, tissues were treated with Alexa Fluor 350-labeled anti-mouse factor H antibodies (1:1,000; Bioss) or Alexa Fluor 350-labeled mouse factor H antibodies (1:1,000; Bioss Antibodies, Woburn, Massachusetts, USA) or Alexa Fluor 488 mouse TER-119 antibodies (1:100; Biolegend), respectively. 0.1% saponin-supplemented PBS was used to dilute the antibodies. Alexa Fluor 594-labeled anti-mouse Pra1 monoclonal antibody (1:100) diluted in 0.1% saponin-supplemented PBS was used for detection of Pra1 deposition in postinfected tissues. Images were captured using LSM 710 with ZEN 2011 (401/421 nm for Alexa Fluor 350 and 561/594 nm for Alexa Fluor 594). The experiments were repeated 3 times.
Statistical Analysis
Differences between 2 groups were analyzed using the unpaired Student t test. Values of * p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001 were considered statistically significant.
Results
Kallikrein Cleaves Complement C3
Kallikrein, plasmin, and thrombin are enzymes that play roles in the activated contact and coagulation systems. To investigate whether kallikrein, like thrombin and plasmin, can cleave C3 to activate the complement system, we incubated kallikrein with C3, followed by Western blot analysis to detect cleavage of C3. Kallikrein cleaved the C3 alpha chain to yield a C3b alpha-like chain (Fig. 1a) and a C3a-like fragment (Fig. 1b). Also, thrombin cleaved C3 into a C3b-like fragment. In contrast to kallikrein, plasmin and neutrophil elastase cleaved C3 into several cleavage products of different sizes. In all cases, C3a-like fragments were generated. Incubation of C3 with kallikrein (180 min) resulted in the complete transformation of C3 into C3b-like proteins (Fig. 1c).
Fig. 1.
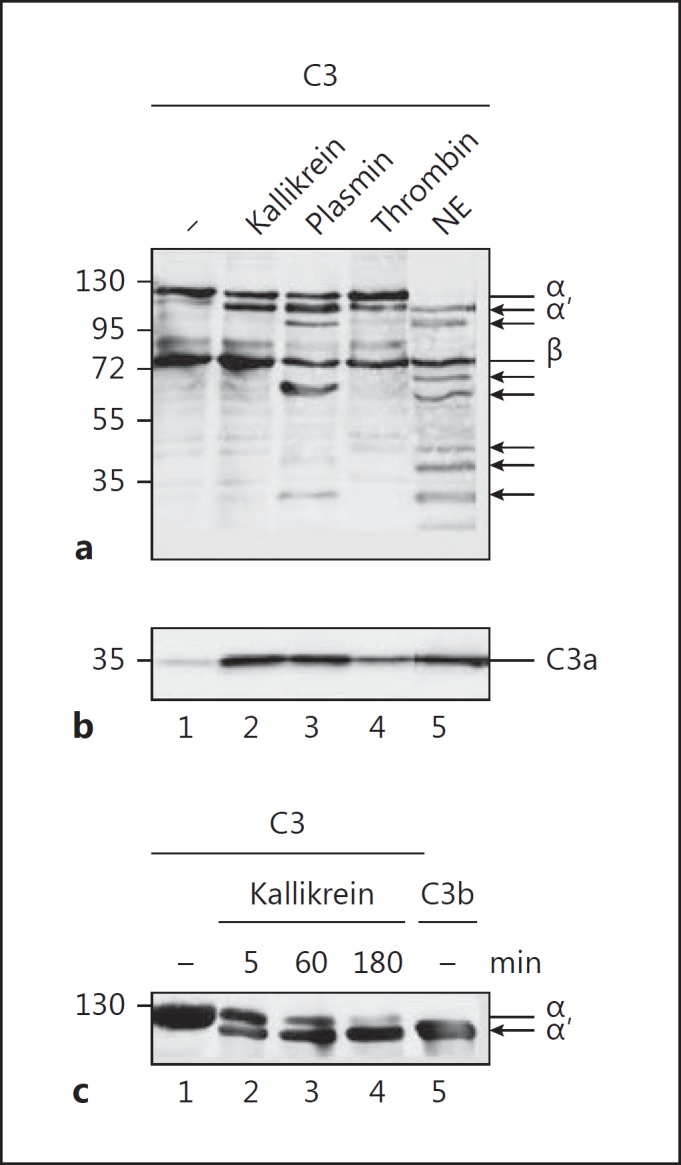
C3 cleavage by proteases. a C3 is cleaved by kallikrein, plasmin, thrombin, and neutrophil elastase (NE). All proteases generate C3b-like (upper panel α′ chain) and C3a-like molecules (b). Incubation of C3 with plasmin (lane 3) and NE (lane 5) generates further cleavage products of C3 as indicated by additional C3 bands in a (arrows) using Western blot analysis. c Kallikrein (100 nM) incubated with C3 (50 nM) for 180 min completely cleaves C3 to C3b-like fragments (lanes 1–4). Purified C3b is shown in lane 5. Proteins were separated by SDS-PAGE and immunoblotted using C3 (a, c) and C3a (b) antibodies. Representative Western blots are shown.
To further characterize C3 cleavage by kallikrein, purified kallikrein was incubated with C3 and cleavage was evaluated by Western blotting. Kallikrein cleaved C3 to yield C3b- and C3a-like molecules, but no further cleavage products were observed, even at higher kallikrein concentrations (100 nM; Fig. 2a). Incubating C3b with kallikrein did not result in any cleavage products (Fig. 2a), confirming that kallikrein targets a single cleavage site. These results demonstrate that kallikrein cleaves C3 at a single site to yield C3b- and C3a-like fragments. In contrast to kallikrein, incubation of plasmin with C3 or C3b (Fig. 2b) generated C3 cleavage products with molecular weights of 60 and 30 kDa, respectively. To confirm C3 cleavage by kallikrein in plasma, we incubated human plasma with kallikrein and examined C3 cleavage by Western blotting. Kallikrein also cleaved plasma C3 to yield C3b-like and C3a-like molecules. Cleavage was observed in EGTA-treated plasma (which inhibits the classical pathway) and in EDTA-treated plasma (which inhibits all 3 complement pathways; Fig. 2c). In contrast to kallikrein, plasmin cleaved C3 to yield additional fragments. The addition of properdin to plasma resulted in cleavage of plasma C3 to C3b and C3a via the alternative pathway; this was not the case in complement-blocked plasma (Fig. 2c). In summary, kallikrein cleaves C3 in human plasma to yield 2 cleavage products.
Fig. 2.
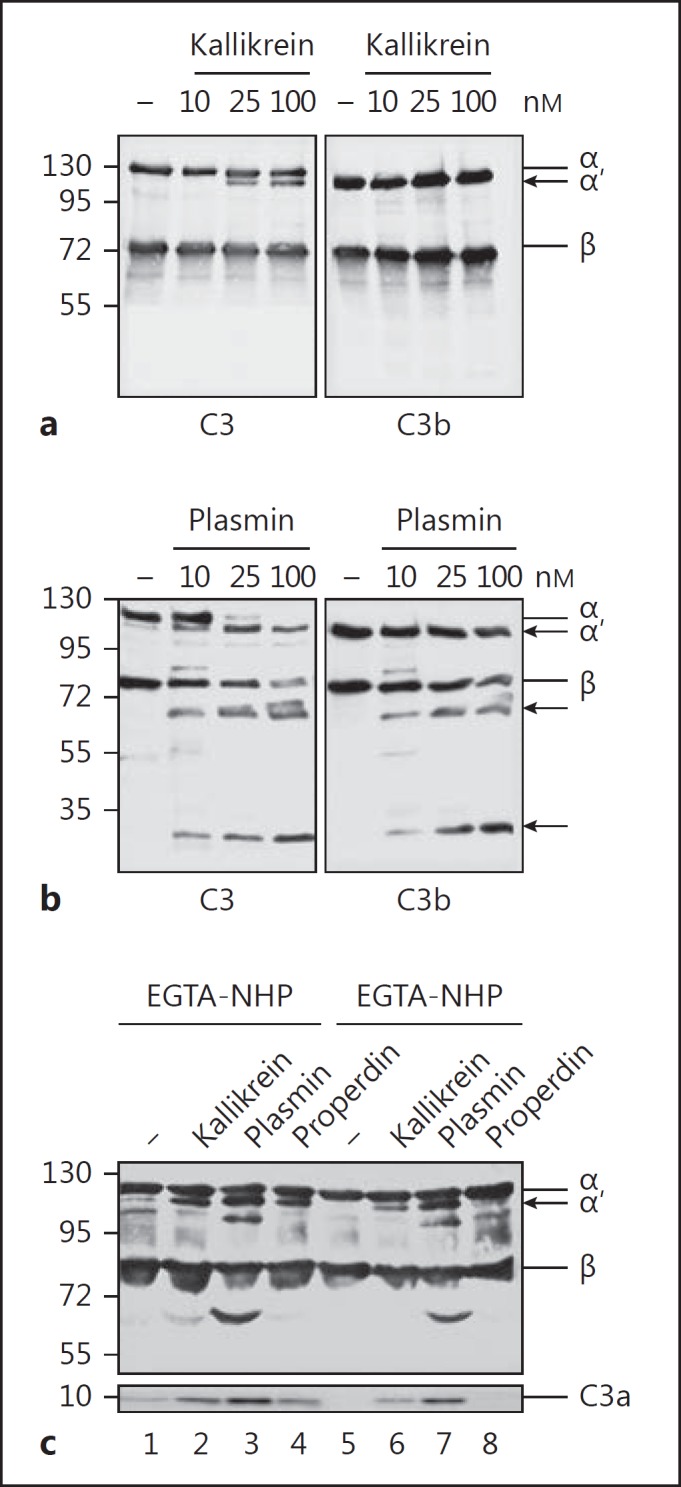
Kallikrein generates C3b-like fragments and plasmin degrades C3 and C3b. a Kallikrein (10, 25, and 100 nM) incubated with C3 (50 nM) for 60 min cleaves C3 to C3b-like fragments (left panel) but has no effect on C3b (right panel), as shown by Western blot analysis. b Plasmin (10, 25, and 100 nM) degrades C3 (left panel) and C3b (right panel), and cleavage products of 60- and 30-kDa appear (arrows). c Kallikrein cleaves C3 in EGTA- (inhibitor of the CP) and EDTA- (inhibitor of AP, CP, and LP) buffered human plasma (1%), and C3a-like fragments are generated (lanes 2 and 6), as shown by Western blot analysis. Also, the addition of properdin (350 nM) to EGTA-treated plasma generates active convertases (lane 4), but the addition of properdin to EDTA-treated plasma (lane 8) does not initiate C3 convertases. In contrast to kallikrein, plasmin cleaves C3 to C3b- and C3a-like fragments, but degrades C3 to further fragments of about 100 and 60 kDa (lanes 3 and 7). Representative immune blots from 3 independent experiments are shown (a–c).
Kallikrein Cleaves C3 in a Manner Similar to the Alternative Pathway C3 Convertase
To identify the kallikrein cleavage site in C3, we incubated C3 with kallikrein, separated the cleavage products by SDS-PAGE and transferred them to a PVDF membrane (Fig. 3a). After staining the proteins with Coomassie blue, the C3b-like cleavage product was isolated from the membrane and the N-terminal amino acid sequence was determined. The sequence was N'749-SNLDEDIIAEENIVSRS-C'765 (Fig. 3a), with the kallikrein cleavage site in C3 located between amino acids R748 and S749. This cleavage site is identical to that targeted by the C3 convertase C3bBb (N'739-ARASHLGLARSNLDEDIIAEENIVSRS-C'765) [24]. The identical cleavage sites suggest that kallikrein-cleaved C3 generates a functional C3b molecule that binds to cell surfaces and forms C3 convertases.
Fig. 3.

Kallikrein cleaves C3 like the C3 convertase. a N-terminal sequencing of the kallikrein-generated C3b-like cleavage product (left panel) revealed cleavage of C3 between the amino acids arginine (R) and serine (S), as indicated by the arrow (right panel). The cleavage site by the C3 convertase is indicated. Cleavage of C3 by kallikrein is shown by Coomassie blue staining (left panel). Identified amino acids are boxed. b Immobilized C3b preincubated with factor B and factor D forms a convertase that cleaves C3 to C3b (lane 4) and C3a. Coated C3b preincubated with kallikrein still forms C3 convertases (lane 2), as seen by the appearance of the C3bα′ chain. No C3 convertases formed when coated C3b was preincubated with plasmin (lane 3). The immune blot is representative for 3 independent experiments. The arrow indicates the sequence of incubation. c Kallikrein cleaves C3 more efficiently than similar amounts of thrombin. C3 cleavage was followed over time by the generation of C3a using Western blot, followed by densitometric analysis. Data represent mean values ± SE of 3 independent experiments. One-tailed t test, * p < 0.05.
C3b Incubated with Kallikrein Still Forms an Active C3 Convertase
We next queried whether preincubating kallikrein with C3b generates active C3 convertases. C3b was immobilized on a microtiter plate and then incubated with either kallikrein or plasmin, followed by a wash step and addition of factor B and factor D to allow formation of a C3 convertase, C3bBb. Functional activity of the formed convertase was examined by adding the substrate C3 and detection of C3b by Western blotting. Preincubating coated C3b with kallikrein led to formation of a C3 convertase and cleavage of C3 to C3b (Fig. 3b). Similarly, incubation of C3b with factor B and factor D formed a C3 convertase that cleaved C3. In contrast to kallikrein, plasmin pretreated C3b failed to form an active convertase, as seen by the lack of C3b formation. (Fig. 3b). These experiments demonstrate that kallikrein allows C3b to form active convertases, whereas plasmin probably degrades C3b into inactive products. Kallikrein cleaves more C3 molecules and forms more C3a over time as compared to similar amounts of thrombin (Fig. 3c).
Kallikrein Cleaves C3 to Yield the Functional Cleavage Products C3b and C3a
To confirm activity of kallikrein-generated C3b, we preincubated NHS with kallikrein and then added it to HK2 cells. Kallikrein and plasmin were used to confirm the activity of the generated C3b. Deposition of kallikrein-generated C3b on the HK2 cell surfaces was determined by flow cytometry. HK2 cells were used because these kidney proximal tubular endothelial cells are sensitive to complement (unpubl. data). The amount of C3b deposited on the HK2 cell surface in the presence of NHS/kallikrein was significantly greater than that deposited in the presence of NHS/plasmin or NHS alone (control; Fig. 4a). These data demonstrate that kallikrein-generated C3b acts as an opsonin that attaches to the surface of HK2 cells. To show that kallikrein is activated upon contact with C. albicans, C. albicans cells were incubated in NHS and activation of kallikrein was followed by cleavage of a kallikrein-specific chromogenic substrate (S-2302). C. albicans activated the contact system in NHS and kallikrein was formed as measured by enhanced cleavage of the substrate (Fig. 4b). Next, we examined the activity of the kallikrein-derived C3a. C3a exerts antimicrobial activity [25]; therefore, we preincubated C3 with kallikrein and added it to E. coli expressing a fluorescent protein in the cytoplasm. The antimicrobial activity of the newly formed C3a was determined by measuring the fluorescent signal in the supernatant (A = 450 nm), which is a marker for lysed E. coli. We observed significantly more fluorescence in the supernatant of E. coli incubated with kallikrein plus C3 (Fig. 4c) than in that of E. coli incubated with kallikrein alone. These data confirmed that kallikrein cleaves C3 to yield functional C3b and C3a molecules.
Fig. 4.
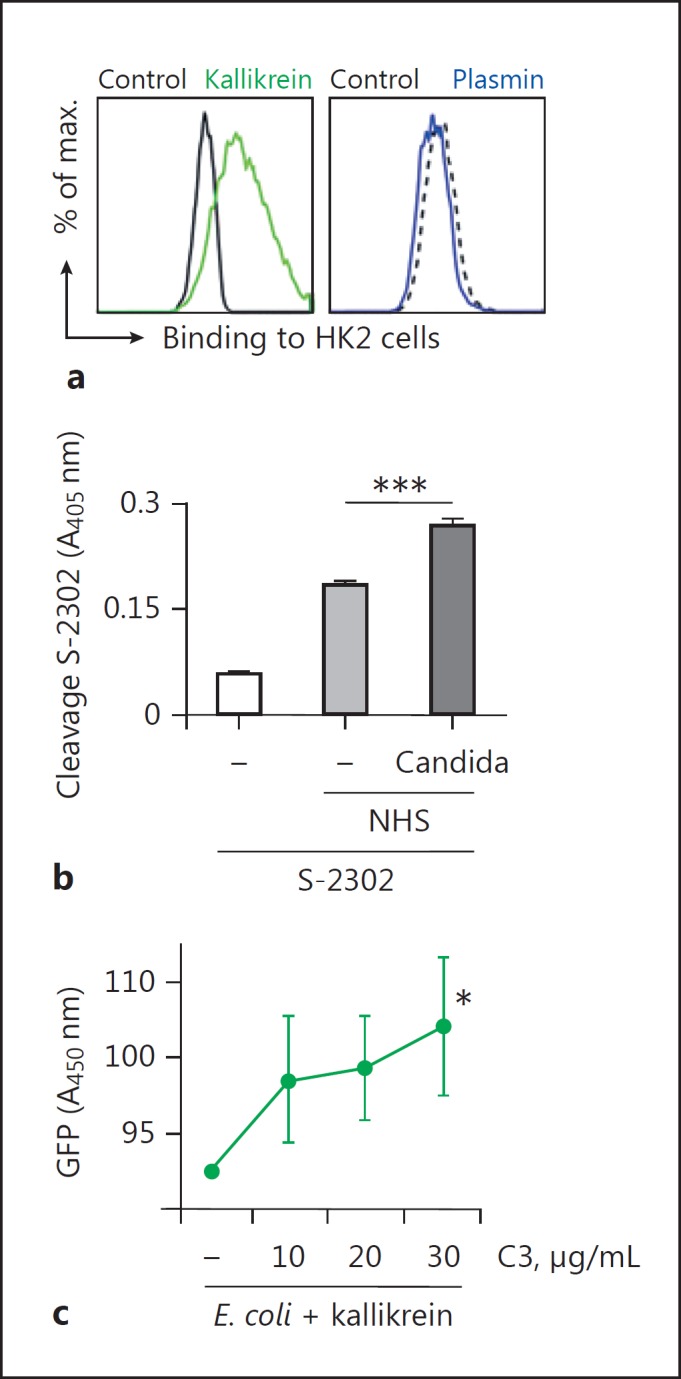
Kallikrein generated C3b deposits on cells. a C3b generated from C3 by kallikrein deposits on HK2 cells. In contrast, plasmin degrades C3. A representative flow cytometry result of 3 independent experiments is shown. bC. albicans activates the contact system. C. albicans was incubated in NHS and kallikrein formation was followed by cleavage of the chromogenic substrate Chromogenix S-2302 by kallikrein. c C3a generated by kallikrein displays antimicrobial action and kills E. coli. GFP-labelled E. coli cells were incubated with C3 and kallikrein, and GFP released by dead E. coli was determined by absorbance. Data in b and c represent mean values ± SE of 3 independent experiments. One-tailed t test, * p < 0.05, *** p < 0.001.
Kallikrein Cleaves Factor B to Yield Active Bb
Kallikrein is a serine protease that cleaves complement factor B [14]. To examine whether cleavage is enhanced when factor B binds C3b, we incubated kallikrein with factor B in the presence of increasing concentrations of C3b. Cleavage of factor B was then determined by Western blotting. The amount of factor B cleavage increased with the concentration of C3b. Thus, factor B was cleaved by kallikrein, a process that was enhanced in the presence of C3b (Fig. 5a). In addition, the factor B cleavage products generated by kallikrein had molecular weights of 63 and 37 kDa, similar to factor Bb and factor Ba, respectively, generated by factor D (Fig. 5a). However, when C3b-bound factor B was incubated with plasmin, we detected several fragments of 80, 60, 58, 48, 40, and 30 kDa (Fig. 5b). Thus, kallikrein cleaves factor B, whereas plasmin degrades factor B.
Fig. 5.

Kallikrein cleaves factor B like factor D. a Kallikrein incubated with factor B and C3b (5, 25, 50, or 100 nM) in the presence of NiCl2 reveals the cleavage of factor B to fragments Bb and Ba (lanes 2–5), as demonstrated by immune blotting. Similarly, factor D incubated with C3b and factor B cleaves factor B to Bb and Ba. Factor B cleavage increases with C3b concentrations (lanes 3–5). b Incubation of plasmin with C3b and factor B results in the degradation of factor B (lanes 2–5) and additional bands appear (arrows). c Kallikrein cleaves factor B in a dose-dependent manner, as determined by ELISA, in contrast to plasmin. Data represent mean values ± SD of 3 independent experiments. One-tailed t test, ** p < 0.01, *** p < 0.001. d Kallikrein forms active C3 convertases upon incubation with C3 and factor B. Kallikrein was incubated with C3 and factor B in the presence of NiCl2. Convertase formation was followed by the generation of C3b (lanes 3 and 4, arrow) and in parallel C3a using immunoblotting (e; lanes 3 and 4). Assembly of a C3 convertase by C3b, factor B, factor D, and C3 also shows C3a formation (lane 2).
Next, we asked whether the kallikrein-generated factor Bb forms active C3 convertases when bound to C3b. To answer this, we immobilized C3b on a microtiter plate and incubated it with factor B and kallikrein or with factor B and plasmin. Factor Bb generation in the presence of C3b was then detected by a specific monoclonal antibody. The amount of C3b-bound Bb increased along with the kallikrein concentration (Fig. 5c). As expected, plasmin did not result in Bb formation. To confirm the activity of the kallikrein-generated C3 convertase, we added the substrate C3 to the washed convertases and detected cleavage of C3 to C3b by Western blot analysis. Preconvertase C3bB yielded C3b when factor B was cleaved by kallikrein or factor D, but not by plasmin (Fig. 5d). Similarly, kallikrein-generated convertases yielded C3a (Fig. 5e).
Factor H Regulates Kallikrein-Generated Convertases
To find out whether factor H also regulates kallikrein-generated convertases, we incubated C3 and factor B with kallikrein to generate C3 convertases. Factor H was added to these C3 convertases at increasing concentrations and factor B binding was assayed using factor B antiserum. Factor H but not FHR1 dissociated factor B from kallikrein-cleaved C3b, as seen by reduced binding of factor B upon addition of factor H (Fig. 6a). This effect was dose dependent. Similarly, factor H dissociated the alternative pathway C3 convertase generated by factor D (Fig. 6b). In contrast, addition of plasmin did not form convertases. Kallikrein-generated C3b was also incubated with factor H and factor I, and degradation of C3b was detected by Western blotting. Both factor H- and factor I-degraded kallikrein converted C3b to iC3b (Fig. 6c). Similar C3b cleavage products (86, 46, and 43 kDa) were generated by kallikrein and C3 convertases. Taken together, these data suggest that factor H decays kallikrein-generated C3 convertases and acts as a cofactor for factor I during the degradation of kallikrein-generated C3b.
Fig. 6.

Factor H regulates kallikrein-formed convertases. a Factor H but not FHR1 dissociates kallikrein-formed active C3 convertases. Kallikrein-formed convertases were incubated with increasing amounts of factor H or FHR1, and factor Bb binding was determined by ELISA. One representative experiment out of 3 is shown. b Factor H dissociates kallikrein- and factor D-generated convertases. Plasmin fails to create convertases. Data represent mean values ± SD of 3 independent experiments. One-tailed t test, ** p < 0.01, *** p < 0.001. c Factor H and factor I form iC3b from kallikrein-formed C3b molecules (lane 4). IC3b generation from C3 convertase-formed C3b is shown in lane 8. A representative Western blot of 3 independent experiments is shown.
C. albicans Triggers the Contact Activation System
The contact system is spontaneously activated upon recognition by and attachment of factor XII (FXII) to negatively charged surfaces [26, 27]. To examine whether C. albicans triggers the contact activation system, we incubated C. albicans cells with purified FXII and detected binding of FXII to the surface by whole-cell ELISA. Purified FXII bound to the C. albicans surface in a dose-dependent manner (Fig. 7a). Next, we measured FXII binding to C. albicans in vivo in tissues from C. albicans-infected mice. Liver sections were stained with antibodies specific for FXII and the fungal surface protein Pra1 (pH-regulated protein 1). Binding of FXII was assessed by laser scanning microscopy. FXII was detected on the surface of C. albicans in liver tissue, indicating in vivo activation of the contact activation system (Fig. 7b). Purified kallikrein added to NHS enhanced C3b deposition on C. albicans, showing the complement-enhancing effect of this enzyme (Fig. 7c). Also, C3b deposited onto C. albicans in complement inhibited NHS and serine protease inhibitors AEBSF and PMSF reduced C3b deposition (Fig. 7d). Thus, the contact system is activated on C. albicans in human plasma and supports C3b opsonization. As shown here, factor H regulates the kallikrein-generated convertases. Both here and previously, we demonstrated in vitro recruitment of factor H to C. albicans in the presence of NHS (Fig. 7e) [21]. To find out whether C. albicans recruits factor H to the surface also in vivo, we stained liver sections from mice infected with C. albicans for 6 h for both C. albicans and mouse complement factor H. Factor H was present on C. albicans infecting the liver tissue, thereby confirming recruitment of the complement regulator in vivo (Fig. 7f).
Fig. 7.

C. albicans activates the contact system and recruits factor H. a Purified factor XII binds to C. albicans in a dose-dependent manner. Data represent mean values ± SE of 3 independent experiments. One-tailed t test, **p < 0.01, *** p < 0.001. b FXII is also recruited to the surface of C. albicans in liver tissue sections derived from C. albicans-infected mice (BALB/c). Blue, FXII; red, fungal protein Pra1. cC. albicans activates complement upon incubation in NHS and C3b is deposited on the surface. The addition of kallikrein enhances C3b deposition. d Inhibition of serine proteases in complement-inactive NHS reduces C3b deposition on C. albicans cells. C. albicans was incubated in complement-inhibited NHS(NHSEDTA) with or without the serine protease inhibitors AEBSF or PMSF. C3b deposition on C. albicans cells was measured by flow cytometry. The data represent means ± SEM of 3–4 independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001. e Purified factor H binds to the surface of C. albicans cells. Red, factor H; blue, DNA (DAPI). f Factor H is recruited to C. albicans in liver tissues. Green, erythrocytes; red, fungal protein Pra1; blue, mouse factor H. Images were taken with an LSM710 microscope (Zeiss) fitted with a ×40 1.4 NA oil-immersion lens. Scale bars, 10 µm.
Discussion
Bacterial and fungal surfaces are immediately recognized by the host complement and contact systems. Once activated, both systems trigger a coordinated immune response that comprises opsonization, phagocytosis, acute and chronic inflammation, vascular permeability, pain, and fever [1, 2, 5, 7]. Here, we have shown that kallikrein (generated from prekallikrein by the induced contact system) activates the complement system by cleaving C3 and factor B, with subsequent formation of an active complement C3 convertase. Damage to host cells caused by this convertase is regulated by factor H, which decays kallikrein-generated C3 convertase and assists factor I-mediated degradation of C3b. The human pathogenic fungus, C albicans, activates both the complement and contact systems in human serum. However, C. albicans expresses microbial proteins that bind factor H to the surface [21], thereby conferring protection from complement components generated by the alternative pathway and contact system. C3 degradation products on C. albicans may also help to adhere via complement receptors to endothelial cells.
Kallikrein cleaves plasma C3 at the amide bond between R748 and S749 to generate active C3b and C3a. The cleavage site in C3 is identical to that recognized by the C3 convertase. In contrast to plasmin, kallikrein cleaves C3 exclusively at this position and no further cleavage products appear. Mature human prekallikrein, a precursor of plasma kallikrein, comprises 619 amino acids [28] and activation of prekallikrein to kallikrein is induced by cleavage at the amide bond between R371 and I372, resulting in production of a heterodimer comprising an N-terminal heavy chain and a C-terminal light chain containing a C364-C484 bond [28]. The light chain includes a serine protease domain harboring a triad of catalytic residues. The main substrate of kallikrein is kininogen, and cleavage of the latter generates kallidin (lysil-bradykinin), which induces vascular permeability and promotes nitric oxide and prostaglandin release by endothelial cells, resulting in vasodilation [29, 30, 31]. Kallikrein also cleaves factor B; the cleavage patterns of kallikrein and factor D are indistinguishable. These results confirm previous observations by DiScipio [14], who demonstrated magnesium ion-dependent cleavage of factor B by kallikrein. Thus, kallikrein induces the C3 convertase amplification loop in the absence of factor D, and links the contact activation and complement systems to enhance opsonization, vascular permeability, and recruitment of inflammatory cells to generate a local immune response. Kallikrein can also activate the classical complement pathway [32]. Activation of both of these systems (contact and complement) causes signs and symptoms often seen in those with septic shock and hereditary angioedema. Hereditary angioedema is a condition caused by a deficiency or defect in the C1 inhibitor [33, 34]. Similar to kallikrein, FXII can activate C1 of the classical pathway [35] and factor B of the alternative pathway [14].
Upon contact with NHS, C. albicans triggers the contact system. Factor XII then binds strongly to the fungal surface and activates kallikrein and kinins. A recent study suggests that kinins released at the C. albicans cell wall promote host colonization by the pathogen, followed by the development of infection via increased vascular permeability and further dissemination of C. albicans[36]. Similarly, C. albicans and bacterial proteases activate upstream components (zymogens) of the host plasma kinin-forming system [37, 38]. However, kallikrein cleaves HMWK to yield bradykinin, which is an important proinflammatory mediator that recruits immune cells such as neutrophils to the sites inhabited by the infectious microbe. As shown herein, kallikrein also activates complement to boost the immune response. Since kallikrein also cleaves complement component C5, it also activates the terminal complement pathway [12]. In contrast to thrombin, kallikrein is a strong activator of C3 and so is more likely to play an “activating role” under physiological conditions. Low thrombin C3 cleavage activity was previously also described by Foley et al. [39].
Kallikrein-generated C3 convertases are inactivated by complement factor H. Factor H binds to stressed human cell surfaces and blocks the complement cascade. In the presence of low levels of factor H (as in patients harboring mutated or dysfunctional protein), the endothelium is less well protected, resulting in diseases such as hemolytic uremic syndrome [8]. C. albicans and a number of other microbes recruit factor H to the cell surface where it inactivates C3b and C3 convertases. Bound factor H then dissociates the C3 convertases generated by complement and kallikrein. Here, we also showed that plasmin (in contrast to kallikrein) inactivates C3 convertases. C. albicans expresses several receptor molecules that recruit plasminogen to the surface [21] where it is cleaved to plasmin. Taken together, these data suggest that C. albicans escapes the different complement activation pathways by recruiting diverse host regulators to the cell surface.
Previous reports show that the contact system is activated by bacterial cell walls [40]. Similar to C. albicans, bacterial pathogens also use a variety of strategies, such as secreted proteinases, to increase kinin generation, thereby facilitating dissemination via vasodilation. However, these pathogens also recruit and exploit factor H [41]. The full relevance of kallikrein-mediated complement activation to host defense in vivo remains unclear; however, some individuals with a severe deficiency of contact factors are highly susceptible to bacterial infections [14]. Our findings support a role of kallikrein in the defense response to infections. Similarly, overactivation of the contact system is expected to also activate the complement system. Furthermore, complement activation via kallikrein may need to be considered in respect to therapeutic complement inhibition.
Disclosure Statement
The authors declare that they have no conflicts of interests.
Author Contributions
C.S. designed and supervised the study. S.I., N.D., L.H., E.J., and I.K. performed experiments and discussed the data. S.L. isolated C3 cleavage products and provided aa sequencing data. I.J. and C.D. performed mouse infection experiments and provided tissue samples. H.S. and S.L. provided antibodies and discussed the data. N.B. provided monoclonal Pra1 antibodies and read the manuscript. C.S. and P.F.Z. discussed the data and wrote the manuscript.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, Sk 46) and within Collaborative Research Center CRC124 FungiNet Projects C4 (C.S., S.L.), C5 (I.J.), C6 (N.B., P.F.Z.), and A5 (H.S.). S.I. and E.J. are doctoral researchers at the Jena School of Microbial Communications (JSMC) and L.H. at the International Leibniz Research School (ILRS), Jena, Germany.
References
- 1.Nickel KF, Renné T. Crosstalk of the plasma contact system with bacteria. Thromb Res. 2012;130:S78–S83. doi: 10.1016/j.thromres.2012.08.284. [DOI] [PubMed] [Google Scholar]
- 2.Maas C, Oschatz C, Renné T. The plasma contact system 2.0. Semin Thromb Hemost. 2011;37:375–381. doi: 10.1055/s-0031-1276586. [DOI] [PubMed] [Google Scholar]
- 3.Zerleder S. C1-inhibitor: more than a serine protease inhibitor. Semin Thromb Hemost. 2011;37:362–374. doi: 10.1055/s-0031-1276585. [DOI] [PubMed] [Google Scholar]
- 4.Derkx FH, Bouma BN, Schalekamp MP, Schalekamp MA. An intrinsic factor XII- prekallikrein-dependent pathway activates the human plasma renin-angiotensin system. Nature. 1979;280:315–316. doi: 10.1038/280315a0. [DOI] [PubMed] [Google Scholar]
- 5.Ekdahl KN, Teramura Y, Hamad OA, Asif S, Duehrkop C, Fromell K, Gustafson E, Hong J, Kozarcanin H, Magnusson PU, Huber-Lang M, Garred P, Nilsson B. Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol Rev. 2016;274:245–269. doi: 10.1111/imr.12471. [DOI] [PubMed] [Google Scholar]
- 6.Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. 2009;104:115–149. doi: 10.1016/S0065-2776(08)04004-2. [DOI] [PubMed] [Google Scholar]
- 7.Ghebrehiwet B. The complement system: an evolution in progress. F1000Research. 2016;5:2840. doi: 10.12688/f1000research.10065.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 9.Wiegner R, Chakraborty S, Huber-Lang M. Complement-coagulation crosstalk on cellular and artificial surfaces. Immunobiology. 2016;221:1073–1079. doi: 10.1016/j.imbio.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amara U, Flierl MA, Rittirsch D. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010;185:5628–5636. doi: 10.4049/jimmunol.0903678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 13.Ghebrehiwet B, Randazzo BP, Dunn JT, Silverberg M, Kaplan AP. Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J Clin Invest. 1983;71:1450–1456. doi: 10.1172/JCI110898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiScipio RG. The activation of the alternative pathway C3 convertase by human plasma kallikrein. Immunology. 1982;45:587–595. [PMC free article] [PubMed] [Google Scholar]
- 15.Hiemstra PS, Daha MR, Bouma BN. Activation of factor B of the complement system by kallikrein and its light chain. Thromb Res. 1985;38:491–503. doi: 10.1016/0049-3848(85)90182-3. [DOI] [PubMed] [Google Scholar]
- 16.Yaseen S, Demopulos G, Dudler T, Yabuki M, Wood CL, Cummings WJ, Tjoelker LW, Fujita T, Sacks S, Garred P, Andrew P, Sim RB, Lachmann PJ, Wallis R, Lynch N, Schwaeble WJ. Lectin pathway effector enzyme mannan-binding lectin-associated serine protease-2 can activate native complement C3 in absence of C4 and/or C2. FASEB J. 2017;31:2210–2219. doi: 10.1096/fj.201601306R. [DOI] [PubMed] [Google Scholar]
- 17.Krarup A, Wallis R, Presanis JS, Gál P, Sim RB. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One. 2007;2:e623. doi: 10.1371/journal.pone.0000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 19.Brown GD, Denning DW, Levitz SM. Tackling human fungal infections. Science. 2012;336:647. doi: 10.1126/science.1222236. [DOI] [PubMed] [Google Scholar]
- 20.Renné T, Schmaier AH, Nickel KF, Blombäck M, Maas C. In vivo roles of factor XII. Blood. 2012;120:4296–4303. doi: 10.1182/blood-2012-07-292094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo S, Skerka C, Kurzai O, Zipfel PF. Complement and innate immune evasion strategies of the human pathogenic fungus Candida albicans. Mol Immunol. 2013;56:161–169. doi: 10.1016/j.molimm.2013.05.218. [DOI] [PubMed] [Google Scholar]
- 22.Gillum AM, Tsay EY, Kirsch DR. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol Gen Genet. 1984;198:179–182. doi: 10.1007/BF00328721. [DOI] [PubMed] [Google Scholar]
- 23.Eberhardt HU, Buhlmann D, Hortschansky P, Chen Q, Böhm S, Kemper MJ, Hartmann A, Hallström T, Zipfel PF, Skerka C. Human factor H-related protein 2 (CFHR2) regulates complement activation. PLoS One. 2013;8:e78617. doi: 10.1371/journal.pone.0078617. 1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janssen BJC, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444:213–216. doi: 10.1038/nature05172. [DOI] [PubMed] [Google Scholar]
- 25.Sonesson A, Ringstad L, Nordahl EA, Malmsten M, Mörgelin M, Schmidtchen A. Antifungal activity of C3a and C3a-derived peptides against Candida. Biochim Biophys Acta. 2007;1768:346–353. doi: 10.1016/j.bbamem.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 26.Morrison DC, Cochrane CG. Direct evidence for Hageman factor (factor XII) activation by bacterial lipopolysaccharides (endotoxins) J Exp Med. 1974;140:797–811. doi: 10.1084/jem.140.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalter ES, van Dijk WC, Timmerman A, Verhoef J, Bouma BN. Activation of purified human plasma prekallikrein triggered by cell wall fractions of Escherichia coli and Staphylococcus aureus. J Infect Dis. 1983;148:682–691. doi: 10.1093/infdis/148.4.682. [DOI] [PubMed] [Google Scholar]
- 28.Chung DW, Fujikawa K, McMullen BA, Davie EW. Human plasma prekallikrein, a zymogen to a serine protease that contains four tandem repeats. Biochemistry. 1986;25:2410–2417. doi: 10.1021/bi00357a017. [DOI] [PubMed] [Google Scholar]
- 29.Wuepper KD, Cochrane CG. Effect of plasma kallikrein on coagulation in vitro. Proc Soc Exp Biol Med. 1972;141:271–276. doi: 10.3181/00379727-141-36757. [DOI] [PubMed] [Google Scholar]
- 30.Hong SL. Effect of bradykinin and thrombin on prostacyclin synthesis in endothelial cells from calf and pig aorta and human umbilical cord vein. Thromb Res. 1980;18:787–795. doi: 10.1016/0049-3848(80)90201-7. [DOI] [PubMed] [Google Scholar]
- 31.Woodruff RS, Sullenger B, Becker RC. The many faces of the contact pathway and their role in thrombosis. J Thromb Thrombolysis. 2011;32:9–20. doi: 10.1007/s11239-011-0578-5. [DOI] [PubMed] [Google Scholar]
- 32.Ghebrehiwet B, Silverberg M, Kaplan AP. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med. 1981;153:665–676. doi: 10.1084/jem.153.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tosi M. Molecular genetics of C1 inhibitor. Immunobiology. 1998;199:358–365. doi: 10.1016/S0171-2985(98)80040-5. [DOI] [PubMed] [Google Scholar]
- 34.Mayilyan KR. Complement genetics, deficiencies, and disease associations. Prot Cell. 2012:3487–3496. doi: 10.1007/s13238-012-2924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmaier AH. The elusive physiologic role of Factor XII. J Clin Invest. 2008;118:3006–3009. doi: 10.1172/JCI36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karkowska-Kuleta J, Kozik A, Rapala-Kozik M. Binding and activation of the human plasma kinin-forming system on the cell walls of Candida albicans and Candida tropicalis. Biol Chem. 2010;391:97–103. doi: 10.1515/BC.2009.145. [DOI] [PubMed] [Google Scholar]
- 37.Kaminishi H, Tanaka M, Cho T, Maeda H, Hagihara Y. Activation of the plasma kallikrein-kinin system by Candida albicans proteinase. Infect Immun. 1990;58:2139–2143. doi: 10.1128/iai.58.7.2139-2143.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frick IM, Björck L, Herwald H. The dual role of the contact system in bacterial infectious disease. Thromb Haemost. 2007;98:497–502. [PubMed] [Google Scholar]
- 39.Foley JH, Walton BL, Aleman MM, O'Bryne AM, Lei V, Harrasser M, Wolberg AS, Conway EM. Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMedicine. 2016;5:175–182. doi: 10.1016/j.ebiom.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben Nasr AB, Herwald H, Müller-Esterl W, Björck L. Human kininogens interact with M protein, a bacterial surface protein and virulence determinant. Biochem J. 1995;305:173–180. doi: 10.1042/bj3050173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imamura T, Potempa J, Travis J. Activation of the kallikrein-kinin system and release of new kinins through alternative cleavage of kininogens by microbial and human cell proteinases. Biol Chem. 2004;385:989–996. doi: 10.1515/BC.2004.129. [DOI] [PubMed] [Google Scholar]