

Abstract
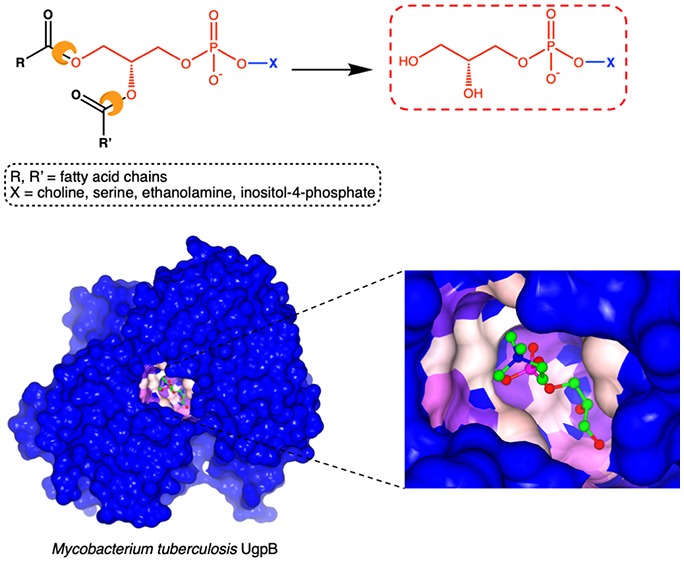
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB) and has evolved an incredible ability to survive latently within the human host for decades. The Mtb pathogen encodes for a low number of ATP-binding cassette (ABC) importers for the acquisition of carbohydrates that may reflect the nutrient poor environment within the host macrophages. Mtb UgpB (Rv2833c) is the substrate binding domain of the UgpABCE transporter that recognizes glycerophosphocholine (GPC), indicating that this transporter has a role in recycling glycerophospholipid metabolites. By using a combination of saturation transfer difference (STD) NMR and X-ray crystallography, we report the structural analysis of Mtb UgpB complexed with GPC and have identified that Mtb UgpB not only recognizes GPC but is also promiscuous for a broad range of glycerophosphodiesters. Complementary biochemical analyses and site-directed mutagenesis precisely define the molecular basis and specificity of glycerophosphodiester recognition. Our results provide critical insights into the structural and functional role of the Mtb UgpB transporter and reveal that the specificity of this ABC-transporter is not limited to GPC, therefore optimizing the ability of Mtb to scavenge scarce nutrients and essential glycerophospholipid metabolites via a single transporter during intracellular infection.
Bacterial pathogens have evolved a wide range of strategies to survive and thrive within their host environment. The ability to assimilate nutrients is vital, and pathogens have evolved diverse strategies to uptake and scavenge the scarce energy sources that are available to them. In the context of intracellular microbial infections, there is growing evidence that in a nutrient limited environment the interplay between the host and the pathogen is important. This is manifested through the ability of bacterial pathogens to utilize discrete nutrient sources with dedicated transport machinery for import. Glycerophosphodiester metabolites that are released by the action of phospholipases on host phospholipids represent an important nutrient source for the supply of carbon and phosphate.
Mycobacterium tuberculosis (Mtb) is a major human pathogen and is now the leading cause of death from a single infectious agent worldwide, resulting in more deaths each year than HIV and malaria combined.1Mtb is a highly evolved pathogen that is able to persist and survive intracellularly within macrophages for decades.2 However, the essential nutrients that are available to Mtb within the stringent environment of the human host and acquisition systems are poorly understood.3,4 Understanding the molecular mechanisms that enable Mtb to survive within this niche environment and the nutrients that are assimilated is critical to understand this major global pathogen and for the development of new therapeutic approaches.
The sugars that are available within the nutrient-limited macrophage environment are unknown; however, Mtb is equipped with five putative importers of carbohydrate substrates: four members of the ATP-binding cassette (ABC) transporter family and one belonging to the major facilitator superfamily.3,4 Until recently, the substrates for these transporters were unresolved; however, recent studies have demonstrated a role for the ABC-transporters in the recycling of components from the complex Mtb cell wall. Trehalose is recycled from the Mtb cell envelope glycolipid trehalose monomycolate and taken up by the LpqY-SugABC-transporter, which plays a critical role in the virulence of the Mtb pathogen.5 The Mtb UspABC-transporter has been found to recognize amino-sugars with a potential role in the uptake of Mtb cell wall peptidoglycan fragments.6
The role of the UgpABCE ABC-transporter is less clear; however, studies of its substrate binding domain Mtb UgpB (Rv2833c) indicate its importance for Mtb survival and pathogenesis, and in vivo Mtb UgpB has been found to be upregulated during infection.7Mtb UgpB has been shown to bind the glycerophosphocholine (GPC) headgroup of the membrane phospholipid phosphatidylcholine, and metabolomic profiling by NMR of intact lung tissue at various stages of Mtb infection has revealed that the GPC metabolite increases significantly as infection progresses, with a concomitant decrease in phosphatidylcholine.8 However, despite the essential role of this Mtb transporter, the molecular mechanisms that dictate how GPC is recognized and whether other glycerophosphodiester metabolites are substrates for this ABC-transporter are currently unknown. The only crystal structure of Mtb UgpB is of the protein in an open conformation without substrate bound (PDB 4MFI).9 Some mechanistic understanding of substrate recognition can be obtained from the crystal structure of a homologue from E. coli with low sequence identity (25%) in complex with glycerol-3-phosphate (G3P) (PDB 4AQ4).10 However, Mtb UgpB does not bind G3P. Comparison of the closed G3P-bound E. coli UgpB with the open Mtb UgpB in the absence of substrate (PDB 4MFI) reveals notable differences in the binding sites of these homologous proteins, indicating that these UgpB ABC-transporters, belonging within the same structural classification (cluster D),11 have diverged to have different substrate specificities. This may reflect the nutritional requirements of the specific organism within different host environments and also the ability of bacteria to produce G3P extracellularly through the action of secreted glycerophosphodiesterases that hydrolyze glycerophopshodiesters.12 Other microorganisms that import GPC have evolved to use either permeases or proton symporters that belong to the major facilitator superfamily indicating that glycerophosphodiester uptake is not limited to ABC-transporters.13,14 It is likely that the divergence of transport systems for the import of glycerophosphodiesters reflects the evolutionary divergence and intracellular lifestyle of the pathogen and the metabolites available within its niche environment.
In this study, we report a detailed functional and structural characterization of the Mtb UgpB substrate binding domain of the ABC-transporter using a combination of biochemical and biophysical approaches. We report the first crystal structure of Mtb UgpB in complex with GPC and identify, in both solid and solution state, the molecular determinants of binding and critical features for glycerophosphodiester recognition. Structure guided mutagenesis has revealed the crucial role of binding-site residues that underpin substrate binding and function. Moreover, we show that Mtb UgpB has a broad selectivity for glycerophosphodiesters, which highlights that the Mtb UgpABCE transporter uptakes metabolites derived from various glycerophospholipids. Thus, Mtb has evolved to use a broad spectrum of nutrients via a single ABC-transporter that enables it to adapt and assimilate essential nutrients during intracellular infection.
Results and Discussion
Production of Mtb UgpB
An N-terminal truncated Mtb UgpB, corresponding to removal of residues 1–34 predicted to form a trans-membrane anchor-helix, was cloned into the pYUB1062 vector with a C-terminal hexa-histidine affinity tag and expressed in Mycobacterium smegmatis mc24517. Soluble Mtb UgpB protein was obtained and purified to apparent homogeneity using Co2+-affinity, anion exchange, and size-exclusion chromatography (Figure S1). The identity of the Mtb UgpB protein was confirmed by using in-gel trypsin digestion and analysis of the peptides by mass spectrometry.
Co-Crystal Structure of Mtb UgpB with GPC
Initial attempts to crystallize Mtb UgpB in the presence of GPC routinely resulted in crystals of UgpB in an open conformation with no ligand bound. Therefore, to overcome this, we chemically modified the surface of Mtb UgpB through reductive methylation, and this resulted in crystals of UgpB in complex with GPC. The UgpB protein co-crystallized with GPC with four molecules in the asymmetric unit. Phases for the structure were determined by molecular replacement using each of the two domains from the apo-structure of Mtb UgpB (PDB 4MFI) as separate search models, and the structure was refined at a resolution of 2.3 Å and to a Rwork of 20.6% and Rfree of 25.6%; see Table S1 for the data collection and refinement statistics. Structural superposition of each molecule of Mtb UgpB using PDBeFOLD15 indicates that each molecule within the asymmetric unit is equivalent, aligning with a rmsd of 0.35–0.44 Å for 394–395 residues. The crystal packing and analysis of the packing interfaces using PDBePISA16 does not suggest that Mtb UgpB forms dimers or higher oligomers and is consistent with our analytical gel filtration studies where the protein behaves as a monomer in solution with an apparent molecular weight of 44 kDa (Figure S1D). It is therefore likely that the monomer is the biologically relevant unit, consistent with substrate binding domains of other ABC-transporters.17,18
Overall Structure of the Mtb UgpB–GPC Complex
Mtb UgpB comprises two α/β domains (Figure 1). Domain I (residues 1–154 and 307–365) consists of a five-stranded β-sheet surrounded by 11 α-helices and domain II (residues 155–306 and 366–436), of a four-stranded β-sheet enclosed by 9 α-helices. The two domains, or globular lobes, are connected via two flexible hinges that are formed between residues Arg152-Pro155 and Ala290-Ala307. Relative to the apo-crystal structure, there is a 22° rotation of domain I relative to domain II about the interdomain screw axis with three hinge/binding regions identified from DynDom analysis19 (residues 152–153, 304–306, and 362–372 (Table S2)). This bending movement results in an almost 2-fold reduction in the volume of the cavity from 1986 to 791 Å3, as determined by CAVER,20 which is in-line with the “Venus Fly-trap mechanism” for other substrate-binding proteins17,18 that close when the substrate is bound. Interdomain bridging and stabilization of this closed conformation of the protein is centered around Arg385, which forms interdomain hydrogen bonds with Asp102 from domain I and Gln381 from domain II. The individual domains of Mtb UgpB apo- and GPC cocomplex structures align with rmsd values of 0.57 and 0.75 Å for domains I and II, respectively (over 178 atoms, domain I; over 216 atoms, domain II, PDBeFOLD16). In comparison, superposition of Mtb UgpB apo- and GPC cocomplex structures align with a rmsd of 2.2 Å (over 385 residues), highlighting the importance of an interdomain conformational change mechanism for substrate recognition by Mtb UgpB.
Figure 1.
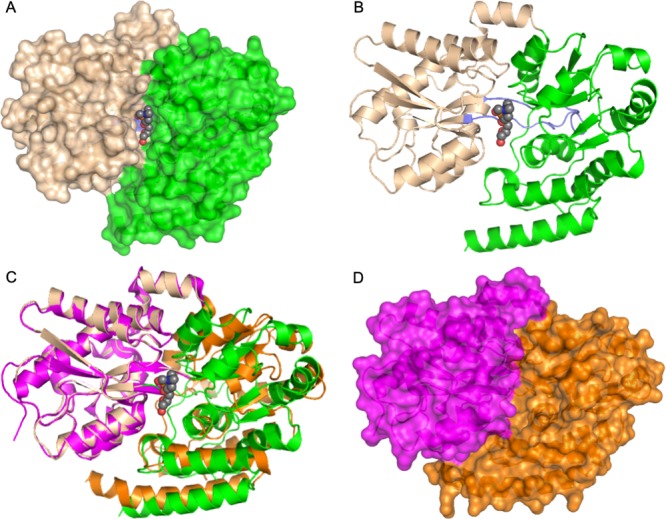
Crystal structure of Mtb UgpB. (A) Surface representation of Mtb UgpB in complex with GPC. The two domains are highlighted: domain I (brown) and domain II (green). The GPC ligand is represented as spheres with dark gray carbon atoms. (B) Cartoon representation of Mtb UgpB in complex with GPC identifying the secondary structure elements. Domain I (brown) and domain II (green). The two hinge regions are highlighted in blue. The GPC ligand is represented as spheres with dark gray carbon atoms. (C) Superposition of domain I of GPC Mtb UgpB cocomplex (brown/green) with domain I of apo-Mtb UgpB (PDB 4MFI) (magenta/orange). (D) Surface representation of the unliganded Mtb UgpB (PDB 4MFI) with the two domains colored magenta (domain I) and orange (domain II).
Ligand-Binding Site of Mtb UgpB
Well-defined electron density for the GPC ligand in all Mtb UgpB molecules within the crystal unit was observed, enabling the GPC ligand to be modeled in the Mtb UgpB binding site (Figure S2A). The GPC ligand is found in an identical position and orientation in each subunit (Figure S2B). Notably, the electrostatic surface shows that GPC is buried in the prominent, acidic interface that is formed between the two domains of UgpB and makes contact with both. The GPC is precisely orientated within the binding cleft such that the glycerol moiety is buried at the base of the cavity, in close proximity to the flexible-hinge region centered around Arg385, while the choline moiety extends outward towards the solvent exposed channel entrance (Figure 2).
Figure 2.

GPC binding site in Mtb UgpB. (A) Illustration showing GPC with dark gray carbon atoms and selected Mtb UgpB amino acid residues in stick representation (colored brown for residues within domain I and green for residues with domain II). (B) Schematic diagram of the interactions of Mtb UgpB with GPC. Dashed lines (black) represent hydrogen bonding, and the thick dotted line (red) represents hydrophobic interactions
The glycerol moiety is located between the side chains of Leu205 and Trp208 from domain II (Figure 2). The ring system of Trp208 lies approximately parallel to the C1, C2, and 2-hydroxy group of the glycerol moiety enabling π-stacking interactions, while Leu205 is orientated perpendicular to this plane and provides additional stabilization. There is an important network of hydrogen bonding interactions that anchors GPC in the binding pocket. The side chain of Asp102, from domain I, is orientated to enable direct hydrogen bonding to both the 1- and 2-hydroxy groups of the glycerol moiety. Two residues that comprise the flexible-hinge linkages are able to directly interact with GPC through the formation of additional hydrogen bond interactions between the side chain of Arg385 and the 1-hydroxy group and the backbone amide nitrogen atom of Gly306 with the 2-hydroxy group, respectively. The direct interaction of these flexible-hinge linkages with the GPC ligand may help to stabilize the UgpB–GPC complex in the closed conformation. The phosphate group of GPC is stabilized through hydrogen bond interactions with the side chains of Tyr78 and Tyr345 (domain I), Ser153 (domain I), Ser272 (domain II), and the backbone amide of Gly306. It is striking that there are no direct or charged interactions between Mtb UgpB and the positively charged choline moiety, though this moiety is well-defined in the electron density.
Comparison with the Binding Site of E. coli UgpB
The comparison with UgpB from E. coli(10) indicates that the overall architecture of these two periplasmic binding proteins in complex with substrate is similar, with a rmsd of 2.1 Å (PDBeFold,15 394 target residues, 25% sequence identity (Figure S3), PDB code 4AQ4), Figure 3. While Mtb was crystallized with GPC, the E. coli protein was crystallized with G3P, which we, as well as previous studies,10 show does not bind to Mtb UgpB. It is interesting to note that the binding mode of the G3P core of GPC resembles the situation found in the E. coli UgpB–G3P complex,10 even though Mtb UgpB is unable to bind or recognize this smaller G3P ligand (Figure 3B). However, while the substrate binding pocket of Mtb UpgB resembles that of E. coli UgpB, there are several important differences. Notably, there are substitutions of critical residues involved in substrate binding. Leu205 is specific to Mtb and is replaced by a larger indole side chain from a tryptophan residue (Trp169) in E. coli UgpB. In addition, Mtb UgpB Asp102 is replaced in E. coli UgpB by a glutamic acid residue (Glu66) (Figure 3C). In this instance, the difference in the length of these acidic side chains may influence substrate selectivity between the different organisms. Intriguingly, while the interaction with an arginine residue is conserved between Mtb and E. coli, the arginine residues in the two proteins originate from different regions of the protein, indicating an evolutionary divergence of these substrate-binding proteins. In addition, a narrowing of the E. coli UgpB binding cleft results from two different loop regions. One loop region (Gly221–Asp230) in domain II of E. coli UgpB linking α-helices 10 and 11 narrows the substrate binding cavity as a result of a 5 Å translational shift. The difference in position of a second loop comprised of residues His8–Gly12 results in the translation of the first α-helix of E. coli UgpB (residues 12–30) located in domain I by approximately 6 Å toward α-helix 11 of domain II, which further narrows the E. coli UgpB substrate binding channel (Figure 3D,E). The comparison of the region at the entrance to the binding cleft reveals an expanded pocket for Mtb UgpB. It is of interest to note that in chain B of Mtb UgpB we observe an additional glycerol molecule located in this expanded pocket that is within 4 Å of the choline moiety of GPC (Figure S4). A glycerol molecule is also present in the E. coli UgpB–G3P complex, though at a different position, indicating that for both proteins the binding pockets are larger than the recognized GPC substrate.10 This may be functionally significant in substrate recognition and have an important role in the accommodation and binding of alternative phosphodiester substrates.
Figure 3.

Comparison of Mtb UgpB with E. coli UgpB. (A) Superposition of the Mtb UgpB GPC complex structure (blue) with E. coli UgpB in complex with G3P (PDB 4AQ4) (brown). Loop regions that differ are highlighted in yellow and magenta. (B) Close-up illustration showing the binding orientation of the GPC ligand and G3P ligand in stick representation (dark gray carbon atoms, GPC; cyan carbon atoms, G3P). (C) Close-up of the overlay of the binding sites of GPC (Mtb) and G3P (E. coli). Selected residues are shown as sticks (Mtb, blue; E. coli, brown), and the font is labeled in black (Mtb) and blue (E. coli). (D) Surface representation of the Mtb UgpB GPC binding pocket with the GPC ligand in stick representation. (E) Surface representation of the E. coli UgpB G3P binding pocket in the same orientation as D with the G3P ligand in stick representation.
Solution Saturation Transfer Difference NMR of Mtb UgpB with Glycerophosphocholine
Given the apparent discrepancy between the lack of interactions formed between the choline moiety and its importance in binding and given that G3P lacking the choline moiety does not bind, we investigated binding in the solution state. We employed saturation transfer difference (STD) NMR to obtain quantitative maps of the ligand–protein complex in solution (Figure 4).21 Binding was detected for GPC, and binding epitope mapping was obtained and analyzed as described in the Methods section.22 The STD NMR signals and the GPC binding epitope and maps obtained are shown in Figure 4. From the epitope map, the glycerol moiety of GPC is identified as the main recognition element showing the highest STD normalized values. In particular, the highest STD intensity values were observed for the protons in positions 1 and 2 (H1G and H2G) of the glycerol moiety (Figure 4A), with slightly lower intensity values for the protons in position 3 (H3G). The STD values decrease from the glycerol moiety to the choline group, indicating that the ligand–protein contacts are closer to the glycerol group than to choline. Intermediate and low STD NMR intensity values were observed for the protons in positions 1 and 2 (H1C and H2C), while low intensity values were observed for the methyl groups from the choline moiety. A quantitative comparison of the NMR solution data with the X-ray structure of the complex was carried out using CORCEMA-ST calculations23 as well as the newly developed method DEEP-STD NMR,24 and the results are summarized in Figure 4. An NOE R-factor25 of 0.25 was obtained when comparing the CORCEMA-ST calculated STD NMR intensities using the crystal structure with the experimentally obtained solution data. This indicates a very good agreement of the complex in the solution state with the crystal structure. In order to probe for additional structural information in the solution state, we then utilized differential epitope mapping by STD NMR (DEEP-STD NMR). This methodology allows us to gain information about the orientation of the ligand within the architecture of the binding site and indirectly gives us information about the type of amino acids (aromatic, polar, or apolar residues) surrounding the ligand in the bound state.26 The DEEP-STD NMR factors clearly identified that the protons in position 3 of the glycerol moiety of GPC are orientated toward aliphatic amino acids, while the protons in position 1 in the choline moiety are oriented toward aromatic residues (Figure 4C). On the basis of the crystal structure of Mtb UgpB, these residues can be mapped to Leu205, Tyr78, and Tyr345, respectively (Figure 2). Notably, our data shows a strong correlation for the molecular determinants of GPC ligand binding to Mtb UgpB in both solution and the solid state.
Figure 4.

STD NMR for Mtb with GPC. (A) Experimental STD build up curves for the GPC/Mtb UgpB complex and the obtained epitope map of GPC/Mtb UgpB. (B) STD values in the red bars were obtained with a 4 s saturation time, while in the blue bars, the CORCEMA-ST calculated STD values from the 3D crystallographic structure of the Mtb UgpB/GPC complex obtained for the same saturation time are shown. RNOE factor, 0.25. (C) Differential epitope (DEEP)-STD factors showing the type of amino acid that the protons of the GPC ligand are orientated toward. Protons orientated toward aliphatic residues are highlighted in blue, and protons orientated toward aromatic residues are highlighted in magenta.
Substrate Specificity of Mtb UgpB
To establish the importance of both the polar headgroup and the glycerol moiety for substrate recognition binding, we analyzed the binding interactions of Mtb UgpB with G3P, the preferred substrate of E. coli UgpB, and phosphocholine by thermal shift analysis and microscale thermophoresis. In contrast to GPC, no binding interactions were observed for these smaller derivatives. Taken together with our structural studies, these results indicate that, while the glycerol moiety is the main recognition element for Mtb UgpB and there are minimal interactions with the polar headgroup, the entire phosphodiester moiety is critical for substrate recognition and binding. The lack of recognition of G3P by Mtb UgpB is consistent with the intracellular location of two putative Mtb glycerophosphodiesterase enzymes (GlpQ1, Rv3842c; GlpQ2, Rv03127c) that are predicted to degrade glyercophosphodiesters to produce G3P and the corresponding alcohol.27,28 In direct contrast, E. coli secretes glycerophosphodiesterase enzymes to enable the extracellular production of G3P, and this is consistent with the ability of the periplasmic E. coli UgpB to recognize the G3P metabolite.12
Our structural studies in both the solid and solution state revealed that the GPC substrate interacts predominantly with Mtb UgpB through interactions with the glycerol backbone. The lack of specific interactions between the protein and the polar choline headgroup located at the entrance of the substrate binding pocket led us to speculate that Mtb UpgB may recognize alternative glycerophosphodiester analogues. To directly investigate the substrate specificity of Mtb UgpB, we used microscale thermophoresis (MST) to analyze the binding interactions of other phosphodiester products formed from the lipolysis of membrane glycerophospholipids (Figure 5). From the substrates tested in each case, we were able to detect binding for GPC, glycerophosphoserine (GPS), glycerophosphoethanolamine (GPE), glycerophosphoinositol (GPI), and glycerophosphoinositol-4-phosphate (GPI4P) (Table 1, Figure 6). The measured Kd value for GPC was consistent with previous results obtained by isothermal titration calorimetry (ITC).9 Notably, Mtb UgpB also binds and recognizes GPE, GPS, GPI, and GPI4P glycerophosphodiesters with binding affinities in the micromolar range (Table 1) with a preference for positively charged polar head groups. Together, this suggests that Mtb has evolved to have a single ABC-transporter to scavenge a range of glycerophosphodiesters within its nutrient poor intracellular environment. The preference for GPC could suggest that, as phosphatidylcholine is the main glycerophospholipid in human lung tissue,29Mtb UgpB has evolved to recognize the most abundant glycerophosphodiester available within the host environment with the potential to recognize and transport a spectrum of additional glycerophosphodiesters, depending on the growth conditions and nutrient availability during intracellular infection that can subsequently be catabolized by Mtb pathways that are involved in polar headgroup recycling.27 Notably, these glycerophospholipids are also major constituents of the Mtb cell envelope,30,31 and further experiments are underway to elucidate whether the glycerophosphodiesters are derived from host- or Mtb-lipids.
Figure 5.

Structure of glycerophosphodiesters and derivatives probed in this study.
Table 1. Binding Data for Mtb UgpBa.
| enzyme | substrate | Kd (μM) | reference |
|---|---|---|---|
| Mtb UgpB | GPC | 3.6 ± 0.5 | this study |
| Mtb UgpB | GPS | 14.9 ± 1.6 | this study |
| Mtb UgpB | GPE | 74.7 ± 13.9 | this study |
| Mtb UgpB | GPI | 1053.2 ± 313.4 | this study |
| Mtb UgpB | GPI4P | 289.8 ± 54.1 | this study |
| Mtb UgpB | G3P | – | this study |
| Mtb UgpB | phosphocholine | – | this study |
| Mtb UgpB Y78A | GPC | – | this study |
| Mtb UgpB Y78A | GPS | – | this study |
| Mtb UgpB Y78A | GPE | – | this study |
| Mtb UgpB D102A | GPC | – | this study |
| Mtb UgpB D102A | GPS | – | this study |
| Mtb UgpB D102A | GPE | – | this study |
| Mtb UgpB Ser153Ala | GPC | 309.8 ± 56.1 | this study |
| Mtb UgpB S153A | GPS | 102.5 ± 16.4 | this study |
| Mtb UgpB S153A | GPE | – | this study |
| Mtb UgpB L205A | GPC | 161.7 ± 15.9 | this study |
| Mtb UgpB L205A | GPE | 1360 ± 210 | this study |
| Mtb UgpB W208A | GPC | – | this study |
| Mtb UgpB S272A | GPC | – | this study |
| Mtb UgpB Y345A | GPC | – | this study |
| Mtb UgpB R385A | GPC | – | this study |
| Mtb UgpB | GPC | 27.3 ± 2.0 | (9) |
| Mtb UgpB | G3P | – | (9) |
| Mtb UgpB | maltose | – | (9) |
| Mtb UgpB L205W | GPC | – | (9) |
| Mtb UgpB L205W | G3P | – | (9) |
| E. coli UgpB | GPC | 5.1 ± 0.3 | (10) |
| E. coli UgpB | G3P | 0.68 ± 0.02 | (10) |
(−) = no binding detected; standard deviations from at least three independent experiments. GPC: glycerophosphocholine; GPS: glycerophosphoserine; GPE: glycerophosphoethanolamine; GPI: glycerophosphoinositol; GPI4P: glycerophosphoinositol-4-phosphate.
Figure 6.
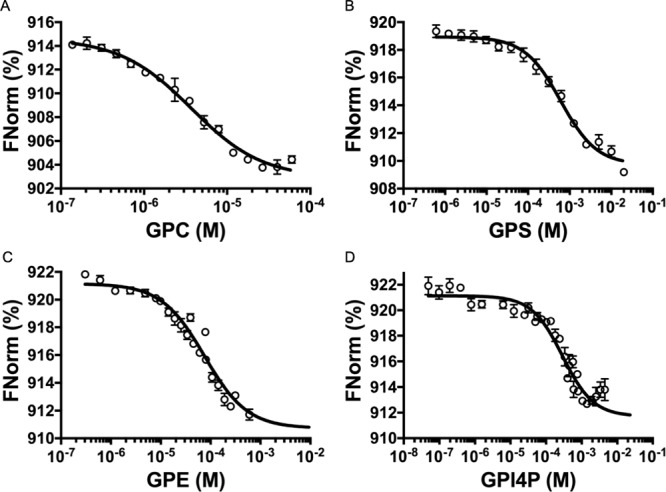
Binding affinities for Mtb UgpB. Binding of (A) GPC, (B) GPS, (C) GPE, and (D) GPI4P to Mtb UgpB measured by microscale thermophoresis (MST). FNorm (%) is the normalized fluorescence signal of the change in MST signal. Error bars represent standard deviations from at least three independent experiments.
As a final evaluation for potential substrate promiscuity, we screened a panel of carbohydrates and amino acids using a thermal shift assay and assessed the binding of putative ligands that resulted in a change in the melting temperature (Tm) of Mtb UgpB, which can be indicative of binding. In total, 37 potential substrates were probed, including trehalose, which is known to be a substrate of the Mtb LpqY-SugABC ABC-transporter,5 and we found that none of the ligands that were screened influenced the melting temperature (Figure S5). It appears that, although Mtb encodes for only five putative carbohydrate importers, each transport system has a defined substrate preference. Interestingly, these data indicate that the substrate binding pocket of Mtb UgpB can efficiently accommodate glycerophosphodiesters, but it is not able to recognize other carbohydrates or amino acids.
STD NMR of Mtb UgpB with GPI4P
Next, to validate some of the MST-binding data, we used STD NMR spectroscopy for a more in-depth investigation of GPI4P binding to Mtb UgpB. Again, the glycerol moiety of GPI4P was the main recognition element with close contacts to Mtb UgpB. High STD NMR intensity values were also observed for the H1 and H2 protons of the inositol ring with intermediate STD NMR values for H3 and H4 protons and low values for H5 and H6 protons (Figure 7A,B). This differs from the situation of the choline headgroup of GPC where instead low STD intensities were observed. Furthermore, the DEEP-STD NMR maps reveal a slight modification in the binding orientation of the glycerol tail of GPI4P compared to GPC as protons in position 3 orientated toward aromatic residues this time. To gain 3D structural insights about this interaction, we carried out docking calculations using Autodock Vina32 followed by validation using CORCEMA-ST calculations. An NOE R-factor of 0.31 was obtained by comparing the CORCEMA-ST calculated STD intensities from the best scored docked structure of GPIP4 bound to Mtb UgpB and the experimental STD values. This indicates a good agreement of the proposed docking structure of the Mtb UgpB/GPIP4 complex with the experimental STD NMR data. From Figure 7, we can observe that the protons in position 3 (H3G) are oriented toward the aromatic residues, which was also determined from DEEP-STD factor analysis. Further, also the protons of inositol–phosphate moiety are in line with the observed orientation from DEEP-STD factor analysis. In fact, protons H4I, H1G, and H2G are oriented toward aliphatic residue Leu205, while protons H1I, H3G, H6I, and H5I are oriented toward the aromatic residues Tyr78 and Tyr345, validating the proposed model structure with the experimental STD and DEEP-STD NMR data. These studies indicate that the size and charge of the glycerophosphodiester headgroup are critical in defining substrate selectivity and the binding orientation of the glycerol tail.
Figure 7.

STD NMR of Mtb UgpB with GPI4P. (A) Experimental STD buildup curve for the Mtb UgpB/GPIP4 complex and the obtained epitope map of GPI4P/Mtb UgpB. (B) Differential epitope (DEEP)-STD factors showing the type of amino acid that the protons of the GPI4P ligand are orientated toward. Protons orientated toward aliphatic residues are highlighted in blue, and protons orientated toward aromatic residues are highlighted in magenta. (C) Docked structure of the GPIP4 in the binding site of Mtb UgpB. GPI4P is in stick representation with the carbon atoms in yellow. The binding orientation of GPC obtained from the crystal structure is shown in stick representation with orange carbon atoms. (D) Close-up overlay of the binding orientations of GPC (cyan carbon atoms) with GPI4P (yellow carbon atoms).
Activity of Sequence Variants
In order to complement our structural studies in both the solution and solid state and assess the significance of individual amino acids that were identified to be important in molecular recognition and binding, we introduced single point mutations in eight individual residues that were suggested to interact with the glycerophosphodiester ligands. In each case, we confirmed that the substituted alanine mutation was not detrimental to the correct folding of the protein by circular dichroism spectroscopy (Figure S6). MST was used to determine the binding affinities of the Mtb UpgB protein with GPC, and complete abrogation of binding was observed when Tyr78, Asp102, Trp208, Ser272, Tyr345, and Arg385 were individually replaced by an alanine, confirming the significance of these residues in substrate selectivity and the importance in binding recognition. In contrast, binding of GPC was still observed when Ser153 and Leu205 were replaced by alanine, with a corresponding 85- and 45-fold reduction in the Kd values, respectively (Table 1), indicating that while these two individual residues are important for binding, they are not critical. Failure of these single-residue mutants to completely abolish binding reflects that multiple amino acids are involved in the interaction with GPC, as observed from the crystal structure. Previous studies that mutated Mtb UgpB Leu205 to a tryptophan residue to mimic the situation found in E. coli UgpB were detrimental for binding of GPC, indicating that the bulky indole side chain cannot be tolerated in Mtb UgpB9 and did not enable recognition of G3P. The distinct glycerophosphodiester recognition of Mtb UgpB compared with E. coli UgpB indicates that the mycobacterial UgpB transporter has evolved to have unique specificity and function that is distinct from other UgpB proteins.
In conclusion, to date, the nutrient requirements of Mtb during infection and the corresponding transport systems have not been fully elucidated. The structural and functional understanding of mycobacterial ABC-transporters that import essential nutrients is an important step to understanding the mechanisms that support intracellular survival. Importantly, we have identified that the essential Mtb UgpABCE importer is linked with glycerophosphodiester uptake with wide substrate selectivity. For the first time, we have established the molecular determinants of the distinct substrate selectivity of the UgpB substrate binding protein from the Mtb pathogen that has important structural and functional differences with E. coli UgpB. We therefore propose a new role for the Mtb UgpABCE transporter in the uptake of glycerophosphodiesters generated from the degradation of membrane phospholipids as a route to scavenge scarce nutrients during intracellular infection.
Methods
Procedures for cloning, protein expression, crystallization, X-ray data collection and refinement, STD NMR experiments, docking, microscale thermophoresis, thermal shift assays, and enzymatic synthesis of substrates in this study are described in the Supporting Information.
Acknowledgments
We would like to thank M. Ahanger for assistance. We thank W. Jacobs (Albert Einstein College of Medicine, USA) for providing expression vector pYUB1062 and the M. smegmatis mc24517 expression system. We acknowledge the contribution of the WPH Proteomics Facility research technology platform, University of Warwick. We thank Diamond Light Source for access to synchrotron beamlines and the staff for support during the experiments. Equipment was supported through the Warwick Integrative Synthetic Biology (WISB) research technology platform (BB/M017982/1). This work was supported by a Sir Henry Dale Fellowship to E.F. jointly funded by the Wellcome Trust and Royal Society (104193/Z/14/Z), a research grant from the Royal Society (RG120405), the MRC for a studentship to J.S.F. (MR/J003964/1), and the EPSRC for funding an Integrate Early Career fellow (J.H.; EP/M027503/1). J.A. and R.N. acknowledge support from the BBSRC through a New Investigator grant awarded to J.A. (BB/P010660/1). We are grateful for the use of the University of East Anglia (UEA) Faculty of Science NMR facility.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00204.
Detailed methods, SDS-PAGE analysis, GPC binding, sequence alignment of UgpB; location of the additional glycerol moiety in the Mtb UgpB binding pocket; thermal shift assay; CD spectra; crystallographic parameters; DynDom analysis; sequence of primers (PDF)
Accession Codes
Coordinates and structure factors for Mtb UgpB have been deposited in the Protein Data Bank under accession code 6R1B.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. (2018) Global Tuberculosis Report 2018, WHO, Geneva, Switzerland. [Google Scholar]
- Getahun H.; Matteelli A.; Chaisson R. E.; Raviglione M. (2015) Latent Mycobacterium tuberculosis infection. N. Engl. J. Med. 372, 2127–2135. 10.1056/NEJMra1405427. [DOI] [PubMed] [Google Scholar]
- Titgemeyer F.; Amon J.; Parche S.; Mahfoud M.; Bail J.; Schlicht M.; Rehm N.; Hillmann D.; Stephan J.; Walter B.; Burkovski A.; Niederweis M. (2007) A genomic view of sugar transport in Mycobacterium smegmatis and Mycobacterium tuberculosis. J. Bacteriol. 189, 5903–5915. 10.1128/JB.00257-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederweis M. (2008) Nutrient acquisition by mycobacteria. Microbiology 154, 679–692. 10.1099/mic.0.2007/012872-0. [DOI] [PubMed] [Google Scholar]
- Kalscheuer R.; Weinrick B.; Veeraraghavan U.; Besra G. S.; Jacobs W. R. Jr. (2010) Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 107, 21761–21766. 10.1073/pnas.1014642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullam E.; Prokes I.; Futterer K.; Besra G. S. (2016) Structural and functional analysis of the solute-binding protein UspC from Mycobacterium tuberculosis that is specific for amino sugars. Open Biol. 6, 160105. 10.1098/rsob.160105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruh N. A.; Troudt J.; Izzo A.; Prenni J.; Dobos K. M. (2010) Portrait of a pathogen: the Mycobacterium tuberculosis proteome in vivo. PLoS One 5, e13938 10.1371/journal.pone.0013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somashekar B. S.; Amin A. G.; Rithner C. D.; Troudt J.; Basaraba R.; Izzo A.; Crick D. C.; Chatterjee D. (2011) Metabolic profiling of lung granuloma in Mycobacterium tuberculosis infected guinea pigs: ex vivo 1H magic angle spinning NMR studies. J. Proteome Res. 10, 4186–4195. 10.1021/pr2003352. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Zhang Q.; Zheng Q.; Zhou H.; Jin J.; Zhou W.; Bartlam M.; Rao Z. (2014) Structural analysis of Mycobacterium tuberculosis ATP-binding cassette transporter subunit UgpB reveals specificity for glycerophosphocholine. FEBS J. 281, 331–341. 10.1111/febs.12600. [DOI] [PubMed] [Google Scholar]
- Wuttge S.; Bommer M.; Jager F.; Martins B. M.; Jacob S.; Licht A.; Scheffel F.; Dobbek H.; Schneider E. (2012) Determinants of substrate specificity and biochemical properties of the sn-glycerol-3-phosphate ATP binding cassette transporter (UgpB-AEC2) of Escherichia coli. Mol. Microbiol. 86, 908–920. 10.1111/mmi.12025. [DOI] [PubMed] [Google Scholar]
- Scheepers G. H.; Lycklama A. N. J. A.; Poolman B. (2016) An updated structural classification of substrate-binding proteins. FEBS Lett. 590, 4393–4401. 10.1002/1873-3468.12445. [DOI] [PubMed] [Google Scholar]
- Larson T. J.; Ehrmann M.; Boos W. (1983) Periplasmic glycerophosphodiester phosphodiesterase of Escherichia coli, a new enzyme of the glp regulon. J. Biol. Chem. 258, 5428–5432. [PubMed] [Google Scholar]
- Grosshennig S.; Schmidl S. R.; Schmeisky G.; Busse J.; Stulke J. (2013) Implication of glycerol and phospholipid transporters in Mycoplasma pneumoniae growth and virulence. Infect. Immun. 81, 896–904. 10.1128/IAI.01212-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton-Vogt J. L.; Henry S. A. (1998) GIT1, a gene encoding a novel transporter for glycerophosphoinositol in Saccharomyces cerevisiae. Genetics 149, 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2256–2268. 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Locher K. P. (2009) Structure and mechanism of ATP-binding cassette transporters. Philos. Trans. R. Soc., B 364, 239–245. 10.1098/rstb.2008.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntsson R. P.; Smits S. H.; Schmitt L.; Slotboom D. J.; Poolman B. (2010) A structural classification of substrate-binding proteins. FEBS Lett. 584, 2606–2617. 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- Hayward S.; Lee R. A. (2002) Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50. J. Mol. Graphics Modell. 21, 181–183. 10.1016/S1093-3263(02)00140-7. [DOI] [PubMed] [Google Scholar]
- Kozlikova B.; Sebestova E.; Sustr V.; Brezovsky J.; Strnad O.; Daniel L.; Bednar D.; Pavelka A.; Manak M.; Bezdeka M.; Benes P.; Kotry M.; Gora A.; Damborsky J.; Sochor J. (2014) CAVER Analyst 1.0: graphic tool for interactive visualization and analysis of tunnels and channels in protein structures. Bioinformatics 30, 2684–2685. 10.1093/bioinformatics/btu364. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. (1999) Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem., Int. Ed. 38, 1784–1788. . [DOI] [PubMed] [Google Scholar]
- Mayer M.; James T. L. (2004) NMR-based characterization of phenothiazines as a RNA binding scaffold. J. Am. Chem. Soc. 126, 4453–4460. 10.1021/ja0398870. [DOI] [PubMed] [Google Scholar]
- Jayalakshmi V.; Krishna N. R. (2002) Complete relaxation and conformational exchange matrix (CORCEMA) analysis of intermolecular saturation transfer effects in reversibly forming ligand-receptor complexes. J. Magn. Reson. 155, 106–118. 10.1006/jmre.2001.2499. [DOI] [PubMed] [Google Scholar]
- Monaco S.; Tailford L. E.; Juge N.; Angulo J. (2017) Differential Epitope Mapping by STD NMR Spectroscopy To Reveal the Nature of Protein-Ligand Contacts. Angew. Chem., Int. Ed. 56, 15289–15293. 10.1002/anie.201707682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Sugar I. P.; Krishna N. R. (1995) A variable target intensity-restrained global optimization (VARTIGO) procedure for determining three-dimensional structures of polypeptides from NOESY data: application to gramicidin-S. J. Biomol. NMR 5, 37–48. 10.1007/BF00227468. [DOI] [PubMed] [Google Scholar]
- Nepravishta R.; Walpole S.; Tailford L.; Juge N.; Angulo J. (2019) Deriving Ligand Orientation in Weak Protein-Ligand Complexes by DEEP-STD NMR Spectroscopy in the Absence of Protein Chemical-Shift Assignment. ChemBioChem 20, 340–344. 10.1002/cbic.201800568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrouy-Maumus G.; Biswas T.; Hunt D. M.; Kelly G.; Tsodikov O. V.; de Carvalho L. P. (2013) Discovery of a glycerol 3-phosphate phosphatase reveals glycerophospholipid polar head recycling in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 110, 11320–11325. 10.1073/pnas.1221597110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza G. A.; Leversen N. A.; Malen H.; Wiker H. G. (2011) Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J. Proteomics 75, 502–510. 10.1016/j.jprot.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Veldhuizen R.; Nag K.; Orgeig S.; Possmayer F. (1998) The role of lipids in pulmonary surfactant. Biochim. Biophys. Acta, Mol. Basis Dis. 1408, 90–108. 10.1016/S0925-4439(98)00061-1. [DOI] [PubMed] [Google Scholar]
- Brennan P. J.; Nikaido H. (1995) The envelope of mycobacteria. Annu. Rev. Biochem. 64, 29–63. 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- Jackson M. (2014) The mycobacterial cell envelope-lipids. Cold Spring Harbor Perspect. Med. 4, a021105. 10.1101/cshperspect.a021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.