

Abstract
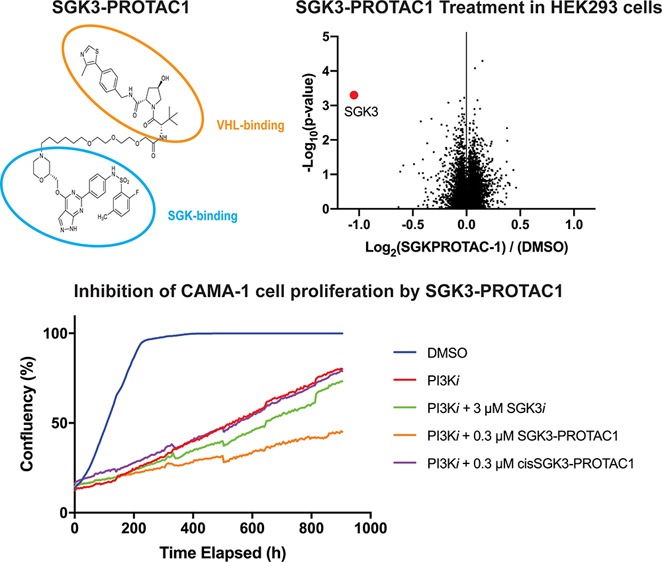
SGK3 is a PX domain containing protein kinase activated at endosomes downstream of class 1 and 3 PI3K family members by growth factors and oncogenic mutations. SGK3 plays a key role in mediating resistance of breast cancer cells to class 1 PI3K or Akt inhibitors, by substituting for the loss of Akt activity and restoring proliferative pathways such as mTORC1 signaling. It is therefore critical to develop tools to potently target SGK3 and obstruct its role in inhibitor resistance. Here, we describe the development of SGK3-PROTAC1, a PROTAC conjugate of the 308-R SGK inhibitor with the VH032 VHL binding ligand, targeting SGK3 for degradation. SGK3-PROTAC1 (0.3 μM) induced 50% degradation of endogenous SGK3 within 2 h, with maximal 80% degradation observed within 8 h, accompanied by a loss of phosphorylation of NDRG1, an SGK3 substrate. SGK3-PROTAC1 did not degrade closely related SGK1 and SGK2 isoforms that are nevertheless engaged and inhibited by 308-R. Proteomic analysis revealed that SGK3 was the only cellular protein whose cellular levels were significantly reduced following treatment with SGK3-PROTAC1. Low doses of SGK3-PROTAC1 (0.1–0.3 μM) restored sensitivity of SGK3 dependent ZR-75-1 and CAMA-1 breast cancer cells to Akt (AZD5363) and PI3K (GDC0941) inhibitors, whereas the cis epimer analogue incapable of binding to the VHL E3 ligase had no impact. SGK3-PROTAC1 suppressed proliferation of ZR-75-1 and CAMA-1 cancer cell lines treated with a PI3K inhibitor (GDC0941) more effectively than could be achieved by a conventional SGK isoform inhibitor (14H). This work underscores the benefit of the PROTAC approach in targeting protein kinase signaling pathways with greater efficacy and selectivity than can be achieved with conventional inhibitors. SGK3-PROTAC1 will be an important reagent to explore the roles of the SGK3 pathway.
The PI3K pathway orchestrates vital cellular processes including metabolism, insulin signaling, and protein synthesis as well as proliferation and growth.1 Hyperactivating mutations in components of the class I PI3K family (p110α, p110β, p110γ, and p110δ) are harbored in the majority of human cancers and drive proliferation and survival of tumors.2 A key downstream component of the class 1 PI3K pathway are isoforms of the serum and glucocorticoid-induced protein kinases (SGK1, SGK2, and SGK3) that are activated by PDK1 and mTORC2.3−5 The kinase domains of SGK isoforms are highly related to intensely studied Akt isoforms that are also activated downstream of class 1 PI3K signaling via the PDK1 and mTORC2 kinases. SGK and Akt isoforms regulate cellular processes by phosphorylating a myriad of overlapping substrates at Ser/Thr residues lying within RXRXXT/S substrate recognition motifs.6,7
SGK3 is the only isoform that possesses an N-terminal phox homology (PX) domain which interacts with high affinity and specificity to PtdIns(3)P, generated by the class III PI3K (hVPS34) at the endosome.8−10 Binding PtdIns(3)P promotes the phosphorylation and activation of SGK3 by PDK1 and mTORC2 kinases.9 In addition, SGK3 can also be activated downstream of class 1 PI3K through a pathway involving activation of mTORC2 and sequential dephosphorylation of PtdIns(3,4,5)P3 to PtdIns(3)P.8 In contrast, SGK1 and SGK2 isoforms lack a phosphoinositide binding domain and are therefore activated in the cytosol downstream of class 1 PI3K through its activation of mTORC2, triggering PDK1 phosphorylation.4,11 Unlike SGK3, Akt isoforms possess an N-terminal PtdIns(3,4,5)P3 binding PH domain. Activation of class 1 PI3K generates PtdIns(3,4,5)P3 at the plasma membrane that in turn promotes recruitment and phosphorylation of Akt isoforms by PDK1 and mTORC2.
Prolonged treatment of various ER+ breast cancer cell lines with class 1 PI3K or Akt inhibitors leads to upregulation and activation of SGK3 through the hVPS34 pathway.12 Under these conditions, SGK3 substitutes for Akt by phosphorylating substrates such as TSC2 to activate mTORC1.12 Moreover, a combination of Akt and SGK protein kinase inhibitors induced a more marked regression of BT-474 breast cancer cell-derived tumors in a xenograft model than observed with Akt inhibitors alone.12 These data support the notion of targeting SGK3 as a therapeutic strategy for counteracting resistance to PI3K/Akt inhibition in cancer treatment. A number of ATP competitive inhibitors that target all SGK isoforms with similar affinity have been reported.13−15 Due to the high homology of their SGK catalytic domains, it has not been possible to elaborate inhibitors that display isoform specificity.16 These compounds could have less toxicity for treating cancer resistance than inhibitors targeting all isoforms.
Proteolysis targeting chimeras (PROTACs) are heterobifunctional small molecules designed to induce rapid proteasome-mediated degradation of a protein of interest.17 They consist of a ligand that binds to the protein of interest, joined via a short linker sequence to an E3 ligase recruitment moiety.18,19 A key advantage of PROTACs is that they can be deployed at much lower doses than conventional inhibitors due to their substochiometric catalytical mode of action efficiently degrading target proteins, minimizing side effects.20−22 The PROTAC approach reduces intracellular protein levels much more rapidly than is achievable with genetic methodologies, which can present other challenges such as lethality or genetic compensation.23 Additionally, PROTACs can be used reversibly and have been demonstrated to display exquisite isoform or paralog specificity that is challenging to achieve by pan-selective inhibitors.21,24−26 A range of PROTAC tool compounds has recently been developed targeting protein kinases, for example, against RIPK2,20 BCR-ABL,27,28 CDK9,29 and PTK2.30,31 As recently reviewed by Ferguson and Gray, PROTACs can evade issues with conventional chemical inhibitors in targeting oncogenic kinases32 and allow targeting both kinase-dependent and kinase-independent protein functions. Targeting of protein kinases for degradation has also allowed for greater isoform specificity, for example, the production of a CDK6-specific PROTAC from a CDK4/6 inhibitor.33
In this study, we describe the optimization and characterization of an SGK3-specific PROTAC termed SGK3-PROTAC1. This compound is a highly selective degrader, targeting for degradation only SGK3 and not the related SGK1 or SGK2 isoform. SGK3-PROTAC1 induces proteasomal-mediated degradation of SGK3 at submicromolar concentrations in a panel of cancer cell lines rendering breast cancer cells more sensitive to PI3K and Akt inhibitors. SGK3-PROTAC1 represents a novel chemical tool to better probe the biological roles of the SGK3 protein kinase.
Results and Discussion
Elaboration of DAT1, a First-Generation SGK3 PROTAC
As no cocrystal structure of the SGK1–3 inhibitors has been disclosed, we inspected the structure–activity relationship (SAR) of a series of structurally related SGK inhibitors reported by Sanofi13 (International Patent WO2014140065), with the aim to identify strategies to elaborate PROTACs. From this series, we have previously characterized compound 14H as an inhibitor of SGK3 and shown that inhibition of SGK3 kinase activity with this compound can reduce PI3K-Akt inhibitor resistant cell growth.12 However, this compound lacks isoform specificity and also has some potency against S6K1. Inhibitor 14H possesses an IC50 of 4 nM for SGK3, 10 nM for SGK1, and 76 nM for S6K1.12 A series of inhibitors that have a pyrazolopyrimidine scaffold appeared to tolerate aliphatic and cyclic substituents at position 4 of the pyrazolopyrimidine core, suggesting that such a portion of the molecule could be solvent exposed. Two SGK inhibitors, termed 290 and 308 (Figure 1B), were judged particularly amenable for linker conjugation, as the morpholine ring can be selectively N-alkylated by means of reductive amination protocols. As the inhibitors described in the patent were racemic, we synthesized both the R and S enantiomers of these compounds and determined by kinase screening that the R forms were marginally more potent (data available at http://www.kinase-screen.mrc.ac.uk/kinase-inhibitors). We determined that the IC50s of these compounds for SGK1 and SGK3 were between 5 and 40 nM (Table 1). The specificity of these compounds was profiled against 140 kinases at a 1 μM concentration, revealing that they were relatively selective (Table S1), with S6K1 being a key off target that was inhibited more potently than SGK isoforms, with an IC50 of 1–10 nM (Table 1).
Figure 1.

Design and cellular activity of first generation SGK3 PROTACs. (A) Structure of compound 14H, previously published by Sanofi as an inhibitor of SGK3. (B) Starting material for SGK PROTACs. PROTACs were derived from SGK inhibitors 308-R or 290-R linked to either VH032 or pomalidomide to target the VHL or cereblon E3 ligases, respectively. (C) Structures of first generation SGK PROTACs. SGK and E3 ligase targeting motifs were joined by a 3xPEG linker to produce the PROTACs. (D) HEK293 cells were treated for 48 h with increasing concentrations of each PROTAC compound from 0.1 nM to 10 μM. Cell lysates were subjected to immunoblot analysis with the indicated antibodies, and SGK3 protein levels were quantified in Image Studio Lite software (Licor).
Table 1. IC50 Values and Degradation Efficiency of SGK Inhibitors and First Generation PROTACsa.
| SGK3 IC50 (nM) | SGK1 IC50 (nM) | S6K1 IC50 (nM) | SGK3 degradation efficiency | |
|---|---|---|---|---|
| 290-R | 35 | 19 | 10 | |
| 308-R | 5 | 10 | 1 | |
| DAT1 | 440 | 1600 | 160 | + |
| DAT2 | 190 | 400 | 180 | – |
| DAT3 | 640 | 1000 | 240 | – |
| DAT4 | 540 | 600 | 190 | – |
For the first generation of SGK PROTAC molecules, we linked 290-R and 308-R to the well characterized VHL ligand VH03234,35 and to the cereblon ligand pomalidomide.36 A medium length linker composed of three PEG units was used in the first instance (Figure 1B and C). We next tested whether the resulting compounds reduced endogenous SGK isoform or S6K1 expression when administered to HEK293 cells for 48 h at concentrations of up to 10 μM (Figure 1D). After 48 h of PROTAC treatment, cell lysates were analyzed by immunoblot analysis, and protein expression under each condition was quantified relative to the DMSO-only control. Only one of the four compounds, in which 308-R was conjugated to the VHL ligand VH032 (termed DAT1), markedly reduced SGK3 expression. Levels of SGK1, SGK2, or S6K1 were not significantly impacted (Figure 1D). DAT1 reduced SGK3 expression by 60% at 1 μM and 75% at 10 μM after 48 h (Figure 1D). Treatment of 1 μM DAT1 maximally reduced SGK3 expression by 16 h (Figure S1A). A dose response analysis revealed that 2 μM DAT1 maximally reduced SGK3 expression at an 8 h time point (Figure S1B). As expected, inhibition of CUL2 neddylation by means of the inhibitor MLN4924 (3 μM) or proteasome inhibition (MG132, 50 μM) blocked DAT1-induced degradation of SGK3 (Figure S1C). This suggests that DAT1-mediated degradation of SGK3 is neddylation and proteasome dependent.
Conjugation of 308-R to VH032 or pomalidomide markedly decreased the inhibitory activity for SGK isoforms and S6K1 in biochemical kinase assays (5 nM to 440 nM for SGK3 and 1 nM to 160 nM for S6K1; Table 1). We found no correlation between the potency of PROTACs for inhibiting SGK3 and the ability to induce degradation of SGK3 in cells. For example, DAT2, which does not impact SGK3 protein levels in HEK293 cells, inhibited SGK1 and SGK3 2–4-fold more potently than DAT1.
Elaboration of DAT8 (SGK3-PROTAC1), a Second Generation SGK3 PROTAC
We next generated further analogues of DAT1 by shortening the linker length from three to two PEG units (DAT5) or extending the linker length to four and five PEG units (DAT6,7; Figure 2A). The ability of each of these compounds to decrease SGK3 expression was evaluated in HEK293 cells (Figure 2C). We found that the optimal linker length was four PEG units (13 atoms), with DAT6 displaying significant improvements both in terms of potency and the amount of reduction of SGK3 observed compared to the original DAT1 (Figure 2C). We then changed the composition of the linkers for more lipophilic alkylic moieties of 13 (DAT8) and 16 atoms (DAT9,10), and for this set of compounds we also conjugated the inhibitor 290-R (DAT11,12; Figure 2A). The best compound was DAT8, which we have renamed SGK3-PROTAC1 (Figure 2D). SGK3-PROTAC1 possesses three oxygen atoms in the linker motif, the same number as DAT1, but one less than DAT6 (Figure 2A). At a concentration of 0.1 μM, SGK3-PROTAC1 reduced SGK3 levels by 65% without effecting SGK1, SGK2, or S6K1 (Figure 2D). At higher concentrations of 1–10 μM, moderate degradation of S6K1 was also observed. SGK3-PROTAC1-mediated degradation of SGK3 was prevented by the neddylation inhibitor MLN4924 as well as the MG132 proteasome inhibitor (Figure S2). In biochemical assays, SGK3-PROTAC1 inhibited SGK3 with an IC50 of 300 nM and S6K1 with an IC50 of 1800 nM (Table 2). As before, little correlation was observed between compound IC50 and degradation efficiency. The IC50 of SGK3-PROTAC1 was similar to that of DAT1 (300 from 440 nM), whereas the IC50 against S6K1 increased approximately 10-fold (0.16 μM to 1.8 μM). The specificity of SGK3-PROTAC1 at 1 μM was assessed in a panel of 140 kinases. This revealed a surprising increase in specificity over the original 308-R compound, with SGK1 and S6K1 the only kinases most potently inhibited by this compound in vitro (Table S1).
Figure 2.

Design and cellular evaluation of second and third generation SGK PROTACs. (A) Chemical structure of compounds DAT5–12, expanding upon DAT1. (B) Chemical structure of cisSGK3-PROTAC1. (C) HEK293 cells were treated for 48 h with increasing concentrations of each PROTAC compound from 1 nM to 10 μM. Cell lysates were subjected to immunoblot analysis with the indicated antibodies, and SGK3 protein levels quantified in Image Studio Lite software. (D) HEK293 cells were treated for 48 h with increasing concentrations of each PROTAC compound from 1 nM to 10 μM. Cell lysates were subjected to immunoblot analysis with the indicated antibodies, and SGK3 and S6K1 protein levels were quantified in Image Studio Lite software.
Table 2. IC50 Values and Degradation Efficiency of Second Generation of SGK PROTACsa.
| SGK3 IC50 (μM) | SGK1 IC50 (μM) | S6K1 IC50 (μM) | SGK3 degradation efficiency | |
|---|---|---|---|---|
| DAT1 | 0.44 | 1.6 | 0.16 | + |
| DAT5 | >10 | >10 | >10 | + |
| DAT6 | >10 | >10 | >10 | ++ |
| DAT7 | >10 | >10 | >10 | ++ |
| DAT8 (SGK3-PROTAC1) | 0.3 | 0.22 | 1.8 | ++++ |
| cisSGK3-PROTAC1 | 0.6 | 1.4 | 1.7 | – |
| DAT9 | 1.79 | 8.99 | 4.53 | ++ |
| DAT10 | >10 | >10 | >10 | ++ |
| DAT11 | 0.73 | 1.34 | 1.80 | +++ |
| DAT12 | >10 | >10 | >10 | ++ |
Characterization of SGK3-PROTAC1
To generate a control compound for SGK3-PROTAC1 that would bind and inhibit SGK3, but not induce recruitment of the CUL2-VHL E3 ligase complex, we synthesized a version of SGK3-PROTAC1 termed cisSGK3-PROTAC1 that contains a hydroxyl epimer of the VH032 moiety (Figure 2B). Previous work has shown that this epimer ablates binding to VHL.20,21,35 As expected, in biochemical assays, cisSGK3-PROTAC1 inhibited our panel of kinases similarly to SGK3-PROTAC1 (Table S1). We also undertook IC50 measurements of the cisSGK3-PROTAC1 against SGK3, SGK1, and S6K1 and found that values were very similar to SGK3-PROTAC1 (Table 2). Cellular degradation assays confirmed that cisSGK3-PROTAC1 failed to induce degradation of SGK3 even at concentrations of up to 3 μM under conditions in which 0.1 μM SGK3-PROTAC1 markedly reduced SGK3 expression (Figure 3A). At doses of up to 1 μM SGK3-PROTAC1, no degradation of S6K1 was observed.
Figure 3.

Characterization of cellular activities of SGK3-PROTAC1 and cisSGK3-PROTAC1. (A) HEK293 cells were first treated for 8 h with 0.03–3 μM SGK3-PROTAC1 and cisSGK3-PROTAC1. One hour before lysis, cells were treated with 3 μM AZD5363. Lysates were subjected to immunoblot analysis with the indicated antibodies. (B) HEK293 cells were treated in the presence or absence of AZD5363 (3 μM), with SGK3-PROTAC1 (0.3 μM), cisSGK3-PROTAC1 (0.3 μM), 14H (1 μM), or 308-R (1 μM) for 8 h prior to lysis. Lysates were subjected to immunoblot analysis with the indicated antibodies. (C) HEK293 cells were treated for up to 48 h with SGK3-PROTAC1 (0.3 μM), and lysates were analyzed by immunoblot analysis using the indicated antibodies. (D) HEK293 cells were treated for 24 h with SGK3-PROTAC1. Cells were washed three times with DMEM to wash out the compound, and recovery of SGK3 expression protein was analyzed by immunoblot analysis. (E–G) As in A except that CAMA-1 (E), ZR-75-1 (F), and JIMT-1 (G) cells were employed rather than HEK293 cells.
To study the impact of SGK3-PROTAC1 on phosphorylation of a physiological substrate, we monitored phosphorylation of NDRG1 at Thr346.37 This site is also phosphorylated by Akt; therefore we treated cells with increasing doses of SGK3-PROTAC1 and cisSGK3-PROTAC1 for 8 h, with the addition of an Akt inhibitor (3 μM AZD5363) 1 h before lysis, to remove the impact of Akt phosphorylation of NDRG1. Concentrations of 0.1 μM SGK3-PROTAC1 reduced NDRG1 phosphorylation under conditions where cisSGK3-PROTAC1 had no effect (Figure 3A). Concentrations of cisSGK3-PROTAC1 of above 1 μM were required to lower NDRG1 phosphorylation. Under basal conditions with no inhibition of Akt, targeting SGK3 with either 0.3 μM SGK3-PROTAC1 or conventional SGK kinase inhibitors (1 μM 14H or 1 μM 308-R) for 8 h had minimal effect on pNDRG1 phosphorylation (Figure 3B). In the presence of the Akt inhibitor (3 μM AZD5363), treatment with 0.3 μM SGK3-PROTAC1 or 1 μM 14H or 1 μM 308-R markedly suppressed NDRG1 phosphorylation (Figure 3B). This is consistent with previous work in HEK293 cells showing that inhibition of both Akt and SGK3 is required to block NDRG1 phosphorylation.8,37
A time course analysis of 0.3 μM SGK3-PROTAC1 revealed 50% degradation within 2 h, with maximum degradation after 8 h (Figure 3C). This degradation is also reversible, as washout of SGK3-PROTAC1 after 24 h of treatment resulted in increased SGK3 expression after 1 h, with protein levels returning to normal levels after 10 h (Figure 3D).
We also examined the impact of SGK3-PROTAC1 and cisSGK3-PROTAC1 in two SGK3 dependent breast cancer cell lines, CAMA-1 (Figure 3E) and ZR-75-1 (Figure 3F).12 In these cells, concentrations of >0.1 μM SGK3-PROTAC1 induced degradation of SGK3, but not SGK1 or S6K1. cisSGK3-PROTAC1 had no impact. To confirm that SGK3-PROTAC1 does not induce degradation of endogenous SGK1, we studied a breast cancer cell line termed JIMT1 that has been shown previously to possess high levels of endogenous SGK1.37 In these cells, concentrations of up to 3 μM SGK3-PROTAC1 failed to induce degradation of SGK1, under conditions where SGK3 expression was reduced (Figure 3G).
Striking Specificity of SGK3-PROTAC1
To establish the specificity of SGK3-PROTAC1 employing an unbiased approach, we performed quantitative Tandem–Mass–Tag (TMT)-labeled global proteomic analysis of HEK293 cells treated in the presence or absence of 0.3 μM SGK3-PROTAC1 compared to either 0.3 μM cisSGK3-PROTAC1 or DMSO for 8 h. Experiments were undertaken in triplicate, and analysis in Proteome Discoverer v2.2 using the Mascot search engine allowed relative quantification of 8766 proteins. This analysis revealed that SGK3-PROTAC1 was remarkably selective with only SGK3 expression being significantly reduced (p value <10–3; Figure 4 and Table S2).
Figure 4.
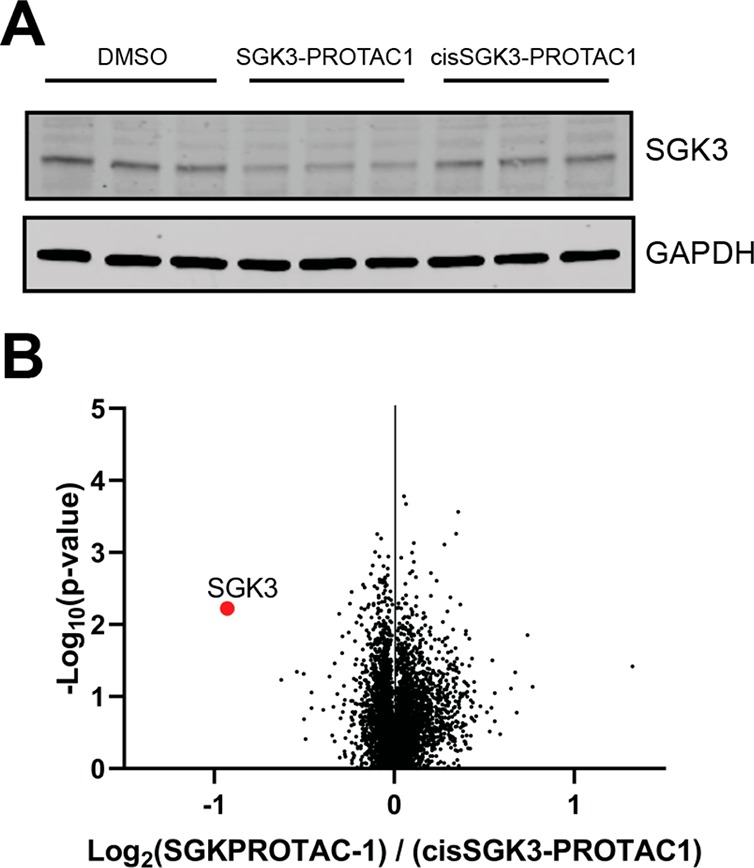
TMT proteomic analysis of HEK293 cells treated with SGK3-PROTAC1. (A) Cells were treated with DMSO (control), SGK3-PROTAC1 (0.3 μM), or cisSGK3-PROTAC1 (0.3 μM) for 8 h and lysed. Lysates were subjected to immunoblot analysis with the indicated antibodies. (B) Lysates were examined by quantitative proteomics. Volcano plot demonstrating global proteomic changes induced by SGK3-PROTAC1 treatment versus cisSGK3-PROTAC1 in HEK23 cells.
Effect of SGK3-PROTAC1 on SGK3-Dependent mTORC1 Activation
Previous work has demonstrated that prolonged treatment of breast cancer cell lines such as CAMA-1 and ZR-75-1 with PI3K or Akt inhibitors resulted in upregulation of SGK3, leading to the activation of mTORC1 signaling, mediated by SGK3 phosphorylating TSC2 at the same sites as Akt.12 We therefore aimed to investigate the effect that degradation of SGK3 would have on this resistance pathway and whether SGK3-PROTAC1 treatment could reverse inhibitor resistance. To investigate the effect of SGK3-PROTAC1 under these conditions, we treated CAMA-1 (Figure 5A,C) or ZR-75-1 (Figure 5B,D) for 5 days with either a class 1A PI3K inhibitor (GDC0941 1 μM;38Figure 5A,B) or an Akt inhibitor (AZD5363, 1 μM;39Figure 5C,D). These cells were treated with SGK3-PROTAC1 or cisSGK3-PROTAC1 (0.3 μM) for either 5 days in combination with the inhibitors or 8 h before lysis. SGK3-PROTAC1 substantially reduced SGK3 expression in both the CAMA-1 and ZR-75-1 cells. Consistent with SGK3-PROTAC1 blocking mTORC1 activation, we found that it also suppressed mTORC1-mediated phosphorylation of S6K1 at Thr389, resulting in a moderate reduction in S6 protein phosphorylation. In contrast, cisSGK3-PROTAC1 had no significant effect on SGK3 protein level or phosphorylation of TSC2, S6K1, or S6 protein. Moreover, we also observed that, in both CAMA-1 and ZR-75-1 cells, SGK3-PROTAC1 inhibited mTORC1 signaling to a greater extent than an SGK inhibitor that does not induce degradation of SGK3 (14H 1 μM;13Figure 5).
Figure 5.

SGK3-PROTAC1-mediated degradation of SGK3 inhibits Akt-independent activation of mTORC1 in cancer cell lines treated with Akt or PI3K inhibitors. CAMA-1 (A, C) or ZR-75-1 (B, D) were treated for 5 days with 1 μM GDC0941 (A, B) or 1 μM AZD5363 (C, D). Cells were treated with the compounds indicated for either 5 days or 8 h before lysis; cells were treated with the compounds indicated. Cell lysates were subject to immunoblot analysis with the indicated antibodies.
Effect of SGK3-PROTAC1 on Proliferation of CAMA-1 and ZR-75-1 Cells Treated with PI3K-Akt Pathway Inhibitors
Given the impact of SGK3-PROTAC1 treatment on Akt-independent mTORC1 activation, we expected to observe a similar effect of SGK3 degradation on cell proliferation in the context of PI3K-Akt pathway inhibitors. We therefore studied the effect that SGK3-PROTAC1 and cisSGK3-PROTAC1 had on the growth of CAMA-1 (Figure 6A,C) or ZR-75-1 (Figure 6B,D) cells in the absence or presence of the class 1A PI3K inhibitor (GDC0941, 1 μM; Figure 6A,B) or Akt inhibitor (AZD5363, 1 μM; Figure 6C,D). Treatment of CAMA-1 or ZR-75-1 cells with SGK3-PROTAC1 alone had no effect on growth (Figure S3), consistent with previous work undertaken with high doses of conventional SGK inhibitors.12,40 As expected, treatment with GDC0941 or AZD5363 substantially reduced growth of these cells (Figure 6). Including SGK3-PROTAC1 (0.3 μM) further reduced the growth of these cells under conditions where cisSGK3-PROTAC1 (0.3 μM) had a minimal effect (Figure 6). We also studied the effect of the conventional SGK3 inhibitor (14H, 3 μM) on the growth curves, in order to compare the effects of degradation and kinase inhibition. As reported previously,12 treatment with 14H further suppressed cell growth in the context of PI3K class I or Akt inhibition. In the presence of the PI3K inhibitor (GDC0941 1 μM), SGK3-PROTAC1 (0.3 μM) inhibited cell growth to a greater extent than treatment with 14H (3 μM; Figure 6A,B). When combined with the Akt inhibitor (AZD5363 1 μM), 14H (3 μM) and SGK3-PROTAC1 (0.3 μM) suppressed growth to a similar extent (Figure 6C,D).
Figure 6.

SGK3-PROTAC1-mediated degradation of SGK3 further inhibiting the growth of cancer cell lines treated with PI3K-Akt pathway inhibitors. CAMA-1 (A, C) and ZR-75-1 (B, D) were treated with compounds as indicated either as monotherapy or in combination, and confluency was measured on the Incucyte S3 every 4 h for up to 4 weeks.
Conclusion
We describe the design and elaboration of a potent, highly specific PROTAC targeted against the SGK3 protein kinase. SGK3-PROTAC1 induced potent degradation within 2 h, with a DC50 below 100 nM. Quantitative mass spectrometry revealed degradation of SGK3 to be highly selective, with SGK3 being the most downregulated protein. It is particularly interesting that SGK3-PROTAC1 was revealed to be remarkably specific for SGK3 and does not degrade the highly related SGK1 and SGK2 isoforms or any other protein in HEK293 cells. This was even observed in cell lines such as JIMT-1 which express high levels of SGK1 and relatively low levels of SGK3. Recent studies attempting to develop isoform specific chemical inhibitors of SGK3 have been unsuccessful, due to the high structural similarity and sequence identity of the SGK1 and SGK3 kinase domains, in particularly in the ATP binding site.16 This ability to generate selective isoform specific degraders has previously been observed in PROTACs derived from the pan-BET inhibitor JQ121,24,41 and PROTACs derived from the promiscuous kinase inhibitor Foretinib42 or TAE-684.43 It has recently been shown that specificity and potency of PROTACs can be dictated by the differential cooperativity and stability of ternary complexes that form protein–protein contacts in relatively less conserved regions outside the width of the active site of the target protein19,24,44 as well as the geometry/orientation of the recruited E3 ligase.45 Future work will investigate the extent to which these molecular recognition features contribute to the exquisite selectivity of SGK3-PROTAC1 for SGK3 induced degradation. Measurement of the stability of the ternary complexes of SGK3 PROTAC compounds with SGK/S6K isoforms and the VHL E3 ligase would be important to better understand the potency and specificity of these PROTACs.44 Furthermore, it would be interesting to explore the effects that SGK3 PROTAC degraders have on selective SGK3 substrates such as STX7 and STX12 that have recently been described that are not phosphorylated by Akt isoforms.46 The finding that growth of SGK3 dependent cancer cell lines is suppressed more efficiently by SGK3-PROTAC1 than achieved by the 14H non-PROTAC inhibitor provides a further example of the benefit of the PROTAC approach in targeting protein kinase signaling pathways with greater efficacy and selectivity than can be achieved with conventional inhibitors. Other examples include the recent finding that a BCR-ABL degrader displays more sustained inhibition of chronic myelogenous leukemia cell growth than can be achieved by a conventional ABL kinase inhibitor.28 SGK3-PROTAC1 will be an important addition to our armory of chemical probes to decipher the biological roles of the SGK3 signaling pathway including in mediating resistance to PI3K and Akt inhibitor therapy in cancer.
Methods
Biology
Materials
Triton X-100, EDTA, EGTA, sodium orthovanadate, sodium glycerophosphate, sodium fluoride, sodium pyrophosphate, 2-mercaptoethanol, sucrose, benzamidine, Tween 20, Tris-HCl, and sodium chloride were from Sigma. Tissue culture reagents, Novex 4–12% Bis–Tris gels, and NuPAGE LDS sample buffer were from Invitrogen.
Cell Culture and Lysis
ZR-75-1, CAMA-1, and JIMT-1 cell lines were sourced as described previously.39 HEK293 cells were purchased from the American Tissue Culture Collection and cultured in DMEM supplemented with 10% (v/v) fetal bovine serum, 2 mM l-glutamine, 100 U/mL penicillin, and 0.1 mg mL–1 streptomycin. The cells were lysed in a buffer containing 50 mM Tris–HCl (pH 7.5), 1 mM EGTA, 1 mM EDTA, 1% (v/v) Triton X-100, 1 mM sodium orthovanadate, 50 mM NaF, 5 mM sodium pyrophosphate, 0.27 M sucrose, 10 mM sodium 2-glycerophosphate, 0.2 mM phenylmethylsulfonyl fluoride, and 1 mM benzamidine. Lysates were clarified by centrifugation at 16 000g for 10 min at 4 °C. Protein concentration was calculated using the Bradford assay (Thermo Scientific).
Cell Treatments and Immunoblot
All cell treatments were carried out as described in figure legends, to a final DMSO concentration of 0.1% (v/v). Lysates were quantified. Immunoblotting was performed using standard procedures, described in brief below. Lysate concentration was quantified by Bradford Assay, and 10 μg of lysate was loaded in the LDS sample buffer for SDS-PAGE electrophoresis on Novex 4–12% Bis–Tris gels. Proteins were electrophoretically transferred onto nitrocellulose membranes (Amersham Protran 0.45 μm NC; GE Healthcare) at 80 V for 80 min on ice in transfer buffer. Transferred membranes were blocked with 5% (w/v) nonfat dry milk dissolved in TBS-T [20 mM Tris/HCl, pH 7.5, 150 mM NaCl, and 0.1% (v/v) Tween 20] at RT for 30 min, before incubation with the primary antibody overnight at 4 °C. The signal was produced with near-infrared secondary antibodies and detected using a Licor Biosciences Odyssey System, and the signal was quantified in Image Studio Lite.
Antibodies
The following antibodies were raised in sheep, by the MRC–PPU Reagents and Services team (https://mrcppureagents.dundee.ac.uk/) and affinity purified against the indicated antigens: anti-SGK3 (S848D, sixth bleed; raised against human SGK3 PX domain comprising residues 1–130 of SGK3; DU2034), anti-S6K1 (S417B, second bleed; raised against residues 25–44 of human S6K1: AGVFDIDLDQPEDAGSEDEL), anti-SGK1 (S062D, third bleed), anti-NDRG1 (S276B third bleed; raised against full-length human NDRG1; DU1557), anti-Akt1 (S695B, third bleed; raised against residues 466–480 of human Akt1: RPHFPQFSYSASGTA).
Anti-GAPDH was from Santa-Cruz (sc-32233). Antiphospho-Akt Ser473 (#9271), antiphospho-NDRG1 Thr346 (#5482), antiphospho-TSC2 Ser939 (#3615), anti-TSC2 (#3612), antiphospho-S6K1 Thr389 (#9205), antiphospho-rpS6 Ser240/244 (#2215), and anti-rpS6 (#2217) antibodies were purchased from Cell Signaling Technology. Total anti-SGK antibody was from Sigma (#5188).
Secondary antibodies coupled to IRDye680LT or IRDye800CW were obtained from Licor Biosciences. Secondary antibodies coupled to horseradish peroxidase (HRP) were obtained from Thermo Scientific. Secondary antibodies coupled to Alexa Fluor 488 and Alexa Fluor 594 were obtained from Thermo Scientific.
Protein Kinase Profiling
Protein kinase profiling against a Dundee panel of 140 protein kinases was undertaken at the International Centre for Protein Kinase Profiling. The result for each kinase was presented as a mean kinase activity of the reaction taken in triplicate relative to a control sample treated with DMSO. Assay conditions and abbreviations are available at http://www.kinase-screen.mrc.ac.uk.
IC50 determination was performed at the MRC PPU International Centre for Protein Kinase Profiling, according to the protocol previously described47,48
Determination Cell Growth in Vitro
For growth assays, ZR-75-1 cells were seeded in 96-well plates at a density of 6000 cells/well and left to adhere overnight. Cells were then treated with compounds as described in the figure legends and imaged every 4 h on the Incucyte S3 (Essen Bioscience) for up to 4 weeks to give a measure of cell confluency. Media were refreshed every 4–5 days.
Supplementary Methods
Full chemistry and mass spectrometry methods are provided in the Supporting Information
Acknowledgments
We thank the excellent technical support of the MRC–Protein Phosphorylation and Ubiquitylation Unit (PPU) DNA Sequencing Service, the MRC PPU tissue culture team (coordinated by E. Allen), and MRC PPU Reagents and Services antibody purification teams (coordinated by H. McLauchlan and J. Hastie).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00505.
Accession Codes
The Mass spectrometry proteomics data have been deposited to the ProteomeXchange consortium via the PRIDE partner repository with the data set identifier PXD013779.
This work was supported by the Medical Research Council [grant number MC_UU_12016/2 (to D.R.A.)]; the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP7/2007–2013) as a Starting Grant to A.C. [grant agreement No. ERC–2012–StG–311460 DrugE3CRLs]; and the pharmaceutical companies supporting the Division of Signal Transduction Therapy Unit (Boehringer Ingelheim, GlaxoSmithKline, and Merck KGaA, to D.R.A.). H.T. is supported by an AstraZeneca BBSRC Studentship.
The authors declare the following competing financial interest(s): The A.C. laboratory receives or has received sponsored research support from Boehringer Ingelheim, Esiai Co., Ltd., Ono Pharmaceuticals and Nurix, Inc. A.C. is a scientific founder, director, and shareholder of Amphista Therapeutics, a company that is developing targeted protein degradation therapeutic platforms. C.C. is an employee of AstraZeneca. The other authors declare no conflict of interest.
Supplementary Material
References
- Vanhaesebroeck B.; Stephens L.; Hawkins P. (2012) PI3K Signalling: The Path to Discovery and Understanding. Nat. Rev. Mol. Cell Biol. 13, 195–203. 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- Sheppard K.; Kinross K. M.; Solomon B.; Pearson R. B.; Phillips W. A. (2012) Targeting PI3 Kinase/AKT/MTOR Signaling in Cancer. Crit. Rev. Oncog. 17, 69–95. 10.1615/CritRevOncog.v17.i1.60. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Cohen P. (1999) Activation of Serum- and Glucocorticoid-Regulated Protein Kinase by Agonists That Activate Phosphatidylinositide 3-Kinase Is Mediated by 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) and PDK2. Biochem. J. 339, 319–328. 10.1042/bj3390319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez J. M.; Alessi D. R. (2008) MTOR Complex 2 (MTORC2) Controls Hydrophobic Motif Phosphorylation and Activation of Serum- and Glucocorticoid-Induced Protein Kinase 1 (SGK1). Biochem. J. 416, 375–385. 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Vasudevan K. M.; Barbie D. A.; Davies M. A.; Rabinovsky R.; McNear C. J.; Kim J. J.; Hennessy B. T.; Tseng H.; Pochanard P.; Kim S. Y.; Dunn I. F.; Schinzel A. C.; Sandy P.; Hoersch S.; Sheng Q.; Gupta P. B.; Boehm J. S.; Reiling J. H.; Silver S.; Lu Y.; Stemke-Hale K.; Dutta B.; Joy C.; Sahin A.; Gonzalez-Angulo A.; Lluch A.; Rameh L.; Jacks T.; Root D.; Lander E.; Mills G.; Hahn W.; Sellers W.; Garraway L. (2009) AKT-Independent Signaling Downstream of Oncogenic PIK3CA Mutations in Human Cancer. Cancer Cell 16, 21–32. 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J. T.; Cummings L. A.; Bloomberg G. B.; Cohen P. (2005) Identification of Different Specificity Requirements between SGK1 and PKBα. FEBS Lett. 579, 991–994. 10.1016/j.febslet.2004.12.069. [DOI] [PubMed] [Google Scholar]
- Faes S.; Dormond O. (2015) PI3K and AKT: Unfaithful Partners in Cancer. Int. J. Mol. Sci. 16, 21138–21152. 10.3390/ijms160921138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N.; Macartney T.; Hornberger A.; Anderson K. E.; Tovell H.; Prescott A. R.; Alessi D. R. (2018) Mechanism of Activation of SGK3 by Growth Factors via the class 1 and class 3 PI3Ks. Biochem. J. 475, 117–135. 10.1042/BCJ20170650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bago R.; Malik N.; Munson M. J.; Prescott A. R.; Davies P.; Sommer E.; Shpiro N.; Ward R.; Cross D.; Ganley I. G.; Alessi D. R. (2014) Characterization of VPS34-IN1, a Selective Inhibitor of Vps34, Reveals That the Phosphatidylinositol 3-Phosphate-Binding SGK3 Protein Kinase Is a Downstream Target of class III Phosphoinositide 3-Kinase. Biochem. J. 463, 413–427. 10.1042/BJ20140889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier M.; Woodgett J. R. (2006) Role of the Phox Homology Domain and Phosphorylation in Activation of Serum and Glucocorticoid-Regulated Kinase-3. J. Biol. Chem. 281, 23978–23989. 10.1074/jbc.M604333200. [DOI] [PubMed] [Google Scholar]
- Biondi R. M.; Kieloch A.; Currie R. A.; Deak M.; Alessi D. R. (2001) The PIF-Binding Pocket in PDK1 Is Essential for Activation of S6K and SGK, but Not PKB. EMBO J. 20, 4380–4390. 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bago R.; Sommer E.; Castel P.; Crafter C.; Bailey F. P.; Shpiro N.; Baselga J.; Cross D.; Eyers P. A.; Alessi D. R. (2016) The HVps34-SGK3 Pathway Alleviates Sustained PI3K/Akt Inhibition by Stimulating MTORC1 and Tumour Growth. EMBO J. 35, 1902–1922. 10.15252/embj.201693929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halland N.; Schmidt F.; Weiss T.; Saas J.; Li Z.; Czech J.; Dreyer M.; Hofmeister A.; Mertsch K.; Dietz U.; Strubing C.; Nazare M. (2015) Discovery of N-[4-(1H-Pyrazolo[3,4-b]Pyrazin-6-Yl)-Phenyl]-Sulfonamides as Highly Active and Selective SGK1 Inhibitors. ACS Med. Chem. Lett. 6, 73–78. 10.1021/ml5003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherk A. B.; Frigo D. E.; Schnackenberg C. G.; Bray J. D.; Laping N. J.; Trizna W.; Hammond M.; Patterson J. R.; Thompson S. K.; Kazmin D.; Norris J. D.; McDonnell D. P. (2008) Development of a Small-Molecule Serum- and Glucocorticoid-Regulated Kinase-1 Antagonist and Its Evaluation as a Prostate Cancer Therapeutic. Cancer Res. 68, 7475–7483. 10.1158/0008-5472.CAN-08-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann T. F.; Boini K. M.; Beier N.; Scholz W.; Fuchss T.; Lang F. (2011) EMD638683, a Novel SGK Inhibitor with Antihypertensive Potency. Cell. Physiol. Biochem. 28, 137–146. 10.1159/000331722. [DOI] [PubMed] [Google Scholar]
- Gong G. Q.; Wang K.; Dai X.-C.; Zhou Y.; Basnet R.; Chen Y.; Yang D.-H.; Lee W.-J.; Buchanan C. M.; Flanagan J. U.; Shepherd P. R.; Chen Y.; Wang M.-W. (2018) Identification, Structure Modification, and Characterization of Potential Small-Molecule SGK3 Inhibitors with Novel Scaffolds. Acta Pharmacol. Sin. 39, 1902–1912. 10.1038/s41401-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M.; Crews C. M. (2019) Proteolysis TArgeting Chimeras (PROTACs) — Past, Present and Future. Drug Discovery Today: Technol. 31, 15–27. 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S.; Cui D.; Chen X.; Xiong X.; Zhao Y. (2018) PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. BioEssays 40, e1700247. 10.1002/bies.201700247. [DOI] [PubMed] [Google Scholar]
- Hughes S. J.; Ciulli A. (2017) Molecular Recognition of Ternary Complexes: A New Dimension in the Structure-Guided Design of Chemical Degraders. Essays Biochem. 61, 505–516. 10.1042/EBC20170041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J.; Casillas L.; Votta B.; Den Besten W.; Famm K.; Kruidenier L.; Carter P.; Harling J.; Churcher I.; Crews C. (2015) Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol. 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K.-H.; Ciulli A. (2015) Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. (2015) Drug Development. Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Birsoy K.; Hughes N. W.; Krupczak K. M.; Post Y.; Wei J. J.; Lander E. S.; Sabatini D. M. (2015) Identification and Characterization of Essential Genes in the Human Genome. Science 350, 1096–1101. 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.-H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. (2017) Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniaci C.; Hughes S. J.; Testa A.; Chen W.; Lamont D. J.; Rocha S.; Alessi D. R.; Romeo R.; Ciulli A. (2017) Homo-PROTACs: Bivalent Small-Molecule Dimerizers of the VHL E3 Ubiquitin Ligase to Induce Self-Degradation. Nat. Commun. 8, 830. 10.1038/s41467-017-00954-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M.; Jiang B.; Bauer S.; Donovan K. A.; Liang Y.; Wang E. S.; Nowak R. P.; Yuan J. C.; Zhang T.; Kwiatkowski N.; Muller A. C.; Fischer E. S.; Gray N. S.; Winter G. E. (2019) Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 26, 300–306. 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; e9.
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. (2016) Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 55, 807–810. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N.; Shimokawa K.; Nagai K.; Ohoka N.; Hattori T.; Miyamoto N.; Ujikawa O.; Sameshima T.; Nara H.; Cho N.; Naito M. (2018) Pharmacological Difference between Degrader and Inhibitor against Oncogenic BCR-ABL Kinase. Sci. Rep. 8, 13549. 10.1038/s41598-018-31913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson C. M.; Jiang B.; Erb M. A.; Liang Y.; Doctor Z. M.; Zhang Z.; Zhang T.; Kwiatkowski N.; Boukhali M.; Green J. L.; Haas W.; Nomanbhoy T.; Fischer E. S.; Young R. A.; Bradner J. E.; Winter G. E.; Gray N. S. (2018) Pharmacological Perturbation of CDK9 Using Selective CDK9 Inhibition or Degradation. Nat. Chem. Biol. 14, 163–170. 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J.; Arnhof H.; Bader G.; Berger H.; Ciulli A.; Covini D.; Dank C.; Gmaschitz T.; Greb P.; Karolyi-Özguer J.; Koegl M.; McConnell D. B.; Pearson M.; Rieger M.; Rinnenthal J.; Roessler V.; Schrenk A.; Spina M.; Steurer S.; Trainor N.; Traxler E.; Wieshofer C.; Zoephel A.; Ettmayer P. (2019) Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J. Med. Chem. 62, 2508–2520. 10.1021/acs.jmedchem.8b01826. [DOI] [PubMed] [Google Scholar]
- Cromm P. M.; Samarasinghe K. T. G.; Hines J.; Crews C. M. (2018) Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 140, 17019–17026. 10.1021/jacs.8b08008. [DOI] [PubMed] [Google Scholar]
- Ferguson F. M.; Gray N. S. (2018) Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discovery 17, 353. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Rana S.; Bendjennat M.; Kour S.; King H. M.; Kizhake S.; Zahid M.; Natarajan A. (2019) Selective Degradation of CDK6 by a Palbociclib Based PROTAC. Bioorg. Med. Chem. Lett. 29, 1375–1379. 10.1016/j.bmcl.2019.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. (2014) Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 57, 8657–8663. 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost J.; Galdeano C.; Soares P.; Gadd M. S.; Grzes K. M.; Ellis L.; Epemolu O.; Shimamura S.; Bantscheff M.; Grandi P.; Read K. D.; Cantrell D. A.; Rocha S.; Ciulli A. (2016) Potent and Selective Chemical Probe of Hypoxic Signalling Downstream of HIF-α Hydroxylation via VHL Inhibition. Nat. Commun. 7, 13312. 10.1038/ncomms13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. (2015) Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer E. M.; Dry H.; Cross D.; Guichard S.; Davies B. R.; Alessi D. R. (2013) Elevated SGK1 Predicts Resistance of Breast Cancer Cells to Akt Inhibitors. Biochem. J. 452, 499–508. 10.1042/BJ20130342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkes A. J.; Ahmadi K.; Alderton W. K.; Alix S.; Baker S. J.; Box G.; Chuckowree I. S.; Clarke P. A.; Depledge P.; Eccles S. A.; Friedman L. S.; Hayes A.; Hancox T. C.; Kugendradas A.; Lensun L.; Moore P.; Olivero A. G.; Pang J.; Patel S.; Pergl-Wilson G.; Raynaud F.; Robson A.; Saghir N.; Salphati L.; Sohal S.; Ultsch M.; Valenti M.; Wallweber H.; Wan C.; Wiesmann C.; Workman P.; Zhyvoloup A.; Zvelebil M.; Shuttleworth S. (2008) The Identification of 2-(1H-Indazol-4-Yl)-6-(4-Methanesulfonyl-Piperazin-1-Ylmethyl)-4-Morpholin-4-Yl-t Hieno[3,2-d]Pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 51, 5522–5532. 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- Davies B. R.; Greenwood H.; Dudley P.; Crafter C.; Yu D.-H.; Zhang J.; Li J.; Gao B.; Ji Q.; Maynard J.; Ricketts S.-A.; Cross D.; Cosulich S.; Chresta C. C.; Page K.; Yates J.; Lane C.; Watson R.; Luke R.; Ogilvie D.; Pass M. (2012) Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 11, 873–887. 10.1158/1535-7163.MCT-11-0824-T. [DOI] [PubMed] [Google Scholar]
- Castel P.; Ellis H.; Bago R.; Toska E.; Razavi P.; Carmona F. J.; Kannan S.; Verma C. S.; Dickler M.; Chandarlapaty S.; Brogi E.; Alessi D. R.; Baselga J.; Scaltriti M. (2016) PDK1-SGK1 Signaling Sustains AKT-Independent MTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell 30, 229–242. 10.1016/j.ccell.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa A.; Lucas X.; Castro G. V.; Chan K.-H.; Wright J. E.; Runcie A. C.; Gadd M. S.; Harrison W. T. A.; Ko E.-J.; Fletcher D.; Ciulli A. (2018) 3-Fluoro-4-Hydroxyprolines: Synthesis, Conformational Analysis, and Stereoselective Recognition by the VHL E3 Ubiquitin Ligase for Targeted Protein Degradation. J. Am. Chem. Soc. 140, 9299–9313. 10.1021/jacs.8b05807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. (2018) Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 25, 78–87. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; e5.
- Huang H.-T.; Dobrovolsky D.; Paulk J.; Yang G.; Weisberg E. L.; Doctor Z. M.; Buckley D. L.; Cho J.-H.; Ko E.; Jang J.; Shi K.; Choi H. G.; Griffin J. D.; Li Y.; Treon S. P.; Fischer E. S.; Bradner J. E.; Tan L.; Gray N. S. (2018) A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-Kinase Degrader. Cell Chem. Biol. 25, 88–99. 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; e6.
- Roy M. J.; Winkler S.; Hughes S. J.; Whitworth C.; Galant M.; Farnaby W.; Rumpel K.; Ciulli A. (2019) SPR-Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 14, 361–368. 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. (2019) Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 10, 131. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N.; Nirujogi R. S.; Peltier J.; Macartney T.; Wightman M.; Prescott A. R.; Gourlay R.; Trost M.; Alessi D. R.; Karapetsas A. (2019) Phosphoproteomics Reveals That the HVPS34 Regulated SGK3 Kinase Specifically Phosphorylates Endosomal Proteins Including Syntaxin-7, Syntaxin-12, RFIP4 and WDR44. bioRxiv 741652. 10.1101/741652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J.; Plater L.; Elliott M.; Shpiro N.; Hastie C. J.; McLauchlan H.; Klevernic I.; Arthur J. S. C.; Alessi D. R.; Cohen P. (2007) The Selectivity of Protein Kinase Inhibitors: A Further Update. Biochem. J. 408, 297–315. 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie C. J.; McLauchlan H. J.; Cohen P. (2006) Assay of Protein Kinases Using Radiolabeled ATP: A Protocol. Nat. Protoc. 1, 968–971. 10.1038/nprot.2006.149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.