

Abstract
Cardiovascular disease remains the number one cause of death and disability worldwide despite significant improvements in diagnosis, prevention, and early intervention efforts. There is an urgent need for improved understanding of cardiovascular processes responsible for disease development in order to develop more effective therapeutic strategies. Recent knowledge gleaned from the study of epigenetic mechanisms in the vasculature has uncovered new potential targets for intervention. Herein, we provide an overview of epigenetic mechanism, and review recent findings related to epigenetics in vascular diseases, highlighting classical epigenetic mechanism such as DNA methylation and histone modification as well as the newly discovered non-coding RNA mechanisms.
Keywords: DNA methylation, Histone modifications, microRNAs, long non-coding RNAs, cardiovascular diseases
Graphical Abstract
1. Introduction
All vasculature-associated pathologies, including atherosclerosis, stenosis, peripheral artery diseases (PAD) and aortic aneurysm, belong to the vast category of cardiovascular diseases (CVDs). CVDs are responsible for 31% of global deaths, and are considered the main cause of death and disability worldwide (https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)). This has pushed the vascular biology research field to identify the molecular mechanisms behind these pathological events and to develop innovative therapeutic approaches.
Regardless of the specific vascular pathology, altered gene expression is central to disease initiation and progression. Many factors contribute to the manifestation of these complex events including mechanical, inflammatory, autonomic, and neuroendocrine. However, for diseases of the vessel to initiate and progress, cells resident to the vascular wall, either endothelial, smooth muscle, or fibroblast, must engage stimulus coupled mechanisms to alter gene expression and activate pathologic phenotypes [1, 2]. These processes are often primed for dysregulation in diabetes and other metabolic disorder.
Events that modulate gene transcription are often dependent on the re-organization of chromatin. These processes are defined as epigenetic mechanisms which classify heritable changes that, independently from the DNA sequence, alter gene expression by inducing chemical modification of nucleotides, and histone proteins [3]. Recently, the definition of epigenetics has expanded to include non-coding RNA-dependent mechanisms [4, 5].
Although current knowledge regarding epigenetic mechanisms in CVD development is still limited, the dramatic improvement in DNA/RNA sequencing capacity has facilitated the definition of the epigenetic landscape for several key epigenetic marks and associated proteins in multiple pathological contexts. We expect that these discoveries will translate to the clinic in the form of improved diagnostics and therapeutics [6]. In this review, we will focus on both chemical alterations of DNA and histones, as well as epigenetic mechanisms mediated by non-coding RNAs. Furthermore, for readers new to the field of epigenetics, we highly recommend a visit to www.thesgc.org for additional resources.
2. Basic principles of epigenetics
2.1. DNA methylation and hydroxymethylation
Methylation is a common chemical modification of DNA, which occurs on the fifth carbon of the cytosine DNA base (5-mC) in CpG islands. This modified carbon faces the major groove of the DNA double helix, which is a hot spot for transcription factor binging. The presence of 5-mC in promoter and enhancer DNA is associated with gene silencing. This is most often attributed to 5-mC reader domains, CXXC and methyl-CpG-binding domain (MBD), in proteins of various silencing complexes [7, 8]. However, there is also an apparent biophysical effect of DNA methylation as in vitro studies indicate that more force is required to separate DNA strands containing 5-mC [9]. A current conundrum in this field is the fact that 5-mC is often enriched in the gene bodies of actively expressed genes (e.g. [10, 11]).
DNA methylating enzymes (DNA Methyl transferases or DNMTs) have been well studied in recent years and excellent reviews are available (e.g. [3, 12]). Similarly, much is known regarding the processes that remove methyl marks from DNA [13]. The Ten-Eleven Translocation (TET) family of dioxygenases catalyse the oxidation of 5-mC to 5-hydroxymethyl cytosine (5-hmC). TET enzymes further catalyse oxidation of 5-hmC to 5-formyl cytosine (5-fC) and 5-carboxyl cytosine (5-caC) which can then be converted to un-methylated cytosine by Thymine DNA Glycosylase (TDG) in base excision repair [14]. Remarkably, these intermediate oxidation states of 5-mC appear to have dramatically different effects on gene expression (e.g. [15]). For instance, 5-mC reader proteins such as MBD1/2 and MeCp-2 Protein (MECP2) that repress transcription at methylated CpG islands, do not bind 5-hmC [16]. 5-hmC, 5-fC and 5-caC are found in the promoters of highly expressed genes and 5-fC has been shown to distort the double helix structure of DNA, leading to recruitment of DNA binding proteins [17]. Several key questions regarding 5-hmC remain unanswered. What regulatory mechanisms are at work to govern the stability of these oxidized forms (5-hmC, 5-fC, and 5-caC), are they dynamic, and what are the potential “readers” of these oxidized forms? In addition, the DNA base Adenine has also been found to be methylated robustly in lower level organisms, though few mammalian examples have surfaced (e.g. [18]).
2.2. Histone post-translational modifications (HPTMs)
Active roles for dynamic histone acetylation and methylation in the regulation of transcription were first proposed in the 1960s [19]. A number of chemical modifications including acetylation, mono-di-and tri-methylation, phosphorylation, SUMOylation, and ubiquitination, have been shown to have functional roles on key amino acid residues in the unstructured N- or C-Terminal tails of core histone proteins [20]. With improvements in mass spectrometry, data analysis capabilities, and the desire to look, there has been rapid expansion of identified post-translational modifications found on histones. These include many larger acyl molecules and intermediates of metabolism such as crotonylation, succinylation, butyrylation, beta-hydroxybutyrylation, and manyolation, all of which are also carried by Coenzyme A (CoA) [21, 22]. Several recent reports indicate that these modifications facilitate biologically relevant gene expression changes, particularly in response to changes in metabolism (e.g. [23, 24]). More study is necessary to determine importance of these mechanisms in the context of animal biology and disease, though current findings are exciting and molecular mechanisms that differentiate them from other modifications are being uncovered (e.g. [25]). Acetylation and methylation are by far the most thoroughly studied histone modifications and have typically opposing effects on gene expression (Figure 1).
Figure 1. Illustration of histone H3 lysine modification effect on gene expression.

The nucleosome is composed of double stranded DNA wrapped around a “hairy hockey puck” comprised of core histone proteins and their unstructured N- and C- terminal tails (H3 tail is exaggerated for illustration purposes). Acetylation and methylation of lysine residues on core histone proteins are mutually exclusive modifications on the same residue of the same subunit. Histone lysine acetylation is associated with increased gene expression at the indicated residues. Histone lysine methylation is associated with gene activation when found at H3K79, H3K36 and H3K4 but is generally associated with gene silencing at other residues. In the boxes, a list of writers and erases is included.
Histone Lysine Acetylation:
Histone acetyl transferases (HATs, e.g. p300) catalyse the addition of two-carbon acetyl groups to lysine residues from acetyl-CoA [26]. Histone deacetylases (HDACs) catalyse removal of acetyl groups from lysine residues and are distributed among four classes (Class I, IIa, IIb, III, and IV) [27]. Class III HDACs are also known as sirtuins and will not be major component of this review. It is important to note that though we discuss class specific HDAC inhibitors in subsequent sections, Class IIa HDACs (HDACs 4/5/7/9) have very week deacetylase activity compared to other HDACs and the field is unsettled regarding target proteins of class IIa HDAC catalytic activity. The notion that the catalytic domain actually functions as a reader domain has gained traction in recent years. Several important interacting partners have been established for class IIa HDACs including Myocyte Enhancer Factor-2 (MEF2), Serum Response Factor (SRF), as well as Class I HDACs found in transcription repressive complexes (reviewed in [28]).
The positive charge found on lysine residues under physiological conditions facilitates histone - DNA interaction as DNA is negatively charged. Addition of an acetyl group to the lysine residue masks the positive charge and relaxes the histone - DNA interaction, causing more accessible DNA. This mechanism of gene activation has been overshadowed in recent biomedical studies due to the characterization of acetyl-lysine reader domains and improved small molecule inhibitors thereof (e.g. [29]).
BROMO, YEATS, and some PHD domains are specialized three-dimensional pockets in a number of proteins that bind to acetyl-lysine residues, allowing for assembly of large transcription regulating complexes based on the presence of histone lysine acetylation [30]. One such complex includes the bromodomain and extra-terminal domain containing (BET) protein, BRD4. BRD4 binds acetyl-lysine residues on chromatin and recruits components of the positive transcription elongation factor β (P-TEFβ) complex, which facilitates phosphorylation and activation of RNA Polymerase II for mRNA synthesis [31]. Some acetyl-lysine binding proteins have additional reader domains for other modifications and can also have enzymatic activity (e.g. P300). This enables intricate cross talk, feed-forward waves of chromatin modification, and the ability of methylated or acetylated nuclear proteins (e.g. transcription factors) to target epigenetic readers and modifiers to specific locations in the genome (Figure 2).
Figure 2. Illustration of feed-forward chromatin remodeling to activate or silence expression.

Signal dependent PTMs on transcription factors and catalytic enzymes cooperate with histone PTMs to govern complex assembly. Chromatin associated complexes and select enzymes can facilitate feedforward expansion of activating or repressing PTM patterns. The HAT, p300 serves as an example whereby acetyl-lysine residues recruit p300 via its bromodomain to multiply local histone acetylation. The resultant chromatin landscape facilitates gene activation by recruiting another acetyl-reader, BRD4, to activate RNA Pol II. Conversely, EZH2, a KMT, adds repressive methyl marks to H3K27 and is found in the PRC complex which contains methy-lysine readers and DNMTs that coordinate histone and DNA methylation.
It is important to note that in the limited study to date, larger and/or more negatively charged modifications (e.g. succinylation) have been associated with more robust gene activation than acetylation [32]. This may highlight the importance of the previously mentioned gene activation mechanism via masking of the lysine positive charge. This positive charge is completely reversed (−1) by succinylation, potentially causing an even more relaxed histone - DNA association.
Histone Lysine Methylation:
Three forms of methylation are found on lysine residues; mono-, di-, and tri-methylation [33]. Enzymes that catalyse the addition of methyl groups are known as lysine methyl transferases (KMTs). KMTs include the EZH, SETD, PRDM, PRMT, METTL, and MLL enzyme families. Enzymes that catalyse removal of methyl marks are termed lysine demethylases (KDMs) and include members of UTX/Y, JARID1, JMJD, LSD, PHF and FBXL enzyme families (though many have been renamed to a KDM nomenclature, e.g. JMJD2A=KDM4A). Generally, there is more diversity in the regulators and readers of methyl-lysine than acetyl-lysine as is illustrated by the fact that MBT, PHD, PWWP, BAH, CW, SPINDLIN, TUDOR, and CHROMO domain containing proteins can bind methyl-lysine residues vs BROMO, YEATS, and some PHD domains for acetyl-lysine.
Importantly, if a lysine residue is methylated it cannot be acetylated, at least on the same histone subunit. Hence, methylated lysine residues are typically associated with silencing and implying that KMT activity is recruited to silence genes and KDM activity is recruited to activate genes. Notable exceptions to this general pattern are K4, K36, K79 residues of histone H3. Mono-, di-, and tri-methylated lysine 4 residues of H3 (or H3K4me, H3K4me2, H3K4me3) are associated with active genes and these marks are found in the promoters, gene bodies and enhancers of activated genes [34]. H3K4 methylation enrichment at super enhancers also occurs, with H3K4me3 being found at broad peak super enhancers associated with cell lineage specific and tumour suppressor genes (e.g. [35]). Key players in H3K4 methylation include, writers: SET1/7, MLL1–4, SMYD1/2 and PRDM9, readers: CHD1, BPTF, TAF3, ING4 and erasers: LSD1/2, NO66, and JARID1A-D. For a comprehensive review of methyl-lysine regulation, see [34].
“Epigenetic memory” is a concept in biology that is dependent on the mechanisms already introduced, though hinges on the longevity of DNA and histone modifications, and is important for vascular biology in several ways. First, proliferating cells must be able to “remember” what type of cell they are to be as they complete DNA replication, and hence what genes they should express. The initial testing of BET protein inhibition with JQ1 as a potential therapeutic in cancer was guided by this concept. By blocking the ability of BRD4 to bind acetyl lysine residues that were retained or immediately restored following cell division, could one make the cells “forget” they were an aggressive cancer cell [29]? Though subsequent study has elucidated multiple mechanisms by which BET protein inhibition elicits benefit, multiple successful preclinical studies and preliminary results from clinical trials of BET inhibition in cancer indicate the answer was a clear, yes [36]. Similar logic has been successfully applied to proliferating cells in aggravated blood vessels [37–39]. Second, exposure to environmental factors that result in gene expression changes via epigenetic mechanisms, even transiently, may predispose cells to more rapid or lower threshold activation upon subsequent insult [40]. This is most often considered in the context of former smokers [41] and diabetics [42], but also has implications for other individuals. Here also, previous exposure to antigens, inflammatory milieu, or hyperglycaemia are clearly important in immune cell activation and immune surveillance, which are major components of atherosclerosis. Finally, the concept of epigenetic memory is crucial in determining what genes are and are not accessible for expression in a given cell. This is largely determined during the normal course of development and is dependent on transcription factor expression and DNA and histone modifications. An elegant study recently showed that DNA CpG hypomethylation at previously activated enhancers served as a long-lived tag for gene reactivation even though histone methyl and acetyl modifications had been lost [43].
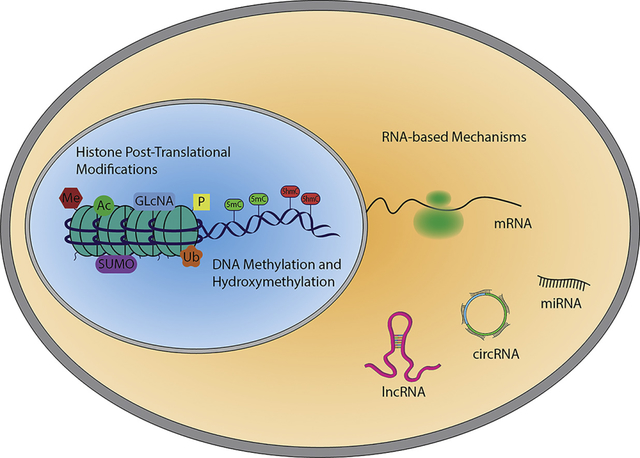
2.3. Non-coding RNAs as epigenetic modulators
Non-coding RNAs (ncRNAs) are able to modulate gene expression through many different mechanisms. In term of classification, the easiest organization is based on their length, therefore two major groups are distinguished: small (<200nt) and long ncRNAs (>200nt). Small ncRNAs includes regulatory elements, whose expression is spatial and temporally regulated, such as microRNAs (miRNAs) and PIWI RNAs (piRNAs) and the more stably expressed housekeeping ncRNAs, such as small nucleolar RNAs (snoRNAs) and transfer RNAs (tRNAs) [44]. The classification of long ncRNAs is more complex and can be based on genomic localization or specific function [45].
The best-characterized small ncRNA elements are miRNAs [46]. miRNAs are single-stranded molecules of approximatively 22 nucleotides, and are well conserved across species. They are generally involved in the negative regulation of gene expression by binding to the 3’ un-translated region (3’UTR) of target genes [47]. miRNAs are transcribed by RNA polymerase II into a primary miRNA (pri-miRNAs) of around 1000bps, that might contain multiple miRNAs [48]. In the nucleus, the pri-miRNA is then cleaved by the RNAse III Drosha into a precursor miRNA (pre-miRNA) of 70 nucleotides, that is sub-sequentially transported into the cytoplasm by the Exportin 5 system [49]. Here, the pre-miRNA is further digested by the RNAse III Dicer in a 22bp double-stranded miRNA, and then opened as a single-stranded mature miRNA that is finally loaded into the Argonaute (Ago) protein. Together with the target messenger RNA (mRNA), this forms the RNA-induced silencing complex (RISC) [5]. The role of miRNAs in cardiovascular system has been thoroughly studied [50], allowing the definition of several pre-clinical applications, that hopefully, will be soon translated to the clinic [51].
piRNAs are another family of small regulatory ncRNAs. These elements of around 30 nucleotides are enriched in mammals and show less species conservation than miRNAs [52]. In terms of mechanism of action, the available information is very limited: it is known that piRNAs are able to bind a specific sub-group of Ago proteins. These protein-piRNAs complexes are then able to interfere with the activity of transposable elements in particular germ line cells [53]. piRNA research is still in its infancy, and more study is needed to evaluate any potential role in vascular diseases.
Another important class of ncRNAs is that of the long ncRNAs [54]. Beside their length, long ncRNAs differ from small ncRNA, and in particular from miRNAs, because they are generally phylogenetically very poorly conserved. To better classify these molecules, a nomenclature based on their genomic location is preferred (Figure 3):
Figure 3. Schematic classification of the different types of lncRNAs.

LncRNAs are defined according to their genomic position in relation to adjacent coding genes and other genomic elements: LncRNAs are represented in pink, enhancer in orange, promoter in light blue, exon in green, while intron in red. Arrows indicate transcription starting sites.
Sense lncRNAs: located and transcribed from the sense strand of a protein-coding gene;
Antisense lncRNAs: located and transcribed from the antisense strand of a protein-coding gene;
Exonic lncRNAs: located in exons of protein-coding genes:
intronic lncRNAs: located in introns of protein-coding genes;
enhancer lncRNA (eRNAs) : derived by enhancer region of specific protein-coding genes;
intergenic lncRNAs (LincRNAs): located between two genes with an independent transcription regulation;
bidirectional promoter lncRNAs: regulated by the same promoter of a gene but transcribed in the opposite direction;
Circular lncRNA (circRNAs): derived by exonic or intronic sequences that circularize following an alternative splicing of linear transcripts.
Long ncRNAs are transcribed, similarly to a normal gene, by RNA polymerase II or III [55]. Following transcription, long ncRNAs are then often 5’-capped, spliced and their final mature form characterized by the attachment of a polyadenylated tail. They are expressed at lower levels than protein coding mRNAs and usually, with few exceptions [56], do not contain an open reading frame (ORF). Long ncRNAs are present in both the cytoplasm and nucleus [57]. In the nucleus, long ncRNAs are able to regulate both silencing and activation of target genes, via modulation of other epigenetic machinery [58]. Cytosolic long ncRNA are able to regulate mRNA translation and stability [59], and modulate protein localization [60] (Figure 4). Although several studies have been published describing the roles of specific long ncRNAs in vascular diseases, the field is clearly in its infancy. Several classes of long ncRNAs have only been studied at the level of one or two specific members out of the thousands that exist. Additional RNA related epigenetic mechanisms have emerged, though are less well characterized in the vasculature. For a pertinent review on RNA methylation and RNA-editing, see [61].
Figure 4. Cellular functions of lncRNAs.

LncRNAs modulate gene expression by different mechanisms: in the nucleus lncRNAs might guide transcription factors (TFs) to specific sites in the genome (1) or sponge TFs repressing their function (2); They can induce histone modifications, guiding chromatin remodeling complexes in specific genomic loci (3) or influence enhancer activity (4); LncRNAs can regulate nucleo-cytoplasmic movement of both nuclear factor of activated T cells (NFAT) (5) or modulate pre-mRNAs splicing (6); In the cytoplasm, lncRNAs can influence mRNA stability (7), can sponge miRNAs (8), can control translational events (9), or act as a scaffold in proteins complexes (10); They might also function in the stabilization of ribonucleoprotein (RNP) complexes (11); a new class of lncRNAs, named circular RNAs (circRNAs), can act as miRNA sponges, or regulate the maturation of ribosomal RNAs (12); Finally, some lncRNAs facilitate cell-to-cell communication (13).
3. Epigenetic regulation of vascular diseases
Endothelial cell (EC) dysfunction as well as vascular smooth muscle cell (VSMC) dedifferentiation, proliferation, and migration are critical components to CVD manifestation. Each of these phenotype changes is resultant to broad changes in gene expression. Given the coordinated and interacting roles of DNA/histone methylation and histone acetylation in regulating gene expression, it is not surprising that inhibition of DNMTs, TETs, KMTs, KDMs, HDACs, or HATs, can prevent many of the cell phenotype changes associated with CVD. However, establishing efficacy in intact animal models has been less successful due to the complexity of disease. The remainder of this review focuses on epigenetic studies employed in intact animal models of cardiovascular diseases.
3.1. Atherosclerosis
Atherosclerosis is a major cause of morbidity and mortality in the Western world. It’s pathological roots involve imbalanced lipid metabolism together with a maladaptive immune response and alterations of vascular cells within the arterial wall. Clinically, atherosclerosis is characterized by a chronic inflammatory response occurring at predilection sites with altered laminar flow, such as branch points [62], at which we observe a progressive reduction of the vessel lumen diameter due to the buildup of plaques, mainly formed by three cell types: ECs, VSMCs and inflammatory cells. During atherosclerosis development, ECs undergo a series of molecular modifications that trigger the formation of plaques: expression of adhesion molecules such as vascular adhesion molecule-1 (VCAM1), intracellular adhesion molecule-1 (ICAM1) and E-selectin, that help the recruitment of leukocyte to the vessel wall [63]. VSMCs with a mature contractile phenotype contribute to the maintenance of vascular function and tone, while in pathological conditions, they undergo to a phenotypic switch acquiring migratory/proliferative capacities [64]. Atherosclerosis development is dependent on the recruitment of circulating monocytes in response to the endothelium activation. Once monocytes transvasate, they differentiate into macrophages, which accumulate lipids and can create a chronic inflammatory environment [65].
Acetylation related studies:
Dynamic regulation of histone posttranslational modifications in atherosclerosis progression has been suspected for some time (e.g. [66]). The role of HDACs and HATs have been robustly studied in the regulation of SRF and Myocardin (MYOCD) dependent VSMC gene activation [67–69]. Despite anti-proliferative and anti-migratory effects in cultured VSMCs, systemic panHDAC inhibitor (targeting Class I, II, IV HDACs) treatment exacerbated atherosclerosis in LDL receptor knockout mice fed a western diet [70]. It is though this exacerbation was due to altered macrophage cholesterol uptake and scavenging through CD36. Another study also demonstrated that targeting of class I HDACs would not be beneficial in atherosclerosis. Here, HDAC3 was selectively disrupted in endothelial cells, which resulted in larger atherosclerotic plaques [71]. Strong preclinical studies have led to HDAC inhibition being proposed as a possible therapeutic strategy for human heart failure (e.g. [72, 73]). Though HDAC inhibitors are already in the clinic for treatment of certain cancers and also Duchenne Muscular Dystrophy, whether or not HDAC inhibition in humans might worsen atherosclerosis remains an open question. A large caveat to this potential concern is that the animal models used to investigate HDACs in atherosclerosis are severe and designed to generate measurable disease. However, it may be necessary to determine if atherosclerotic effects of pan and class I HDAC inhibition 1) do occur in humans and 2) can be overcome by anti-atherosclerotic therapies such as statins. Given confusion regarding the benefits of statin therapy in systolic heart failure (e.g. [74, 75]), perhaps diastolic heart failure is the preferred patient population for initial trials of HDAC inhibition in such context as statin therapy appears to be exclusively associated with benefit in this population [76].
Specifically targeting non-class I HDACs may be a useful strategy for treating atherosclerosis. For instance, mice lacking the class IIa HDAC, HDAC9, were protected from atherosclerosis, potentially through a mechanism also tied to macrophage phenotype and cholesterol handling [77].
Disruption of acetyl-lysine and bromodomain protein-protein interactions with a small molecule inhibitor, JQ1, prevented atherosclerosis in LDL receptor deficient mice through anti-inflammatory action in endothelial cells and macrophages [37]. Targeting this family of BET proteins (acetyl-lysine readers), especially BRD4, has dramatically reduced NF- κB and AP-1 dependent inflammation signalling in multiple contexts (e.g. [78, 79]). It is important to note that JQ1 and most available small molecule BET protein inhibitors act on both of the bromodomains in this protein family, which are characterized by two tandem n-terminal bromodomains.
Specific inhibition of only the second Bromodomain in BET proteins via RVX-208 also showed promising anti-atherosclerotic preclinical results via its ability to increase expression of ApoA-1 and improve lipid profiles [80]. However, RVX-208 failed to provide benefit in the clinic when assessing apoA-1, HDL-C, or atheroma volume [81]. Post hoc analysis has revealed a significant reduction in major adverse cardiovascular events (MACE) for patients randomized to the RVX-208 treatment group and a phase-III trail (BETonMACE) is underway. Several studies indicate the potential MACE protective effects of RVX-208 may be mediated through modulation of vascular calcification, kidney function, and the complement cascade [82–84]. BD1 and BD2 of BRD4 do exhibit differential binding affinities to acetyl-histone proteins as well as acetyl-lysine residues on non-histone proteins. This may explain unique phenotype effects of selective bromodomain inhibition (reviewed in [85]), though more work is required in this area to fully characterize what BET protein mechanisms are engaged or not by selective bromodomain inhibition.
Methylation related studies:
Histone methylation likely also plays a key role in atherosclerosis. Over expression of the histone methyl transferase, Enhancer of Zeste Homolog 2 (EZH2) in ApoE deficient mice caused significantly more atherosclerosis and inhibited cholesterol efflux via regulation of the ABCA1 gene [86]. EZH2 is known to methylate H3K27 residues, leading to gene silencing. Protein expression of the H3K4 demethylating enzyme KDM5D (JARID1D) was significantly up regulated (log2 fold change > 6) in aortas from three animal models of atherosclerosis and was doubled in human aorta samples from patients with atherosclerosis relative to controls. Importantly, H3K4me3 was reduced in the same diseased human samples with a significant negative correlation between KDM5D expression and H3K4me3 abundance [87]. Despite EZH2 being a methyl transferase and KDM5D being a demethylase, the end result of their activities given different substrate preferences results in gene silencing. Can this be taken to indicate that increased gene silencing results in atherosclerosis? Likely not, as dedifferentiation of VSMCs at least is associated with broad gene activation. However, much work remains in understanding how these and other epigenetic modifying enzymes are target to specific gene loci in response to specific stimuli.
Many DNA CpG islands are hypermethylated in human atherosclerotic plaques [88, 89]. A more recent report also indicates hypomethylation at select loci. Genes associated with hypomethylation vs hypermethylation in these samples were linked to different functional associations [90]. This indicates a mechanism for target specificity in recruitment and activation of DNA methylation regulators that correspond with phenotypic impact of particular gene sets.
Endothelial cell DNA methytransferase (e.g. DNMT1) expression and activity is increased in response to atherogenic insults such as oxidized low-density lipoproteins. This corresponds to increased CpG methylation in the promoters of key protective genes such as Krüppel-like Factor 2 (KLF2), leading to cell proliferation and inflammatory signalling [91]. Similarly, in VSMCs, treatment with platelet growth factor (PDGF-BB), fibroblast growth factor (FGF) or 10% FBS (all de-differentiating stimuli) leads to increased expression of Ubiquitin Like with PHD And Ring Finger Domains 1 (UHRF1) through suppression of a microRNA. UHRF1, a partner protein of DNMT1 (also up regulated by these treatments), stimulates DNA methylation by DNMT1 at key pro-differentiation genes. Importantly, UHRF1 expression was also increased in atherosclerosis. When UHRF1 was genetically targeted with shRNA encoding virus, neointima was reduced in an ApoE deficient mouse restenosis model [92]. Given the VSMC phenotype effect, this axis of potential therapeutic targets is likely to be relevant in atherosclerosis, restenosis, and potentially aneurysm.
Macrophage DNA methylation patterns are also altered in atherosclerosis, highlighting DNA methylation regulation as an attractive target in atherosclerosis given similar involvement in all three key cell types. DNMT1 over expression specifically in macrophages stimulates inflammation and atherosclerosis via apparent suppression of Peroxisome Proliferator-Activated Receptor (PPAR) [93] and Krüppel-like Factor 4 (KLF4) [94]. Similarly, chemical inhibition of DNMTs via 5-Axa-2’-deoxycytidine blocks atherosclerosis formation [77]. Also, strong evidence supports the role of a DNA demethylating enzyme, TET2, in activating VSMC pro-differentiation genes MYOCD, SRF, and Myosin Heavy Chain 11 (MYH11) [95].
ncRNA studies:
Multiple ncRNAs have been implicated in activation of the endothelium, including miRNAs. For instance, miR-17–3p targets VCAM1, while miR-31 targets E-selectin, though specific roles for these miRNAs are not well defined in atherosclerosis [96]. miR-126, the most abundant miRNA expressed by ECs, was initially identified as a direct regulator of VCAM1 [97]. Subsequent studies have also demonstrated the role of miR-126 in controlling endothelial flow-dependent angiogenesis in atherogenic conditions [98, 99]. EC cytokine secretion is mainly regulated by pathways controlled by the NF-κb transcription factor [100]. miR-181b is a critical regulator of this pathway via its ability to directly control expression of the NF-κb nuclear transporter, importin-a3 in ECs [101, 102]. Another miRNA involved in EC-specific cytokine communication is miR-146, which targets ELAV Like RNA Binding Protein 1 (ELAVL1). This short ncRNA is activated by tumor necrosis factor-a (TNF-α) and interleukin-1 (IL-1) in both humans [103] and mice [104].
Similar to miRNAs, long ncRNAs play important roles in atherosclerosis development. Retinal ncRNA 3 (RNCR3) is upregulated in ECs of atherosclerotic plaques both in mice and humans [105]. In murine models, targeting of RNCR3 aggravates atherosclerosis development in the aorta, and increases systemic inflammation.
Another important long ncRNA involved in atherosclerosis development is an antisense noncoding RNA found in the Inhibitors of Cyclin Dependent Kinase 4 (INK4) locus known as ANRIL. ANRIL acts locally to bind to the Polycomb repression complexes 1 and 2 (PRC1 and 2), inducing H3K27 trimethylation and silencing of the INK4 locus [106]. Interestingly, ANRIL is expressed by all three-principal cell components of atherosclerotic plaques: ECs, VSCMs, and monocyte-derived macrophages [107]. Therefore, the level of ANRIL expression has been directly associated with the severity of atherosclerosis [108].
miRNA involvement in VSMC de-differentiation, proliferation and migration, in the context of atherosclerosis has been well studied. miR-143 and miR-145 are considered pivot regulators of VSMC phenotypic switching. In human and mouse pathological vessels both miRNAs are down-regulated [109], and miR-143 and −145 knockout mice exhibit reduced VSMC contractility and function [109–113]. In line with these studies, ectopic delivery of miR-145 through a lentiviral system reduces atherosclerotic plaques in ApoE deficient mice [114]. Opposing the miR-143/145 cluster, miR-221 and miR-222 expression is induced by vascular injury, atherosclerosis [115], and PDGF treatment, leading to decreased VSMC-specific contractile gene expression [116]. Accordingly, the acute loss of miR-221 and miR-222 has been directly associated with plaque rupture as disarrayed VSMCs within the plaque likely regain contractile function [117].
Compared to miRNAs, knowledge regarding the role of VSMC long ncRNAs in atherosclerosis is very limited. Recently, next-generation sequencing experiments, identified a VSMC-enriched lncRNA named Smooth Muscle Enriched Long Noncoding RNA (SMILR), for its ability to modulate VSMC proliferation. SMILR is deregulated in unstable human carotid plaques, and it was also found in plasma of atherosclerotic patients. Plasma SMILR content correlated with the level of circulating C-Reactive Protein, a marker for the inflammatory status of unstable plaques [118].
Again, recent work with a relatively unstudied class of lncRNAs named circRNAs has further illuminated ncRNA mediated mechanisms in VSMC biology. These elements do not possess strand polarities, being generated by a back-splicing of linear RNAs [119]. Although they were discovered over 40 years ago [120], it was thought that these RNA circles derived from mis-splicing events and had no biological function [121]. However, multiple labs have demonstrated that circRNAs derive from guided splicing events and play fundamental roles in the development of pathological conditions including those of the cardiovascular system [122–124]. Though circRNAs can have multiple functions, they are perhaps most well known as endogenous miRNA sponges [125, 126]. In this regard, Hall et al. identified a specific circRNA conserved in human and mouse, highly expressed by VSMCs that is able to specifically inhibit the activity of miR-145. This circRNA derives from alternate splicing of the Lipoprotein Receptor 6 (LRP6) gene, and through comprehensive in vitro and in vivo approaches, the authors showed its role VSMC phenotypic switching and its involvement in vascular disease, including atherosclerosis [127].
Multiple miRNAs have been identified that regulate macrophage mediated cholesterol engulfment. For instance, miR-27a/b family regulates macrophage cholesterol homeostasis by directly targeting proteins involved in cholesterol esterification (Acetyl-CoA Acetyltransferase 1, Low-density lipoprotein receptor and scavenger receptor class B member 3), and efflux (ATP-binding cassette A1) [128]. miR-146 and miR-125a-5p regulate macrophage cytokine production and cholesterol uptake, directly targeting Toll-like receptor 4 (TLR4) [129] and Oxysterol binding protein-like 9 [130]. Furthermore, miR-145 is able to control atherosclerosis development by controlling the ATP-binding cassette A1 cholesterol transporter and therefore macrophages infiltration in atherosclerotic plaques [131]. Inflammation and innate immune responses in atherosclerosis are also regulated by long ncRNAs. Recently, a lncRNA proximal to the Cox2 gene (lncCox2) has been identified through whole-transcriptome analysis. In this study, mouse bone marrow-derived macrophages were stimulated with TLR ligands and lncCox2 was the most significantly induced transcript. Very interestingly, lncCox2 was found in chromosomal regions in which there was a higher expression of other immune genes, such as Prostaglandin-Endoperoxide Synthase 2 (Cox2), strongly indicating that these transcripts were co-regulated [132].
3.2. Vascular stenosis
Vascular stenosis/restenosis is a common process that develops following endovascular procedures, including stent placement in the coronary circulation and in bypass grafts. Stenosis mainly depends on the re-activation/de-differentiation of VSMCs, triggered by EC damage, that migrate to the site of injury and generate neointima [133].
Acetylation related studies:
Complex interactions between critical VSMC differentiation factors such as SRF and MYOCD with class IIa HDACs have been appreciated for some time (e.g. [134]). In 2011, pan HDAC inhibition was shown to impressively block stenosis in an arterial balloon angioplasty model [135]. However, there have been relatively few studies directly targeting specific HDACs in restenosis (e.g. [136] and [137]), that might capitalize on beneficial effects in VSMC pathologic behaviours while avoiding apparent class I HDAC complications in endothelial and macrophage populations mentioned in the discussion of atherosclerosis above. Again, disruption of the acetyl-lysine - BET bromodomain, protein-protein interaction via JQ1 has proven effective at robustly blunting restenosis [37, 39].
Given the central role of VSMCs in restenosis biology [64], many of the histone methylation and DNA methylation strategies mentioned above to target VSMCs in atherosclerosis are also effective in restenosis models. In a carotid artery ligation model, an HDAC9- metastasis-associated lung adenocarcinoma transcript 1 (MALAT1)-EZH2 complex was recently described that mediated silencing of VSMC pro-differentiation genes. Targeting any of the members of this complex resulted in blunted restenosis [138]. Consistent with the pro-differentiation aspect of inhibiting this complex, similar strategies show promise in experimental models of aneurysm [138, 139].
ncRNA mechanisms:
In neointimal formation, the modulation of RNA-mediated pathways is also very important, and indeed several studies have correlated miRNA modulation and neointimal formation in response to vascular damage [140]. Potentially, the up-regulation of miRNAs associated with VSMC differentiation and down-regulation of those modulating VSMC de-differentiation might blunt neointimal formation [141]. Among miRNAs identified in stenosis, miR-143 and miR-145 play a central role. As mentioned above, these miRNAs control the VSMC phenotypic switch, and normalization of their expression in damaged vessels strongly reduces in stenosis formation [109, 142]. miR-21 has been also implicated in promoting VSMC switch toward a proliferative phenotype in response to vascular injuries. Inhibition of miR-21 expression reduces neointimal formation in response to balloon injury in rat carotids [143]. The long ncRNA lincRNA-p21 is also involved in neointimal formation. In vivo down-regulation of linc-RNA-p21increases the size of stenotic formations via regulation of p53-dependent apoptosis in VSMCs [144].However, although much work has been done regarding epigenetics in neointimal formation, no epigenetic-based drugs have been approved for clinical use in this patient population.
3.3. Hypertension
One of the largest risk factor for cardiovascular disease development is hypertension. For many patients, hypertension goes untreated or is ineffectively controlled, which leads to many cardiovascular complications. Hypertension results from complex interactions between genes and environmental factors, and recent evidence implicate epigenetic mechanisms in disease pathology [145, 146]. Many factors contribute to hypertension including renal, autonomic, neuroendocrine, and vessel diameter/compliance. The vast majority of genetically driven hypertension cases for which causal relationships have been determined are associated with renal or neuroendocrine regulation of blood volume [147]. However, other GWAS studies suggest additional association with mechanisms acting in the vessel wall [148].
Acetylation related studies:
HDAC inhibition with valproic acid (VPA) attenuated hypertension in the SHR rat while reducing NF-κB and ATR-1 expression [149]. VPA was also able to reduce blood pressure and hyperglycemia in a rat model of Cushing Syndrome [150]. Here, it was shown that class I HDACs deacetylate the glucocorticoid receptor (GR), potentially de-restricting its transcriptional activity. It should be noted that HDAC mediated regulation of hypertension appears to be model specific, as others have not found HDACi mediated effects on blood pressure (e.g. [73]). Like other vascular pathologies mentioned above where HDAC inhibition and BET inhibition tend to have similar beneficial effects, BET inhibition with JQ1 potently reduced systolic and diastolic pressures in spontaneously hypertensive rats, while also reducing NF-κB signalling [151]. Though it is still not entirely clear why inhibition of deacetylases and inhibition of acetyl lysine reader proteins tend to result in similar effects, two scenarios dominate speculation in the field. First, HDAC mediated de-acetylation of transcription factors like GR may allow increased transcriptional activity. When HDACs are inhibited, so is transcriptional activity of the transcription factor, and when BET proteins are inhibited, particularly BRD4, subsequent RNA Polymerase II activation is also blocked. The second scenario is dependent on the fact that HDAC catalytic activity is required to free BRD4 from acetyl-lysine bound configurations for chromatin redistribution.
Methylation related studies:
Peripheral blood mononuclear cell (PBMC) epigenetic status has been proposed as having a “canary in the coal mine” type reporter quality on the general sta tus of health (either metabolic or inflammatory disease state). This PBMC utility is well illustrated in hypertension, where global and gene specific alterations in DNA methylation are observed [152]. Key genes with altered methylation in PBMCs from hypertensive subjects include Angiotensin II Converting Enzyme 1 (ACE-1) [153], Hydroxysteroid 11-Beta Dehydrogenase 2 (HSD11B2) [154], and Sodium Channel Epithelial 1 Alpha and Beta Subunits (SCNN1A/B) [155]. Additional epigenetic alterations have been seen in target tissues (adrenal, kidney, or aorta) including for Solute Carrier Family 12 Member 2 (Slc12a2), Angiotensin II Receptor Type 1 (AT1bR), ACE-1 and HSD11B2 (reviewed in [152]).
ncRNA mechanisms:
In this puzzle of gene regulation, genes that respond to the renin-angiotensin-aldosterone system (RAAS) have been studied, in particular for their potential to be direct miRNA targets. The angiotensin 1a receptor (Atgr1a) gene presents as a very interesting example given genetic variation among the population in its 3’UTR. miR-155 preferentially binds the A allele of Atgr1a, leading to a reduction of its expression; on the other hand, in individuals carrying the C variation, miR-155 does not control RNA translation of Atgr1a and therefore these patients exhibit a greater pressure in response to angiotensin II [156].
Literature investigating long ncRNAs in the regulation of blood pressure is still limited [157]. One noteworthy study by Leung and colleagues identified a long ncRNA (Lnc-Ang362) that was modulated in VSMCs following angiotensin II stimulation. This long ncRNA is proximal to miR-221 and miR-222, two short ncRNA that, as mentioned above, regulate VSMC proliferation [115]. The three ncRNAs are similarly upregulated by angiotensin II stimulation and knockdown of Lnc-Ang362 reduces the expression of the miR-221 and miR-222, suggesting that they are co-regulated with the lncRNA. Furthermore, the knockdown of Lnc-Ang362 reduces VSMC proliferation, as do miR-221 and miR-222 modulation [158]. In a very recent study, Lin et al. performed a profile of circulating lncRNAs comparing hypertensive versus normotensive individuals [159]. In hypertensive patients, AK098656 was the most upregulated lncRNA. Furthermore, the authors studied the molecular mechanism involving AK098656, finding that it promotes a VSMC synthetic phenotype, by directly regulating the lysosomal degradation of MYH11 and fbronectin-1 (FN1) [159].
3.4. Vascular aneurysm
An aneurysm is defined as a dilation of a vessel to greater than 1.5 times its normal size [160]. All vessels can develop such pathology; however, the aortic aneurysm is the most dangerous condition. Aortic aneurysms are most commonly located in the abdomen (AAA) but can also be found in the thoracic aorta (TAA). Normally the specific vascular enlargement does not cause any symptoms except when a rupture occurs [161]. In the event of rupture however, patients develop massive internal bleeding resulting in death unless immediately treated. AAA and TAA are mainly associated with problems of extracellular matrix (ECM) integrity [162]. For instance, the imbalance between ECM degradation and deposition leads to an increased fragility of the aorta with the consequential dilatation. However, molecular mechanisms of this condition are not well understood. Perhaps this is most clearly illustrated by incomplete understanding of how syndromes driven by transforming growth factor β (TGF- β) signalling loss of function mutations (Loey Dietz Syndrome) and by mutations leading to increased TGF- β bioavailability and signalling (Marfan Syndrome) both cause aortic aneurysm [163].
Acetylation related studies:
Some insights have been gained by focusing on the regulation of SMAD Family Member 2 (SMAD2) expression. SMAD2 expression, phosphorylation and nuclear localization is increased in human and animal model aneurysm samples. This is associated with increased H3K9/14 acetylation and H3K4 methylation at the SMAD2 locus [164] that directed expression of a larger SMAD2 isoform. Differential association between MYC repressive and P53/P300 activating complexes at the SMAD2 locus was also seen in VSMCs from normal vs aneurysm containing thoracic aortas [165]. Interestingly, another group has shown that SMAD3 overexpression in VSMCs can shift TGF-β treatment from a pro-differentiation stimulus to a de-differentiation stimulus in the context of restenosis [166]. It is likely that SMAD protein dosing and/or alternate SMAD transcripts, fine tune gene expression programs downstream of TGF-β stimulation, potentially regulating ECM composition. Nearly 30% of thoracic aortic aneurysms have identified genetic roots [167]. Additional key proteins in aneurysm biology have been identified through GWAS studies and include extracellular matrix proteins and modifiers, VSMC contractile proteins, and TGF-β signalling molecules [168]. Epigenetic mechanisms that regulate the expression the genes encoding these critical proteins will likely also prove dominant determinants for the progression of aneurysm. In addition to targeting the HDAC9- MALAT1-EZH2 complex mentioned in stenosis section above [139], others have also shown benefits of inhibiting HDACs in aneurysm. In this study, a class I HDAC inhibitor (MS-275) and a class IIa HDAC inhibitor (MC-1568) were tested in an experimental model of aortic aneurysm where Angiotensin II was given to ApoE null mice. While both inhibitors reduced aneurysm size, the class IIa inhibitor dramatically improved survival [169].
ncRNA studies:
ncRNAs have been identified as important regulators of ECM cellular metabolism as well. In AAA pathogenesis, the miR-29 family has been thoroughly studied. miR-29b is significantly downregulated in both human and mouse AAA specimens [170], while important component of ECM, direct targets of miR-29b, such as collagens (Col1a1, Col3a1, Col5a1), and elastin (Eln) are up-regulated [170]. Additional miRNAs known to be regulators of aneurysm include: the above-mentioned miR-143 and miR-145, for direct impact on VSMC biology, and they are indeed modulated in AAA human formations [109]; and miR-24, as a key regulator of vascular inflammation in AAA development [171]. Very few publications have investigated the role of long ncRNAs in aneurysm. Those that have been studied include the previously discussed ANRIL [172] and HIF1 alpha-antisense RNA 1 (HIF1A-AS1) which is upregulated by Brahma-related gene 1 (BRG1) over expression [173] and in TAAs [174].
3.5. Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is an expanding public health problem. This pathology is characterized by increased blood pressure in the pulmonary circulation and consequential pathological hypertrophy of the right ventricle. PAH is a very complex disease in which there is a massive remodelling of the pulmonary vasculature due to pulmonary artery smooth muscle cell (PASMC) proliferation, pulmonary artery endothelial cell (PAEC) dysfunction and inflammatory cell infiltration in the arterial wall [175].
Acetylation related studies:
HDAC inhibition, which is potently anti-proliferative [176] and antiinflammatory [177], has been effective at reducing pulmonary artery pressures and right sided cardiac remodeling. This effect has been demonstrated using various HDAC inhibitors including valproic acid, SAHA [178], MGCD0103, MS275 [179], MC1568 [180], and tubastatin A [181]. Valproic acid, MGCD0103, and MS275 are selective for Class I HDACs. MC1568 is a class IIa HDAC inhibitor while Tubastatin A is a Class IIb HDAC inhibitor. SAHA is classified as a pan HDAC inhibitor (excluding Class III/Sirtums), as is the compound Tricostatin A (TSA). Unlike other HDAC inhibitors employed in PAH models, TSA treatment has failed to provide beneficial effects [182, 183]. It is quite remarkable that inhibition of these HDAC classes independently resulted in reduced PAH and it remains unclear why two pan HDAC inhibitors produced opposing results, though one reason could be potential toxicity of high dose TSA. Again, related to histone acetylation, inhibition of BET bromodomains with JQ1 reduced pulmonary and RV pressures while reducing pulmonary vessel diameters in a Sugen-5416/hypoxia PAH model [38].
Methylation related studies:
Investigation of histone methylation regulators in PAH has been very limited. In a hypoxia model of PAH, it was shown that the Myocardin Related Transcription Factor A (MRTF-A) targeted H3K4 methyl transferases, ASH2 and WDR5, to activate pro-inflammatory genes. When ASH2 or WDR5 were disrupted selectively in endothelial cells, pulmonary hypertension was reduced along with a corresponding reduction in medial layer thickness of pulmonary vessels [184]. Critical roles for H2O2 regulating superoxide dismutase-2 (SOD2) and H202 responsive Hypoxia Inducible Factor 1 Subunit Alpha (HIF1α) have been established in PAH, particularly those forms driven by hypoxia. DNA methylation has been shown to be a potent silencer of Superoxide Dismutase 2 (SOD2) gene expression in PAH which is likely linked to increased expression of DNMT1 and DNMT3B. The DNMT inhibitor, 5-aza-2’-deoxycytidine has been shown to increase SOD2 expression and slow PASMC proliferation [185].
ncRNA studies:
In PAH, the interaction between PASMCs and PAECs is fundamental [186], and miRNAs play a critical role in this communication. For instance, miR-143 and miR-145 are physically transferred from PASMCs to PAECs via multiple mechanisms [187, 188]. miR-143 and miR-145 can be packaged into exosomes for secretion mediated transfer, or directly transferred through cell protrusions called tunnelling nanotubes. Tunnelling nanotube mediated transfer is stimulated by TGF-β , allowing the modulation of the angiogenic and proliferative capacities of ECs, and therefore regulating their physio-pathological behaviour [189].
At least two long ncRNAs are known modifiers of PAH. One is MALAT1, which regulates EC physiology [190]. This long ncRNAs is induced by hypoxia, and its pharmacological inhibition reduces blood flow recovery and capillary density following hind limb ischemia. Furthermore, a single nucleotide polymorphism (SNP) located in the MALAT1 sequence is able to influence the risk of PAH development in humans [191]. Another EC-enriched long ncRNA named ncRNA n342419 (MANTIS), may also be important in the pathogenesis of PAH. MANTIS is downregulated in patients with idiopathic PAH and in rats treated with monocrotaline. It is thought this lncRNC regulates endothelial cell phenotype via its ability to interact with the chromatin remodelling protein, BRG1 [192].
3.6. Vascular complication of diabetes
Diabetes mellitus collectively describes a number of metabolic diseases characterized by hyperglycemia. Over 300 millions of persons worldwide are affected by diabetes: 10% of those patients have type 1 diabetes (insulin-dependent), while 90% of patients have type 2 defined also as noninsulin-dependent diabetes [193]. It is likely that the number of people diagnosed with type 2 diabetes will rise in the next decades as a consequence of lifestyle patterns contributing to obesity [194]. The main causes of death and disability in diabetic patients are vascular diseases. Chronic hyperglycemia contributes to trigger endothelial dysfunction, diabetic nephropathy, cardiomyopathy, and retinopathy as well as atherosclerotic. Indeed, in patients with diabetes the incidence/prevalence of, and mortality from, atherosclerosis-induced cardiovascular diseases are 2–8-fold higher than in those without diabetes [195, 196].
Acetylation related studies:
The acetylation of histone and non-histone proteins is clearly affected by hyperglycemia. Remarkably, at the homogenate level, activity of Zn+ dependent HDACs (e.g. [197]) and multiple HATs (e.g. [198]) can be increased in these conditions. Outcomes of the activation of these opposing forces is theoretically dependent upon many factors, including enzyme targeting subcellular domains and regulatory complex association. Generally, however, much work remains to understand how individual HATs and HDACs are specifically directed to target proteins in cardiovascular systems. Differential O-GlcNAcalation of epigenetic enzymes in diabetes has been proposed to modify their activity and regulatory complex assembly, including for HDACs [199]. Another biochemical consequence of prolonged hyperglycemia is the accumulation of advanced glycation endproducts (AGEs) [200]. It is thought that AGEs decrease the expression of class III HDACs, SIRT1 and SIRT3 ([201, 202). Class III HDACs/Sirtuins are intimately coupled to cellular metabolism via NAD+ regulation and there role in diabetes associated diseases have been thoroughly reviewed in [203] and [204]. Hyperglycemia induced histone acetyl transferase activity has been linked to increased lysine acetylation and activation of inflammatory genes in monocytes, including from human diabetic patients (e.g. [205] and [198]). Also, expression the 66-kDa Src Homology domain containing protein (p66Shc) has been strongly associated with diabetes induced ROS and inflammation in endothelial cells and monocytes. Acetylation of histone proteins in the p66shc locus and of the p66shc protein itself have been linked to hyperglycemia induced regulation of epigenetic modifier enzymes ([206, 207]).
In terms of epigenetic mechanism regulating diabetes, HDACs have been a major focus of research. Overwhelming evidence supports HDAC inhibition as enhancing insulin secretion, decreasing inflammation, and improving insulin sensitivity via mechanisms that include up regulation of glucose transporters (reviewed in [208]). Interestingly, BET protein inhibition appears to promote insulin production ([209, 210]) and Beta cell differentiation, though may suppress insulin gene expression in chronic treatment settings ([211]).
Methylation related studies:
Given the increased inflammation associated with diabetes, it is not surprising that hyperglycemia/T2D causes increased NF-κB dependent gene expression and increased expression of NF- κB (e.g. p65 subunit). This is associated with increased H3K4 methylation (activating) in the p65 locus. Genetic targeting of the histone methyl transferase, Set7, blunted local H3K4 methylation and blocked hyperglycemia induced p65 expression in vitro and in vivo ([212, 213]). Additional methylation related studies have been conducted in diabetic retinopathy models and are reviewed in [203].
ncRNA studies:
Among the genes modulated by diabetes, ncRNAs represent a large class. For instance, miR-221 and −222, that we have already discussed for vascular restenosis [115], are also upregulated in VSMCs and arteries of diabetic mice [214]. Furthermore, abnormal VSMC proliferation in response to arterial injury in diabetic rats is blunted by the knock-down of both miRNAs [214]. EC miRNAs modulated in hyperglycemic conditions have also been identified. MiR-146 is upregulated in early diabetic retinopathy in rats, through a mechanism involving NF- κB as a direct miR-146 target [215]. On the other hand, hyperglycemia causes the downregulation of miR-200b, by directing controlling the activity of PRC2 and therefore the level of H3 lysine-27 trimethylation mark on the miRNA promoter [216].
The role of lncRNA-myocardial infarction-associated transcript (MIAT), a very well conserved lncRNA, has been investigated in diabetes mellitus-induced microvascular dysfunction [217]. In this study, Yan et al demonstrated that MIAT is expressed in several retinal layers in humans and rats. Furthermore, MIAT expression was positively modulated in ECs exposed to high glucose and in diabetic retinas. Interestingly, inhibition of MIAT was able to improve diabetes mellitus-induced vascular leakage as well as visual function in diabetic rats [217].
Another lncRNA studied in diabetes is the maternally expressed gene (Meg3), which is involved in diabetes-related microvascular dysfunction. ECs exposed to high glucose and oxidative stress downregulated MEG3 expression, and a similar modulation has been also observed in retinas of streptozotocin-induced diabetic mice [218]. Meg3 knock-down activates EC proliferation in vitro by modulating the Phosphoinositide 3-kinases (PI3K)/Protein Kinase B (AKT) pathway, while in vivo this worsens vascular leakage in the retina of mice [218].
These examples highlight several ncRNA mechanisms in diabetes aggravated vascular conditions, however further studies are needed to definitively establish whether ncRNAs might be considered as therapeutic options in such pathological settings.
4. Innovative therapeutic approaches.
Accumulating evidence for the role of ncRNAs in vascular disease development has triggered interest in developing RNA targeting therapeutic approaches to treat such pathologies. While drugs based on chemical inhibitors against epigenetic enzymes have been discussed above, here we will consider innovative therapeutics designed on ncRNAs. The translation of ncRNA pre-clinical results to humans has been limited by hurdles in delivery, organ selectivity and long-term safety.
For miRNA-based therapeutics, the possibilities are to use miRNA inhibitors or mimics, in order to, respectively, reduce or restore the ncRNA level. Inhibition might be achieved by different technologies :
small molecule inhibitors;
anti-miRNA oligodeoxyribonucleotides.
The first approach is based on the use of low molecular-weight compounds, designed to target the miRNA machinery at different levels, including maturation and/or degradation. For instance, small molecules can impair Dicer activity, inhibiting miRNA maturation and its subsequent processing. The first proof of concept for this approach was accomplished by Gumireddy et al. who demonstrated the capacity of the small molecule diazobenzene 1 to directly inhibit miR-21 action [219].
However, the most widely used methods to inhibit miRNA expression are based on direct anti-miRNA approaches. This technology relies on the use of short antisense oligonucleotides complementary to the specific miRNA. Annealing of the antisense sequence to the mature target miRNA induces the formation of a stable duplex, impairing the function of the ncRNA by its direct degradation [220]. Over the last decade, the chemistry behind such molecules has been extensively improved in order to increase their cellular uptake, nuclease resistance, and binding specificity [221, 222]. For instance, addition of a 2’-O-methyl or 2’-O-methoxyethyl group to the 2’-ribose of the RNA backbone stabilizes the oligonucleotide. This prevents its nuclease degradation outside of the cell as well as the consequent endonucleolytic activity of the RISC complex, thus leading to an irreversible inhibition of the miRNA [223]. Beside this antagomir molecule design, another important anti-miRNA technology includes that of locked-nucleotides (LNAs). This chemical modification relies on the formation of a methylene bridge between the 2 oxygen and the 4 carbon of the ribose ring. This modified nucleotide structure is able to profoundly increase the affinity towards a complementary single-stranded RNA molecule [222]. The high specificity of LNA oligonucleotides allows lower anti-miRNA doses, therefore limiting possible side effects. Thus, the first clinical trial on human was based on an anti-miR-122 molecule based on LNA chemistry [224].
A large amount of work has shown that the level of miRNAs in pathological conditions is reduced [50]. In line with such consideration, therapeutic strategies aimed at miRNA restoration were required. For this purpose, miRNA mimics are the most widely utilized approach. Mimics are synthetic double- or triple-stranded RNA molecules with one strand identical to the mimicked miRNA [225]. In term of chemical modifications, strategies exist to improve miRNA mimic properties, despite restrictions in the available modifications relative to antagomirs.
Given the importance of lncRNAs in vascular diseases, they hold potential as novel therapeutic targets despite several significant translation hurdles. The principal limit is that the majority of lncRNAs are not evolutionarily conserved between human and the studied animal models. Thus, directly testing human lncRNAs is, in many cases, challenging. In addition, information regarding molecular kinetics of lncRNAs is still limited. The available methods to target lncRNAs are based on therapeutic silencing using RNAi with antisense oligos and GapmeRs. The first approach is mainly used to knock-down cytoplasmic RNAs, while GapmeRs are more suitable for lncRNAs located in the nucleus. In terms of chemistry, the antisense oligos, except the length, is identical to what we have described for miRNAs. However, GapmeRs are hybrid molecules: they are LNA/DNA antisense oligonucleotides with perfect sequence complementary to their lncRNA target. Once in cells, they sequester their target within a highly stable DNARNA heteroduplexes that leads to RNase H mediated target degradation.
Safe and effective in vivo delivery is necessary before consideration as a therapeutic strategy [226]. Unfortunately, several limitations exist here as well, including natural barriers such as the circulatory system, kidney filtration, immune cell surveillance, and non-specific tissue uptake. Alternate strategies include delivery via viral vectors for both loss- and gain-of function approaches, such as lentivirus and adeno-associated virus (AAV). However, this approach may raise safety concerns due to potential random insertion of the viral genome into the host genome as well as the persistent expression of the inserted gene [227]. A very powerful alternative approach may be the association of the above mentioned technologies with advancing nanomedicine delivery technologies. This approach involves the use of nanoparticles (NPs), which are any particulate material with a size between 1 and 100 nm. NPs can be loaded with various chemical compounds depending on hydrophobicity of the compound and the nanoparticle. Since these molecules can be also linked to specific superficial elements, such as antibodies or aptamers [228], they can become “smart” nanocarriers designed to target specific types of diseased cells, and thus release the therapeutic drug within the diseased region. This combined approach has great potential for drug delivery and circumvents the limitations of conventional medicine. However, despite use of such technology in the cancer field, examples of NP-delivered drugs or nucleic acids for the treatment of the cardiovascular diseases has been limited [229], but it is rapidly becoming a hot topic for future clinical applications in this area [230].
5. Concluding remarks
Epigenetic mechanisms play a central role in the development of vascular diseases. This involves multiple mechanisms, driven by external stimuli, that sum to modulate gene expression. Among those, we have discussed classical epigenetics mechanisms, such as DNA methylation and histone modifications, and also more recently uncovered paradigms based on the biology of ncRNAs. Extreme enhancements in next generation sequencing methodology have driven more complete understanding of complex regulatory systems in which DNA and histone modifications are critical. Similar ingenuity cleared technical and conceptual hurdles in the word of ncRNAs, which has rapidly yielded near exponential expansion and continuing discovery of new classes of these players. From a research perspective, the number of unstudied and uncharacterized ncRNAs is astounding. Also, given the diversity of effects seen with specific HDAC and BET protein inhibitors in vascular disease, there is a clear need for additional class and/or isoform specific inhibitors of other epigenetic readers, writers and eraser proteins. Moreover, a better understanding of how these modifiers are targeted to specific genes and how these mechanisms of targeting might differ based on cell type and physiologic state is needed. We are optimistic that epigenetic therapies will soon enter the clinic for prevention and treatment of cardiovascular disease.
Highlights :
Cardiovascular disease remains the leading cause of death worldwide and novel therapeutic strategies are needed.
Epigenetic based therapies for cardiovascular disease show promise in preclinical models but remain under-developed clinically.
Additional class and isoform specific small molecule inhibitors for epigenetic proteins are needed.
The field has only scratched the surface of long non-coding RNA research in cardiovascular disease.
Sources of Funding
This work was supported by the following grants: Ministry of Health (#GR2013_02355011) to L.E. and NIH AG056858 to M.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: none declared
Reference:
- [1].Tirziu D, Giordano FJ, Simons M, Cell communications in the heart, Circulation 122(9) (2010) 928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Billaud M, Lohman AW, Johnstone SR, Biwer LA, Mutchler S, Isakson BE, Regulation of cellular communication by signaling microdomains in the blood vessel wall, Pharmacol Rev 66(2) (2014) 513–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Elia L, Condorelli G, The involvement of epigenetics in vascular disease development, Int J Biochem Cell Biol 107 (2019) 27–31. [DOI] [PubMed] [Google Scholar]
- [4].Egger G, Liang G, Aparicio A, Jones PA, Epigenetics in human disease and prospects for epigenetic therapy, Nature 429(6990) (2004) 457–63. [DOI] [PubMed] [Google Scholar]
- [5].Quintavalle M, Condorelli G, Elia L, Arterial remodeling and atherosclerosis: miRNAs involvement, Vascul Pharmacol 55(4) (2011) 106–10. [DOI] [PubMed] [Google Scholar]
- [6].van der Harst P, de Windt LJ, Chambers JC, Translational Perspective on Epigenetics in Cardiovascular Disease, J Am Coll Cardiol 70(5) (2017) 590–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xu C, Bian C, Lam R, Dong A, Min J, The structural basis for selective binding of non-methylated CpG islands by the CFP1 CXXC domain, Nat Commun 2 (2011) 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M, The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties, Nucleic Acids Res 31(6) (2003) 1765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Severin PM, Zou X, Schulten K, Gaub HE, Effects of cytosine hydroxymethylation on DNA strand separation, Biophys J 104(1) (2013) 208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F, Matarese F, Stunnenberg HG, Oliviero S, Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs, Cell 155(1) (2013) 121–34. [DOI] [PubMed] [Google Scholar]
- [11].Arechederra M, Daian F, Yim A, Bazai SK, Richelme S, Dono R, Saurin AJ, Habermann BH, Maina F, Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer, Nat Commun 9(1) (2018) 3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lyko F, The DNA methyltransferase family: a versatile toolkit for epigenetic regulation, Nat Rev Genet 19(2) (2018) 81–92. [DOI] [PubMed] [Google Scholar]
- [13].Kinney SR, Pradhan S, Ten eleven translocation enzymes and 5-hydroxymethylation in mammalian development and cancer, Adv Exp Med Biol 754 (2013) 57–79. [DOI] [PubMed] [Google Scholar]
- [14].Neri F, Incarnato D, Krepelova A, Rapelli S, Anselmi F, Parlato C, Medana C, Dal Bello F, Oliviero S, Single-Base Resolution Analysis of 5-Formyl and 5-Carboxyl Cytosine Reveals Promoter DNA Methylation Dynamics, Cell Rep 10(5) (2015) 674–683. [DOI] [PubMed] [Google Scholar]
- [15].Greco CM, Kunderfranco P, Rubino M, Larcher V, Carullo P, Anselmo A, Kurz K, Carell T, Angius A, Latronico MV, Papait R, Condorelli G, DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy, Nat Commun 7 (2016) 12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC, Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2), Nucleic Acids Res 32(14) (2004) 4100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Raiber EA, Murat P, Chirgadze DY, Beraldi D, Luisi BF, Balasubramanian S, 5- Formylcytosine alters the structure of the DNA double helix, Nat Struct Mol Biol 22(1) (2015) 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, Liu Y, Byrum SD, Mackintosh SG, Zhong M, Tackett A, Wang G, Hon LS, Fang G, Swenberg JA, Xiao AZ, DNA methylation on N(6)-adenine in mammalian embryonic stem cells, Nature 532(7599) (2016) 329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Allfrey VG, Faulkner R, Mirsky AE, Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis, Proc Natl Acad Sci U S A 51 (1964) 786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang H, Sabati BR, Garcia BA, Allis CD, Zhao Y, SnapShot: histone modifications, Cell 159(2) (2014) 458–458 el. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rousseaux S, Khochbin S, Histone Acylation beyond Acetylation: Terra Incognita in Chromatin Biology, Cell J 17(1) (2015) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sabari BR, Zhang D, Allis CD, Zhao Y, Metabolic regulation of gene expression through histone acylations, Nat Rev Mol Cell Biol 18(2) (2017) 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, Dai L, Shimada M, Cross JR, Zhao Y, Roeder RG, Allis CD, Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation, Mol Cell 58(2) (2015) 203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, Qi S, Li J, Colak G, Chen Y, Xia C, Peng C, Ruan H, Kirkey M, Wang D, Jensen LM, Kwon OK, Lee S, Pletcher SD, Tan M, Lombard DB, White KP, Zhao H, Li J, Roeder RG, Yang X, Zhao Y, Metabolic Regulation of Gene Expression by Histone Lysine beta-Hydroxybutyrylation, Mol Cell 62(2) (2016) 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, Wan L, Huang H, Tang Z, Zhao Y, Roeder RG, Shi X, Allis CD, Li H, Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain, Mol Cell 62(2) (2016) 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Carrozza MJ, Utley RT, Workman JL, Cote J, The diverse functions of histone acetyltransferase complexes, Trends Genet 19(6) (2003) 321–9. [DOI] [PubMed] [Google Scholar]
- [27].McKinsey TA, The biology and therapeutic implications of HDACs in the heart, Handb Exp Pharmacol 206 (2011) 57–78. [DOI] [PubMed] [Google Scholar]
- [28].Wright LH, Menick DR, A class of their own: exploring the nondeacetylase roles of class IIa HDACs in cardiovascular disease, Am J Physiol Heart Circ Physiol 311(1) (2016) H199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains, Nature 468(7327) (2010) 1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Greschik H, Schule R, Gunther T, Selective targeting of epigenetic reader domains, Expert Opin Drug Discov 12(5) (2017) 449–463. [DOI] [PubMed] [Google Scholar]
- [31].Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription, Mol Cell 19(4) (2005) 523–34. [DOI] [PubMed] [Google Scholar]
- [32].Smestad J, Erber L, Chen Y, Maher LJ 3rd, Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism, iScience 2 (2018) 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cornett EM, Dickson BM, Vaughan RM, Krishnan S, Trievel RC, Strahl BD, Rothbart SB, Substrate Specificity Profiling of Histone-Modifying Enzymes by Peptide Microarray, Methods Enzymol 574 (2016) 31–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hyun K, Jeon J, Park K, Kim J, Writing, erasing and reading histone lysine methylations, Exp Mol Med 49(4) (2017) e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, Flores ER, Northcott PA, Rajaram V, Li W, Shilatifard A, Sillitoe RV, Chen K, Lee MG, MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes, Mol Cell 70(5) (2018) 825–841 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Stathis A, Bertoni F, BET Proteins as Targets for Anticancer Treatment, Cancer Discov 8(1) (2018) 24–36. [DOI] [PubMed] [Google Scholar]
- [37].Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, Yang T, Wang H, Luscinskas FW, Croce K, Bradner JE, Plutzky J, NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis, Mol Cell 56(2) (2014) 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils-Bonnet S, Tremblay E, Nadeau V, Lambert C, Paradis R, Provencher S, Bonnet S, Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension, Circ Res 117(6) (2015) 525–35. [DOI] [PubMed] [Google Scholar]
- [39].Wang B, Zhang M, Takayama T, Shi X, Roenneburg DA, Kent KC, Guo LW, BET Bromodomain Blockade Mitigates Intimal Hyperplasia in Rat Carotid Arteries, EBioMedicine 2(11) (2015) 1650–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, Xavier RJ, Trained immunity: A program of innate immune memory in health and disease, Science 352(6284) (2016) aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Guida F, Sandanger TM, Castagne R, Campanella G, Polidoro S, Palli D, Krogh V, Tumino R, Sacerdote C, Panico S, Severi G, Kyrtopoulos SA, Georgiadis P, Vermeulen RC, Lund E, Vineis P, Chadeau-Hyam M, Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation, Hum Mol Genet 24(8) (2015) 2349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R, Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes, Proc Natl Acad Sci U S A 105(26) (2008) 9047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jadhav U, Cavazza A, Banerjee KK, Xie H, O’Neill NK, Saenz-Vash V, Herbert Z, Madha S, Orkin SH, Zhai H, Shivdasani RA, Extensive Recovery of Embryonic Enhancer and Gene Memory Stored in Hypomethylated Enhancer DNA, Mol Cell 74(3) (2019) 542–554 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Beermann J, Piccoli MT, Viereck J, Thum T, Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches, Physiol Rev 96(4) (2016) 1297–325. [DOI] [PubMed] [Google Scholar]
- [45].St Laurent G, Wahlestedt C, Kapranov P, The Landscape of long noncoding RNA classification, Trends Genet 31(5) (2015) 239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Elia L, Condorelli G, RNA (Epi)genetics in cardiovascular diseases, J Mol Cell Cardiol 89(Pt A) (2015) 11–6. [DOI] [PubMed] [Google Scholar]
- [47].Bartel DP, MicroRNAs: genomics, biogenesis, mechanism, and function, Cell 116(2) (2004) 281–97. [DOI] [PubMed] [Google Scholar]
- [48].Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E, Deadenylation is a widespread effect of miRNA regulation, RNA 15(1) (2009) 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ha M, Kim VN, Regulation of microRNA biogenesis, Nat Rev Mol Cell Biol 15(8) (2014) 50924. [DOI] [PubMed] [Google Scholar]
- [50].Barwari T, Joshi A, Mayr M, MicroRNAs in Cardiovascular Disease, J Am Coll Cardiol 68(23) (2016) 2577–2584. [DOI] [PubMed] [Google Scholar]
- [51].Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS, Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine, Mol Ther Nucleic Acids 8 (2017) 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rajan KS, Velmurugan G, Gopal P, Ramprasath T, Babu DD, Krithika S, Jenifer YC, Freddy A, William GJ, Kalpana K, Ramasamy S, Abundant and Altered Expression of PIWI-Interacting RNAs during Cardiac Hypertrophy, Heart Lung Circ 25(10) (2016) 1013–20. [DOI] [PubMed] [Google Scholar]
- [53].Siomi MC, Sato K, Pezic D, Aravin AA, PIWI-interacting small RNAs: the vanguard of genome defence, Nat Rev Mol Cell Biol 12(4) (2011) 246–58. [DOI] [PubMed] [Google Scholar]
- [54].Mihailescu R, Gene expression regulation: lessons from noncoding RNAs, RNA 21(4) (2015) 695–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Quinn JJ, Chang HY, Unique features of long non-coding RNA biogenesis and function, Nat Rev Genet 17(1) (2016) 47–62. [DOI] [PubMed] [Google Scholar]
- [56].Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, Olson EN, A micropeptide encoded by a putative long noncoding RNA regulates muscle performance, Cell 160(4) (2015) 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigo R, The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression, Genome Res 22(9) (2012) 1775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang CP, Dorn GW 2nd, Thum T, Heymans S, Cardiolinc n., Long noncoding RNAs in cardiac development and ageing, Nat Rev Cardiol 12(7) (2015) 415–25. [DOI] [PubMed] [Google Scholar]
- [59].Rashid F, Shah A, Shan G, Long Non-coding RNAs in the Cytoplasm, Genomics Proteomics Bioinformatics 14(2) (2016) 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mercer TR, Mattick JS, Structure and function of long noncoding RNAs in epigenetic regulation, Nat Struct Mol Biol 20(3) (2013) 300–7. [DOI] [PubMed] [Google Scholar]
- [61].Dorn LE, Tual-Chalot S, Stellos K, Accornero F, RNA epigenetics and cardiovascular diseases, J Mol Cell Cardiol 129 (2019) 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Moore KJ, Tabas I, Macrophages in the pathogenesis of atherosclerosis, Cell 145(3) (2011) 34155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Libby P, Ridker PM, Hansson GK, Progress and challenges in translating the biology of atherosclerosis, Nature 473(7347) (2011) 317–25. [DOI] [PubMed] [Google Scholar]
- [64].Owens GK, Regulation of differentiation of vascular smooth muscle cells, Physiol Rev 75(3) (1995) 487–517. [DOI] [PubMed] [Google Scholar]
- [65].Moore KJ, Sheedy FJ, Fisher EA, Macrophages in atherosclerosis: a dynamic balance, Nat Rev Immunol 13(10) (2013) 709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kawahara K, Watanabe S, Ohshima T, Soejima Y, Oishi T, Aratani S, Nakata M, Shibata M, Inoue K, Amano T, Fujii R, Yanai K, Hagiwara M, Fukamizu A, Maruyama I, Nakajima T, Hypernuclear acetylation in atherosclerotic lesions and activated vascular smooth muscle cells, Biochem Biophys Res Commun 266(2) (1999) 417–24. [DOI] [PubMed] [Google Scholar]
- [67].Cao D, Wang Z, Zhang CL, Oh J, Xing W, Li S, Richardson JA, Wang DZ, Olson EN, Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin, Mol Cell Biol 25(1) (2005) 364–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Manabe I, Owens GK, CArG elements control smooth muscle subtype-specific expression of smooth muscle myosin in vivo, J Clin Invest 107(7) (2001) 823–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Xu X, Ha CH, Wong C, Wang W, Hausser A, Pfizenmaier K, Olson EN, McKinsey TA, Jin ZG, Angiotensin II stimulates protein kinase D-dependent histone deacetylase 5 phosphorylation and nuclear export leading to vascular smooth muscle cell hypertrophy, Arterioscler Thromb Vasc Biol 27(11) (2007) 2355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Choi JH, Nam KH, Kim J, Baek MW, Park JE, Park HY, Kwon HJ, Kwon OS, Kim DY, Oh GT, Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice, Arterioscler Thromb Vasc Biol 25(11) (2005) 2404–9. [DOI] [PubMed] [Google Scholar]
- [71].Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, Pepe AE, Wang G, Habi O, deFalco E, Cockerill G, Mason JC, Hu Y, Xu Q, Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow, Circulation 121(1) (2010) 13242. [DOI] [PubMed] [Google Scholar]
- [72].Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA, Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy, Proc Natl Acad Sci U S A 108(10) (2011) 4123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, Monzani V, Saripalli C, Mascagni P, Reece TB, Ambardekar AV, Granzier HL, Dinarello CA, McKinsey TA, Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism, Sci Transl Med 10(427) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sola S, Mir MQ, Lerakis S, Tandon N, Khan BV, Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure, J Am Coll Cardiol 47(2) (2006) 332–7. [DOI] [PubMed] [Google Scholar]
- [75].Krum H, Ashton E, Reid C, Kalff V, Rogers J, Amarena J, Singh B, Tonkin A, Double-blind, randomized, placebo-controlled study of high-dose HMG CoA reductase inhibitor therapy on ventricular remodeling, pro-inflammatory cytokines and neurohormonal parameters in patients with chronic systolic heart failure, J Card Fail 13(1) (2007) 1–7. [DOI] [PubMed] [Google Scholar]
- [76].Liu G, Zheng XX, Xu YL, Ru J, Hui RT, Huang XH, Meta-analysis of the effect of statins on mortality in patients with preserved ejection fraction, Am J Cardiol 113(7) (2014) 1198–204. [DOI] [PubMed] [Google Scholar]
- [77].Cao Q, Rong S, Repa JJ, St Clair R, Parks JS, Mishra N, Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development, Arterioscler Thromb Vasc Biol 34(9) (2014) 1871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA, Haldar SM, BET bromodomain inhibition suppresses innate inflammatory and profbrotic transcriptional networks in heart failure, Sci Transl Med 9(390) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Stratton MS, Lin CY, Anand P, Tatman PD, Ferguson BS, Wickers ST, Ambardekar AV, Sucharov CC, Bradner JE, Haldar SM, McKinsey TA, Signal-Dependent Recruitment of BRD4 to Cardiomyocyte Super-Enhancers Is Suppressed by a MicroRNA, Cell Rep 16(5) (2016) 1366–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Jahagirdar R, Zhang H, Azhar S, Tobin J, Attwell S, Yu R, Wu J, McLure KG, Hansen HC, Wagner GS, Young PR, Srivastava RA, Wong NC, Johansson J, A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice, Atherosclerosis 236(1) (2014) 91–100. [DOI] [PubMed] [Google Scholar]
- [81].Nicholls SJ, Puri R, Wolski K, Ballantyne CM, Barter PJ, Brewer HB, Kastelein JJ, Hu B, Uno K, Kataoka Y, Herrman JP, Merkely B, Borgman M, Nissen SE, Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial, Am J Cardiovasc Drugs 16(1) (2016) 55–65. [DOI] [PubMed] [Google Scholar]
- [82].Kulikowski E, Halliday C, Johansson J, Sweeney M, Lebioda K, Wong N, Haarhaus M, Brandenburg V, Beddhu S, Tonelli M, Zoccali C, Kalantar-Zadeh K, Apabetalone Mediated Epigenetic Modulation is Associated with Favorable Kidney Function and Alkaline Phosphatase Profile in Patients with Chronic Kidney Disease, Kidney Blood Press Res 43(2) (2018) 449–457. [DOI] [PubMed] [Google Scholar]
- [83].Gilham D, Tsujikawa LM, Sarsons CD, Halliday C, Wasiak S, Stotz SC, Jahagirdar R, Sweeney M, Johansson JO, Wong NCW, Kalantar-Zadeh K, Kulikowski E, Apabetalone downregulates factors and pathways associated with vascular calcification, Atherosclerosis 280 (2019) 75–84. [DOI] [PubMed] [Google Scholar]
- [84].Wasiak S, Gilham D, Tsujikawa LM, Halliday C, Calosing C, Jahagirdar R, Johansson J, Sweeney M, Wong NC, Kulikowski E, Downregulation of the Complement Cascade In Vitro, in Mice and in Patients with Cardiovascular Disease by the BET Protein Inhibitor Apabetalone (RVX-208), J Cardiovasc Transl Res 10(4) (2017) 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Liu Z, Wang P, Chen H, Wold EA, Tian B, Brasier AR, Zhou J, Drug Discovery Targeting Bromodomain-Containing Protein 4, J Med Chem 60(11) (2017) 4533–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Lv YC, Tang YY, Zhang P, Wan W, Yao F, He PP, Xie W, Mo ZC, Shi JF, Wu JF, Peng J, Liu D, Cayabyab FS, Zheng XL, Tang XY, Ouyang XP, Tang CK, Histone Methyltransferase Enhancer of Zeste Homolog 2-Mediated ABCA1 Promoter DNA Methylation Contributes to the Progression of Atherosclerosis, PLoS One 11(6) (2016) e0157265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Mokou M, Klein J, Makridakis M, Bitsika V, Bascands JL, Saulnier-Blache JS, Mullen W, Sacherer M, Zoidakis J, Pieske B, Mischak H, Roubelakis MG, Schanstra JP, Vlahou A, Proteomics based identification of KDM5 histone demethylases associated with cardiovascular disease, EBioMedicine 41 (2019) 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramirez-Ruz J, Gomez A, Goncalves I, Moran S, Esteller M, DNA methylation map of human atherosclerosis, Circ Cardiovasc Genet 7(5) (2014) 692–700. [DOI] [PubMed] [Google Scholar]
- [89].Valencia-Morales Mdel P, Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramirez-Ruz J, Gomez A, Moran S, Lund G, Rodriguez-Rios D, Lopez-Gonzalez G, Ramirez-Nava M, de la Rocha C, Sanchez-Flores A, Esteller M, The DNA methylation drift of the atherosclerotic aorta increases with lesion progression, BMC Med Genomics 8 (2015) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lacey M, Baribault C, Ehrlich KC, Ehrlich M, Atherosclerosis-associated differentially methylated regions can reflect the disease phenotype and are often at enhancers, Atherosclerosis 280 (2019) 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kumar A, Kumar S, Vikram A, Hoffman TA, Naqvi A, Lewarchik CM, Kim YR, Irani K, Histone and DNA methylation-mediated epigenetic downregulation of endothelial Kruppel-like factor 2 by low-density lipoprotein cholesterol, Arterioscler Thromb Vasc Biol 33(8) (2013) 1936–42. [DOI] [PubMed] [Google Scholar]
- [92].Elia L, Kunderfranco P, Carullo P, Vacchiano M, Farina FM, Hall IF, Mantero S, Panico C, Papait R, Condorelli G, Quintavalle M, UHRF1 epigenetically orchestrates smooth muscle cell plasticity in arterial disease, J Clin Invest 128(6) (2018) 2473–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yu J, Qiu Y, Yang J, Bian S, Chen G, Deng M, Kang H, Huang L, DNMT1-PPARgamma pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice, Sci Rep 6 (2016)30053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Tang RZ, Zhu JJ, Yang FF, Zhang YP, Xie SA, Liu YF, Yao WJ, Pang W, Han LL, Kong W, Wang YX, Zhang T, Zhou J, DNA methyltransferase 1 and Kruppel-like factor 4 axis regulates macrophage inflammation and atherosclerosis, J Mol Cell Cardiol 128 (2019) 11–24. [DOI] [PubMed] [Google Scholar]
- [95].Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, Hwa J, Yu J, Martin KA, Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity, Circulation 128(18) (2013) 2047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Suarez Y, Wang C, Manes TD, Pober JS, Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation, J Immunol 184(1) (2010) 21–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ, MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1, Proc Natl Acad Sci US A 105(5) (2008) 1516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Nicoli S, Standley C, Walker P, Hurlstone A, Fogarty KE, Lawson ND, MicroRNA-mediated integration of haemodynamics and Vegf signalling during angiogenesis, Nature 464(7292) (2010) 1196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Schober A, Nazari-Jahantigh M, Wei Y, Bidzhekov K, Gremse F, Grommes J, Megens RT, Heyll K, Noels H, Hristov M, Wang S, Kiessling F, Olson EN, Weber C, MicroRNA-126–5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1, Nat Med 20(4) (2014) 368–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hoesel B, Schmid JA, The complexity of NF-kappaB signaling in inflammation and cancer, Mol Cancer 12 (2013) 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Registry M, Blackwell TS, Baron RM, Feinberg MW, MicroRNA-181b regulates NF-kappaB-mediated vascular inflammation, J Clin Invest 122(6) (2012) 1973–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sun X, He S, Wara AKM, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, Croce K, Feinberg MW, Systemic delivery of microRNA-181b inhibits nuclear factor-kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice, Circ Res 114(1) (2014) 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Raitoharju E, Lyytikainen LP, Levula M, Oksala N, Mennander A, Tarkka M, Klopp N, Illig T, Kahonen M, Karhunen PJ, Laaksonen R, Lehtimaki T, miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study, Atherosclerosis 219(1) (2011) 211–7. [DOI] [PubMed] [Google Scholar]
- [104].Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI, Fish JE, MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways, EMBO Mol Med 5(7) (2013) 1017–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Shan K, Jiang Q, Wang XQ, Wang YN, Yang H, Yao MD, Liu C, Li XM, Yao J, Liu B, Zhang YY, Y. J, B. Yan, Role of long non-coding RNA-RNCR3 in atherosclerosis-related vascular dysfunction, Cell Death Dis 7(6) (2016) e2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L Mujtaba S, Gil J, Walsh MJ, Zhou MM, Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a, Mol Cell 38(5) (2010) 662–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, Finstermeier K, Stahringer A, Wilfert W, Beutner F, Gielen S, Schuler G, Gabel G, Bergert H, Bechmann I, Stadler PF, Thiery J, Teupser D, Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks, PLoS Genet 9(7) (2013) e1003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Holdt LM, Beutner F, Scholz M, Gielen S, Gabel G, Bergert H, Schuler G, Thiery J, Teupser D, ANRIL expression is associated with atherosclerosis risk at chromosome 9p21, Arterioscler Thromb Vasc Biol 30(3) (2010) 620–7. [DOI] [PubMed] [Google Scholar]
- [109].Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G, The knockout of miR-143 and −145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease, Cell Death Differ 16(12) (2009) 1590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel- Duby R, Olson EN, MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury, Genes Dev 23(18) (2009) 2166–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T, Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster, J Clin Invest 119(9) (2009) 2634–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Quintavalle M, Elia L, Condorelli G, Courtneidge SA, MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro, J Cell Biol 189(1) (2010) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Norata GD, Pinna C, Zappella F, Elia L, Sala A, Condorelli G, Catapano AL, MicroRNA 143–145 deficiency impairs vascular function, Int J Immunopathol Pharmacol 25(2) (2012) 467–74. [DOI] [PubMed] [Google Scholar]
- [114].Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta N, Steer BM, Ingram AJ, Gupta M, Al-Omran M, Teoh H, Marsden PA, Verma S, MicroRNA-145 targeted therapy reduces atherosclerosis, Circulation 126(11 Suppl 1) (2012) S81–90. [DOI] [PubMed] [Google Scholar]
- [115].Liu X, Cheng Y, Yang J, Xu L, Zhang C, Cell-specific effects of miR-221/22 in vessels: molecular mechanism and therapeutic application, J Mol Cell Cardiol 52(1) (2012) 245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A, Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype, J Biol Chem 284(6) (2009) 3728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bazan HA, Hatfield SA, O’Malley CB, Brooks AJ, Lightell D Jr., Woods TC, Acute Loss of miR-221 and miR-222 in the Atherosclerotic Plaque Shoulder Accompanies Plaque Rupture, Stroke 46(11) (2015) 3285–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Ballantyne MD, Pinel K, Dakin R, Vesey AT, Diver L, Mackenzie R, Garcia R, Welsh P, Sattar N, Hamilton G, Joshi N, Dweck MR, Miano JM, McBride MW, Newby DE, McDonald RA, Baker AH, Smooth Muscle Enriched Long Noncoding RNA (SMILR) Regulates Cell Proliferation, Circulation 133(21) (2016) 2050–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Li X, Yang L, Chen LL, The Biogenesis, Functions, and Challenges of Circular RNAs, Mol Cell 71(3) (2018) 428–442. [DOI] [PubMed] [Google Scholar]
- [120].Hsu MT, Coca-Prados M, Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells, Nature 280(5720) (1979) 339–40. [DOI] [PubMed] [Google Scholar]
- [121].Cocquerelle C, Mascrez B, Hetuin D, Bailleul B, Mis-splicing yields circular RNA molecules, FASEB J 7(1) (1993) 155–60. [DOI] [PubMed] [Google Scholar]
- [122].Cheng X, Joe B, Circular RNAs in rat models of cardiovascular and renal diseases, Physiol Genomics 49(9) (2017) 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, Kohlmaier A, Herbst A, Northoff BH, Nicolaou A, Gabel G, Beutner F, Scholz M, Thiery J, Musunuru K, Krohn K, Mann M, Teupser D, Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans, Nat Commun 7 (2016) 12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Werfel S, Nothjunge S, Schwarzmayr T, Strom TM, Meitinger T, Engelhardt S, Characterization of circular RNAs in human, mouse and rat hearts, J Mol Cell Cardiol 98 (2016) 103–7. [DOI] [PubMed] [Google Scholar]
- [125].Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F, Rajewsky N, Circular RNAs are a large class of animal RNAs with regulatory potency, Nature 495(7441) (2013) 333–8. [DOI] [PubMed] [Google Scholar]
- [126].Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J, Natural RNA circles function as efficient microRNA sponges, Nature 495(7441) (2013) 384–8. [DOI] [PubMed] [Google Scholar]
- [127].Hall IF, Climent M, Quintavalle M, Farina FM, Schorn T, Zani S, Carullo P, Kunderfranco P, Civilini E, Condorelli G, Elia L, Circ_Lrp6, a Circular RNA Enriched in Vascular Smooth Muscle Cells, Acts as a Sponge Regulating miRNA-145 Function, Circ Res 124(4) (2019) 498–510. [DOI] [PubMed] [Google Scholar]
- [128].Zhang M, Wu JF, Chen WJ, Tang SL, Mo ZC, Tang YY, Li Y, Wang JL, Liu XY, Peng J, Chen K, He PP, Lv YC, Ouyang XP, Yao F, Tang DP, Cayabyab FS, Zhang DW, Zheng XL, Tian GP, Tang CK, MicroRNA-27a/b regulates cellular cholesterol effux, influx and esterification/hydrolysis in THP-1 macrophages, Atherosclerosis 234(1) (2014) 54–64. [DOI] [PubMed] [Google Scholar]
- [129].Yang K, He YS, Wang XQ, Lu L, Chen QJ, Liu J, Sun Z, Shen WF, MiR-146a inhibits oxidized low-density lipoprotein-induced lipid accumulation and inflammatory response via targeting toll-like receptor 4, FEBS Lett 585(6) (2011) 854–60. [DOI] [PubMed] [Google Scholar]
- [130].Chen T, Huang Z, Wang L, Wang Y, Wu F, Meng S, Wang C, MicroRNA-125a-5p partly regulates the inflammatory response, lipid uptake, and ORP9 expression in oxLDL-stimulated monocyte/macrophages, Cardiovasc Res 83(1) (2009) 131–9. [DOI] [PubMed] [Google Scholar]
- [131].Sala F, Aranda JF, Rotllan N, Ramirez CM, Aryal B, Elia L, Condorelli G, Catapano AL, Fernandez-Hernando C, Norata GD, MiR-143/145 deficiency attenuates the progression of atherosclerosis in Ldlr−/−mice, Thromb Haemost 112(4) (2014) 796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, O’Neill LA, Moore MJ, Caffrey DR, Fitzgerald KA, A long noncoding RNA mediates both activation and repression of immune response genes, Science 341(6147) (2013) 789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Indolfi C, Stabile E, Perrino C, Chiariello M, Mechanisms of restenosis after angioplasty and approach to therapy (Review), Int J Mol Med 2(2) (1998) 143–148. [PubMed] [Google Scholar]
- [134].McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK, Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo, J Clin Invest 116(1) (2006) 3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D, Bruemmer D, Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition, Arterioscler Thromb Vasc Biol 31(4) (2011) 851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Zhang M, Urabe G, Little C, Wang B, Kent AM, Huang Y, Kent KC, Guo LW, HDAC6 Regulates the MRTF-A/SRF Axis and Vascular Smooth Muscle Cell Plasticity, JACC Basic Transl Sci 3(6) (2018) 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Lino Cardenas CL, Kessinger CW, Chou EL, Ghoshhajra B, Yeri AS, Das S, Weintraub NL, Malhotra R, Jaffer FA, Lindsay ME, HDAC9 complex inhibition improves smooth muscle-dependent stenotic vascular disease, JCI Insight 4(2) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lino Cardenas CL, Kessinger CW, MacDonald C, Jassar AS, Isselbacher EM, Jaffer FA, Lindsay ME, Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm, JCI Insight 3(5) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Lino Cardenas CL, Kessinger CW, Cheng Y, MacDonald C, MacGillivray T, Ghoshhajra B, Huleihel L, Nuri S, Yeri AS, Jaffer FA, Kaminski N, Ellinor P, Weintraub NL, Malhotra R, Isselbacher EM, Lindsay ME, An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm, Nat Commun 9(1) (2018) 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Elia L, Quintavalle M, Epigenetics and Vascular Diseases: Influence of Non-coding RNAs and Their Clinical Implications, Front Cardiovasc Med 4 (2017) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Gareri C, De Rosa S, Indolii C, MicroRNAs for Restenosis and Thrombosis After Vascular Injury, Circ Res 118(7) (2016) 1170–84. [DOI] [PubMed] [Google Scholar]
- [142].Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C, MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation, Circ Res 105(2) (2009) 158–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C, MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation, Circ Res 100(11) (2007) 1579–88. [DOI] [PubMed] [Google Scholar]
- [144].Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, Cai Y, Huang H, Yang Y, Liu Y, Xu Z, He D, Zhang X, Hu X, Pinello L, Zhong D, He F, Yuan GC, Wang DZ, Zeng C, LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity, Circulation 130(17) (2014) 1452–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Wise IA, Charchar FJ, Epigenetic Modifications in Essential Hypertension, Int J Mol Sci 17(4) (2016) 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Wang J, Gong L, Tan Y, Hui R, Wang Y, Hypertensive epigenetics: from DNA methylation to microRNAs, J Hum Hypertens 29(10) (2015) 575–82. [DOI] [PubMed] [Google Scholar]
- [147].Friso S, Carvajal CA, Fardella CE, Olivieri O, Epigenetics and arterial hypertension: the challenge of emerging evidence, Transl Res 165(1) (2015) 154–65. [DOI] [PubMed] [Google Scholar]
- [148].Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, Kelly TN, Saleheen D, Lehne B, Leach IM, Drong AW, Abbott J, Wahl S, Tan ST, Scott WR, Campanella G, Chadeau-Hyam M, Afzal U, Ahluwalia TS, Bonder MJ, Chen P, Dehghan A, Edwards TL, Esko T, Go MJ, Harris SE, Hartiala J, Kasela S, Kasturiratne A, Khor CC, Kleber ME, Li H, Yu Mok Z, Nakatochi M, Sapari NS, Saxena R, Stewart AFR, Stolk L, Tabara Y, Teh AL, Wu Y, Wu JY, Zhang Y, Aits I, Da Silva Couto Alves A, Das S, Dorajoo R, Hopewell JC, Kim YK, Koivula RW, Luan J, Lyytikainen LP, Nguyen QN, Pereira MA, Postmus I, Raitakari OT, Scannell Bryan M, Scott RA, Sorice R, Tragante V, Traglia M, White J, Yamamoto K, Zhang Y, Adair LS, Ahmed A, Akiyama K, Asif R, Aung T, Barroso I, Bjonnes A, Braun TR, Cai H, Chang LC, Chen CH, Cheng CY, Chong YS, Collins R, Courtney R, Davies G, Delgado G, Do LD, Doevendans PA, Gansevoort RT, Gao YT, Grammer TB, Grarup N, Grewal J, Gu D, Wander GS, Hartikainen AL, Hazen SL, He J, Heng CK, Hixson JE, Hofman A, Hsu C, Huang W, Husemoen LLN, Hwang JY, Ichihara S, Igase M, Isono M, Justesen JM, Katsuya T, Kibriya MG, Kim YJ, Kishimoto M, Koh WP, Kohara K, Kumari M, Kwek K, Lee NR, Lee J, Liao J, Lieb W, Liewald DCM, Matsubara T, Matsushita Y, Meitinger T, Mihailov E, Milani L, Mills R, Mononen N, Muller-Nurasyid M, Nabika T, Nakashima E, Ng HK, Nikus K, Nutile T, Ohkubo T, Ohnaka K, Parish S, Paternoster L, Peng H, Peters A, Pham ST, Pinidiyapathirage MJ, Rahman M, Rakugi H, Rolandsson O, Ann Rozario M, Ruggiero D, Sala CF, Sarju R, Shimokawa K, Snieder H, Sparso T, Spiering W, Starr JM, Stott DJ, Stram DO, Sugiyama T, Szymczak S, Tang WHW, Tong L, Trompet S, Turjanmaa V, Ueshima H, Uitterlinden AG, Umemura S, Vaarasmaki M, van Dam RM, van Gilst WH, van Veldhuisen DJ, Viikari JS, Waldenberger M, Wang Y, Wang A, Wilson R, Wong TY, Xiang YB, Yamaguchi S, Ye X, Young RD, Young TL, Yuan JM, Zhou X, Asselbergs FW, Ciullo M, Clarke R, Deloukas P, Franke A, Franks PW, Franks S, Friedlander Y, Gross MD, Guo Z, Hansen T, Jarvelin MR, Jorgensen T, Jukema JW, Kahonen M, Kajio H, Kivimaki M, Lee JY, Lehtimaki T, Linneberg A, Miki T, Pedersen O, Samani NJ, Sorensen TIA, Takayanagi R, Toniolo D, B. consortium, C.A. GRAMplusCD, S. LifeLines Cohort, C. InterAct, Ahsan H, Allayee H, Chen YT, Danesh J, Deary IJ, Franco OH, Franke L, Heijman BT, Holbrook JD, Isaacs A, Kim BJ, Lin X, Liu J, Marz W, Metspalu A, Mohlke KL, Sanghera DK, Shu XO, van Meurs JBJ, Vithana E, Wickremasinghe AR, Wijmenga C, Wolffenbuttel BHW, Yokota M, Zheng W, Zhu D, Vineis P, Kyrtopoulos SA, Kleinjans JCS, McCarthy MI, Soong R, Gieger C, Scott J, Teo YY, He J, Elliott P, Tai ES, van der Harst P, Kooner JS, Chambers JC, Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation, Nat Genet 47(11) (2015) 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Cardinale JP, Sriramula S, Pariaut R, Guggilam A, Mariappan N, Elks CM, Francis J, HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats, Hypertension 56(3) (2010) 437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Lee HA, Kang SH, Kim M, Lee E, Cho HM, Moon EK, Kim I, Histone deacetylase inhibition ameliorates hypertension and hyperglycemia in a model of Cushing’s syndrome, Am J Physiol Endocrinol Metab 314(1) (2018) E39–E52. [DOI] [PubMed] [Google Scholar]
- [151].Yang YM, Shi RH, Xu CX, Li L, BRD4 expression in patients with essential hypertension and its effect on blood pressure in spontaneously hypertensive rats, J Am Soc Hypertens 12(12) (2018) e107–e117. [DOI] [PubMed] [Google Scholar]
- [152].Stoll S, Wang C, Qiu H, DNA Methylation and Histone Modification in Hypertension, Int J Mol Sci 19(4) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Riviere G, Lienhard D, Andrieu T, Vieau D, Frey BM, Frey FJ, Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation, Epigenetics 6(4) (2011) 478–89. [DOI] [PubMed] [Google Scholar]
- [154].Friso S, Pizzolo F, Choi SW, Guarini P, Castagna A, Ravagnani V, Carletto A, Pattini P, Corrocher R, Olivieri O, Epigenetic control of 11 beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension, Atherosclerosis 199(2) (2008) 323–7. [DOI] [PubMed] [Google Scholar]
- [155].Zhong Q, Liu C, Fan R, Duan S, Xu X, Zhao J, Mao S, Zhu W, Hao L, Yin F, Zhang L, Association of SCNN1B promoter methylation with essential hypertension, Mol Med Rep 14(6) (2016) 5422–5428. [DOI] [PubMed] [Google Scholar]
- [156].Sethupathy P, Borel C, Gagnebin M, Grant GR, Deutsch S, Elton TS, Hatzigeorgiou AG, Antonarakis SE, Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3’ untranslated region: a mechanism for functional single-nucleotide polymorphisms related to phenotypes, Am J Hum Genet 81(2) (2007) 405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Marques FZ, Booth SA, Charchar FJ, The emerging role of non-coding RNA in essential hypertension and blood pressure regulation, J Hum Hypertens 29(8) (2015) 459–67. [DOI] [PubMed] [Google Scholar]
- [158].Leung A, Trac C, Jin W, Lanting L, Akbany A, Saetrom P, Schones DE, Natarajan R, Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells, Circ Res 113(3) (2013) 266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Jin L, Lin X, Yang L, Fan X, Wang W, Li S, Li J, Liu X, Bao M, Cui X, Yang J, Cui Q, Geng B, Cai J, AK098656, a Novel Vascular Smooth Muscle Cell-Dominant Long Noncoding RNA, Promotes Hypertension, Hypertension 71(2) (2018) 262–272. [DOI] [PubMed] [Google Scholar]
- [160].Mortality results for randomised controlled trial of early elective surgery or ultrasonographic surveillance for small abdominal aortic aneurysms. The UK Small Aneurysm Trial Participants, Lancet 352(9141) (1998) 1649–55. [PubMed] [Google Scholar]
- [161].Kent KC, Clinical practice. Abdominal aortic aneurysms, N Engl J Med 371(22) (2014) 2101–8. [DOI] [PubMed] [Google Scholar]
- [162].Rabkin SW, The Role Matrix Metalloproteinases in the Production of Aortic Aneurysm, Prog Mol Biol Transl Sci 147 (2017) 239–265. [DOI] [PubMed] [Google Scholar]
- [163].Takeda N, Hara H, Fujiwara T, Kanaya T, Maemura S, Komuro I, TGF-beta Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections, Int J Mol Sci 19(7) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Gomez D, Coyet A, Ollivier V, Jeunemaitre X, Jondeau G, Michel JB, Vranckx R, Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms, Cardiovasc Res 89(2) (2011) 446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Gomez D, Kessler K, Michel JB, Vranckx R, Modifications of chromatin dynamics control Smad2 pathway activation in aneurysmal smooth muscle cells, Circ Res 113(7) (2013) 881–90. [DOI] [PubMed] [Google Scholar]
- [166].Tsai S, Hollenbeck ST, Ryer EJ, Edlin R, Yamanouchi D, Kundi R, Wang C, Liu B, Kent KC, TGF-beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation, Am J Physiol Heart Circ Physiol 297(2) (2009) H540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Rizzo JA, Elefteriades JA, Familial thoracic aortic aneurysms and dissections--incidence, modes of inheritance, and phenotypic patterns, Ann Thorac Surg 82(4) (2006) 1400–5. [DOI] [PubMed] [Google Scholar]
- [168].Verhagen JMA, Kempers M, Cozijnsen L, Bouma BJ, Duijnhouwer AL, Post JG, Hilhorst-Hofstee Y, Bekkers S, Kerstjens-Frederikse WS, van Brakel TJ, Lambermon E, Wessels MW, Loeys BL, Roos-Hesselink JW, van de Laar I, National BAV Working Group on, Taa, Expert consensus recommendations on the cardiogenetic care for patients with thoracic aortic disease and their first-degree relatives, Int J Cardiol 258 (2018) 243–248. [DOI] [PubMed] [Google Scholar]
- [169].Galan M, Varona S, Orriols M, Rodriguez JA, Aguilo S, Dilme J, Camacho M, Martinez-Gonzalez J, Rodriguez C, Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: therapeutic potential of HDAC inhibitors, Dis Model Mech 9(5) (2016) 541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ, McConnell MV, Dalman RL, Spin JM, Tsao PS, Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development, J Clin Invest 122(2) (2012) 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Maegdefessel L, Spin JM, Raaz U, Eken SM, Toh R, Azuma J, Adam M, Nakagami F, Heymann HM, Chernogubova E, Jin H, Roy J, Hultgren R, Caidahl K, Schrepfer S, Hamsten A, Eriksson P, McConnell MV, Dalman RL, Tsao PS, miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development, Nat Commun 5 (2014) 5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Congrains A, Kamide K, Katsuya T, Yasuda O, Oguro R, Yamamoto K, Ohishi M, Rakugi H, CVD-associated non-coding RNA ANRIL, modulates expression of atherogenic pathways in VSMC, Biochem Biophys Res Commun 419(4) (2012) 612–6. [DOI] [PubMed] [Google Scholar]
- [173].Wang S, Zhang X, Yuan Y, Tan M, Zhang L, Xue X, Yan Y, Han L, Xu Z, BRG1 expression is increased in thoracic aortic aneurysms and regulates proliferation and apoptosis of vascular smooth muscle cells through the long non-coding RNA HIF1A-AS1 in vitro, Eur J Cardiothorac Surg 47(3) (2015) 439–46. [DOI] [PubMed] [Google Scholar]
- [174].He Q, Tan J, Yu B, Shi W, Liang K, Long noncoding RNA HIF1A-AS1A reduces apoptosis of vascular smooth muscle cells: implications for the pathogenesis of thoracoabdominal aorta aneurysm, Pharmazie 70(5) (2015) 310–5. [PubMed] [Google Scholar]
- [175].Rabinovitch M, Molecular pathogenesis of pulmonary arterial hypertension, J Clin Invest 122(12) (2012) 4306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Cantoni S, Galletti M, Zambelli F, Valente S, Ponti F, Tassinari R, Pasquinelli G, Galie N, Ventura C, Sodium butyrate inhibits platelet-derived growth factor-induced proliferation and migration in pulmonary artery smooth muscle cells through Akt inhibition, FEBS J 280(9) (2013) 2042–55. [DOI] [PubMed] [Google Scholar]
- [177].Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ, Histone deacetylases as regulators of inflammation and immunity, Trends Immunol 32(7) (2011) 335–43. [DOI] [PubMed] [Google Scholar]
- [178].Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, Wilkins MR, Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid, Circulation 126(4) (2012) 455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, Weiser-Evans MC, Harral J, Irwin DC, Anwar A, Yeager ME, Li M, Watson PA, Nemenoff RA, Buttrick PM, Stenmark KR, McKinsey TA, Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism, Circ Res 110(5)(2012)739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D, Park H, Ju H, McLean DL, Comhair SA, Erzurum SC, Chun HJ, Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension, Circulation 131(2) (2015) 190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Boucherat O, Chabot S, Paulin R, Trinh I, Bourgeois A, Potus F, Lampron MC, Lambert C, Breuils-Bonnet S, Nadeau V, Paradis R, Goncharova EA, Provencher S, Bonnet S, HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension, Sci Rep 7(1) (2017) 4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF, Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats, Am J Respir Crit Care Med 183(10) (2011) 1402–10. [DOI] [PubMed] [Google Scholar]
- [183].De Raaf MA, Hussaini AA, Gomez-Arroyo J, Kraskaukas D, Farkas D, Happe C, Voelkel NF, Bogaard HJ, Histone deacetylase inhibition with trichostatin A does not reverse severe angioproliferative pulmonary hypertension in rats (2013 Grover Conference series), Pulm Circ 4(2) (2014) 237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Chen D, Yang Y, Cheng X, Fang F, Xu G, Yuan Z, Xia J, Kong H, Xie W, Wang H, Fang M, Gao Y, Xu Y, Megakaryocytic leukemia 1 directs a histone H3 lysine 4 methyltransferase complex to regulate hypoxic pulmonary hypertension, Hypertension 65(4) (2015) 821–33. [DOI] [PubMed] [Google Scholar]
- [185].Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J, Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target, Circulation 121(24) (2010) 2661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M, Cellular and molecular pathobiology of pulmonary arterial hypertension, J Am Coll Cardiol 43(12 Suppl S) (2004) 13S–24S. [DOI] [PubMed] [Google Scholar]
- [187].Deng L, Blanco FJ, Stevens H, Lu R, Caudrillier A, McBride M, McClure JD, Grant J, Thomas M, Frid M, Stenmark K, White K, Seto AG, Morrell NW, Bradshaw AC, MacLean MR, Baker AH, MicroRNA-143 Activation Regulates Smooth Muscle and Endothelial Cell Crosstalk in Pulmonary Arterial Hypertension, Circ Res 117(10) (2015) 870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Caruso P, Dempsie Y, Stevens HC, McDonald RA, Long L, Lu R, White K, Mair KM, McClure JD, Southwood M, Upton P, Xin M, van Rooij E, Olson EN, Morrell NW, MacLean MR, Baker AH, A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples, Circ Res 111(3) (2012) 290–300. [DOI] [PubMed] [Google Scholar]
- [189].Climent M, Quintavalle M, Miragoli M, Chen J, Condoreli G, Elia L, TGFbeta Triggers miR-143/145 Transfer From Smooth Muscle Cells to Endothelial Cells, Thereby Modulating Vessel Stabilization, Circ Res 116(11) (2015) 1753–64. [DOI] [PubMed] [Google Scholar]
- [190].Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, Boon RA, Dimmeler S, Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth, Circ Res 114(9) (2014) 1389–97. [DOI] [PubMed] [Google Scholar]
- [191].Zhuo Y, Zeng Q, Zhang P, Li G, Xie Q, Cheng Y, Functional polymorphism of lncRNA MALAT1 contributes to pulmonary arterial hypertension susceptibility in Chinese people, Clin Chem Lab Med 55(1) (2017) 38–46. [DOI] [PubMed] [Google Scholar]
- [192].Leisegang MS, Fork C, Josipovic I, Richter FM, Preussner J, Hu J, Miller MJ, Epah J, Hofmann P, Gunther S, Moll F, Valasarajan C, Heidler J, Ponomareva Y, Freiman TM, Maegdefessel L, Plate KH, Mittelbronn M, Uchida S, Kunne C, Stellos K, Schermuly RT, Weissmann N, Devraj K, Wittig I, Boon RA, Dimmeler S, Pullamsetti SS, Looso M, Miller FJ Jr., Brandes RP, Long Noncoding RNA MANTIS Facilitates Endothelial Angiogenic Function, Circulation 136(1) (2017) 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Amos AF, McCarty DJ, Zimmet P, The rising global burden of diabetes and its complications: estimates and projections to the year 2010, Diabet Med 14 Suppl 5 (1997) S1–85. [PubMed] [Google Scholar]
- [194].Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP, The continuing epidemics of obesity and diabetes in the United States, JAMA 286(10) (2001) 1195–200. [DOI] [PubMed] [Google Scholar]
- [195].Grundy SM, Howard B, Smith S Jr., Eckel R, Redberg R, Bonow RO, Prevention Conference VI: Diabetes and Cardiovascular Disease: executive summary: conference proceeding for healthcare professionals from a special writing group of the American Heart Association, Circulation 105(18) (2002) 2231–9. [DOI] [PubMed] [Google Scholar]
- [196].Howard BV, Rodriguez BL, Bennett PH, Harris MI, Hamman R, Kuller LH, Pearson TA, Wylie-Rosett J, Prevention Conference VI: Diabetes and Cardiovascular disease: Writing Group I: epidemiology, Circulation 105(18) (2002) e132–7. [DOI] [PubMed] [Google Scholar]
- [197].Xu Z, Tong Q, Zhang Z, Wang S, Zheng Y, Liu Q, Qian LB, Chen SY, Sun J, Cai L, Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway, Clin Sci (Lond) 131(15) (2017) 1841–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Yun JM, Jialal I, Devaraj S, Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin, J Nutr Biochem 22(5) (2011) 450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Marsh SA, Collins HE, Chatham JC, Protein O-GlcNAcylation and cardiovascular (patho)physiology, J Biol Chem 289(50) (2014) 34449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ Jr., Chow WS, Stern D, Schmidt AM, Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts, Nat Med 4(9) (1998) 1025–31. [DOI] [PubMed] [Google Scholar]
- [201].Chang M, Zhang B, Tian Y, Hu M, Zhang G, Di Z, Wang X, Liu Z, Gu N, Liu Y, AGEs Decreased SIRT3 Expression and SIRT3 Activation Protected AGEs-Induced EPCs’ Dysfunction and Strengthened Anti-oxidant Capacity, Inflammation 40(2) (2017) 473–485. [DOI] [PubMed] [Google Scholar]
- [202].Li P, Zhang L, Zhou C, Lin N, Liu A, Sirt 1 activator inhibits the AGE-induced apoptosis and p53 acetylation in human vascular endothelial cells, J Toxicol Sci 40(5) (2015) 615–24. [DOI] [PubMed] [Google Scholar]
- [203].Khullar M, Cheema BS, Raut SK, Emerging Evidence of Epigenetic Modifications in Vascular Complication of Diabetes, Front Endocrinol (Lausanne) 8 (2017) 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Kitada M, Ogura Y, Monno I, Koya D, Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function, Front Endocrinol (Lausanne) 10 (2019) 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Miao F, Gonzalo IG, Lanting L, Natarajan R, In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions, J Biol Chem 279(17) (2004) 18091–7. [DOI] [PubMed] [Google Scholar]
- [206].Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, Huang S, Liu JJ, Lu YB, Zhang ZQ, Yang RF, Zhang R, Cai H, Liu DP, Liang CC, Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction, Circ Res 109(6) (2011) 639–48. [DOI] [PubMed] [Google Scholar]
- [207].Kumar S, Kim YR, Vikram A, Naqvi A, Li Q, Kassan M, Kumar V, Bachschmid MM, Jacobs JS, Kumar A, Irani K, Sirtuin1-regulated lysine acetylation of p66Shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction, Proc Natl Acad Sci US A 114(7) (2017) 17141719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Luís MHP, Schütz F, Choudhury Mahua, Chapter 35 - HDACs in Diabetes: A New Era of Epigenetic Drug,, Nutritional and Therapeutic Interventions for Diabetes and Metabolic Syndrome (Second Edition) (2018) 475–486. [Google Scholar]
- [209].Fu W, Farache J, Clardy SM, Hattori K, Mander P, Lee K, Rioja I, Weissleder R, Prinjha RK, Benoist C, Mathis D, Epigenetic modulation of type-1 diabetes via a dual effect on pancreatic macrophages and beta cells, Elife 3 (2014) e04631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Deeney JT, Belkina AC, Shirihai OS, Corkey BE, Denis GV, BET Bromodomain Proteins Brd2, Brd3 and Brd4 Selectively Regulate Metabolic Pathways in the Pancreatic beta-Cell, PLoS One 11(3) (2016) e0151329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Huijbregts L, Petersen MBK, Berthault C, Hansson M, Aiello V, Rachdi L, Grapin-Botton A, Honore C, Scharfmann R, Bromodomain and Extra Terminal Protein Inhibitors Promote Pancreatic Endocrine Cell Fate, Diabetes 68(4) (2019) 761–773. [DOI] [PubMed] [Google Scholar]
- [212].El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M, Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia, J Exp Med 205(10) (2008) 2409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, Calkin AC, Brownlee M, Cooper ME, El-Osta A, Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail, Diabetes 58(5) (2009) 1229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Lightell DJ Jr., Moss SC, Woods TC, Upregulation of miR-221 and −222 in response to increased extracellular signal-regulated kinases 1/2 activity exacerbates neointimal hyperplasia in diabetes mellitus, Atherosclerosis 269 (2018) 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Kovacs B, Lumayag S, Cowan C, Xu S, MicroRNAs in early diabetic retinopathy in streptozotocin-induced diabetic rats, Invest Ophthalmol Vis Sci 52(7) (2011) 4402–9. [DOI] [PubMed] [Google Scholar]
- [216].Ruiz MA, Feng B, Chakrabarti S, Polycomb repressive complex 2 regulates MiR-200b in retinal endothelial cells: potential relevance in diabetic retinopathy, PLoS One 10(4) (2015) e0123987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, Tao ZF, Song YC, Chen Q, Jiang Q, lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA, Circ Res 116(7) (2015) 1143–56. [DOI] [PubMed] [Google Scholar]
- [218].Qiu GZ, Tian W, Fu HT, Li CP, Liu B, Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction, Biochem Biophys Res Commun 471(1) (2016) 135–41. [DOI] [PubMed] [Google Scholar]
- [219].Gumireddy K, Young DD, Xiong X, Hogenesch JB, Huang Q, Deiters A, Small-molecule inhibitors of microrna miR-21 function, Angew Chem Int Ed Engl 47(39) (2008) 7482–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Garzon R, Marcucci G, Croce CM, Targeting microRNAs in cancer: rationale, strategies and challenges, Nat Rev Drug Discov 9(10) (2010) 775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S, MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice, Science 324(5935) (2009) 1710–3. [DOI] [PubMed] [Google Scholar]
- [222].Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjarn M, Hansen HF, Berger U, Gullans S, Kearney P, Sarnow P, Straarup EM, Kauppinen S, LNA-mediated microRNA silencing in non-human primates, Nature 452(7189) (2008) 896–9. [DOI] [PubMed] [Google Scholar]
- [223].Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M, Silencing of microRNAs in vivo with ‘antagomirs’, Nature 438(7068) (2005) 685–9. [DOI] [PubMed] [Google Scholar]
- [224].Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR, Treatment of HCV infection by targeting microRNA, N Engl J Med 368(18) (2013) 1685–94. [DOI] [PubMed] [Google Scholar]
- [225].Rupaimoole R, Slack FJ, MicroRNA therapeutics: towards a new era for the management of cancer and other diseases, Nat Rev Drug Discov 16(3) (2017) 203–222. [DOI] [PubMed] [Google Scholar]
- [226].Chen Y, Zhao H, Tan Z, Zhang C, Fu X, Bottleneck limitations for microRNA-based therapeutics from bench to the bedside, Pharmazie 70(3) (2015) 147–54. [PubMed] [Google Scholar]
- [227].Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M, LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1, Science 302(5644) (2003) 415–9. [DOI] [PubMed] [Google Scholar]
- [228].Jo H, Ban C, Aptamer-nanoparticle complexes as powerful diagnostic and therapeutic tools, Exp Mol Med 48 (2016) e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Wang B, Chen G, Urabe G, Xie R, Wang Y, Shi X, Guo LW, Gong S, Kent KC, A paradigm of endothelium-protective and stent-free anti-restenotic therapy using biomimetic nanoclusters, Biomaterials 178 (2018) 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Miragoli M, Ceriotti P, Iafisco M, Vacchiano M, Salvarani N, Alogna A, Carullo P, Ramirez-Rodriguez GB, Patricio T, Esposti LD, Rossi F, Ravanetti F, Pinelli S, Alinovi R, Erreni M, Rossi S, Condorelli G, Post H, Tampieri A, Catalucci D, Inhalation of peptide-loaded nanoparticles improves heart failure, Sci Transl Med 10(424) (2018). [DOI] [PubMed] [Google Scholar]