

Abstract
The development of drug resistance remains a critical problem for current HIV-1 antiviral therapies, creating a need for new inhibitors of HIV-1 replication. We previously reported on a novel anti-HIV-1 compound, N2-(phenoxyacetyl)-N-[4-(1-piperidinyl-carbonyl)benzyl]glycinamide (14), that binds to the highly conserved phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2)binding pocket of the HIV-1 matrix (MA) protein. In this study, we re-evaluate the hits from the virtual screen used to identify compound 14 and test them directly in an HIV-1 replication assay using primary human peripheral blood mononuclear cells. This study resulted in the identification of three new compounds with antiviral activity; 2-(4-{[3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl]methyl})-1-piperazinyl)-N-(4-methylphenyl)-acetamide (7), 3-(2-ethoxyphenyl)-5-[[4-(4-nitrophenyl)piperazin-1-yl]methyl]-1,2,4-oxadiazole (17), and N-[4-ethoxy-3-(1-piperidinylsulfonyl)phenyl]-2-(imidazo[2,1-b][1,3]thiazol-6-yl)acetamide (18), with compound 7 being the most potent of these hits. Mechanistic studies on 7 demonstrated that it directly interacts with and functions through HIV-1 MA. In accordance with our drug target, compound 7 competes with PI(4,5)P2 for MA binding and, as a result, diminishes the production of new virus. Mutation of residues within the PI(4,5)P2 binding site of MA decreased the antiviral effect of compound 7. Additionally, compound 7 displays a broadly neutralizing anti-HIV activity, with IC50 values of 7.5–15.6 μm for the group M isolates tested. Taken together, these results point towards a novel chemical probe that can be used to more closely study the biological role of MA and could, through further optimization, lead to a new class of anti-HIV-1 therapeutics.
Keywords: antiviral agents; HIV-1; matrix proteins; phosphatidylinositol 4,5-bisphosphates; virtual screening
Graphical Abstract
PIP2-ing HIV-1 to the post! The discovery and characterization of a new small-molecule inhibitor (shown) of HIV-1 replication that targets the HIV-1 matrix (MA) protein is described. This novel agent exhibits a broad therapeutic spectrum, inhibiting all of the group M isolates tested, and functions via the novel mechanism of disrupting the critical phosphatidylinositol 4,5-bisphosphate (PI[4,5]P2)-MA interaction.
Introduction
Owing to the emergence of drug-resistant strains and the cumulative toxicities associated with current therapies, demand remains for new inhibitors of HIV-1 replication. The HIV-1 matrix (MA) protein, encoded as the N-terminal portion of Gag, is critically involved in the late, assembly stages in the life cycle of HIV-1. Firstly, HIV-1 MA is proposed to act as a scaffold that brings together Env and Gag proteins in infected cells. Several lines of evidence support an interaction of MA with the gp41 cytoplasmic tail during assembly and a role for MA in facilitating the incorporation of Env into virus particle.[1] Until recently, this interaction was thought to be direct;[1e,2] however, other studies have implicated the participation of a cellular factor in both the interaction between Env and Gag, and the process of envelope incorporation.[3]
Secondly, the HIV-1 MA protein directs the localization of Gag to the plasma membrane. Specific interaction of MA with cellular transport proteins has been implicated in the trafficking of Gag to the plasma membrane. Once at the membrane, membrane binding of HIV-1 Gag is mediated by two signals in MA: the N-terminal myristic acid and the conserved basic region between amino acids 17 and 31.[4] The myristate moiety is considered to be regulated by a mechanism termed a myristoyl switch, whereby the N-terminal myristate is sequestered in the MA globular domain, but a structural change exposes the myristate and enhances Gag membrane binding.[5] The MA basic domain is also involved in specific localization of Gag to the plasma membrane, with mutations in this domain shifting Gag localization from the plasma membrane to intracellular vesicles in HeLa and T cells.[6] Perhaps most critically, however, the basic domain of MA has been demonstrated to interact with phosphatidylinositol 4,5-bisphosphate (PI[4,5]P2), a minor phospholipid that is concentrated primarily on the cytoplasmic leaflet of the plasma membrane.[5f,7] PI(4,5)P2 binding to HIV-1 MA is thought to serve two functions: inducing the conformational change that triggers myristate exposure[5f] and acting as a site-specific membrane anchor, allowing the targeting of Gag to the plasma membrane.[8,7c]
The structure of the PI(4,5)P2–MA complex has been solved by NMR spectroscopy.[5f] The PI(4,5)P2 binding site resides in a cleft in the HIV-1 MA globular head domain and is composed of a large number of charged residues. The majority of these residues are highly conserved across the HIV-1 subtypes, highlighting the functional importance of the PI(4,5)P2-MA interaction in the HIV-1 replication cycle. Given its functional importance and its high degree of conservation across isolates, we propose that the PI(4,5)P2 binding site might represent a new, attractive antiviral target. Using a combination of virtual and surface plasmon resonance (SPR)-based screening, we have previously identified a compound (Enamine ID T5872535; see Table S1 in the Supporting Information) that interacts with HIV-1 MA in the PI(4,5)P2 binding site and displays broad-range antiviral activity.[9] In this study, we re-evaluated the compounds identified from the initial virtual screening, this time, however, using antiviral efficacy as the primary screen. This approach resulted in the identification of three new compounds with antiviral activity. Mechanistic studies of the lead compound (7), demonstrated that it interacts with the MA protein, disrupts the interaction between MA and PI(4,5)P2, and possesses broad-spectrum anti-HIV-1 activity.
Results and Discussion
Identification of compound 7
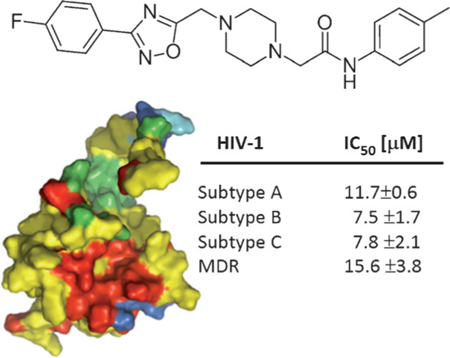
To identify small molecules capable of binding to and inhibiting the functions of HIV-1 MA, we previously coupled a structure-based in silico screening with a surface plasmon resonance (SPR)-based primary screen, which identified one active hit compound (14).[9] Therefore, to identify other potential hits that were not identified in the SPR-based screen, but still represent matrix-targeted inhibitors of HIV-1 replication, we evaluated the 19 compounds identified from the initial virtual screen (Table S1 in the Supporting Information) in a standardized HIV-1 replication assay (Table S2 in the Supporting Information). This led to the discovery of the novel antiviral compounds 7, 17, and 18, as well as confirmation of compound 14 as a genuine hit. The lead compound from this analysis, compound 7, inhibited the replication of the primary isolate HIV-189BZ167 with the lowest IC50 value, showed minimal toxicity, and did not inhibit the replication of a panel of non-retroviruses (Table 1; see alsoTable S3 in the Supporting Information). In addition, compound 7 inhibited the multi-drug-resistant (MDR) strain HIV-1 MDR769,[10] suggesting that it functions in a different manner than drugs currently in use as part of the “highly active antiretroviral therapy” (HAART) cocktail.
Table 1.
Therapeutic spectrum of compound 7 against HIV-1 subtypes.[a]
 | |||
|---|---|---|---|
| HIV-1 group M isolate | IC50 [μm] | Antiviral index (TC50/IC50) |
|
| 92UG031[b] | (clade A) | 11.7 ± 0.6 | > 8.6 |
| 92BR030[b] | (clade B) | 7.5 ± 1.7 | > 13.3 |
| 89BZ167[c] | (clade B) | 12.3 ± 2.9 | > 8.1 |
| MDR769[e] | (clade B) | 15.6 ± 3.8 | > 6.4 |
| 92BR025[b] | (clade C) | 7.8 ± 2.1 | > 12.8 |
| 93IN101[b] | (clade C) | 11.0 ± 2.8 | > 9.1 |
| 92UG024[c] | (clade D) | 8.5 ± 0.8 | > 11.8 |
| CMU08[c] | (clade E) | 11.7 ± 6.2 | > 8.6 |
| 93BR020[d] | (clade F) | 13.0 ± 0.1 | > 7.7 |
| G3[b] | (clade G) | 12.5 ± 6.8 | > 8.0 |
TC50 values for all compounds were determined to be > 100 μm in this study. IC50 values are the mean ± standard deviation (SD) of three replicates.
CCR5-tropic.
CXCR4-tropic.
CCR5/CXCR4 dual-tropic.
Clinical isolate presumed to be group M.
The genetic diversity of HIV can pose certain problems for the development of new antiviral drugs. Therefore, in order to be clinically viable, a compound must be able to inhibit the replication of genetically diverse isolates, especially isolates from the most globally prevalent subtypes (A, B, and C). Therefore, the antiviral efficacy of compound 7 was further evaluated in a standardized PBMC-based anti-HIV-1 assay with a panel of HIV-1 clinical isolates from different geographic locations that included HIV-1 group M subtypes A–G (Table 1). The panel included CCR5-tropic (R5), CXCR4-tropic (X4), and dual-tropic (R5X4) viruses. Compound 7 inhibited the replication of all the group M viruses tested, further highlighting the potential of targeting the MA protein.
Compound 7 exerts its action in the late, assembly stages of the HIV-1 life cycle
To determine the stage at which compound 7 exerts its antiviral effect, we made use of a single-round infection assay. The modular nature of this assay allows the determination of whether a compound acts on the early, preintegration events, or on the late, assembly events in the virus life cycle. Effects on assembly were identified by incubating the viral producer cells in the absence or presence of various concentrations of compound 7. Supernatants containing HIV-1HxBc2 envelope-pseudotyped virus (which encodes for firefly luciferase as a reporter gene) were then diluted tenfold and used to infect U87–CD4–CXCR4 target cells. Compound-induced effects are manifested as a decrease in infectivity in the target cells, compared with those infected with virus from untreated cells. Effects on early-stage events were determined by using virus produced in the absence of compound for infection of target cells in the absence or presence of various concentrations of compound 7. As can be seen in Figure 1, compound 7 displays effects only on the late, assembly stages of the HIV-1 replication cycle. Quantification of the virus produced in the presence of 100 mM of the compound indicates that it functions by decreasing the amount of virus released from the cells, as judged by p24 content (see theSupporting Information). These observations are in line with the proposed action of compound 7 through interaction with the HIV-1 MA protein.
Figure 1.
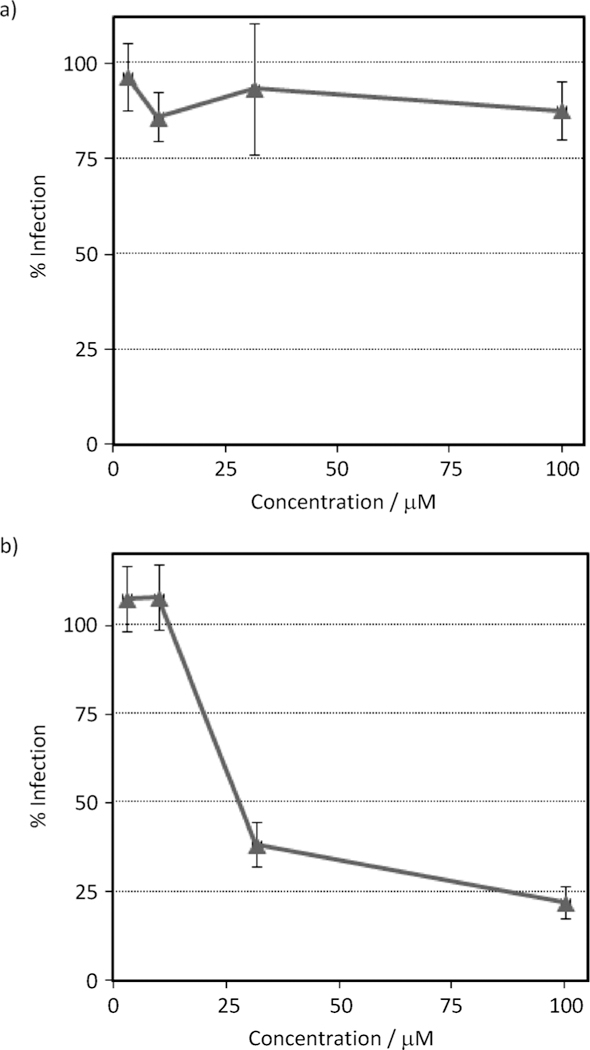
Compound 7 functions in the late stages of HIV-1 replication. a) Effects of compound 7 on early-stage events were determined by producing virus via transfection of 293T cells (as described in the Experimental Section) for infection of U87.CD4.CXCR4 target cells in the absence or presence of various concentrations of compound 7. b) To assess the effect of compound 7 on late-stage events, recombinant virus was produced from 293T cells either in the absence or presence of compound 7, and the culture supernatants containing pseudotype stocks were diluted tenfold and then used to infect target cells in the absence of test compound. Compound-induced effects are manifested as a decrease in infectivity in the target cells (measured as luciferase activity), normalized against the infectivity of virus from untreated cells. Data are the mean ± standard error of the mean (SEM) from at least four independent experiments (each performed in triplicate). Compound 7 (early stages): IC50 = not applicable; Compound 7 (late stages): IC50 = 21.2 μM. The antiviral effect of compound 7 was determined to be due to a specific effect on the virus as the compound was not cytotoxic to 293T cells up to a test concentration high of 1 mM (cf. Figure S2 in the Supporting Information), giving a therapeutic index in this assay of >47.
Compound 7 binds to HIV-1 MA and competes with PI(4,5)P2
Having determined that compound 7 has antiviral activity and exerts its effect in the late stages of the viral replication cycle by decreasing the total amount of virus produced, we next sought to investigate the proposed interaction of compound 7 with the HIV-1 MA protein. To explore the interaction of compound 7 with HIV-1 MA, we employed SPR. Using SPR and a novel HaloTag method of orientation, compound 14 was previously shown to be capable of interacting with HIV-1 MA in the PI(4,5)P2 binding site.[9] However, the HaloTag ligand (chloroalkane) can nonspecifically interact with compounds that display hydrophobic characteristics.[9] Therefore, we decided to immobilize HIV-1 MA directly on the sensor chip surface. In order to preserve the PI(4,5)P2 binding site and create a more active protein surface, the immobilization of the MA protein was performed in the presence of compound 14. As can be seen in Figure 2a, compound 7 interacts with HIV-1 MA with an equilibrium dissociation constant (KD) of 22.6 (± 3.1) μM. This value is in remarkable agreement with the IC50 value derived from the single-round infection assay (IC50 = 21.2 μM).
Figure 2.
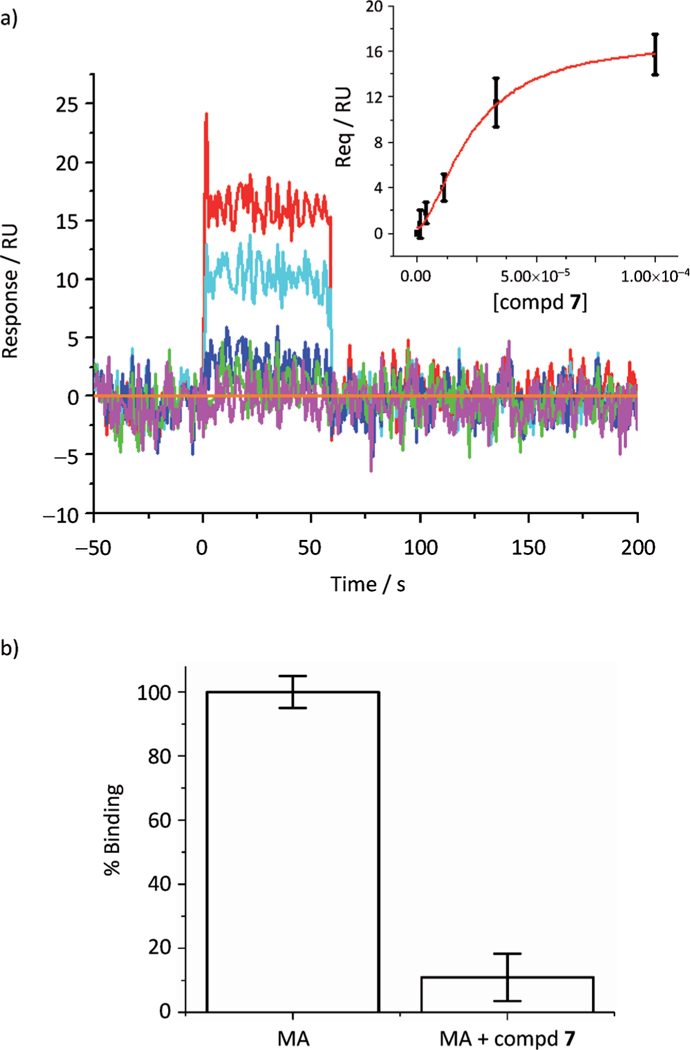
a) Sensorgrams depicting the interaction of compound 7 with immobilized HIV-1|A| MA. Compound concentrations of 0, 6.25, 12.5, 25, 50, and 100 mM were tested and are represented as orange, magenta, green, blue, cyan and red lines, respectively. The insert shows the equilibrium plot used to derive the equilibrium dissociation constant (KD). Compound 7–MA interaction: KD = 22.6 (± 3.1) μM. b) Compound 7 competes with PI(4,5)P2 for binding to HIV-1 MA. MA protein (1 μM) in the presence or absence of compound 7 (75 μM) was passed over a PI(4,5)P2 surface, and the response was recorded. To allow comparison, the responses were normalized with respect to the degree of free MA.
Having demonstrated that compound 7 interacts with HIV-1 MA, we next sought to establish whether compound 7 can compete with PI(4,5)P2 for binding to MA. To accomplish this, we established an SPR assay that allowed monitoring of the interaction of HIV-1 MA with a soluble form of PI(4,5)P2. The soluble, biotinylated PI(4,5)P2 was attached to a sensor chip, and purified MA protein (1 μM) in the presence or absence of compound 7 (75 μM) was passed over this surface. Using the affinity of the compound 7–MA interaction, we established the fraction of MA bound to compound 7 under the experimental conditions and normalized the results to the unbound fraction (for details, see the Experimental Section). As can be seen in Figure 2b, the presence of compound 7 decreased the ability of MA to interact with PI(4,5)P2 by 89% under the assay conditions. This result, coupled with the decrease in the amount of virus produced in the presence of compound 7, suggests a mechanism of action whereby compound 7 disrupts the interaction of Gag with PI(4,5)P2 at the plasma membrane, resulting in a failure of the new virions to bud correctly.
Mutation of residues in the PI(4,5)P2-binding site of HIV-1 MA reduces the efficacy of compound 7
Having demonstrated that compound 7 binds to HIV-1 MA and competes with PI(4,5)P2 for binding, we next sought to demonstrate that the antiviral effect is mediated through this interaction. Changes to residues within the PI(4,5)P2 binding site can render the virus nonfunctional. However, mutations in L21 have been previously described to have no effect on virus particle production,[6a] and a virus harboring the T81A mutation is replication competent.[11] As both of these residues are in proximity to compound 7 in the original docking model,[9] we changed the codons in the gag gene present in the proviral backbone that code for residues L21 and T81 to code for alanine residues and produced the mutant viruses in the presence or absence of compound 7 (100 μM). The effect of compound 7 on the production of both wild-type (WT) and the L21A and T81A mutant viruses was assessed by p24 content in the supernatants after 48 h. Figure 3a shows the results of this analysis. Compound 7 decreased the amount of WT virus produced to approximately 39± 2.5% of the control with no compound (DMSO only). However, changing residues L21 and T81 to alanine completely abrogated the effects of compound 7.
Figure 3.
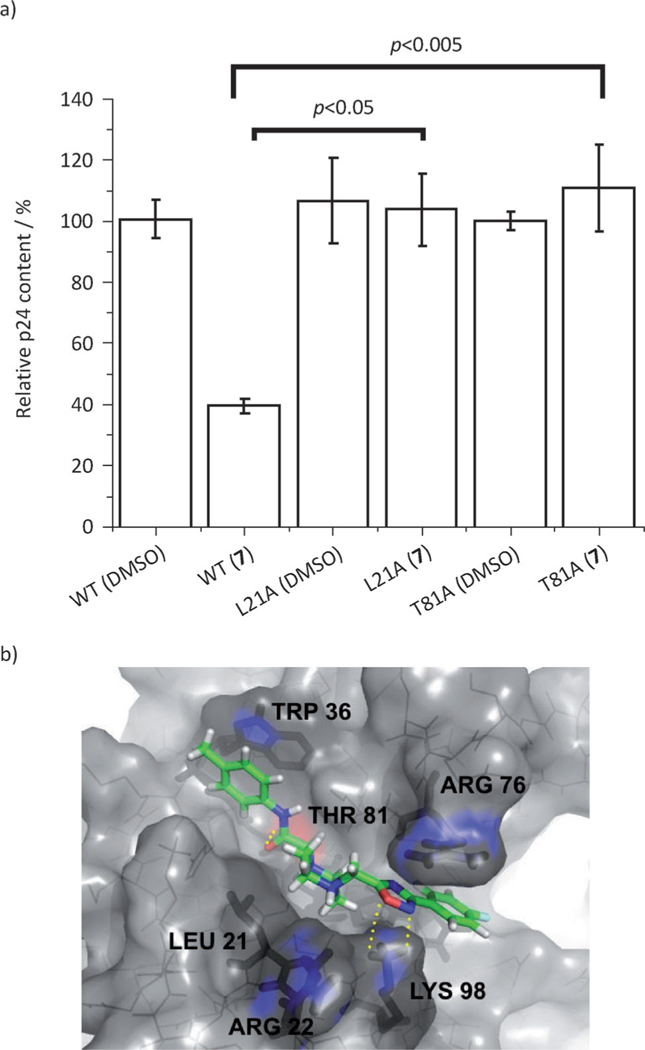
a) Effect of compound 7 on the production of HIV-1 wild-type and mutant pseudotypes as judged by supernatant p24 content. 293T cells were transfected for production of luciferase-reporter pseudotypes (as described in the Experimental Section) in the absence or in the presence of compound 7 (100 μM), and the culture supernatants containing pseudotype stocks were then analyzed by p24 ELISA. Shown are the mean values of four independent productions, with error bars depicting the standard error of the mean (SEM). The indicated p value was derived from a two-tailed paired t-test analysis. b) Refined predicted binding mode of compound 7 in the PI(4,5)P2 binding site of HIV-1 MA. Binding site residues (L21, R22, W36, R76, T81, and K98) are represented as dark sticks, whereas compound 7 is shown in green sticks. Residues highlighted in blue are thought to be critical for compound binding. Yellow dashed lines represent hydrogen bonds.
We next sought to refine the docking model and to identify the most probable binding mode of compound 7 to see whether or not the model could offer any insight into the mutational results. Exhaustive dockings to multiple conformations of HIV-1 MA resulted in the binding pose shown in Figure 3b. This docking model suggests plausible explanations of why mutation of residues L21 and T81 residues abolishes the antiviral activity compound 7. In this model, the carbonyl oxygen of the acetamide linker of compound 7 points towards the hydroxy group of T81 in the base of the binding groove to form a hydrogen bond, whereas L21 forms a weak hydrophobic contact with the aliphatic carbons of the piperazine moiety of compound 7. Disrupting either of these interactions would result in decreased affinity of compound 7 for the target MA protein and the concomitant decrease in potency observed in the single round infection assay. Future structural studies on compound 7 or higher affinity analogues in complex with MA will ultimately determine the validity of this proposed model.
Conclusions
We have identified a novel compound (7) that binds to and functions through the conserved PI(4,5)P2 binding site on HIV-1 MA, inhibits the production of new virus, and exhibits broad-spectrum anti-HIV activity with IC50 values in the range of 7.5 to 15.6 μM for the group M isolates tested (Table 1). This compound provides a novel chemical probe to investigate the roles of MA in the HIV-1 replication cycle and could serve as a good starting point for the development of a new class of HIV therapeutics through optimization by medicinal chemistry approaches.
Experimental Section
Virtual screening
The in silico screening was previously reported.[9] Refinement of the docking model of compound 7 was performed with QUANTUM software utilities (Quantum Pharmaceuticals, Moscow, Russia)[12] on a total of 40 different conformations of the MA protein derived from the Protein Data Bank (PDB) and from molecular dynamic simulations. In addition, several analogues of compound 7 that have antiviral activity (Cocklin et al., unpublished data) were similarly docked into the 40 protein conformers and consensus positions were used to establish the most probable binding mode of compound 7.
Chemistry
All chemicals were purchased from Enamine Ltd and were 95% pure or greater. Compound 7 was resynthesized to 98% purity using the route shown in Scheme 1. Briefly, diisopropylethylamine (208 mg, 1.612 mmol) was added dropwise to a mixture of piperazine I (342 mg, 1.465 mmol) and compound II (312 mg, 1.465 mmol) in dry DMF (1.5 mL). The reaction mixture was sonicated in an ultrasonic bath at 60°C for 3 h. The mixture was then diluted with water (10 mL), and the resultant precipitate was isolated by filtration and washed with EtOH (1 mL) to give compound III (513 mg, 86%).
Scheme 1.
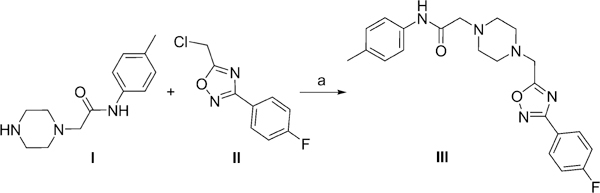
Synthesis of compound III : Reagents and conditions: N,N-diisopropylethylamine (1.1 equiv), DMF, 60°C, 3 h, 86%.
Biology
Anti-HIV efficacy evaluation in human peripheral blood mononuclear cells:
All viral isolates were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), US National Institutes of Health (NIH), and used to perform antiviral evaluations in human PBMCs as described previously.[9,13]
Determination of the antiviral spectrum of MA-targeted compounds:
To determine the spectrum of antiviral activity of compound 7, cytopathic effect assays against a panel of viruses from different classes were performed as described previously.[13]
Cells:
Human embryonic kidney 293T cells were cultured in Dulbecco′s modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), L-glutamine, and antibiotics (penicillin/streptomycin). Human astroglioma U87 cells stably transfected for the expression of CD4 and CXCR4 (obtained from Prof. Hongkui Deng (Peking University) and Prof. Dan Littman (New York University, USA), through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH)[14] were cultured in DMEM supplemented with 10% FBS, L-glutamine, penicillin streptomycin, puromycin and G418 (Gibco BRL Life Technologies, Grand Island, NY, USA).
Production of pseudotypes:
Single-round infectious envelope-pseudotyped luciferase-reporter viruses were produced in 293T cells co-transfected by calcium phosphate precipitation (Profection Mammalian Transfection System) with the envelope-deficient HIV-1 NL4–3 vector (pNL4–3-LucR+E−; a gift from Prof. Nathaniel R. Landau, New York University, USA),[15] which carries the luciferase-reporter gene, and the HxBc2 envelope-expressing vector (obtained from Dr. Kathleen Page (University of Southern California, USA) and Prof. Littman through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH).[16] All MA mutations within the pNL4–3-LucR+E− vector were synthesized and validated by Genescript (Piscataway, NJ, USA). After 6 h of incubation at 37 °C, the DNA-containing medium was removed, cells were washed, and fresh medium with or without compound 7 (at concentrations between 3.1 and 100 μm) was added. Supernatants containing the envelope-pseudotyped viruses were collected two days later, clarified by centrifugation, aliquoted, and stored at −80°C until use. Pseudotype stocks were quantified by p24gag content.
Single-round infection assays:
To determine whether compound 7 affected early stages of the viral life cycle, 50 μL medium alone or containing compound 7 at twice the desired final concentration (between 3.1 and 100 μm) was added to the U87.CD4.CXCR4 target cells plated in 96-well plates, and 50 μL of pseudotype stocks (not treated during production in 293T cells) was added immediately. Alternatively, to study the inhibitory effects of compound 7 during the late stages of the viral life cycle, untreated or compound 7-treated pseudotype stocks were diluted 1:10 with fresh medium, and an aliquot of 100 μL was used for infection of U87.CD4.CXCR4 target cells; this dilution ensured that any effect observed was due to the concentration of the compound present during viral production and not to the residual amount of compound present during infection of target cells. Assessment of the effects of compound 7 on the late stages of infection with pseudotype stocks produced with a luciferase-reporter vector encoding a mutant MA was performed in the same manner. Infections were performed for two days and quantified by measuring luciferase activity in cell lysates (Luciferase Assay System, Promega) using a microplate luminometer (GloMax, Promega).
Compound cytotoxicity:
Supernatants from target cells treated with or without compound 7 to test its effects at early stages, and pseudotype stocks produced with or without compounds to evaluate their effects at late stages, were used for analysis of cytotoxicity. Using a colorimetric assay (Takara Bio Inc., Japan), these effects were estimated through the presence in culture supernatants of lactate dehydrogenase (a stable cytoplasmic enzyme), which would indicate plasma membrane damage.
p24 enzyme-linked immunosorbent assays (ELISAs):
p24 ELISAs were performed to determine the amount of virus produced in the presence or absence of compound 7. The ELISA kits used were obtained from either Zeptometrix Corp. (Buffalo, NY, USA) or Advanced Bioscience Laboratories (Kensington, MD, USA).
Recombinant protein production and purification:
The MA region of the HIV-1 gag gene was amplified from plasmid pLAI (a generous gift from Drs. Evelyn Kilareski and Brian Wigdahl, both Drexel University College of Medicine, Philadelphia, USA) using primers designed to facilitate ligation-independent cloning into the vector pETHSUL.[17] This vector is designed for the insertion of genes of interest in frame with an N-terminal small ubiquitin-related modifier (SUMO) tag.[17] The recombinant pETHSUL plasmid was verified for the presence of MA insert by restriction digestion and sequence analysis (Genewiz, Inc., South Plainfield, NJ, USA). The resultant vector was designated pSUMO-MA. The purification of H6SUMO-MA was achieved via immobilized metal affinity chromatography using a TALON cobalt resin affinity column (ClonTech).
The Escherichia coli strain BL21 (DE3) Codon +-RIL (Stratagene, La Jolla, CA, USA) was used for expression of H6SUMO-MA from pSUMO-MA. Luria-Bertani broth (2 mL) containing 100 μg Ml−1 ampicillin and 50 μg mL−1 chloramphenicol was inoculated with a single transformed colony, and the colony was allowed to grow at 37°C for 9 h. A 100 μL aliquot of the preculture was used to inoculate 100 mL of the autoinducing media ZYP-5052[18] containing 100 μg mL−1 ampicillin and 34 μg mL−1 chloramphenicol. The culture was grown at 30°C for 16 h. Cells were harvested by centrifugation at 1076×g for 20 min at 4 °C, and the pellet was suspended in 30 μL phosphate-buffered saline (PBS) (Roche, Nutley, NJ, USA) containing 2.5 mM imidazole. Cells were lysed by sonication, and the supernatant clarified by centrifugation at 11,952×g (SS-34, Sorvall RC 5C Plus) for 20 min at 4°C. The supernatant was removed and applied to a TALON cobalt resin affinity column (ClonTech), previously equilibrated with PBS.
Loosely bound proteins were removed via seven-column volumes of PBS containing 7.5 mM imidazole. Tightly associated proteins were eluted in three-column volumes of PBS containing 250 mM imidazole. The eluates were then pooled and made 1 mM with respect to ethylenediamine tetraacetic acid (EDTA). To this pooled sample, 10 μg of a recombinant His6-tagged form of the catalytic domain (dtUD1) of the Saccharomyces cerevisiae SUMO hydrolase was added.[17] Cleavage was allowed to proceed for 4 h at 18°C. Following cleavage, the sample was dialyzed at 4°C overnight against 2 L of PBS to remove any imidazole. After dialysis, the dtUD1-catalyzed cleavage reaction was subjected to a second cobalt affinity purification using the TALON cobalt resin affinity column. In this purification step, however, the cleaved MA protein passes straight through the column owing to removal of the His6 tag. Subsequently, the subtractively purified MA was dialyzed overnight at 4°C against 25 mM Tris-HCl, pH 8.0 containing 10% glycerol. This dialyzed sample was then filtered and loaded onto a 5 mL Hi-TrapQ HP column (GE Healthcare, Chalfont, UK). The flowthrough, containing the MA protein, was concentrated, flash frozen in liquid nitrogen, and stored at −80 °C.
Surface plasmon resonance (SPR):
Interaction analyses were performed on a ProteOn XPR36 SPR Protein Interaction Array System (Bio-Rad Laboratories, Hercules, CA, USA). ProteOn GLH sensor chips were preconditioned and equilibrated as previously described.[9] HIV-1LAI, MA was immobilized to the flow cells of a GLH sensor chip to high density (8000 RU) using standard amine coupling. However, in order to preserve the PI(4,5)P2 binding site and create a more active protein surface, the immobilization of the MA protein was performed in the presence of 1.5 mM compound 14. A reference surface was similarly created by immobilizing a nonspecific antibody ARC4033 (antimouse/rat interferon-γ: BioSource; Invitrogen, Carlsbad, CA, USA).
Binding assays were performed with PBS-T with 3% DMSO as the running buffer. Samples of compound 7 were prepared as previously described[9] and injected across the MA and antibody reference surfaces at a flow rate of 100 μL min−1, for a 1 min association phase, followed by a 5 min dissociation phase using the “one-shot kinetics” capability of the Proteon instrument.[19] Specific regeneration of the surfaces between injections was not needed owing to the nature of the interaction. Data were analyzed using the ProteOn Manager software version 3.0 (Bio-Rad). The responses of a buffer injection and the responses from the reference flow cell were subtracted to account for nonspecific binding and injection artifacts. The experimental data could not be adequately fitted to a simple 1:1 binding model as the individual on and off rates were outside of the dynamic range of the instrument. Therefore, the equilibrium dissociation constant (KD) was obtained by fitting equilibrium binding data on the ProteOn Manager software, using the four-parameter Equation (1), where Rhigh is the response value at high analyte concentrations, Rlow is the response at an analyte concentration of zero, A1 is the midrange concentration and is equivalent to the equilibrium constant (KD), and A2 is the slope factor.
| (1) |
For PI(4,5)P2 competition assays, biotin–PI(4,5)P2 (Echelon Biosciences, Inc., Salt Lake City, UT, USA) was captured on the surface of a GLC chip, to which streptavidin had been immobilized using standard amine coupling. A reference surface with captured biotin–PI(3)P was also created. MA protein (1 μm) in the presence or absence of compound 7 (75 μm) was passed over this surface and the response recorded. The average responses from five data sets were used. To normalize the competition data to the degree of free MA, the concentration of free ligand and degree of saturation of the HIV-1 MA were calculated directly using Equations (2) and (3). The relation between the total ligand concentration [X]T and free ligand concentration [X] is given by Equation (2), where KA is the association constant, [P] is the concentration of macromolecule, and n is the stoichiometry. After determining the free concentration of MA, the fraction bound (Fb) was calculated using Equation (3).
| (2) |
| (3) |
Supplementary Material
Acknowledgements
This work was supported in part by the US National Institutes of Health (NIH)/US National Institute of Allergy and Infectious Diseases (NIAID) (1R03AI078790-01A1 and 1R21AI087388-01A1 to S.C.), and by the US NIH/US National Institute of Neurological Disorders and Stroke (NINDS)(R01NS065727 to J.M.-G.). Andrei Vinnik and Peter Fedichev are supported by the Skolkovo Foundation (Russian Federation). The authors thank Diana Winters (Academic Publishing Services, Drexel University College of Medicine) for proofreading the manuscript and Dr. Timothy Pyrkov (QuantumLead, Quantum Pharmaceuticals, Moscow, Russia) for the help in setting up the docking calculations and valuable discussions.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201200577.
References
- [1]a).Dorfman T, Mammano F, Haseltine WA, Göttlinger HG, J. Virol. 1994, 68, 1689–1696; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Freed EO, Martin MA, J. Virol. 1995, 69, 1984–1989; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Freed EO, Martin MA, J. Virol. 1996, 70, 341–351; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Murakami T, Freed EO, J. Virol. 2000, 74, 3548–3554; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wyma DJ, Kotov A, Aiken C, J. Virol. 2000, 74, 9381–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cosson P, EMBOJ. 1996, 15, 5783–5788. [PMC free article] [PubMed] [Google Scholar]
- [3]a).Bauby H, Lopez-Verges S, Hoeffel G, Delcroix-Genete D, Janvier K, Mammano F, Hosmalin A, Berlioz-Torrent C, Traffic 2010, 11, 455–467; [DOI] [PubMed] [Google Scholar]; b) Lopez-Verges S, Camus G, Blot G, Beauvoir R, Benarous R, Berlioz-Torrent C, Proc. Natl. Acad. Sci. USA 2006, 103, 14947–14952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]a).Bryant M, Ratner L, Proc. Natl. Acad. Sci. USA 1990, 87, 523–527; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Göttlinger HG, Sodroski JG, Haseltine WA, Proc. Natl. Acad. Sci. USA 1989, 86, 5781–5785; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhou W, Parent LJ, Wills JW, Resh MD, J. Virol. 1994, 68, 2556–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]a).Hermida-Matsumoto L, Resh MD, J. Virol. 1999, 73, 1902–1908; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ono A, Freed EO, J. Virol. 1999, 73, 4136–4144; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Paillart J-C, Göttlinger HG, J. Virol. 1999, 73, 2604–2612; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Resh MD, Proc. Natl. Acad. Sci. USA 2004, 101, 417–418; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Saad JS, Loeliger E, Luncs-ford P, Liriano M, Tai J, Kim A, Miller J, Joshi A, Freed EO, Summers MF, J. Mol. Biol. 2007, 366, 574–585; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF, Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Spearman P, Horton R, Ratner L, Kuli-Zade I, J. Virol. 1997, 71, 6582–6592; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF, Proc. Natl. Acad. Sci. USA 2004, 101, 517–522; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Zhou W, Resh MD, J. Virol. 1996, 70, 8540 – 8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]a).Freed EO, Orenstein JM, Buckler-White AJ, Martin MA, J. Virol. 1994, 68, 5311–5320; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hermida-Matsumoto L, Resh MD, J. Virol. 2000, 74, 8670 – 8679; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ono A, Freed EO, J. Virol. 2004, 78, 1552–1563; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ono A, Orenstein JM, Freed EO, J. Virol. 2000, 74, 2855–2866; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yuan X, Yu X, Lee TH, Essex M, J. Virol. 1993, 67, 6387–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]a).Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A, J. Virol. 2008, 82, 2405–2417; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M, Biochemistry 2006, 45, 4077–4083; [DOI] [PubMed] [Google Scholar]; c) Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO, Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]a).Inlora J, Chukkapalli V, Derse D, Ono A, J. Virol. 2011, 85, 3802–3810; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Monde K, Chukkapalli V, Ono A, J. Virol. 2011, 85, 3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zentner I, Sierra L-J, Maciunas L, Vinnik A, Fedichev P, Mankowski MK, Ptak RG, Martín-García J, Cocklin S, Bioorg. Med. Chem. Lett. 2013, 23, 1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Palmer S, Shafer RW, Merigan TC, AIDS 1999, 13, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Parry CM, Kolli M, Myers RE, Cane PA, Schiffer C, Pillay D, Anti-microb. Agents Chemother. 2011, 55, 1106–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]a).Timakhov RA, Fedichev PO, Vinnik AA, Testa JR, Favorova OO, Acta Naturae 2011, 3, 47–51; [PMC free article] [PubMed] [Google Scholar]; b) Fedichev P, Timakhov R, Pyrkov T, Getmantsev E, Vinnik A, PLoS Curr. 2011, 3, RRN1253; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Joce C, Stahl JA, Shridhar M, Hutchinson MR, Watkins LR, Fedichev PO, Yin H, Bioorg. Med. Chem. Lett. 2010, 20, 5411–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kortagere S, Madani N, Mankowski MK, Schon A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang X, Jones DM, Stavale E, Krebs FC, Martin-Garcia J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB III, J. Virol. 2012, 86, 8472–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Björndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, Albert J, Scarlatti G, Littman DR, Fenyö EM, J. Virol. 1997, 71, 7478–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Connor RI, Chen BK, Choe S, Landau NR, Virology 1995, 206, 935–944. [DOI] [PubMed] [Google Scholar]
- [16].Page KA, Landau NR, Littman DR, J. Virol. 1990, 64, 5270–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Weeks SD, Drinker M, Loll PJ, Protein Expression Purif. 2007, 53, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Studier FW, Protein Expression Purif. 2005, 41, 207–234. [DOI] [PubMed] [Google Scholar]
- [19].Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG, Anal. Biochem. 2006, 358, 281–288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.