

Abstract
Ozone-induced systemic effects are modulated through activation of the neuro-hormonal stress response pathway. Adrenal demedullation (DEMED) or bilateral total adrenalectomy (ADREX) inhibits systemic and pulmonary effects of acute ozone exposure. To understand the influence of adrenal-derived stress hormones in mediating ozone-induced lung injury/inflammation, we assessed global gene expression (mRNA sequencing) and selected proteins in lung tissues from male Wistar-Kyoto rats that underwent DEMED, ADREX, or sham surgery (SHAM) prior to their exposure to air or ozone (1 ppm), 4 h/day for 1 or 2 days. Ozone exposure significantly changed the expression of over 2300 genes in lungs of SHAM rats, and these changes were markedly reduced in DEMED and ADREX rats. SHAM surgery but not DEMED or ADREX resulted in activation of multiple ozone-responsive pathways, including glucocorticoid, acute phase response, NRF2, and PI3-AKT. Predicted targets from sequencing data showed a similarity between transcriptional changes induced by ozone and adrenergic and steroidal modulation of effects in SHAM but not ADREX rats. Ozone-induced increases in lung Il6 in SHAM rats coincided with neutrophilic inflammation, but were diminished in DEMED and ADREX rats. Although ozone exposure in SHAM rats did not significantly alter mRNA expression of Ifnγ and Il-4, the IL-4 protein and ratio of IL-4 to IFNγ (IL-4/IFNγ) proteins increased suggesting a tendency for a Th2 response. This did not occur in ADREX and DEMED rats. We demonstrate that ozone-induced lung injury and neutrophilic inflammation require the presence of circulating epinephrine and corticosterone, which transcriptionally regulates signaling mechanisms involved in this response.
Keywords: ozone, lung, stress hormones, adrenalectomy, RNAseq
INTRODUCTION
Ozone is a ubiquitous gaseous air pollutant and one of the major components of smog in urban areas (Cooper et al., 2014; Haagen-Smit, 1952). Due to its reactive nature, acute ozone inhalation causes oxidation of biomolecules such as proteins (Hemming et al., 2015; Kim et al., 2010) and lipids (Kadiiska et al., 2013; Thompson et al., 2013) in lung lining fluid and epithelial cells. An imbalance in the lung oxidant/antioxidant ratio has been widely reported after ozone exposure (Bromberg, 2016; Wiegman et al., 2014). Subsequent activation of pro-inflammatory signaling cascades involving mitogen-activated protein kinases (MAPK), phosphoinositide 3-kinase- protein kinase B (PI3-AKT), and nuclear factor erythroid 2-related factor 2 (NRF2) (Yan et al., 2016) have been shown in vitro and in vivo, to increase transcription of cell adhesion molecules and pro-inflammatory cytokines (Bromberg and Koren, 1995; Chen et al., 2007; Montuschi et al., 2002). This activation results in the extravasation of innate immune cells, including neutrophils, to the lungs within hours after ozone exposure (Kim et al., 2011; Kirsten et al., 2013; Cabello et al., 2015; Williams et al., 2007; Auerbach and Hernandez, 2012). Ozone exposure has been shown to alter the balance between Th1 and Th2 phenotype (Steerenberg et al., 1996). Th1-type cytokines are involved in immunity against intracellular pathogens where the effector cytokine IFNγ plays a pivotal role. Th2-type cytokines are involved in the immunity against extracellular parasites as well as development of type 1 hypersensitivity response seen in allergic asthma and are driven mainly by IL4 and IL13 (Berger, 2000). In mice, an ozone-induced Th2 phenotype shift was shown to depend on innate lymphoid cells (Kumagai et al., 2016).
Adverse ozone effects are not restricted to the lungs and multiple extra-pulmonary alterations have also been reported (Watkinson et al., 2001; Thomson et al. 2013). Ozone inhalation activates stress-responsive regions of the brain, including the nucleus tractus solitarius (NTS) where terminal fields of the lung vagal afferents overlap (Gackière et al., 2011). Ozone exposure also induces reflexively-mediated cardiovascular alterations, such as bradycardia and hypothermia, and decreases blood pressure (Akcilar et al., 2015; Uchiyama and Yokoyama, 1989; Gordon et al., 2014), indicating activation of the autonomic nervous system (Watkinson et al., 1996). It is postulated that nociceptive bronchial C-fibers stimulated by ozone inhalation transmit sensory information to the CNS and mediate systemic responses. These C-fibers modulate ozone-induced airway hyperresponsiveness but not hypothermia or bradycardia in rats (Jimba et al., 1995; Taylor-Clark and Undem, 2011).
Acute ozone inhalation induces systemic metabolic alterations including hyperglycemia, glucose intolerance, release of free fatty acids, activation of acute phase response, and muscle protein catabolism associated with a rise in circulating epinephrine and corticosterone levels (Bass et al., 2013; Miller et al., 2015, 2016a, 2016b). Further, performing an adrenal demedullation (DEMED) or adrenalectomy (ADREX) in rats, which diminishes the source of circulating catecholamines (synthesized in adrenal medulla upon sympathetic stimulation) and catecholamines plus steroid hormones (later synthesized in the adrenal cortex upon stimulation of HPA-axis), respectively, inhibits ozone-induced systemic metabolic impairment (Miller et al., 2016c). Importantly, pulmonary injury and inflammation induced by ozone exposure in rats that have undergone sham surgery (SHAM) are also diminished by ADREX (Miller et al., 2016c), highlighting a potential role of stress hormones in mediating ozone-induced pulmonary injury and inflammation. Although the mechanisms by which ozone induces local lung injury and inflammation are fairly well characterized, the role of circulating factors, such as stress hormones, in modulating pulmonary vascular leakage and extravasation of circulating immune cells has not been studied. We hypothesized that analysis of the expressed lung transcriptome in SHAM DEMED and ADREX rats exposed to ozone would elucidate potential mechanisms by which these hormones contribute to inflammation and injury in the lung following ozone exposure. Further, that decreased circulating levels of epinephrine and corticosterone in ADREX and DEMED rats would transcriptionally inhibit inflammatory modulators involved in ozone-induced neutrophilic inflammation while altering the balance of specific cytokine pools involved in immune function.
MATERIALS AND METHODS
Animals, surgeries and exposure
Lung tissue samples from healthy, male Wistar Kyoto (WKY) rats aged 12–13 weeks (Charles River Laboratories Inc., Raleigh, NC) were used from Miller et al, 2016c. SHAM, DEMED and ADREX surgeries were performed using aseptic sterile technique. Briefly, ketamine plus xylazine (50 mg plus 4 mg/kg body weight, i.p.) were used for anesthesia. Prior to surgery, rats received buprenorphine (0.02 mg/kg, subcutaneous) as an analgesic. Charles River Surgeons performed three types of sterile surgeries: control sham surgeries where all procedures were identical to the ADREX surgery except for the removal of adrenal gland (SHAM), bilateral adrenal demedullation where only the medulla portion of the adrenal glands were removed while the cortex was kept in place (DEMED), or bilateral total adrenalectomy where whole adrenal glands were removed (ADREX). Following 4 days of recovery, rats were exposed to either air or 1 ppm ozone, 4 h/day for 1 day or 2 consecutive days (1-D or 2-D) in whole body chambers under controlled flow, temperature and relative humidity. Ozone was generated by a silent arc discharge generator (OREC, Phoenix, Arizona). The chambers flow (Rochester style “Hinners”) was controlled by mass flow controllers (Coastal Instruments Inc., Burgaw, North Carolina) and the ozone concentrations were recorded using photometric ozone analyzers (API model 400, Teledyne Instruments; San Diego, California). Within 1 hour of the final exposure, animals were euthanized via an overdose of pentobarbital (>200 mg/kg, i.p.). Bronchoalveolar lavage (BAL) was performed on the right lungs and left lung tissues were snap frozen in liquid nitrogen for storage at −80oC for later analysis. Cell free BALF aliquots were stored at −80°C for further analysis.
Lung RNA isolation, and quantification
A uniform portion of left lung (10–20 mg) was extracted for RNA isolation using reagents provided in RNeasy mini kits (Qiagen, Valencia, CA). RNA yield was determined using Qubit fluorometric quantitation (Thermo Fisher Scientific Inc., Waltham, MA).
RNA sequencing
mRNA was isolated from total RNA (500 ng each) using prepX polyA mRNA Isolation Kit (Wafergen Biosystems, Fremont, CA). Samples were run using manufacturer’s protocol on the Apollo324 automated sample processing system for mRNA selection and continued on RNA-Seq library prep with Wafergen’s PrepX mRNA 48 Protocol. The resulting cDNA libraries were PCR amplified for 15 cycles with indexing primers according to Wafergen’s protocol. One microliter was taken from each library for quantitation by Qubit dsDNA HS Assay kit (Molecular Probes, Eugene, Oregon). The quality of libraries was checked by Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA) and the molar concentration of each library was estimated by using average molecular sizes from Bioanalyzer data and the concentration from Qubit measurement, and each library was diluted to 4 nM accordingly. The diluted libraries were again checked by Qubit to confirm the working concentrations and pooled to make the sample for sequencing run. The pooled libraries were denatured and diluted according to Illumina NextSeq 500 protocols (Illumina Inc., San Diego, CA). The final concentration for sequencing was 1.8 pM + 5% PhiX from Illumina. The sequencing data were stored in Illumina’s BaseSpace-cloud.
Real-time reverse transcriptase quantitative polymerase chain reaction (RT-qPCR)
For qPCR, each sample was diluted to 10 ng/μL and kept at −80°C until the day of the experiment. One-step qPCR (SuperScript III, Invitrogen, Grand Island, NY) was run on an ABI Prism 7900 HT sequence detection system (Applied Biosystems, Foster City, CA) using 50 ng of RNA. Primers containing a 6-carboxy-fluorescein (FAM dye) label at the 5’ end were purchased from Applied Biosystems (Foster City, CA) for the following genes: β-actin (Rn00667869_m1), tumor necrosis factor alpha (Tnfα, Rn99999017_m1), interleukin 6 (IL-6, Rn01410330_m1), interleukin 1 beta (IL-1β, Rn00580432_m1), interleukin 4 (IL-4, Rn01456866_m1), interferon gamma (Ifnγ, Rn00594078_m1), interleukin 5 (IL-5, Rn01459975_m1), and interleukin 13 (IL-13, Rn00587615_m1). Data were analyzed using ABI sequence detection software, version 2.2 using β-actin as an endogenous control. Expression of each group was calculated as relative fold change over air-SHAM group at each time point (1-D or 2-D) using the 2-ΔΔCT method.
Cytokine protein quantification
BALF levels of cytokine proteins (IL-1β, IL-4, IL-5, IL-6, IL-10, IFN-γ, KC-GRO, TNF-α) were quantified using the V-PLEX proinflammatory panel 2 (rat) kit per manufacturer’s protocol (Meso Scale Discovery, Gaithersburg, MD). The resulting electrochemiluminescence signals for each target protein in sample wells were detected using Meso Scale Discovery® electrochemiluminescence (MSD-ECL) platform (Mesoscale Discovery Inc., Rockville, MD). The values below the limit of detection were substituted with the lowest quantified value for each cytokine. Measurement of BALF inflammatory mediator proteins was restricted to 2-D only since ozone-induced inflammation peaks at this time point (Ward et al. 2015).
Statistics
Analysis of lung mRNA sequencing data
For mRNA sequencing, sequenced reads were mapped to the rat genome (rn6) using ensemble release 83 in the Partek® Flow suite. R version 3.2.3 (2015–12-10) was used for subsequent analyses of gene expression changes. The count matrix was rounded to the nearest integer and assessed for differential gene expression using DESeq2 (v. 1.10.1) (Love et al., 2014). Gene expression was considered different when fold change (FC) was more than 1.5 (upregulation) or less than 0.667 (downregulation) and the adjusted p value was ≤ 0.05. Pathway analysis was conducted using the log2 (fold change) and adjusted p-values from DESeq2 with Ingenuity Pathways Analysis® (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) using an adjusted p-value cut-off of 0.10 (Krämer et al., 2014). Pathway heat maps of normalized counts from DESeq2 were generated with the heatmap.2 function of the g-plots package (Warnes et al., 2015). For glucocorticoid responsive genes, fold changes in ozone-exposed rats relative to air control are reported for each surgery group. To validate qPCR and RNAseq data, correlations between selected genes analyzed using qPCR are shown in supplementary Fig. 1.
Analysis of RT-qPCR and BALF cytokine protein data
Relative gene expression and BALF cytokine data were analyzed using a two-way analysis of variance (ANOVA) with the 1-D and 2-D exposure groups treated as independent experiments. The two independent variables were exposure (air or 1 ppm ozone) and surgery (SHAM, DEMED or ADREX). The Holm-Sidak’s test was used to correct for all multiple comparisons and significant differences were considered when a p-value of ≤ 0.05 was achieved. All data (n=4–6 animals/group; 4 for SHAM air and ozone groups and 5–6 for ADREX and DEMED groups) are expressed as mean ± SEM. GraphPad prism 6.07 software was used for statistical analysis.
RESULTS
Ozone-induced transcriptome changes in SHAM rats are diminished in DEMED and ADREX rats
To determine the impact of DEMED and ADREX-induced depletion of stress hormones on ozone-induced global transcriptional changes, total RNA from lung tissue was assessed with RNA sequencing (RNAseq). For 1-D time point, air and ozone-exposed SHAM, DEMED and ADREX groups were analyzed, while for 2-D time point only air and ozone-exposed SHAM and ADREX groups were analyzed. DEMED groups for 2-D were not analyzed due to constraints in the number of wells in the plate design. Ozone exposure at 1-D time point induced significant changes (P≤0.05) in the expression of 2337 genes in SHAM rats (Fig. 1A). The number of genes changed by ozone were dropped to over 5-fold in DEMED (461 genes) and ADREX (463 genes) rats exposed for 1 day (1-D). Of 1452 genes uniquely changed by ozone in SHAM rats at 2-D time point, 716 were upregulated and 736 were downregulated. Ozone exposure in ADREX rats at 2-D time point resulted in 74 unique significantly changed genes (36 upregulated and 38 downregulated). The number of genes changed by ozone were dropped to over 15-fold ADREX rats exposed for 2 days (2-D). Only 26 genes were commonly changed by ozone between SHAM and ADREX rats at 2-D time point (Fig. 1B). Changes in lung gene expression of SHAM rats after ozone exposure were reflective of changes in inflammatory, oxidative stress, steroid metabolism and cell cycle control processes known to be altered after ozone exposure (Ward and Kodavanti, 2015).
Figure 1:

Venn diagrams showing the number of significantly changed genes in lungs of SHAM, DEMED and ADREX rats after ozone exposure. Gene expression differences were considered significant when the log2 Fold Change (FC) was ≥ 0.585 or ≤ −0.585 [if FC was higher than 1.5 (upregulation) or lesser than 0.667 (downregulation)] and the adjusted p value was ≤ 0.05. A) SHAM, DEMED and ADREX rats were assessed for 1-D time point while B) only SHAM and ADREX were assessed at 2-D time point.
Ozone-induced expression changes were greatly diminished in DEMED and ADREX rats (Supplementary materials tables 1 and 2). Genes that were induced after ozone exposure in SHAM rats were either not induced or only moderately induced in DEMED and ADREX rats, whereas those that were inhibited in SHAM rats were not inhibited or moderately inhibited in DEMED and ADREX rats. The relative fold change in expression of forty most induced and forty most inhibited genes in SHAM rats exposed to ozone, and their relative expression changes in DEMED and ADREX rats are shown in Supplementary Materials, tables 1A and 1B. These genes are reflective of cellular changes related to oxidative stress, acute phase response, inflammatory cell signaling and metabolism processes in SHAM rats.
To assess ozone-induced gene changes not affected by DEMED and ADREX, we examined unique and shared genes (Supplementary materials Table 2 A-D) within the Venn diagram (Fig.1). At 1-D, there were 235 genes uniquely changed by ozone in DEMED rats (145 upregulated and 90 down regulated) and 250 genes in ADREX rats (53 upregulated and 197 down regulated). Genes upregulated in DEMED by ozone included those involved in DNA replication processes while genes downregulated in ADREX by ozone were related to processes involved in acquired immunity (Supplementary materials, table 2). There were 193 genes changed by ozone in common between SHAM and DEMED rats, and 180 genes were shared between SHAM and ADREX rats. Those in common with SHAM and DEMED included genes increased in expression related to IL-1 signaling and those involved in temperature and hypoxia or mechanical stimulation response. Whereas, those in common between SHAM and ADREX included genes with increased expression of those involved in protein metabolic processes and decreased expression of those in immune processes (Supplementary materials, table 2). Only 86 genes changed by ozone were shared between, DEMED and ADREX rats whereas 53 significant ozone -induced gene changes were shared by all; SHAM, DEMED and ADREX rats.
Using IPA software, canonical pathways differentially regulated by ozone in SHAM, DEMED and ADREX rats (Tables, 1 and 2; Supplementary Materials, table 3 and 4) were identified. The top 20 significantly up-regulated (activation score > 0) and down-regulated (activation score < 0) canonical pathways after ozone exposure in SHAM rats and their relative changes in DEMED and ADREX rats are presented in Tables 1A and 1B. Notable pathways activated by ozone in SHAM rats on 1-D and /or 2-D included ERK5 signaling, acute phase response, cell cycle, ceramide signaling, p38 MAPK signaling and Notch signaling while those inhibited include interferon signaling, mTOR signaling, aryl hydrocarbon receptor signaling, iNOS signaling, JAK/Stat signaling, and IL-2 signaling (Table 1A and 1B; Supplementary materials, tables 3 and 4). In most cases pathway alterations induced by ozone in SHAM rats were abolished or mitigated by both DEMED and ADREX surgeries at 1-D (Table 1A) or at 2-D time points (Table 1B). Gene specific responses in the selected pathways known to be upregulated by ozone such as acute phase response, NRF-2 mediated oxidative stress and PI3/AKT pathways are displayed as heat maps in Figures 2A-C to get insights into how adrenergic and steroidal hormones regulate expression of specific genes within a given pathway. The individual changes in SHAM rats for specific genes in these pathways are in agreement with our previously published data on ozone effects determined using Affymetrix platform (Ward et al., 2015). Visual display of expression changes for each animal in genes related to specific functional category indicates that the pattern of genes changed by ozone exposure in SHAM rats are lost in DEMED and ADREX rats (Fig. 2A-C).
Table 1A.
Twenty most up-regulated and twenty most down-regulated canonical pathways after ozone exposure in SHAM, DEMED and ADREX rats (1-D time point).
|
ACTIVATION SCORE |
|||
|---|---|---|---|
| Canonical Pathway | SHAM O3 / SHAM AIR |
DEMED O3 / DEMED AIR |
ADREX O3 / ADREX AIR |
| Ephrin Receptor Signaling | 2.65 | 0.00 | 0.24 |
| ERK5 Signaling | 2.40 | 1.90 | 1.90 |
| Acute Phase Response Signaling | 2.31 | 0.00 | −0.78 |
| VDR/RXR Activation | 2.12 | 0.00 | 0.00 |
| 14–3-3-mediated Signaling | 2.11 | 0.00 | 0.00 |
| PCP pathway | 1.89 | 0.00 | 0.00 |
| Actin Nucleation by ARP-WASP Complex | 1.88 | 0.00 | 0.00 |
| Ceramide Signaling | 1.76 | 0.00 | −1.60 |
| p38 MAPK Signaling | 1.76 | 2.00 | 0.26 |
| CD27 Signaling in Lymphocytes | 1.70 | 0.00 | −1.13 |
| NRF2-mediated Oxidative Stress Response | 1.68 | 0.26 | 0.00 |
| Agrin Interactions at Neuromuscular Junction | 1.63 | 0.00 | 0.00 |
| nNOS Signaling in Neurons | 1.63 | 1.00 | 0.00 |
| Gαi Signaling | 1.46 | 0.00 | 0.00 |
| Apoptosis Signaling | 1.44 | 0.00 | 1.29 |
| Ephrin B Signaling | 1.41 | 0.00 | 0.00 |
| Hypoxia Signaling in the Cardiovascular System | 1.41 | 0.00 | −0.45 |
| PI3K/AKT Signaling | 1.41 | −0.24 | −1.40 |
| Cell Cycle Regulation by BTG Family Proteins | 1.26 | 2.00 | 0.00 |
| PTEN Signaling | 1.26 | 0.00 | 0.24 |
| P2Y Purigenic Receptor Signaling Pathway | −1.95 | 0.00 | 0.26 |
| Macropinocytosis Signaling | −2.00 | 0.00 | 1.26 |
| PEDF Signaling | −2.00 | 0.00 | −0.26 |
| Fc Epsilon RI Signaling | −2.06 | 0.00 | 0.00 |
| Growth Hormone Signaling | −2.12 | 0.00 | 0.90 |
| Role of NFAT in Regulation of the Immune Response | −2.12 | 0.00 | −0.73 |
| IL-9 Signaling | −2.13 | 0.00 | 0.00 |
| Role of Pattern Recognition Receptors in Recognition of Bacteria and Viruses | −2.14 | 0.00 | −1.21 |
| Role of NFAT in Cardiac Hypertrophy | −2.17 | 0.00 | 0.00 |
| VEGF Family Ligand-Receptor Interactions | −2.20 | 0.00 | 0.28 |
| Type II Diabetes Mellitus Signaling | −2.24 | 0.00 | −0.50 |
| FcγRIIB Signaling in B Lymphocytes | −2.27 | 0.00 | 0.00 |
| TREM1 Signaling | −2.27 | 0.00 | −2.89 |
| Neuropathic Pain Signaling In Dorsal Horn Neurons | −2.29 | 0.00 | 0.00 |
| Glioma Signaling | −2.38 | −0.83 | 0.50 |
| Tec Kinase Signaling | −2.39 | 0.00 | −0.89 |
| ErbB4 Signaling | −2.47 | 0.00 | 0.58 |
| eNOS Signaling | −2.48 | 0.00 | 0.00 |
| NF-κB Activation by Viruses | −2.59 | 0.00 | −1.15 |
| iCOS-iCOSL Signaling in T Helper Cells | −2.92 | 0.00 | −1.21 |
Values indicate the activation score for canonical pathways and were ranked by significant changes determined in SHAM rats. When the p values were not significant the activation score indicated is zero.
Table 2.
Identification of 10 most predictive targets changed by ozone exposure (1-D) in SHAM, DEMED and ADREX rats using Ingenuity Upstream Regulator Analysis.
|
ACTIVATION SCORE |
|||
|---|---|---|---|
| Predicted Targets | SHAM O3 / SHAM AIR |
DEMED O3 / DEMED AIR |
ADREX O3 / ADREX AIR |
| forskolin | 5.84 | 3.82 | 1.60 |
| methylprednisolone | 4.97 | 1.81 | −0.74 |
| CREB1 | 4.84 | 2.02 | 1.62 |
| PGR | 4.57 | 1.68 | 1.27 |
| XBP1 | 4.54 | 1.23 | −1.86 |
| PDGF BB | 4.51 | 3.40 | 0.78 |
| dexamethasone | 4.51 | 0.03 | 2.35 |
| gentamicin | 4.49 | 2.97 | −0.64 |
| NUPR1 | 4.45 | −1.12 | 1.80 |
| Salmonella enterica serotype abortus equi lipopolysaccharide | 4.41 | 4.07 | 0.00 |
Values indicate the activation score for predicted upstream targets determined by IPA. This analysis is based on the previous knowledge/published database of how known targets act as a transcription regulator considering the direction of change. Values were ranked for the magnitude of the change in SHAM rats and when the p values were not significant or unchanged the activation score indicated is zero
Table 1B.
Twenty most up-regulated and twenty most down-regulated canonical pathways after ozone exposure in SHAM and ADREX rats (2-D time point).
|
ACTIVATION SCORE |
||
|---|---|---|
| Canonical Pathway | SHAM O3 / SHAM AIR |
ADREX O3 / ADREX AIR |
| LPS/IL-1 Mediated Inhibition of RXR Function | 2.18 | 0 |
| Role of BRCA1 in DNA Damage Response | 2.11 | 0 |
| ERK5 Signaling | 1.96 | 0 |
| Acute Phase Response Signaling | 1.62 | 0 |
| CD27 Signaling in Lymphocytes | 1.39 | 0 |
| Cell Cycle: G1/S Checkpoint Regulation | 1.39 | 0 |
| Ceramide Signaling | 1.34 | 0 |
| Mitotic Roles of Polo-Like Kinase | 1.29 | 0 |
| Ephrin Receptor Signaling | 0.85 | 0 |
| p38 MAPK Signaling | 0.76 | 0 |
| Notch Signaling | 0.71 | 0 |
| CDK5 Signaling | 0.69 | −0.45 |
| Cdc42 Signaling | 0.65 | 0 |
| p53 Signaling | 0.65 | 0 |
| GNRH Signaling | 0.65 | 0 |
| Rac Signaling | 0.60 | 0 |
| B Cell Receptor Signaling | 0.59 | 0 |
| Toll-like Receptor Signaling | 0.53 | 0 |
| ILK Signaling | 0.51 | 0 |
| TGF-β Signaling | 0.45 | 0 |
| Regulation of Cellular Mechanics by Calpain Protease | −1.73 | 0 |
| Macropinocytosis Signaling | −1.79 | 0 |
| PI3K/AKT Signaling | −1.80 | 0 |
| Growth Hormone Signaling | −1.88 | 0 |
| iCOS-iCOSL Signaling in T Helper Cells | −1.88 | 0 |
| IL-2 Signaling | −1.89 | 0 |
| JAK/Stat Signaling | −1.89 | 0 |
| eNOS Signaling | −1.98 | 0 |
| Gαq Signaling | −2.00 | 0 |
| iNOS Signaling | −2.00 | 0 |
| Fc Epsilon RI Signaling | −2.12 | 0 |
| UVA-Induced MAPK Signaling | −2.13 | −1.34 |
| mTOR Signaling | −2.19 | 1 |
| Aryl Hydrocarbon Receptor Signaling | −2.27 | 0 |
| IL-9 Signaling | −2.31 | 0 |
| Antiproliferative Role of Somatostatin Receptor 2 | −2.32 | 0 |
| Leukocyte Extravasation Signaling | −2.34 | 0 |
| Tec Kinase Signaling | −2.40 | 0 |
| NF-κB Activation by Viruses | −2.45 | 0 |
| Interferon Signaling | −3.46 | 0 |
Values indicate the activation score for canonical pathways and were ranked for significant changes determined in SHAM rats. When the p values were not significant the activation score indicated is zero.
Figure 2:
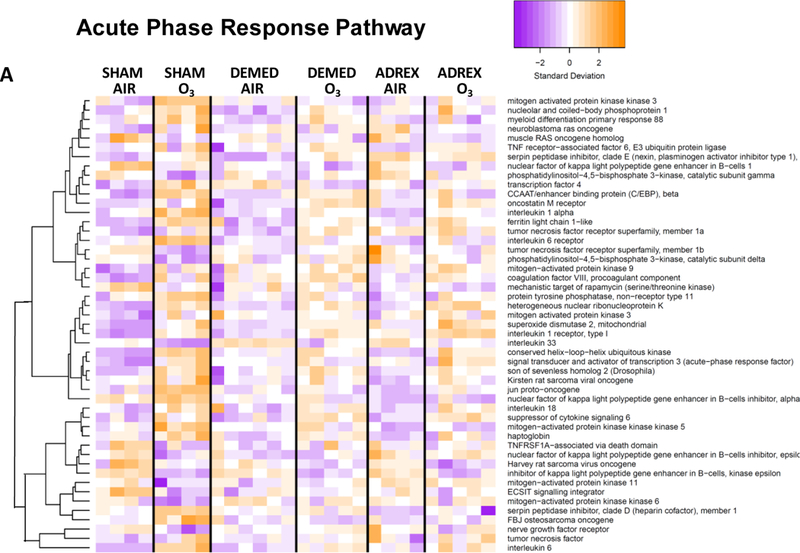
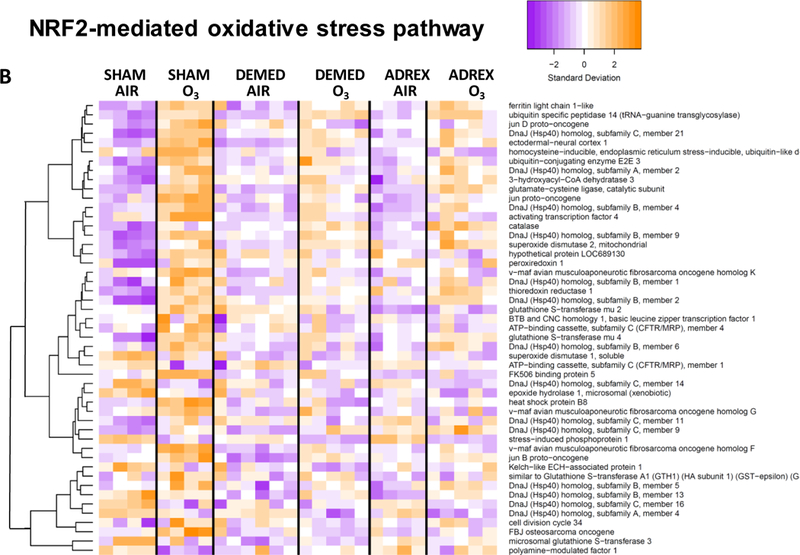
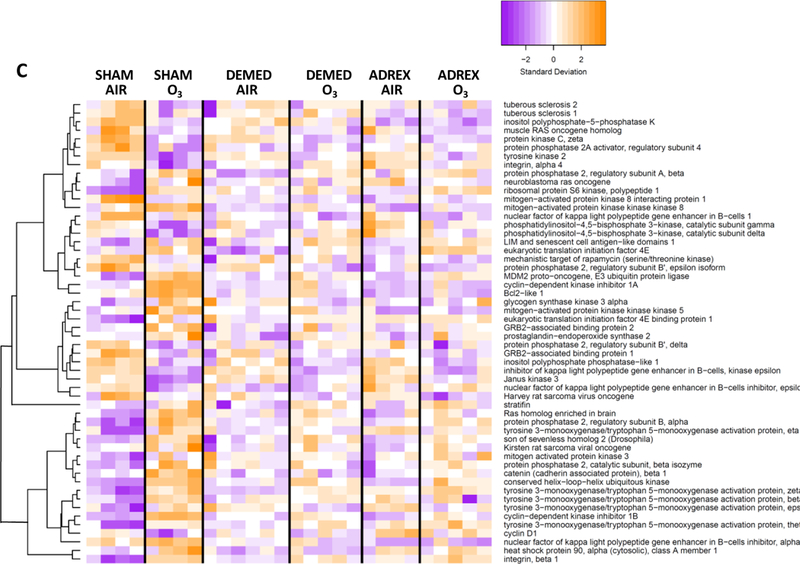
Heat maps showing hierarchical clustering of selected genes within given signaling pathways after 1-D air or ozone exposure in SHAM, DEMED or ADREX rats. A master gene list was prepared for genes showing significant change in at least one of the comparison among all possible comparisons between groups. From this master list, genes pertinent to a given pathway that were significantly impacted by ozone in SHAM rats were used for heat map. IPA version 01–07 was used to identify biological pathways significantly impacted by surgery or ozone exposure. Pathway heat maps of normalized counts from DESeq2 were generated with the heatmap.2 function of the g-plots package. Increased and decreased expression of selected genes are shown by orange and purple color combination, respectively. Heat maps were for A) Acute phase response genes, B) NRF2-mediated oxidative stress response genes and C) PI3-AKT pathways genes.
Using the IPA comparison of gene expression signatures with previously known xenobiotics in its database, xenobiotics known to cause similar expression changes were compared to our experiment. Changes in gene expression determined in 1-D ozone exposure in SHAM rats were similar to those induced, among others, by methylprednisolone, dexamethasone, forskolin, and CREB-1 (Table 2), suggesting the significant contribution of glucocorticoids and adrenergic mechanisms in ozone-induced changes in lung tissue gene expression. The predicted activation scores in 1-D ozone vs. air exposed SHAM rats for these compounds were attenuated in animals that underwent either DEMED and ADREX surgeries (Table 2).
Ozone exposure increased expression of glucocorticoid responsive genes in SHAM but not ADREX rats.
RNA sequencing results were useful in determining if glucocorticoid responsive genes were changed after ozone exposure. A list of 8 upregulated by glucocorticoid receptor activation were selected (Bhlhe40, Tsc22d3, Thbd, Sdpr, Slc19a2, Gem, Plk2 and Srgn) (Wang et al., 2004) to determine if the lungs were an active target of the increased glucocorticoid activity driven by ozone-induced increases in circulating corticosterone (Miller et al., 2016a; 2016c). SHAM rats exposed to ozone (1-D) compared with air exposed animals displayed significantly increased lung expression (in all but Thbd (Table 3)). These ozone-induced increases were not apparent in ADREX rats demonstrating that ozone activates the expression of glucocorticoid responsive genes in SHAM but not in ADREX rats. As expected, ADREX in air-exposed rats did not change the expression of these genes. On day 2 of exposure, although the induction of glucocorticoid responsive genes in SHAM rats was reduced when compared to day 1 changes, the increases in some of these genes still persisted in ozone-exposed SHAM rats and these effects were diminished in ADREX rats (Table 3).
Table 3.
Ozone-induced fold change in the expression of glucocorticoid responsive genes in SHAM, DEMED and ADREX rats.
| 1-D | 2-D | ||||
|---|---|---|---|---|---|
| SHAM Ozone / SHAM Air |
DEMED Ozone / DEMED Air |
ADREX Ozone / ADREX Air |
SHAM Ozone / SHAM Air |
ADREX Ozone / ADREX Air |
|
| Tsc22d3 | 2.32* | 1.34 | 1.16 | 1.25 | 0.77 |
| Thbd | 1.23 | 0.92 | 1.16 | 1.04 | 0.98 |
| Sdpr | 1.24* | 1.05 | 1.2* | 0.93 | 0.92 |
| Slc19a2 | 1.38* | 1.21 | 1.25 | 1.76* | 1.11 |
| Gem | 2.73* | 1.80* | 1.10 | 1.88* | 0.82 |
| Plk2 | 2.17* | 1.56* | 1.3* | 1.20 | 0.91 |
| Srgn | 1.49* | 1.11 | 1.03 | 1.10 | 1.08 |
| Bhlhe40 | 2.07* | 1.34 | 1.32 | 1.53* | 0.80 |
Glucocorticoid responsive genes were identified from the master list of ozone-induced differentially expressed genes in the lung. TSC22 domain family protein 3, Tsc22d3 also known as Gilz; thrombomodulin, Thbd; serum deprivation-response protein, Sdpr; thiamine transporter 1, Slc19a2; GTP-binding protein, Gem; serine/threonine-protein kinase PLK2, Plk2; serglycin. Srgn; and class E basic helix-loop-helix protein 40, Bhlhe40. These genes have been shown to be transcriptionally up-regulated after activation of glucocorticoid receptors (Wang et al. 2004). Values indicate mean fold change by ozone when compared to air.
indicate significant ozone effect (adjusted p value<0.05)
Modulation of lung innate immune response genes (qPCR) and BALF proteins by ozone in SHAM, DEMED and ADREX rats
In order to validate the results obtained using RNAseq, we performed qPCR for innate immune genes known to be induced by ozone exposure. Inflammatory gene expression changes in the lung were determined at both time points, 1-D and 2-D. Scatter plots of relative expression by qPCR and RNAseq in all samples in general, showed significant correlation (Supplementary Materials, Fig. 1), indicating that the expression levels obtained using RNAseq are reflective of the actual changes in the gene expression. In general, the qPCR of key inflammatory genes in ozone-exposed SHAM rats was reflective of changes noted previously in other publications (Ward et al., 2015) (Fig. 3). There was a trend for increased expression of Tnfα after ozone exposure in SHAM rats (p=0.21) at 2-D time point (Fig. 3A). Il-6 was markedly induced by ozone exposure in SHAM rats at both time points (Fig. 3B) and for Il1-β at the 2-D time point (Fig. 3C). These ozone-induced changes were maintained for Tnfα (2-D) in DEMED rats, while for Il-6 and Il-1β the changes were not observed in DEMED or ADREX rats.
Figure 3:

the expression of selected inflammatory cytokine genes in lungs of SHAM, DEMED, and ADREX rats after exposure to air or ozone as determined using qPCR. Values represent mean ± SE of relative fold change from each day SHAM-air control (n=3–4 animals for SHAM air or ozone, and n= 5–6 for other groups). Comparisons between air and ozone under each surgery condition and the comparisons between different surgeries under the same exposure condition are shown in graphs (* indicate p value ≤ 0.05 with respect to surgery or ozone exposure). A) tumor necrosis factor alpha (Tnfα), B) interleukin 6 (Il-6), C) interleukin 1 beta (Il-1β), D) interleukin 4 (Il-4), E) interferon gamma (Ifnγ), F) interleukin 5 (Il-5) and G) interleukin 13 (Il-13).
Expression of Tnfα, Ifnγ and Il-5 (Fig. 3A, E, F) as determined using qPCR were increased at 2-D time point, while the expression of Il-13 was increased at both time points in ADREX rats exposed to air (Fig. 3G). The expression of Il4, Ifnγ, Il-5 and Il-13 (Fig. 3D, E, F, G) was increased in ADREX rats exposed to ozone (2-D only). DEMED did not have significant effect on the expression of any of these genes in air- and ozone-exposed rats.
BALF proteins were analyzed only for 2-D time point since ozone-induced inflammation peaks at this time. While neither ozone nor ADREX or DEMED changed the levels of IL-1β and IFN-γ (Fig. 4C, E), compared to air, ozone exposure resulted in increased levels of IL-6, IL-4, IL-5 and IL-13 proteins in BALF of SHAM rats (Fig. 4B, D, F, G). Significant ozone-induced increases of IL-6, IL-4, and IL-13 proteins were not observed in ADREX or DEMED rats (Fig. 4B, D, G). Ozone-induced IL-5 increase was not affected by DEMED. In ADREX rats increases in IL-5 were noted in both air and ozone groups (Fig. 4F). ADREX also increased BALF protein levels of TNFα (Fig. 4A, F).
Figure 4:
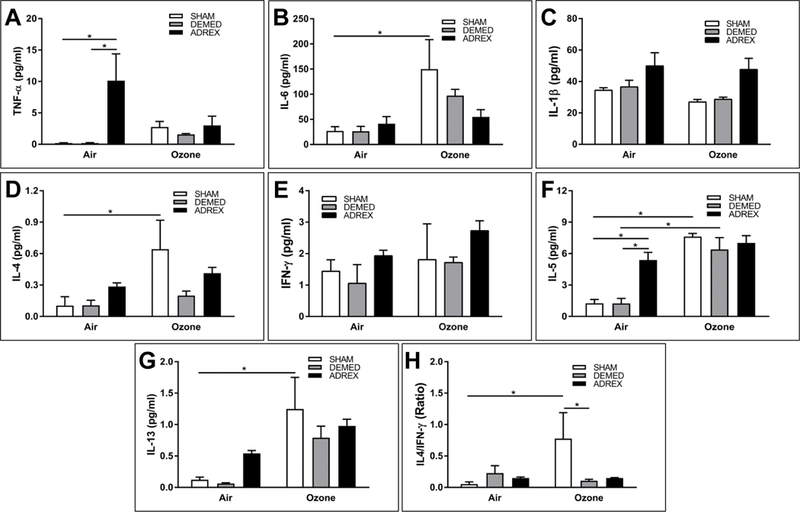
Inflammatory cytokine proteins in bronchoalveolar lavage fluid (BALF) of SHAM, DEMED and ADREX rats after exposure to air or ozone. Values represent mean ± SE (n=4 animals for SHAM air or ozone, and n= 5–6 for other groups). Comparisons between air and ozone under each surgery condition and the comparisons between different surgeries under the same exposure condition are shown in graphs (* indicate p value ≤ 0.05 with respect to surgery or ozone exposure). A) Tumor necrosis factor alpha (TNFα), B) interleukin 6 (IL-6), C) interleukin 1 beta (IL-1β), D) interleukin 4 (IL-4), E) interferon gamma (IFNγ), F) interleukin 5 (IL-5), G) interleukin 13 (IL-13) and H) IL4 / IFNγ ratio.
To explore changes in Th1 and Th2 modulation after ozone exposure in SHAM, DEMED and ADREX rats, BALF protein levels of IFNγ and IL4 and their ratio were examined. Increased expression of IFNγ preferentially drives a Th1 response while increased IL4 drives a Th2 response (Serrano et al., 1997). Although IFNγ protein levels were not significantly changed after ozone exposure in SHAM rats, the BALF IL-4/IFNγ ratio was increased (Fig. 4H) suggesting that ozone appears to preferentially drive a Th2 response over Th1. This effect was not observed in DEMED and ADREX rats.
DISCUSSION
We have recently shown that circulating stress hormones are increased after ozone exposure in rats and humans, and are linked to both systemic and pulmonary effects of ozone exposure (Bass et al., 2013; Miller et al., 2015; 2016a; 2016b, 2016c). In this study, a transcriptional approach was used to understand the molecular underpinnings of ozone-induced acute lung injury and inflammation in SHAM, DEMED and ADREX rats. The goal was to elucidate potential mechanisms and roles of adrenergic and steroidal hormones in ozone-induced lung injury and inflammation. Ozone-induced changes in expression of number of genes in the lungs of SHAM rats were markedly diminished in DEMED and ADREX rats (5-fold decrease from over 2300 genes). Moreover, pathways involved in ozone-induced inflammatory and oxidative stress responses, such as NRF2, acute phase response, PI3/AKT and p38 MAP-kinase were up-regulated after ozone exposure in SHAM rats but were not changed in DEMED or ADREX rats. Genes regulating the neutrophilic innate immune response and associated proteins favoring TH2 phenotype induced after ozone exposure in SHAM rats were diminished in DEMED and ADREX rats. When predictive pathways were examined, it was evident that ozone-induced transcriptional changes were similar to those induced by glucocorticoid-like compounds such as dexamethasone and prednisolone and to those induced by forskolin. Ozone exposure was associated with increases in the expression of glucocorticoid-responsive genes in the lungs of SHAM rats but not DEMED or ADREX rats. Together, these findings show that adrenergic and steroidal hormones modulate ozone-induced global gene expression changes in the lung. Hormonal regulation of air pollution-induced injury and inflammation has not been well studied even though steroidal and adrenergic interventions are widely used in treatment of lung diseases (Barnes, 2013; Fuso et al., 2013).
Gene expression outputs obtained by RNAseq were adequate in quantifying the magnitude of changes induced by ozone and were generally consistent with those reported in previous publications (Gohil et al., 2003; Ward et al., 2015). Eighty percent of the top ozone-induced changed transcripts in the lung as measured by an Affymetrix rat panel (Ward et al., 2015) were altered in the same direction in SHAM rats. The diminution of ozone-induced lung transcriptome changes in the absence of circulating stress hormones in DEMED and ADREX rats could be characterized using RNAseq. Since we were not able to remove the cortex while keeping the medulla intact in these animals, it was not possible to determine if corticosterone plus mineralocorticoids and epinephrine played independent roles in ozone-induced changes. Future work with pharmacological interventions is planned to address the role of individual stress hormones.
Oxidative stress responsive pathways have been shown to be upregulated in the lungs after ozone exposure including NRF2 (Kim et al. 2004) and acute phrase response (Bass et al., 2013; Laskin et al., 1994) which are presumed to counter ozone-induced lung injury and inflammation through induction of genes involved in these pathways (Cho et al. 2013). Moreover, it has been shown that steroid-induced NRF2 activation is a key event induced by oxidant injury and enhances airway epithelial barrier integrity (Shintani et al., 2015). Adrenal-derived stress hormones have been shown to independently increase plasma levels of acute phase proteins (Eastman et al., 1996; Merchant et al., 2010; Schade et al., 1987). The upregulation of NRF2 and acute phase pathways in ozone-exposed SHAM rats but their inhibition in DEMED and ADREX rats suggests that NRF2 nuclear translocation and activation of acute phase response in the lung cells may require the presence of circulating epinephrine and/or corticosterone.
We have shown that ozone exposure induces systemic metabolic changes (Miller et la., 2015). PI3/AKT is an intracellular signaling pathway involved in metabolism, cell cycle control, and proliferation (Hassan et al., 2013). The activation of selected genes in PI3/AKT pathway by ozone in SHAM rats at 1-D suggests that the transcriptional changes observed after ozone exposure are not only restricted to inflammatory mechanisms, but also involve other signaling processes including metabolic (Miller et al., 2015; Ward and Kodavanti, 2015). The activation of adrenergic receptors has been shown to induce PI3/AKT pathway (Nakaoka et al. 2015) while blockade of these receptors by propranolol has shown downregulation (Pan et al., 2015). Thus, the diminution of the pathway activation score in DEMED and ADREX rats emphasizes the contribution of stress hormones in interactively modulating multiple biological processes.
Based on known glucocorticoid responsive genes (Wang et al. 2004), we identified gene signatures in ozone-exposed SHAM, DEMED and ADREX rats. Although ozone did not change the expression of the glucocorticoid receptor Nr3c1 in SHAM rats (data not shown), the expression of glucocorticoid target genes - Tcs22d3 (also known as Gilz), Bhlhe40, Srgn, Plk2 and Gem - were significantly increased by ozone. These ozone-induced changes in glucocorticoid responsive genes have been shown previously (Thomson et al., 2016) and suggest that the lung is an active target for corticosterone action. The magnitude of this effect was greater on day 1 than on day 2, perhaps suggesting a degree of adaptation to the ozone exposure as has been described for lung injury and inflammation (Kirschvink, et al., 2002; Iwasaki et al. 1998).
Although the cellular signaling induced by epinephrine is mediated primarily by posttranslational events, some signature changes known to be mediated by adrenergic receptor activation were found. The predicted gene signature included the activation of Forskolin pathway in ozone-exposed SHAM but not in DEMED or ADREX rats. Forskolin raises the levels of cAMP which is also downstream of β adrenergic receptor activation (Wallukat, 2002). The diminution of ozone effects in DEMED rats supports the role of epinephrine as a modulator of ozone-induced lung injury/inflammation.
Ozone-induced innate immune response has been shown to be associated with increases in the neutrophil chemo-attractant IL6 (Gabehart et al., 2015; Johnston et al., 2003; Krishna et al., 1998). We noted that Il6 mRNA and proteins were up-regulated in SHAM but not in ADREX and DEMED rats, suggesting that adrenergic and steroidal hormones transcriptionally regulate innate immune response normally activated by ozone. Increased levels of epinephrine, and cortisol favors the development of the Th2 response (Spellberg and Edwards, 2001). Ozone exposure has also been shown to promote Th2 phenotype shift in nasal and airway tissues (Kumagai et al., 2016; Wu et al., 2014). Similarly, we observed that there was an increase in BALF IL4/IFN-γ ratio in SHAM but not in DEMED and ADREX rats exposed to ozone suggesting that stress hormones may be involved in preferentially mediating the Th2 shift.
Since stress hormones play a fundamental role in homeostatic balance, ADREX and DEMED surgeries likely change some of the basic physiological processes in tissues where stress hormone receptors are expressed. It is also likely that the lack of circulating epinephrine, mineralocorticoids and glucocorticoids in ADREX rats may impact lung epithelial integrity. ADREX increased the expression of several lung cytokines, including Il-5, Il-13, and Tnfα, as determined using qPCR and immunoassays, in air and ozone exposed animals. ADREX was associated with inhibition of many genes regulating acquired immunity even in air-exposed animals suggesting a key role of adrenal-derived hormones in regulation of immune response upon encountering injury. Surprisingly, DEMED alone, associated with depletion of only epinephrine (Miller et al., 2016b), did not alter the expression of these genes, suggesting that the lack of glucocorticoids but not epinephrine, likely contributes to immune homeostasis. Increased expression of beta-2 adrenergic receptors in alveolar type II cells has been shown to increase alveolar fluid clearance (McGraw et al., 2001), while glucocorticoids have been reported to improve epithelial barrier function (Kielgast et al., 2016).
Ozone remains a potential health hazard as its levels could exceed >0.2 ppm in areas with hot climate and heavy industry (The Royal Society, 2008). Human clinical studies often use 0.2–0.4 ppm concentration with intermittent exercise which is comparable to 1.0 ppm exposure in the rats during rest (Hatch et al., 1994, 2013). The level of ozone used in our study (1.0 ppm) is much higher than what is expected in the polluted air. However, our goal was to achieve detectable lung injury and inflammation in SHAM rats, such that any potential protective effects of DEMED and ADREX could be reliably detected.
This study did not allow assessment of contribution of individual hormones in ozone-induced lung injury and inflammation since it is not possible to surgically remove only the cortex. Although, the role of stress hormones in the development and magnitude of immunological responses has been extensively studied (Dhabhar et al., 2012), it has not been examined in the context of inhaled pollutants. The temporality of sequential events after the activation of a stress response was not taken into account and the role of ozone-induced hormonal changes in this context will need to be further examined.
In summary, ozone-induced transcriptional changes in the lung are greatly mitigated by both DEMED and ADREX surgeries demonstrating the key roles played by circulating adrenergic as well as steroidal hormones in the ozone-induced injury and inflammation. Ozone-induced increases in innate immune genes and proteins were markedly attenuated, including an increase in the ratio of IL-4/IFN-γ, in DEMED and ADREX rats suggesting the immune modulation by stress hormones. Immune, metabolic and oxidative stress pathways were induced in the lungs by ozone in SHAM rats while ADREX and DEMED conferred protection against these changes. In addition, upstream analysis by IPA showed that global gene changes induced by ozone in the lungs of SHAM rats were similar to steroidal chemicals and adrenergic influence. From the public health perspective, glucocorticoid and adrenergic receptors are widely targeted to combat pulmonary chronic disease conditions such as asthma and COPD. The dynamic role of environmental stressors and the use of therapeutic approaches targeting steroidal and adrenergic mechanisms should be investigated in the treatment of lung inflammatory conditions.
Supplementary Material
Acknowledgements:
The authors would like to thank Drs. Colette Miller, Stephen Gavett and Ian Gilmour of the US EPA for their critical review of the manuscript, and Ms. Beena Vallanat for her help in RNAseq experiment. This work was supported in part by the Fulbright (CONICYT) to A.H. and the EPA-UNC Center for Environmental Medicine, Asthma and Lung Biology Cooperative Agreement as well as EPA-UNC Cooperative Training Agreement (CR-83515201).
Footnotes
Disclaimer: The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency, and approved for publication. Approval does not signify that the contents necessarily reflect the views and policies of the Agency, nor does the mention of trade names of commercial products constitute endorsement or recommendation for use.
Bibliography
- Akcilar R, Akçer S, Şimşek H, Akcilar A, Bayat Z, and Genç O (2015). The effect of ozone on blood pressure in DOCA-salt-induced hypertensive rats. Int. J. Clin. Exp. Med. 8, 12783–12791. [PMC free article] [PubMed] [Google Scholar]
- Auerbach A, and Hernandez ML (2012). The effect of environmental oxidative stress on airway inflammation. Curr. Opin. Allergy Clin. Immunol. 12, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (2013). Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 131, 636–645. [DOI] [PubMed] [Google Scholar]
- Bass V, Gordon CJ, Jarema KA, MacPhail RC, Cascio WE, Phillips PM, Ledbetter AD, Schladweiler MC, Andrews D, Miller D, et al. (2013). Ozone induces glucose intolerance and systemic metabolic effects in young and aged Brown Norway rats. Toxicol. Appl. Pharmacol. 273, 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger A (2000). Th1 and Th2 responses: what are they? BMJ 321, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg PA (2016). Mechanisms of the acute effects of inhaled ozone in humans. Biochim. Biophys. Acta 1860, 2771–2781. [DOI] [PubMed] [Google Scholar]
- Bromberg PA, and Koren HS (1995). Ozone-induced human respiratory dysfunction and disease. Toxicol. Lett. 82, 307–316. [DOI] [PubMed] [Google Scholar]
- Cabello N, Mishra V, Sinha U, DiAngelo SL, Chroneos ZC, Ekpa NA, Cooper TK, Caruso CR, and Silveyra P (2015). Sex differences in the expression of lung inflammatory mediators in response to ozone. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L1150–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Arjomandi M, Balmes J, Tager I, and Holland N (2007). Effects of chronic and acute ozone exposure on lipid peroxidation and antioxidant capacity in healthy young adults. Environ. Health Perspect. 115, 1732–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H-Y, Gladwell W, Yamamoto M, and Kleeberger SR (2013). Exacerbated airway toxicity of environmental oxidant ozone in mice deficient in Nrf2. Oxid. Med. Cell. Longev. 2013, 254069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper OR, Parrish DD, Ziemke J, Balashov NV, Cupeiro M, Galbally IE, Gilge S, Horowitz L, Jensen NR, Lamarque J-F, et al. (2014). Global distribution and trends of tropospheric ozone: An observation-based review. Elem. Sci. Anthr. 2, 29. [Google Scholar]
- Dhabhar FS, Malarkey WB, Neri E, and McEwen BS (2012). Stress-induced redistribution of immune cells--from barracks to boulevards to battlefields: a tale of three hormones--Curt Richter Award winner. Psychoneuroendocrinology 37, 1345–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman HB, Fawcett TW, Udelsman R, and Holbrook NJ (1996). Effects of perturbations of the hypothalamic-pituitary-adrenal axis on the acute phase response: altered C/EBP and acute phase response gene expression in lipopolysaccharide-treated rats. Shock Augusta Ga 6, 286–292. [DOI] [PubMed] [Google Scholar]
- Fuso L, Mores N, Valente S, Malerba M, and Montuschi P (2013). Long-acting beta-agonists and their association with inhaled corticosteroids in COPD. Curr. Med. Chem. 20, 1477–1495. [DOI] [PubMed] [Google Scholar]
- Gabehart K, Correll KA, Loader JE, White CW, and Dakhama A (2015). The lung response to ozone is determined by age and is partially dependent on toll-Like receptor 4. Respir. Res. 16, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gackière F, Saliba L, Baude A, Bosler O, and Strube C (2011). Ozone inhalation activates stress-responsive regions of the CNS. J. Neurochem. 117, 961–972. [DOI] [PubMed] [Google Scholar]
- Gohil K, Cross CE, and Last JA (2003). Ozone-induced disruptions of lung transcriptomes. Biochem. Biophys. Res. Commun. 305, 719–728. [DOI] [PubMed] [Google Scholar]
- Gordon CJ, Johnstone AF, Aydin C, Phillips PM, MacPhail RC, Kodavanti UP, Ledbetter AD, and Jarema KA (2014). Episodic ozone exposure in adult and senescent Brown Norway rats: acute and delayed effect on heart rate, core temperature and motor activity. Inhal. Toxicol. 26, 380–390. [DOI] [PubMed] [Google Scholar]
- Haagen-Smit AJ (1952). Chemistry and Physiology of Los Angeles Smog. Ind. Eng. Chem. 44, 1342–1346. [Google Scholar]
- Hassan B, Akcakanat A, Holder AM, and Meric-Bernstam F (2013). Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg. Oncol. Clin. N. Am. 22, 641–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch GE, Slade R, Harris LP, Mcdonnell WF, Devlin RB, Koren HS, Costa DL, and Mckee J. (1994). Ozone dose and effect in humans and rats. A comparison using oxygen-18 labeling and bronchoalveolar lavage. Am J Respir Crit Care Med 150:676–683. [DOI] [PubMed] [Google Scholar]
- Hatch GE, McKee J, Brown J, McDonnell W, Seal E, Soukup J, Slade R, Crissman K, and Devlin R. (2013). Biomarkers of Dose and Effect of Inhaled Ozone in Resting versus Exercising Human Subjects: Comparison with Resting Rats. Biomark Insights. 8, 3–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming JM, Hughes BR, Rennie AR, Tomas S, Campbell RA, Hughes AV, Arnold T, Botchway SW, and Thompson KC (2015). Environmental Pollutant Ozone Causes Damage to Lung Surfactant Protein B (SP-B). Biochemistry (Mosc.) 54, 5185–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki T, Takahashi M, Saito H, and Arito H (1998). Adaptation of extrapulmonary responses to ozone exposure in conscious rats. Ind. Health 36, 57–60. [DOI] [PubMed] [Google Scholar]
- Jimba M, Skornik WA, Killingsworth CR, Long NC, Brain JD, and Shore SA (1995). Role of C fibers in physiological responses to ozone in rats. J. Appl. Physiol. Bethesda Md 1985 78, 1757–1763. [DOI] [PubMed] [Google Scholar]
- Johnston RA, Schwartzman IN, and Shore SA (2003). Macrophage inflammatory protein-2 levels are associated with changes in serum leptin concentrations following ozone-induced airway inflammation. Chest 123, 369S–70S. [PubMed] [Google Scholar]
- Kadiiska MB, Basu S, Brot N, Cooper C, Saari Csallany A, Davies MJ, George MM, Murray DM, Jackson Roberts L 2nd, Shigenaga MK, et al. (2013). Biomarkers of oxidative stress study V: Ozone exposure of rats and its effect on lipids, proteins, and DNA in plasma and urine. Free Radic. Biol. Med. 61C, 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielgast F, Schmidt H, Braubach P, Winkelmann VE, Thompson KE, Frick M, Dietl P, Wittekindt OH (2016). Glucocorticoids Regulate Tight Junction Permeability of Lung Epithelia by Modulating Claudin 8. Am. J. Respir. Cell Mol. Biol. 54, 707–717. [DOI] [PubMed] [Google Scholar]
- Kim CS, Alexis NE, Rappold AG, Kehrl H, Hazucha MJ, Lay JC, Schmitt MT, Case M, Devlin RB, Peden DB, et al. (2011). Lung Function and Inflammatory Responses in Healthy Young Adults Exposed to 0.06 ppm Ozone for 6.6 Hours. Am. J. Respir. Crit. Care Med. 183, 1215–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HI, Kim H, Shin YS, Beegle LW, Jang SS, Neidholdt EL, Goddard WA, Heath JR, Kanik I, and Beauchamp JL (2010). Interfacial reactions of ozone with surfactant protein B in a model lung surfactant system. J. Am. Chem. Soc. 132, 2254–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Song KS, Park GH, Chang SH, Kim HW, Park JH, Jin H, Eu KJ, Cho HS, Kang G, et al. (2004). B6C3F1 mice exposed to ozone with 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone and/or dibutyl phthalate showed toxicities through alterations of NF-kappaB, AP-1, Nrf2, and osteopontin. J. Vet. Sci. 5, 131–137. [PubMed] [Google Scholar]
- Kirschvink N, Fiévez L, Bureau F, Degand G, Maghuin-Rogister G, Smith N, Art T, and Lekeux P (2002). Adaptation to multiday ozone exposure is associated with a sustained increase of bronchoalveolar uric acid. Free Radic. Res. 36, 23–32. [DOI] [PubMed] [Google Scholar]
- Kirsten A, Watz H, Pedersen F, Holz O, Smith R, Bruin G, Koehne-Voss S, Magnussen H, and Waltz DA (2013). The anti-IL-17A antibody secukinumab does not attenuate ozone-induced airway neutrophilia in healthy volunteers. Eur. Respir. J. 41, 239–241. [DOI] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinforma. Oxf. Engl. 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna MT, Chauhan AJ, Frew AJ, and Holgate ST (1998). Toxicological mechanisms underlying oxidant pollutant-induced airway injury. Rev. Environ. Health 13, 59–71. [PubMed] [Google Scholar]
- Kumagai K, Lewandowski R, Jackson-Humbles DN, Li N, Van Dyken SJ, Wagner JG, and Harkema JR (2016). Ozone-Induced Nasal Type 2 Immunity in Mice Is Dependent on Innate Lymphoid Cells. Am. J. Respir. Cell Mol. Biol. Online 54, 782–791. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Pendino KJ, Punjabi CJ, Rodriguez del Valle M, and Laskin JD (1994). Pulmonary and hepatic effects of inhaled ozone in rats. Environ. Health Perspect. 102 Suppl 10, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw DW, Fukuda N, James PF, Forbes SL, Woo AL, Lingrel JB, Witte DP, Matthay MA, and Liggett SB (2001). Targeted transgenic expression of beta(2)-adrenergic receptors to type II cells increases alveolar fluid clearance. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L895–903. [DOI] [PubMed] [Google Scholar]
- Merchant S, Huang N, and Korbelik M (2010). Expression of complement and pentraxin proteins in acute phase response elicited by tumor photodynamic therapy: the engagement of adrenal hormones. Int. Immunopharmacol. 10, 1595–1601. [DOI] [PubMed] [Google Scholar]
- Miller DB, Karoly ED, Jones JC, Ward WO, Vallanat BD, Andrews DL, Schladweiler MC, Snow SJ, Bass VL, Richards JE, et al. (2015). Inhaled ozone (O3)-induces changes in serum metabolomic and liver transcriptomic profiles in rats. Toxicol. Appl. Pharmacol. 286, 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DB, Ghio AJ, Karoly ED, Bell LN, Snow SJ, Madden MC, Soukup J, Cascio WE, Gilmour MI, and Kodavanti UP (2016a). Ozone Exposure Increases Circulating Stress Hormones and Lipid Metabolites in Humans. Am. J. Respir. Crit. Care Med. 193(12):1382–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DB, Snow SJ, Henriquez A, Schladweiler MC, Ledbetter AD, Richards JE, Andrews DL, and Kodavanti UP (2016b). Systemic metabolic derangement, pulmonary effects, and insulin insufficiency following subchronic ozone exposure in rats. Toxicol. Appl. Pharmacol. 306, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DB, Snow SJ, Schladweiler MC, Richards JE, Ghio AJ, Ledbetter AD, and Kodavanti UP (2016c). Acute Ozone-Induced Pulmonary and Systemic Metabolic Effects are Diminished in Adrenalectomized Rats. Toxicol. Sci. 150(2):312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montuschi P, Nightingale JA, Kharitonov SA, and Barnes PJ (2002). Ozone-induced increase in exhaled 8-isoprostane in healthy subjects is resistant to inhaled budesonide. Free Radic. Biol. Med. 33, 1403–1408. [DOI] [PubMed] [Google Scholar]
- Nakaoka M, Iwai-Kanai E, Katamura M, Okawa Y, Mita Y, and Matoba S (2015). An alpha-adrenergic agonist protects hearts by inducing Akt1-mediated autophagy. Biochem. Biophys. Res. Commun. 456, 250–256. [DOI] [PubMed] [Google Scholar]
- Pan W-K, Li P, Guo Z-T, Huang Q, and Gao Y (2015). Propranolol induces regression of hemangioma cells via the down-regulation of the PI3K/Akt/eNOS/VEGF pathway. Pediatr. Blood Cancer 62, 1414–1420. [DOI] [PubMed] [Google Scholar]
- Schade R, Göhler K, Bürger W, and Hirschelmann R (1987). Modulation of rat C-reactive protein serum level by dexamethasone and adrenaline--comparison with the response of alpha 2-acute phase globulin. Agents Actions 22, 280–287. [DOI] [PubMed] [Google Scholar]
- Serrano D, Ghiotto F, Roncella S, Airoldi I, Cutrona G, Truini M, Burgio VL, Baroni CD, Ferrarini M, and Pistoia V (1997). The patterns of IL2, IFN-gamma, IL4 and IL5 gene expression in Hodgkin’s disease and reactive lymph nodes are similar. Haematologica 82, 542–549. [PubMed] [Google Scholar]
- Shintani Y, Maruoka S, Gon Y, Koyama D, Yoshida A, Kozu Y, Kuroda K, Takeshita I, Tsuboi E, Soda K, et al. (2015). Nuclear factor erythroid 2-related factor 2 (Nrf2) regulates airway epithelial barrier integrity. Allergol. Int. Off. J. Jpn. Soc. Allergol. 64 Suppl, S54–63. [DOI] [PubMed] [Google Scholar]
- Spellberg B, and Edwards JE (2001). Type 1/Type 2 immunity in infectious diseases. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 32, 76–102. [DOI] [PubMed] [Google Scholar]
- Steerenberg PA, Garssen J, van Bree L, van Loveren H (1996). Ozone alters T-helper cell mediated bronchial hyperreactivity and resistance to bacterial infection. Exp. Toxicol. Pathol. 48, 497–499. [DOI] [PubMed] [Google Scholar]
- Taylor-Clark TE, and Undem BJ (2011). Sensing pulmonary oxidative stress by lung vagal afferents. Respir. Physiol. Neurobiol. 178, 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Royal Society. 2008. Ground-level ozone in the 21st century: Future trends, impacts and policy implications. Science Policy Report 15/08. [Google Scholar]
- Thomson EM, Pal S, Guénette J, Wade MG, Atlas E, Holloway AC, Williams A, and Vincent R (2016). Ozone inhalation provokes glucocorticoid-dependent and independent effects on inflammatory and metabolic pathways. Toxicol. Sci. 152(1):17–28. [DOI] [PubMed] [Google Scholar]
- Thomson EM, Vladisavljevic D, Mohottalage S, Kumarathasan P, and Vincent R (2013). Mapping acute systemic effects of inhaled particulate matter and ozone: multiorgan gene expression and glucocorticoid activity. Toxicol. Sci. 135, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KC, Jones SH, Rennie AR, King MD, Ward AD, Hughes BR, Lucas COM, Campbell RA, and Hughes AV (2013). Degradation and rearrangement of a lung surfactant lipid at the air-water interface during exposure to the pollutant gas ozone. Langmuir ACS J. Surf. Colloids 29, 4594–4602. [DOI] [PubMed] [Google Scholar]
- Uchiyama I, and Yokoyama E (1989). Effects of short- and long-term exposure to ozone on heart rate and blood pressure of emphysematous rats. Environ. Res. 48, 76–86. [DOI] [PubMed] [Google Scholar]
- Wallukat G (2002). The beta-adrenergic receptors. Herz 27, 683–690. [DOI] [PubMed] [Google Scholar]
- Wang J-C, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, and Yamamoto KR (2004). Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc. Natl. Acad. Sci. U. S. A. 101, 15603–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WO, and Kodavanti UP (2015). Pulmonary transcriptional response to ozone in healthy and cardiovascular compromised rat models. Inhal. Toxicol. 27 Suppl 1, 93–104. [DOI] [PubMed] [Google Scholar]
- Ward WO, Ledbetter AD, Schladweiler MC, and Kodavanti UP (2015). Lung transcriptional profiling: insights into the mechanisms of ozone-induced pulmonary injury in Wistar Kyoto rats. Inhal. Toxicol. 27 Suppl 1, 80–92. [DOI] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, et al. (2015). “gplots: Various R Programming Tools for Plotting Data.” [Google Scholar]
- Watkinson WP, Highfill JW, Slade R, and Hatch GE (1996). Ozone toxicity in the mouse: comparison and modeling of responses in susceptible and resistant strains. J. Appl. Physiol. Bethesda Md 1985 80, 2134–2142. [DOI] [PubMed] [Google Scholar]
- Watkinson WP, Campen MJ, Nolan JP, and Costa DL (2001). Cardiovascular and systemic responses to inhaled pollutants in rodents: effects of ozone and particulate matter. Environ. Health Perspect. 109 Suppl 4, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegman CH, Li F, Clarke CJ, Jazrawi E, Kirkham P, Barnes PJ, Adcock IM, and Chung KF (2014). A comprehensive analysis of oxidative stress in the ozone-induced lung inflammation mouse model. Clin. Sci. Lond. Engl. 1979 126, 425–440. [DOI] [PubMed] [Google Scholar]
- Williams AS, Leung S-Y, Nath P, Khorasani NM, Bhavsar P, Issa R, Mitchell JA, Adcock IM, and Chung KF (2007). Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia. J. Appl. Physiol. Bethesda Md 1985 103, 1189–1195. [DOI] [PubMed] [Google Scholar]
- Wu D, Tan W, Zhang Q, Zhang X, and Song H (2014). Effects of ozone exposure mediated by BEAS-2B cells on T cells activation: a possible link between environment and asthma. Asian Pac. J. Allergy Immunol. Launched Allergy Immunol. Soc. Thail. 32, 25–33. [DOI] [PubMed] [Google Scholar]
- Yan Z, Jin Y, An Z, Liu Y, Samet JM, and Wu W (2016). Inflammatory cell signaling following exposures to particulate matter and ozone. Biochim. Biophys. Acta. 1860(12):2826–34 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.