

Abstract
The α-synuclein gene is implicated in the pathogenesis of Parkinson's disease. Although α-synuclein function is uncertain, the protein has homology to the chaperone molecule 14-3-3. In addition, α-synuclein can bind to 14-3-3, and both α-synuclein and 14-3-3 bind to many of the same proteins. Because 14-3-3 binds to and activates tyrosine hydroxylase, the rate-limiting enzyme in dopamine (DA) biosynthesis, we explored whether α-synuclein also bound to tyrosine hydroxylase and influenced its activity. Immunoprecipitation revealed an interaction between α-synuclein and tyrosine hydroxylase in brain homogenates and MN9D dopaminergic cells. Colocalization of α-synuclein with tyrosine hydroxylase was confirmed by immunoelectron microscopy. To explore the consequences of the interaction, we measured the effect of recombinant α-synuclein on tyrosine hydroxylase activity in a cell-free system and observed a dose-dependent inhibition of tyrosine hydroxylase by α-synuclein. To measure the impact of α-synuclein on tyrosine hydroxylase in dopaminergic cells, we stably transfected MN9D cells with wild-type or A53T mutant α-synuclein. Overexpression of wild-type or A53T mutant α-synuclein did not significantly alter tyrosine hydroxylase protein levels in our stably transfected cells. However, overexpressing cell lines had significantly reduced tyrosine hydroxylase activity and a corresponding reduction in dopamine synthesis. The reduction in cellular dopamine levels was not caused by increased dopamine catabolism or dopamine efflux. These data suggest that α-synuclein plays a role in the regulation of dopamine biosynthesis, acting to reduce the activity of tyrosine hydroxylase. If so, a loss of soluble α-synuclein, by reduced expression or aggregation, could increase dopamine synthesis with an accompanying increase in reactive dopamine metabolites.
Keywords: 14-3-3, MN9D, Parkinson's disease, phosphorylation, rat brain, tyrosine hydroxylase
Parkinson's disease (PD) is a neurodegenerative disorder that results from the loss of dopamine (DA)-containing neurons projecting from the substantia nigra to the dorsal striatum (Bernheimer et al., 1973; Hornykiewicz and Kish, 1986). Although PD etiology remains unknown, evidence points to a role for α-synuclein, a presynaptic protein of undetermined function (Takeda et al., 1998; Galvin et al., 2001). Familial PD can result from α-synuclein gene mutations, producing A53T (Polymeropoulos et al., 1997) or A30P (Kruger et al., 1998) amino acid changes. Reduced α-synuclein expression is reported in sporadic PD (Neystat et al., 1999), and α-synuclein is a major component in Lewy bodies, a neuropathological hallmark of PD (Spillantini et al., 1997, 1998;Jenner and Olanow, 1998; Markopoulou et al., 1999). α-Synuclein inclusions occur in several animal models (Betarbet et al., 2000; Feany and Bender, 2000; Kowall et al., 2000; Masliah et al., 2000), and cell-free studies confirm that α-synuclein mutations accelerate oligomerization/aggregation (Conway et al., 1998, 2000; Hashimoto et al., 1998; Giasson et al., 1999; Narhi et al., 1999). Moreover, during PD pathogenesis, mutant α-synuclein aggregation appears to be accelerated, because Lewy body pathology is reported in early-onset familial PD (Golbe et al., 1996). Cumulatively, these findings implicate α-synuclein aggregation in PD pathogenesis.
α-Synuclein may be a chaperone protein based on its ability to inhibit thermally induced protein precipitation (Kim et al., 2000;Souza et al., 2000), a hypothesis that is further supported by the finding that α-synuclein has structural homology with and binds to 14-3-3 proteins, a family of molecular chaperones (Ostrerova et al., 1999). The homologous domains of α-synuclein and 14-3-3 contain residues that for 14-3-3 mediate protein-binding interactions (Zhang et al., 1997). Thus, identifying proteins to which α-synuclein binds may help elucidate α-synuclein function.
Early studies revealed that 14-3-3 binds to and activates tyrosine hydroxylase (TH) (Ichimura et al., 1988), the rate-limiting enzyme in catecholamine synthesis. 14-3-3 preferentially binds to phosphoserines (for review, see Muslin and Xing, 2000), and short-term TH activity is regulated by serine phosphorylation (Kumer and Vrana, 1996; Zhang et al., 1997). For example, Ca2+/calmodulin-dependent protein kinase II, which is linked to depolarization-dependent Ca2+ influx, phosphorylates TH on Ser19 and Ser40 (Haycock, 1990; Kumer and Vrana, 1996). Furthermore, 14-3-3 interacts with TH through an association with Ser19 and Ser40 (Kleppe et al., 2001), and the interaction of 14-3-3 with TH Ser19 is required for TH activation (Itagaki et al., 1999). Because 14-3-3 and α-synuclein may affect the activities of proteins to which they both bind (Jenco et al., 1998; Ostrerova et al., 1999; Van Der Hoeven et al., 2000), we hypothesized that α-synuclein, like 14-3-3, may also bind to TH but, in contrast, inhibit its activity. Although the function of α-synuclein remains obscure, we now provide data suggesting a role for α-synuclein as a key regulator of DA synthesis. This finding suggests that a loss of α-synuclein by its aggregation or its decreased expression, as occurs in PD (Neystat et al., 1999), may selectively disrupt DA homeostasis and negatively impact DA neuronal survival.
MATERIALS AND METHODS
Unless stated otherwise, all reagents used were obtained from Sigma/Aldrich (St. Louis, MO).
Cell culture
The MN9D dopaminergic line is a fusion of rostral mesencephalic neurons from embryonic C57BL/6J (embryonic day 14) mice with the N18TG2 neuroblastoma cells (gift from Drs. A. Heller and L. Wong, University of Chicago). These cells express both TH and aromatic amino acid decarboxylase (AADC) (Choi et al., 1991) and produce measurable levels of DA (Choi et al., 1992). Cells were grown on Primaria tissue culture plates (BD-Falcon Biosciences, Lexington, TN) in DMEM (D5648; Sigma), pH 7.2, with 50 U/ml penicillin, 50 μg/ml streptomycin, and 10% fetal bovine serum (Hyclone, Logan, UT) and incubated at 37°C in a 5% CO2 environment. Frozen stocks of low-pass cells (<10) were used to ensure high-level transgene expression. To provide large enough pools of cells for biochemical measurements, we used undifferentiated MN9D cells at 60–80% confluence. To control for clonal variability, we made biclonal pools of wild-type (WT; clones D7 and D10) and A53T mutant α-synuclein (clones A7 and C8) cells by 1:1 platings using equal numbers of cells at the time of plating for each experiment.
Transfection
MN9D cells were stably transfected using pcDNA3.1 constructs for expression of green fluorescent protein (GFP) (gift from Dr. Guodong Cao, University of Pittsburgh), human wild-type α-synuclein, or A53T α-synuclein (gifts from Dr. Yong-Jian Liu, University of Pittsburgh) by CaPO4 precipitation (Perez et al., 1996) with minor modifications (Brunet et al., 1999). Cells in 60 mm Primaria tissue culture plates (BD-Falcon Biosciences) were at 60% confluence when transfected. Polyclonal pools of stably transfected cells and monoclonal lines were selected with 400 μg/ml G418 and maintained in 200 μg/ml G418 added to media.
Cell viability assay
MN9D cells were plated 5000 cells per well on 96-well plates and evaluated at 5 hr after plating and at 24 hr after switching cells to serum-free medium. For sodium 3,3′-{1-[(phenylamino)carbonyl]-3,4-tetrazolium}bis-4-methoxy-6-nitrobenzene sulfonic acid (XTT) assay, media were supplemented with 20 μl/well XTT (Procheck, Intergen, Purchase, NY) 1–2 hr before spectrophotometric evaluation. Conversion of XTT to formazan was measured at 490 nm by microplate spectrophotometry (SpectraMax 340, Molecular Devices, Sunnyvale, CA). Total protein was assessed by bicinchoninic acid (BCA) spectrophotometry (see below) for normalizing data.
Co-immunoprecipitation
For co-immunoprecipitation (co-IP), all steps were performed at 4°C. Adult rat striata were collected, weighed, and homogenized in 5 vol of ice-cold co-IP buffer that contained 50 mm Tris, pH 7.4, 100 mm NaCl, 5 mm EDTA, 0.3% Triton X-100, 10% glycerol plus aprotinin, leupeptin, 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), β-glycerophosphate, and dithiothreitol to inhibit protease and phosphatase activities. High stringency co-IP buffers were identical but also contained 500 mm NaCl and 0.1% SDS. Supernatants were collected after centrifugation at 17,000 × g(Sorvall RC5B; Kendro Laboratory Products, Newtown, CT). A control aliquot of each supernatant was separated and frozen before co-IP for total protein determinations. Samples were precleared for 1 hr with 10 μl 1% BSA plus 25 μl each of protein A and protein G Sepharose beads (Zymed Laboratories, South San Francisco, CA). Primary antibodies included mouse anti-14-3-3 (Transduction Laboratories, Lexington, KY), mouse anti-TH (MAB 318, Chemicon, Temecula, CA), and mouse anti-α-synuclein (Syn-1, Transduction Laboratories). Immunoprecipitating antibodies (5 μg) were coupled to SiezeX beads according to the manufacturer (Pierce, Rockford, IL). Equal aliquots of homogenates (5.0 mg/ml total protein) were incubated with antibodies against α-synuclein, TH, or 14-3-3, or with preabsorbed antibodies against α-synuclein or TH. Immune complexes were eluted, separated on 12–15% Tris-glycine SDS-PAGE gels, transferred to nitrocellulose, reacted with the same primary antibodies described above, and visualized by chemiluminescence. We noted elution of IgG heavy and light chains along with antigens in some experiments. A 45 kDa band of uncertain origin has been reported on immunoblots using the Syn-1 antibody (Payton et al., 2001). MN9D cell extracts were prepared using the same buffer and co-IP conditions described above, and final supernatants containing ∼500 μg/ml total protein per cell line were separated into equal volume samples for IP.
Assays for DA, dihydroxphenylalanine, dihydroxyphenylacetic acid, and homovanillic acid
Assays were performed by HPLC with electrochemical detection as described by Wagner et al. (1979) with minor modifications (Jackson et al., 1993). Briefly, 20–50 μl supernatant samples were injected onto a Symmetry C18 column (3 mm particle size; 3.9 × 150 mm; Waters Symmetry, Milford, MA). The mobile phase consisted of 50 mmH2NaO4P·H2O, 0.72 mm sodium octyl sulfate, 0.075 mmNa2EDTA, and 16% methanol (v/v), pH 2.7. The mobile phase was pumped through the system at 1.2 ml/min using a Shimadzu LC-10AD pump (Shimadzu Scientific Instruments, Columbia, MD). Compounds were detected and quantified with an ESA coulochem detector (model 5100A, ESA, Bedford, MA) equipped with conditioning (model 5010, ESA) and microdialysis cells (model 5014B, ESA) (E1 = +0.26 V, E2 = +0.28 V, and guard cell = +0.4 V). The limits of detection for DA, dihydroxyphenylalanine (DOPA), dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) were 2 pg/20 μl. Peaks were identified by retention times set to known standards. Data were normalized for total protein for each experiment.
Tissue. Cell monolayers were washed with Dulbecco's PBS containing Ca2+ and Mg2+, then covered in 0.1m perchloric acid. Cells were frozen at −80°C and then thawed. Samples were collected and sonicated followed by centrifugation at 15,000 × g for 15 min at 4°C to remove particulates. Samples were assayed immediately or stored at −80°C until assay.
DA efflux. Cells were rinsed one time with artificial CSF (ACSF) containing (in mm): 147 NaCl, 2.7 KCl, 1.2 CaCl2, 1.0 MgCl2), then incubated for 15 min at 37°C to measure basal DA efflux. Samples were acidified, and particulates were eliminated by centrifugation. Samples were assayed the same day or stored at −80°C until assay as described above using 50 μl injections.
DOPA accumulation assay. TH activity was assessed by measuring the accumulation of DOPA within cells treated with the AADC inhibitor n-hydroxybenzylhydrazine dihydrochloride (NSD-1015) using a modification of the technique of O'Hara et al. (1996) and measured by HPLC from 50 μl samples as described above. Using this method, the rate of intracellular DOPA accumulation is known to be proportional to the rate of tyrosine hydroxylation in TH-containing cells (J. Waymire, personal observation). Briefly, triplicate cultures of untransfected MN9D cells, or cells stably transfected with GFP, wild-type α-synuclein, or A53T α-synuclein were grown in 12-well tissue culture plates, washed two times in ACSF, and equilibrated 20 min at 37°C before addition of 200 μm NSD-1015 in ACSF for 30 min. Parallel triplicate cultures in ACSF without NSD-1015 served as controls. Untransfected and GFP-transfected cells were compared with the wild-type α-synuclein or A53T α-synuclein biclonal lines in three to six independent experiments. ACSF samples and tissues were collected and assayed by HPLC as described above. Data were normalized for total protein for each condition.
Immunoelectron microscopy
Cells were fixed in cryofix (2% paraformaldehyde, 0.01% glutaraldehyde in 0.1 m PBS) and stored at 4°C for 1 hr. Cells were pelleted and resuspended in a small amount of 3% gelatin in PBS, solidified at 4°C, then fixed an additional 15 min in cryofix. Gelatin-cell block was cryoprotected in polyvinylpyrrolidone (PVP) cryoprotectant overnight at 4°C (25% PVP, 2.3 m sucrose, 0.055 m Na2CO3, pH 7.4) as described in Tokuyasu (1989). Cell blocks were frozen on ultracryotome stubs under liquid nitrogen and stored in liquid nitrogen until use. Ultrathin sections (70–100 nm) were cut using a Reichert Ultracut U ultramicrotome with a FC4S cryo-attachment, lifted on a small drop of 2.3 m sucrose, and mounted on Formvar-coated copper grids. Sections were washed three times with PBS, then three times with PBS containing 0.5% bovine serum albumin and 0.15% glycine (PBG buffer) followed by a 30 min blocking incubation with 5% normal goat serum in PBG. Sections were labeled with rabbit anti-TH (AB151, Chemicon; 1:500) and mouse anti-synuclein (Syn-1, Transduction Labs; 1:25) in PBG for 1 hr. Sections were washed four times in PBG and labeled with goat anti-rabbit or goat anti-mouse (5 nm) gold-conjugated secondary antibodies (Amersham Biosciences, Piscataway, NJ), each at a dilution of 1:25 for 1 hr. Sections were washed three times in PBG, three times in PBS, then fixed in 2.5% glutaraldehyde in PBS for 5 min, washed two times in PBS, then washed six times in ddH2O. Sections were post-stained in 2% neutral uranyl acetate, for 7 min, washed three times in ddH2O, stained 2 min in 4% uranyl acetate, then embedded in 1.25% methyl cellulose. Labeling was observed on a JEOL JEM 1210 electron microscope (Peabody, MA) at 80 kV. This technique, although superior for immunogold labeling, somewhat compromises ultrastructural detail (D. Beer-Stolz, personal communication).
In situ DA biosynthesis
We measured the conversion ofl-[1-14C]tyrosine to14CO2 essentially as described previously (Waymire et al., 1988). Briefly, cells were grown on 2- well plates, washed with HEPES-buffered saline (HBS) containing (in mm): 25 HEPES, 5 glycine, 1 CaCl2, 150 NaCl, 2.0 MgCl2, 0.1 KH2PO4, at pH 7.1, and incubated with 200 μl HBS containing 50 mCi/mmol radiolabeled tyrosine for 10–60 min at 37°C. Biosynthesis was stopped by the addition of 400 μl 10% w/v trichloroacetic acid. A small well (Kimble/Kontes, Vineland, NJ) was held in place by a rubber stopper above each culture and contained a folded Whatman paper moistened with Soluene 350 (Packard Instrument Co., Meriden, CT) for14CO2 collection. Radioactivity was measured by liquid scintillation spectroscopy (Beckman-Coulter LS6500, Fullerton, CA).
Phosphorylation of TH and phosphorimage analysis
Quadruplicate cultures were grown on 24-well Primaria plates (BD-Falcon Biosciences) in four independent experiments as described above. For phosphorylation, media were replaced with HBS supplemented with 0.5 mCi/ml32PO4-orthophosphate to label endogenous ATP by 1.5–4 hr incubation at 37°C.32P buffer was removed, and cells were washed and equilibrated for 20 min at 37°C in 200 μl/well HBS. Duplicate samples of cells were either unstimulated or exposed to 40 mm KCl for 5 min at room temperature to stimulate TH activity. Cultures were treated with protease inhibitors and frozen at −80°C. Samples were thawed, and lysates were supplemented with phosphatase inhibitors and processed for TH-IP. For lysates, samples were separated by SDS-PAGE. Recombinant TH protein (gift of Dr. Paul Fitzpatrick, Texas A & M University) was used to verify the identity of the TH bands on Coomassie-stained gels used for phosphorimage analysis. Proteins from IP samples were separated by SDS-PAGE and transferred to nitrocellulose membranes. Phosphorylated TH was quantitated using ImageQuant software (Molecular Dynamics) after exposure of gels or membranes on a phosphor plate for 3–24 d. Total TH was determined by immunoblot of the membranes with AB151 anti-TH antibody (Chemicon).
Protein assay, gel electrophoresis, and immunoblotting
Protein concentrations were determined using BCA relative to BSA protein standards according to the manufacturer (Pierce, Rockford, IL). For striatal homogenates and cell lysates, an aliquot of each sample was reserved as a control. Lysates of MN9D cells were prepared from cell cultures in 1% NP-40 single detergent buffer containing leupeptin, aprotinin, and AEBSF at 4°C as described previously (Perez et al., 1996, 1999). Samples for IP were homogenized; all other lysate samples were sonicated for ∼5 sec using three bursts at an intensity setting of 4.0, and particulates were eliminated by centrifugation at 17,000 × g for 20 min at 4°C. Supernatants were assayed immediately or stored at −20°C until assayed. Proteins in 2–4× Laemmli sample buffer were boiled 1 min, separated on Tris-glycine SDS-PAGE (8–15%), and transferred to nitrocellulose membranes. Prestained protein standards (RPN800; Amersham Biosciences) were also used. Equivalent sample loading was verified by 1% Ponceau S staining before blocking in 5–10% nonfat milk in TBS. Immunoblots were incubated with primary antibodies for 2 hr at room temperature or overnight at 4°C. After three TBS washes, blots were incubated with either a peroxidase-coupled anti-mouse or anti-rabbit secondary antibody (Calbiochem, La Jolla, CA) followed by TBS and TBS-Tween (0.05%) washes. Immune complexes were detected by chemiluminescence using SuperSignal (Pierce) or ECL (DuPont NEN, Boston, MA). For some experiments, linear densitometry of the chemiluminescence signal was performed using a Kodak Image Station (440 CF; Kodak, Rochester, NY).
TH activity
Tritiated water release from 3,5-3H-l-tyrosine was measured using modifications of the methods of Nagatsu et al. (1964), Reinhard et al. (1986), Waymire et al. (1991), and Tanji et al. (1994). Using this assay, 1 mol of3H2O is generated for each mole of 3H-l-tyrosine that is converted to DOPA by TH and is thus a direct measure of TH activity. Briefly, rat adrenal gland was homogenized in 5 vol of HBS (see above) containing protease inhibitors and phosphatase inhibitors, as for co-IP buffer, at 4°C. Particulates were pelleted by centrifugation at 17,000 × g, and supernatants were added to an equal volume of 2× assay buffer to bring the final concentrations to 150 mm Tris maleate, 50 μml-tyrosine, 5 mm ascorbate, 0.45 mg/ml catalase, and 0.5 mm 6-MPH4 at pH 6.8. Samples were incubated for 5 min at 37°C, and reactions were stopped on ice. Released3H2O was separated from unreacted3H-l-tyrosine by mixing with 7.5% charcoal/HCl. Released3H2O was assayed from the supernatant by liquid scintillation counting. To measure the impact of α-synuclein on TH activity, 0–100 μm purified recombinant human α-synuclein (gift from Ronald Hamilton, University of Pittsburgh) was added to homogenates before the 5 min incubations at 37°C. BSA at the same concentrations served as a nonspecific control.
Statistical analyses
ANOVA followed by Tukey-Kramer post hoc comparisons for ANOVA data significant at 0.05 or better was performed using Instat3 (Graphpad, San Diego, CA). Data represent the mean ± SEM from three to eight independent experiments using two to four samples for each condition.
RESULTS
α-Synuclein and TH interact in brain
To evaluate a potential binding interaction between TH and α-synuclein, we prepared rat brain homogenates and performed co-IP assays. Rat striatal samples were immunoprecipitated with antibodies to α-synuclein, TH, or preabsorbed antibodies coupled to protein G beads. An aliquot of homogenate was reserved to reveal the relative amount of α-synuclein (Fig.1a, lane 1) or TH (Fig. 1b, lane 1) in the starting samples. Co-IP with the α-synuclein antibody precipitated the expected band at ∼19 kDa (Fig. 1a, lane 2) along with an ∼60 kDa protein identified as TH by immunoblotting using the TH antibody (Fig.1b, lane 2). When the TH antibody was used for co-IP and proteins were immunoblotted, we observed the expected 60 kDa TH band (Fig. 1b, lane 3) as well as an ∼19 kDa band identified as α-synuclein using an α-synuclein-specific antibody (Fig. 1a, lane 3). Furthermore, the association between TH and α-synuclein was not diminished by high-stringency co-IP with 500 mm NaCl and 0.1% SDS in control experiments (data not shown). Preincubation of the α-synuclein antibody with recombinant α-synuclein protein effectively blocked IP of α-synuclein and co-IP of TH (Fig.1a,b, lane 5). Similarly, incubation of homogenate with preabsorbed TH antibody (preincubated with recombinant TH before coupling onto beads) blocked IP of TH and co-IP of α-synuclein (Fig. 1a,b, lane 4). In additional control experiments, we also reconfirmed the association between 14-3-3 and TH (Ostrerova et al., 1999) by co-IP of striatal homogenates (data not shown). These data identify a previously undefined association between TH and α-synuclein in brain as confirmed by co-IP using either a TH-specific or an α-synuclein-specific antibody and appropriate controls for specificity.
Fig. 1.

Interaction of α-synuclein with TH in rat brain. Data from a representative experiment showing Western blots (WB) reacted with the α-synuclein antibody (a) or with the TH antibody (b). a, WB of α-synuclein from rat striatum shows α-synuclein in the initial homogenate (lane 1), the α-synuclein IP (Syn-IP) sample (lane 2), the TH-IP sample (lane 3), a control IP using preabsorbed TH antibody (Abs TH-IP, lane 4), and control IP using preabsorbed α-synuclein antibody (Abs Syn-IP,lane 5). In b the WB reacted with the TH antibody shows TH in the initial homogenate (lane 1), the α-synuclein IP sample (lane 2), the TH IP sample (lane 3), a control IP using preabsorbed TH antibody (lane 4), and control IP using preabsorbed α-synuclein antibody (lane 5). Nonspecific bands, two of which appear to be IgG bands, are evident in lanes with stronger chemiluminescence signal (indicated by asterisks ina and b). Molecular weights in kilodaltons (kDa), determined from prestained standards, are indicated on the left.
α-Synuclein inhibits TH activity in a cell-free assay
Adrenal gland, a rich source of TH, was homogenized and assayed for TH activity in the presence or absence of recombinant wild-type α-synuclein (0–100 μm) and3H-l-tyrosine. Whether this concentration of α-synuclein activity is physiologically relevant is currently unknown. However, the concentrations of α-synuclein were within the range of protein previously used in similar cell-free assays (Tanji et al., 1994). We observed a dose-dependent decrease in TH activity as determined by the ability of TH to form3H2O from3H-l-tyrosine during a 5 min incubation, whereas BSA at the same concentrations did not significantly inhibit TH activity (Fig.2).
Fig. 2.

Inhibition of TH activity by α-synucleinin vitro. Unreacted3H-l-tyrosine was separated from3H2O using an acidic charcoal wash, and3H2O in the resulting supernatants was measured by scintillation counting for 10 min. Release of3H2O from 3H-l-tyrosine was diminished by recombinant α-synuclein in a dose-dependent manner using a cell-free in vitro assay. BSA did not significantly diminish TH activity at any dose. Data represent the mean ± SEM for five independent experiments using triplicate samples for each condition. *p < 0.05; ***p < 0.001.
MN9D dopaminergic cells as a model for evaluating the interaction of α-synuclein with TH
To generate a model for exploring the physiological relevance of the impact of α-synuclein on TH in neuronal cells, we stably transfected the dopaminergic MN9D line with wild-type or mutant A53T α-synuclein constructs. We also generated stably transfected GFP-expressing cell lines as a control. We observed that GFP-MN9D cells expressed endogenous levels of α-synuclein identical to untransfected MN9D cells (Fig. 3a,lanes 1, 2). In contrast, cells stably transfected with wild-type α-synuclein or A53T mutant α-synuclein expressed 5–13 times more α-synuclein than parental MN9D cells or control MN9D cells transfected with GFP as determined using linear-range densitometry of immunoblots. Clones expressing similar levels of α-synuclein were selected for further characterization (Fig. 3a, lanes 3–6). All MN9D cells expressed equivalent high endogenous levels of TH as measured by TH immunoblot analysis (Fig. 3b). Equivalent TH expression among the cell lines was also confirmed by immunoblots from MN9D cell extracts normalized for total protein levels (ANOVA; p> 0.12).
Fig. 3.

Expression of α-synuclein and TH in parental and in stably transfected MN9D clonal cell lines. a, Parental MN9D (lane 1) and GFP-transfected MN9D cells (lane 2) express low endogenous α-synuclein levels compared with clonal cells overexpressing wild-type α-synuclein (WT Syn, lanes 3,4), or A53T mutant α-synuclein (lanes 5, 6) as determined by immunoblot of 20 μg protein per lane reacted with the Syn-1 anti-α-synuclein antibody. b, TH levels are equivalent in all MN9D cell lines whether expressing endogenous α-synuclein (parentalMN9D, lane 1, or GFP-MN9D,lane 2) or overexpressing α-synuclein [wild-type (WT), lanes 3 and4, or A53T, lanes 5 and6] as determined by immunoblot of 20 μg protein per lane reacted with the MAB318 anti-TH antibody.
Because a loss of viability has been reported in some cells that overexpress α-synuclein (Ostrerova et al., 1999; Zhou et al., 2000), we also assessed cell viability in our MN9D cell lines. We measured basal viability using the XTT spectrophotometric assay at equal times after plating using equal numbers of cells for each condition. We compared each condition with its own baseline value obtained at 5 hr to parallel cultures analyzed 24 hr later. We observed that percentage viability, as determined by XTT, was similar for all MN9D lines. Parental MN9D cells served as baseline controls (control set to 1.00, with a SEM of ±0.024% between experiments) for comparison with MN9D GFP (1.05 ± 0.05%), wild-type α-synuclein (1.015 ± 0.04%), and A53T α-synuclein overexpressing lines (1.16 ± 0.16%; ANOVA, p = 0.72). These data indicate that α-synuclein overexpression in our cells did not significantly diminish endogenous TH levels or alter basal cell viability.
An association between α-synuclein and TH occurs in MN9D cells
We next determined whether the TH and α-synuclein interaction observed in brain homogenates was also apparent in MN9D cells as measured by co-IP. Using an α-synuclein-specific antibody, we immunoprecipitated α-synuclein from the various MN9D cell lines. As expected, we were able to immunoprecipitate more α-synuclein from stably transfected α-synuclein MN9D lines than from either parental or GFP transfected control MN9D lines (Fig.4a). When we probed the co-IP samples for TH, we observed that TH was immunoprecipitated along with α-synuclein from all cell lines and that more TH was precipitated with α-synuclein in overexpressing cell lines (Fig. 4b), confirming the association of TH with α-synuclein in MN9D cells. In control experiments we determined that co-IP with a TH-specific antibody also immunoprecipitated α-synuclein from the various cell lines and that co-IP was eliminated by antibody preabsorption (data not shown).
Fig. 4.

Interaction of α-synuclein with TH in MN9D cells. Western blots (WB) from a representative co-IP experiment reacted with the anti-α-synuclein antibody (a) or with the anti-TH antibody (b). Immunoprecipitation of α-synuclein from cell extracts (a) resulted in co-IP of TH (b) from parental MN9D (MN9D), GFP-transfected MN9D (GFP), wild-type (WT) α-synuclein, and A53T α-synuclein (A53T Syn) cells. Immunoelectron microscopy reveals colocalization of TH with α-synuclein that is apparent on mitochondria (c) and vesicles (d,e) in MN9D cells. In c andd, the larger (10 nm) gold particles label α-synuclein and the smaller (5 nm) particles label TH colocalized on the surface of a mitochondrion (c) and at the edge of a vesicular structure (d) in an MN9D cell stably transfected with A53T α-synuclein. In e the large (10 nm) gold particles label TH and the small (5 nm) particles label α-synuclein in the cytoplasm of an MN9D cell stably transfected with wild-type α-synuclein. Arrowheads inc–e point to colocalized small and large gold particles. Arrows in d point to the lipid bilayer of a vesicle decorated with large and small gold particles. m, Mitochondrion; v, vesicle. Scale bars, 100 nm.
To further evaluate the potential association of α-synuclein with TH in MN9D cells, we performed double-label immunoelectron microscopy using antibodies specific for TH and α-synuclein. Parental MN9D, GFP-expressing MN9D, and α-synuclein-overexpressing cells were cryoembedded and sectioned before incubation with both α-synuclein and TH antibodies. We confirmed colocalization of TH with α-synuclein on or near mitochondria (Fig. 4c) and vesicular membranes (Fig. 4d,e) within the cytosol of MN9D cells. This distribution is reminiscent of immunogold-labeled TH observed in rat brain (Glass et al., 2001). In control MN9D cells, which expressed endogenous levels of α-synuclein and TH, TH was also found colocalized with α-synuclein but at lower overall levels, as expected (data not shown). Specificity of the labeling reaction was confirmed in control samples from which the primary antibodies were omitted before incubation with immunogold-labeled secondary antibodies. Taken together, these co-IP and electron microscopy data reconfirm an association of TH with α-synuclein seen in rat brain (Fig. 1) and strengthen the validity of MN9D cells as a model system for our studies.
Transfected MN9D cells that overexpress α-synuclein have reduced DA levels
To assess steady-state levels of DA from the various MN9D cells, we prepared cell extracts and measured DA content by HPLC with electrochemical detection. Parental MN9D cells and GFP control cells had similarly high DA levels compared with either wild-type α-synuclein or A53T α-synuclein clonal lines (Fig.5a). Steady-state DA levels were significantly reduced in both wild-type α-synuclein and A53T α-synuclein clonal lines (Fig. 5a). DA levels were fourfold lower in wild-type α-synuclein lines (5.3 ± 0.98 ng/mg protein) and approximately eightfold lower in A53T mutant α-synuclein lines (1.9 ± 0.37 ng/mg protein) compared with parental MN9D cells (20.8 ± 1.52 ng/mg protein) or GFP cells (22.4 ± 0.75 ng/mg protein; ANOVA; p < 0.0001) (Fig.5a). DA levels were lower in A53T cells compared with wild-type cells in this series of experiments; however, post hoc analyses confirmed that the difference was not statistically significant (p > 0.05). Thus, wild-type and A53T mutant α-synuclein reduced steady-state DA levels similarly, suggesting that both α-synucleins interact with TH to influence DA synthesis.
Fig. 5.

Effect of α-synuclein overexpression on cellular DA and DA efflux from MN9D cells. Supernatants obtained from cell lysates of parental MN9D, GFP, wild-type α-synuclein (WT SYN), and A53T mutant α-synuclein cells were assayed for DA using HPLC with electrochemical detection (a). Although A53T cells had even lower cellular DA levels than wild-type α-synuclein cells in a, the difference was not statistically significant using ANOVA with Tukey-Kramer post hoc analyses (p > 0.05). To measure DA efflux from the various MN9D lines, cells were washed in ACSF and incubated in fresh ACSF for 15 min at 37°C before collection (b). Data, normalized for total protein, represent the mean ± SEM of triplicate samples from two to six independent experiments.Black bar, MN9D; light gray bar, GFP;white bar, wild-type α-synuclein; dark gray bar, A53T α-synuclein. **p < 0.01; ***p < 0.001.
The decrease in DA levels observed in MN9D cells overexpressing α-synuclein is consistent with an inhibition of TH; however, there are other possible explanations. For example, reduced DA levels may have resulted from enhanced DA degradation. To assess this possibility we measured DA metabolites DOPAC and HVA by HPLC from MN9D cell lysates. We observed that although the DOPAC and HVA levels in cell lysates varied somewhat between experiments, DOPAC and HVA were consistently higher in lysates from control MN9D cells than in lysates from stably transfected α-synuclein MN9D lines, where they were only slightly above the limits of detection (data not shown). Thus, increased degradation of DA did not appear to explain the reduced intracellular DA in wild-type or A53T mutant α-synuclein lines.
α-Synuclein binds to lipids and associates with vesicles (Maroteaux et al., 1988; Davidson et al., 1998; Jensen et al., 1998) (Fig.4c,d). Thus, another possible explanation for the reduction in DA levels in cells overexpressing α-synuclein is that the protein increases DA efflux. To evaluate this possibility as a means of reducing cellular DA, we measured DA efflux from the various MN9D lines. In three independent experiments using triplicate cultures for each condition, media were replaced with 37°C Dulbecco's PBS or ACSF, and cells were incubated in these media for 15 min at 37°C. The new media were collected and acidified, and the amount of DA released from cells during the 15 min incubation period was then measured (Fig. 5b). The concentration of DA present in media from wild-type (3.5 ± 0.5 nm) and A53T mutant (3.2 ± 0.2 nm) α-synuclein lines was ∼65% less than in media from untransfected parental MN9D cells (9.3 ± 1.5 nm) or GFP-transfected MN9D cells (8.8 ± 0.95 nm) (Fig. 5b). Similar levels of DA efflux were observed from cells expressing wild-type or A53T mutant α-synuclein, and efflux paralleled intracellular DA levels in each of the cell lines (ANOVA;p < 0.001). Taken together, the data suggest that the differences in DA levels observed from cells overexpressing wild-type or A53T mutant α-synuclein cannot be explained as an increase in DA catabolism or in DA efflux but instead were the result of reduced DA biosynthesis perhaps caused by TH inhibition.
DA synthesis is diminished in MN9D cells stably expressing wild-type or A53T mutant α-synuclein
The above data revealed that MN9D cells expressing endogenous levels of α-synuclein had equivalent levels of DA synthesis but that the rate of DA biosynthesis was reduced in cells overexpressing wild-type or A53T mutant α-synuclein. Because parental MN9D and GFP expressing MN9D lines were essentially identical by all measures, parental MN9D cells were used as controls in subsequent studies. To directly assess DA biosynthesis as a function of α-synuclein expression and TH activity, we used the hydrazine compound NSD-1015 to inhibit AADC, thereby blocking the conversion of DOPA to DA (see model in Fig. 8a). Cells were exposed to 200 μm NSD-1015 for 30 min at 37°C, lysates were collected, and samples were assayed for differences in cellular DOPA after NSD treatment. We observed that parental MN9D cells significantly increased DOPA (78.5 ± 21.5%) (Fig.6a) as compared with untreated cells. However, in marked contrast, wild-type α-synuclein lines increased their DOPA levels by only 9.0 ± 7.0%, and A53T mutant α-synuclein lines increased DOPA by only 4.0 ± 3.0% (ANOVA;p < 0.001) compared with untreated cells for each group. Because DOPA accumulation could only occur if TH actively converted tyrosine to DOPA, these data suggest that TH activity was diminished in cells overexpressing either wild-type or mutant α-synuclein.
Fig. 8.

The potential roles of α-synuclein in dopamine synthesis (a) and neurodegeneration (b). DA is synthesized in a multistep process in which the amino acid tyrosine is converted to DOPA by the activity of phosphorylated TH and aromatic amino acid decarboxylase (AADC). 14-3-3 is known to bind to TH that is phosphorylated and α-synuclein appears to bind to dephospho-TH. 14-3-3 and α-synuclein may differentially regulate TH activity (at1 in a) to maintain optimal DA levels in concert with the activities of the vesicular monoamine transporter (VMAT2, indicated by a small box on vesicle), which normally packages intracellular DA into vesicles, and the dopamine transporter (DAT, indicated by large white box outlined in black on the plasma membrane), which can function bidirectionally. α-Synuclein may directly bind to TH and inhibit TH activity and/or phosphorylation, or α-synuclein may act indirectly by activating a phosphatase (indicated by + in a) or inhibiting a kinase (indicted by − in a) to affect TH phosphorylation. In b, a reduction in free soluble α-synuclein (indicated by gray dashed X over α-syn, at 2) may occur by downregulation of α-synuclein mRNA or by stimuli that induce fibrilization (e.g., environmental toxins, mutation, nitration, or ubiquitination). Disinhibition of TH may lead to elevated cytosolic DA in neurons with subsequent generation of DA-quinone and DA-related ROS (at 3 in b), which can damage proteins, lipids, and DNA and contribute to neurotoxicity.
Fig. 6.
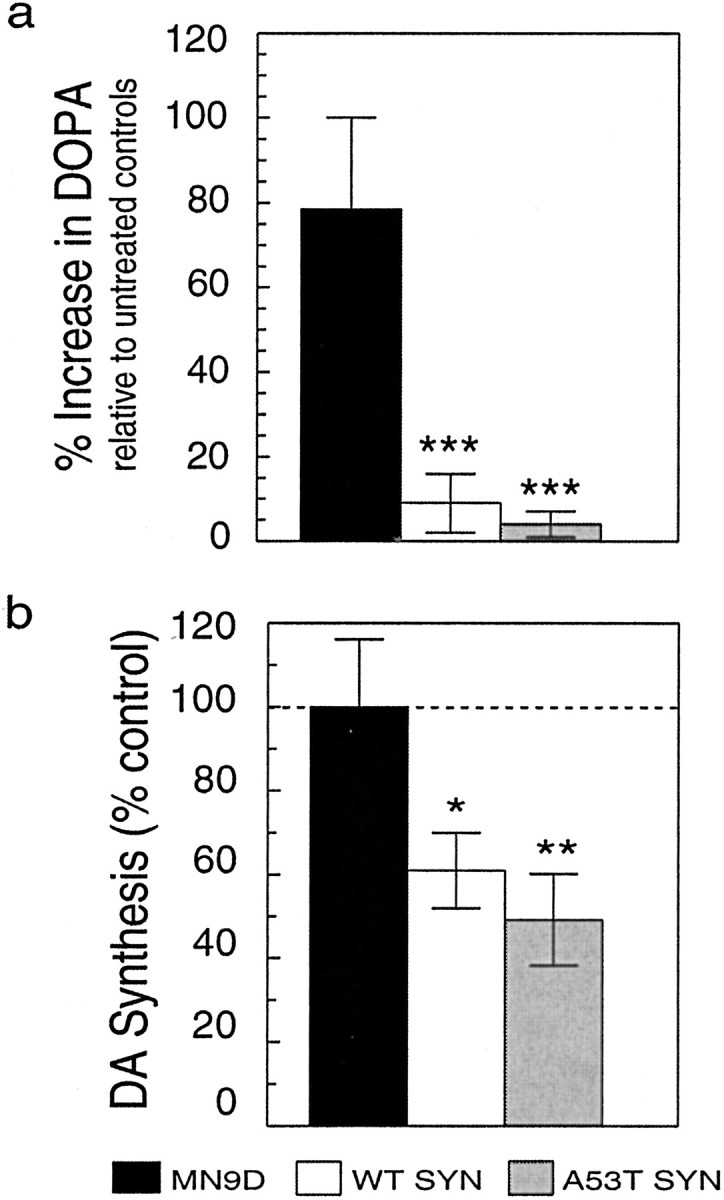
Effect of α-synuclein overexpression on DA synthesis in MN9D cell lines. Cells lysates were prepared from equivalent cultures of MN9D cells incubated in the presence of 200 μm NSD-1015 for 30 min. a, More DOPA accumulated after blocking AADC activity with NSD-1015 in parental MN9D cells than in wild-type (WT) or A53T mutant α-synuclein lines, indicating that TH activity is dramatically reduced in cells that overexpress α-synuclein. b, In situ DA synthesis was significantly greater for parental MN9D cells than for WT or A53T α-synuclein clonal lines incubated withl-[1-14C]tyrosine and compared for their ability to generate 14CO2 from the radioactive precursor. Data are from two to six independent experiments presented as the mean ± SEM for each condition. *p < 0.05; **p < 0.01; ***p < 0.001.
We next assessed DA synthesis in the various MN9D lines by comparing the conversion ofl-[1-14C]tyrosine with14CO2, which requires the activities of both TH and AADC. We observed significantly less 14CO2 generated by wild-type or A53T mutant α-synuclein MN9D lines when compared with parental cells (Fig. 6b). In data from eight independent experiments we observed that wild-type α-synuclein expressing cells produced ∼40% less14CO2 than controls (p < 0.05), and A53T mutant α-synuclein lines produced ∼50% less14CO2 than controls (p < 0.01). Taken together, the data implicate both wild-type and A53T mutant α-synuclein in DA synthesis inhibition.
TH phosphorylation is diminished by overexpression of wild-type or A53T mutant α-synuclein
The above data revealed that DA synthesis was reduced in cells overexpressing either wild-type or A53T mutant α-synuclein. The short-term regulation of TH activity occurs primarily by changes in TH phosphorylation. Because 14-3-3 is known to bind to and can sustain the activity of phosphorylated TH (Itagaki et al., 1999; Muslin and Xing, 2000) and because both 14-3-3 and α-synuclein appear to affect the activities of proteins to which they bind (Ostrerova et al., 1999; Van Der Hoeven et al., 2000), we next measured TH phosphorylation in our MN9D lines to determine whether overexpression of α-synuclein may have altered TH phosphorylation. We labeled cells with32P-orthophosphate and measured TH-phosphorylation using autoradiography and phosphorimage analysis. When we compared TH phosphorylation from cell lysates we observed that cells overexpressing wild-type α-synuclein or A53T mutant α-synuclein had 20–30% less phosphorylated TH compared with controls (ANOVA; p < 0.01) (Fig.7a). Immunoprecipitation of TH from the various cell lines brought down similar amounts of TH (Fig.7b); however, phosphorylated TH levels were significantly reduced in cells that overexpressed α-synuclein (Fig. 7c). Because an equivalent reduction in TH phosphorylation was observed for both wild-type and A53T α-synuclein lines, the data suggest that both forms of α-synuclein were able to inhibit TH phosphorylation similarly.
Fig. 7.
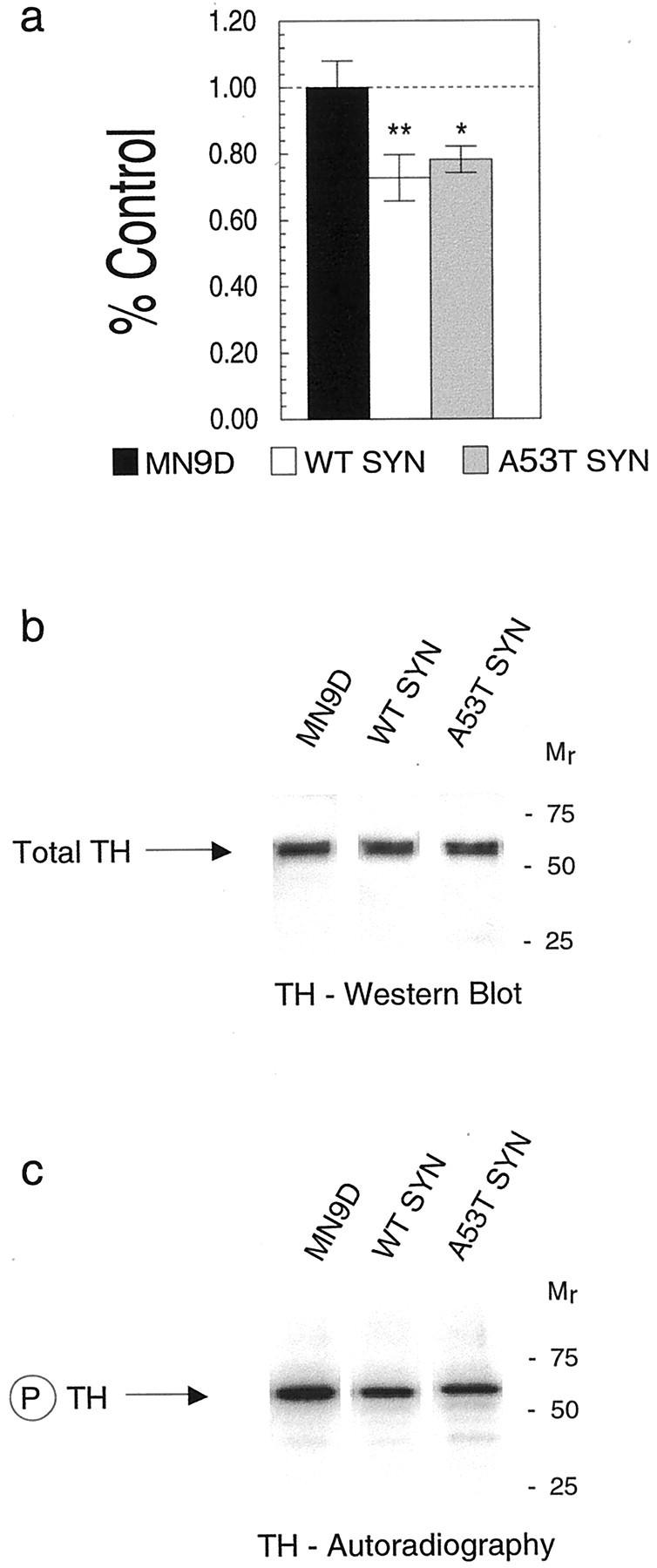
The impact of α-synuclein on TH phosphorylation in MN9D cells. Cells labeled with 32P were evaluated for TH phosphorylation by autoradiography of cell lysates from control MN9D and stably transfected α-synuclein lines separated using SDS-PAGE and analyzed by phosphorimaging (a). After TH immunoprecipitation from cell lysates, total TH levels (atarrow) from a representative experiment appear similar for MN9D, WT, and A53T MN9D lines as determined by Western blot reacted with the AB151 anti-TH antibody and visualized by chemiluminescence (b). Phospho-TH levels (atarrow in c) are significantly reduced in cells overexpressing wild-type α-synuclein or A53T mutant α-synuclein compared with control MN9D cells as seen by autoradiography. In a, black barrepresents MN9D, white bar represents wild-type α-synuclein, and gray bar represents A53T α-synuclein. Molecular weights, determined from prestained standards, are indicated on the right in b andc. *p < 0.05; **p < 0.01.
DISCUSSION
Our studies have allowed us to identify an association between α-synuclein and TH that appears to be functionally significant for DA neurons. This association, first identified in brain homogenates, prompted us to explore the potential impact of the α-synuclein–TH interaction in vitro using a cell-free assay. Addition of recombinant human α-synuclein produced a dose-dependent inhibition of TH activity. To explore the potential effect of α-synuclein on TH in a more physiological model, we compared control dopaminergic cells expressing endogenous levels of α-synuclein with cell lines stably transfected to overexpress wild-type or A53T mutant α-synuclein. We confirmed that TH expression was not significantly altered by α-synuclein overexpression in our stably transfected cell lines and that α-synuclein transgene expression was maintained at high levels during our studies.
The data that we obtained allow us to make three novel observations: (1) α-synuclein interacts with TH in brain as well as in a dopaminergic cell line, (2) α-synuclein inhibits TH activity in a cell-free assay, and (3) overexpression of α-synuclein in a dopaminergic cell line dramatically reduces TH activity, TH phosphorylation, and DA synthesis.
The region of 14-3-3 that is homologous to α-synuclein contains conserved amino acids that mediate the binding of 14-3-3 to diverse ligands (Zhang et al., 1997). Thus, assuming that the 14-3-3 homologous region on α-synuclein, which extends from amino acids 1 to 60 in the amino terminus, subserves protein binding, we anticipated that the A53T mutation of α-synuclein would alter the effect of α-synuclein on TH by altering protein–protein interactions. However, the A53T mutant α-synuclein appeared to behave similarly to wild-type α-synuclein by all measures related to DA synthesis in our studies, strongly suggesting that both forms of α-synuclein interact similarly with TH to inhibit DA synthesis. Furthermore, although not directly evaluated in these studies, our findings suggest that the region of α-synuclein involved in its association with TH may not reside in the first 60 amino acids of α-synuclein, which harbors both A53T and A30P mutations. Stefanis and colleagues (2001) recently reported that overexpressing A53T mutant α-synuclein, but not wild-type α-synuclein, in PC12 cells induced a loss of dense-core secretory granules (DCGs) with a coincident loss of depolarization-evoked DA release. However, because PC12 cells store DA and other catecholamines in DCGs, the loss of DCGs would be anticipated to eliminate DA release. Furthermore, basal DA efflux in the absence of depolarization, as we measured, was not described for PC12 α-synuclein lines, making direct comparison of our DA data with theirs impossible. In addition, the A53T PC12 cells exhibited impaired lysosomal/proteosomal function and reduced cell viability, whereas our α-synuclein-expressing MN9D cell lines were equally viable. Moreover, no loss of nigral DA neurons is reported for A53T overexpressing transgenic mice (van der Putten et al., 2000; Matsuoka et al. 2001), suggesting that not all dopaminergic cells are compromised by A53T α-synuclein overexpression.
α-Synuclein and 14-3-3 are both enriched in the cytoplasm of nerve terminals (Maroteaux et al., 1988; Irizarry et al., 1996; Broadie et al., 1997; Withers et al., 1997; Murphy et al., 2000), the same compartment that contains TH (Pickel et al., 1975; Glass et al., 2001) and in which tyrosine hydroxylation takes place. Once generated, DA is normally packaged into synaptic vesicles by the activity of the vesicular monoamine transporter, which not only provides packaged neurotransmitter available for stimulated release (Nirenberg et al., 1996) but also serves to detoxify the intracellular environment by removing highly reactive DA from the cytoplasm (Liu et al., 1992). Thus, if the activities of α-synuclein and 14-3-3 are necessary to maintain normal DA levels, a loss of α-synuclein by decreased expression (Neystat et al., 1999) or aggregation (for review, see El-Agnaf and Irvine, 2000), should lead to an overproduction of DA and a subsequent increase in the cytoplasmic concentration of this molecule.
On the basis of previous reports from the literature and our new findings regarding α-synuclein inhibition of TH activity and DA synthesis, we propose the following model for α-synuclein in DA homeostasis with possible consequences for neurodegeneration (Fig.8). (1) Under normal circumstances, 14-3-3 binds to phospho-TH to enhance TH activity. Active TH converts tyrosine to DOPA, which in turn is decarboxylated by AADC to produce DA. α-Synuclein, on the other hand, interacts with TH and inhibits its activity by decreasing TH phosphorylation. (2) Soluble α-synuclein levels, however, are reduced in PD. The reduction frees binding partners of α-synuclein, such as TH, to bind to 14-3-3 leading to TH activation and increased DA synthesis. (3) If DA levels exceed the ability of the cells to remove DA from the cytosol, the accumulated cytosolic DA forms DA-quinone and DA-associated oxyradicals leading to DA neuronal cell death.
Support for our model derives from several observations indicating that 14-3-3 binds to phospho-TH (Ichimura et al., 1988; Kumer and Vrana, 1996; Muslin and Xing, 2000; Kleppe et al., 2001), interaction of TH with 14-3-3 enhances TH activation (Itagaki et al., 1999) and phosphorylation (Bevilaqua et al., 2001), cytosolic DA can form DA-quinone and reactive oxygen species (ROS) that are neurotoxic (Graham, 1978; Hastings et al., 1996; Stokes et al., 1999; Lotharius and O'Malley, 2000), α-synuclein itself is modified by DA quinone (Conway et al., 2001), and DA release is enhanced in mice lacking α-synuclein expression as measured by paired-pulse stimulation (Abeliovich et al., 2000). According to our model, the absence of α-synuclein would increase TH activity and thus provide more DA for release at the second pulse, which is the case. However, other findings in the α-synuclein knock-out mice appear less supportive of our model. For example, Abeliovich and colleagues (2000) reported lower peak levels of extracellular DA from amphetamine-stimulated striatal slices. Interestingly, substantia nigra DA levels were not reduced in α-synuclein knock-out mice. Perhaps the reduction in striatal DA occurred secondary to a reduction in the size of the pool of synaptic vesicles as suggested by the work of Murphy and colleagues (2000) using an in vitro model. Additionally, DA release was evoked by amphetamine, which acts to reverse the DA transporter. Because α-synuclein binds to the DA transporter and affects its activity (Lee et al., 2001), this may also have contributed to differences in extracellular DA levels from knock-out mice. Finally, our model proposes that a loss of α-synuclein function would lead to elevated intracellular DA with potential neurotoxic consequences. α-Synuclein knock-out mice have normal numbers of DA neurons at 3–6 weeks (Abeliovich et al., 2000). However, neurotoxicity of DA neurons secondary to oxyradical damage can be delayed for many months (Ueda et al., 2000) and thus may not be present after only a few weeks.
Our findings suggest that the impact of α-synuclein on TH activity may be direct, as suggested by the homology of α-synuclein with 14-3-3, the ability to co-IP TH with α-synuclein from brain and dopaminergic cells, and the colocalization of both proteins in neuronal cells. However, it is feasible that the effects of α-synuclein are indirect. One possibility is that α-synuclein may inhibit a kinase or activate a phosphatase involved in TH phosphorylation/dephosphorylation. Alternatively, α-synuclein and TH may be associated within a larger protein complex, and we are further exploring these possibilities. In any event, our data identify a functional role for α-synuclein in the regulation of DA synthesis that is maintained by the A53T mutant form of α-synuclein. Because α-synuclein is strongly implicated in PD and because DA neurons are the cells selectively damaged during PD pathogenesis, our findings provide a focal point for further exploration into the causes and treatment of the disease. Whether a loss of functional α-synuclein is associated with increased brain DA turnover is not yet known. However, increased DA turnover in residual DA neurons has long been reported in patients with even mild forms of PD (Bernheimer et al., 1973). Elucidation of the potential role of α-synuclein in such a process awaits further analysis.
Footnotes
This work was supported by the Scaife Family Foundation, the National Parkinson Foundation, and the National Institute of Neurological Disorders and Stroke (NS19608). We thank Alfred Heller and Lisa Won for the MN9D cells; Ronald Hamilton and Paul Fitzpatrick for recombinant proteins; Yong-Jian Liu for α-synuclein plasmids; Guodong Cao for the eGFP plasmid; Simon Watkins and Donna Beer-Stolz for help with electron microscopy; Alex Glessner and Susan Slagel for expert technical assistance; and Teresa Hastings for thoughtful critique of this manuscript. This research is dedicated to D. Beyer, J. Cordy, M. J. Fox, and to the memory of Lester “Rusty” Lanelli.
Correspondence should be addressed to Dr. Ruth G. Perez, Department of Neurology, Biomedical Science Tower, S510, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213. E-mail:perezrg@pitt.edu.
REFERENCES
- 1.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 2.Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington: clinical, morphological, and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- 3.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 4.Bevilaqua LR, Graham ME, Dunkley PR, von Nagy-Felsobuki EI, Dickson PW. Phosphorylation of ser19 alters the conformation of tyrosine hydroxylase to increase the rate of phosphorylation of ser40. J Biol Chem. 2001;276:40411–40416. doi: 10.1074/jbc.M105280200. [DOI] [PubMed] [Google Scholar]
- 5.Broadie K, Rushton E, Skoulakis EM, Davis RL. Leonardo, a Drosophila 14-3-3 protein involved in learning, regulates presynaptic function. Neuron. 1997;19:391–402. doi: 10.1016/s0896-6273(00)80948-4. [DOI] [PubMed] [Google Scholar]
- 6.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 7.Choi HK, Won LA, Kontur PJ, Hammond DN, Fox AP, Wainer BH, Hoffmann PC, Heller A. Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res. 1991;552:67–76. doi: 10.1016/0006-8993(91)90661-e. [DOI] [PubMed] [Google Scholar]
- 8.Choi HK, Won L, Roback JD, Wainer BH, Heller A. Specific modulation of dopamine expression in neuronal hybrid cells by primary cells from different brain regions. Proc Natl Acad Sci USA. 1992;89:8943–8947. doi: 10.1073/pnas.89.19.8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 10.Conway KA, Lee SJ, Rochet JC, Ding TT, Harper JD, Williamson RE, Lansbury PT., Jr Accelerated oligomerization by Parkinson's disease linked alpha-synuclein mutants. Ann NY Acad Sci. 2000;920:42–45. doi: 10.1111/j.1749-6632.2000.tb06903.x. [DOI] [PubMed] [Google Scholar]
- 11.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 12.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 13.El-Agnaf OM, Irvine GB. Review: formation and properties of amyloid-like fibrils derived from alpha-synuclein and related proteins. J Struct Biol. 2000;130:300–309. doi: 10.1006/jsbi.2000.4262. [DOI] [PubMed] [Google Scholar]
- 14.Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 15.Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58:186–190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- 16.Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wildtype human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 17.Glass MJ, Huang J, Aicher SA, Milner TA, Pickel VM. Subcellular localization of the α-2A-adrenergic receptors in the rat medial nucleus tractus solitarius: regional targeting and relationship with catecholamine neurons. J Comp Neurol. 2001;433:193–207. doi: 10.1002/cne.1135. [DOI] [PubMed] [Google Scholar]
- 18.Golbe LI, Di Iorio G, Sanges G, Lazzarini AM, La Sala S, Bonavita V, Duvoisin RC. Clinical genetic analysis of Parkinson's disease in the Contursi kindred. Ann Neurol. 1996;40:767–775. doi: 10.1002/ana.410400513. [DOI] [PubMed] [Google Scholar]
- 19.Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- 20.Hashimoto M, Hsu LJ, Sisk A, Xia Y, Takeda A, Sundsmo M, Masliah E. Human recombinant NACP/alpha-synuclein is aggregated and fibrillated in vitro: relevance for Lewy body disease. Brain Res. 1998;799:301–306. doi: 10.1016/s0006-8993(98)00514-9. [DOI] [PubMed] [Google Scholar]
- 21.Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haycock JW. Phosphorylation of tyrosine hydroxylase in situ at serine 8, 19, 31, and 40. J Biol Chem. 1990;265:11682–11691. [PubMed] [Google Scholar]
- 23.Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson's disease. In: Yahr MD, Bergmann KJ, editors. Advances in neurology, Vol 45, Parkinson's disease. Raven; New York: 1986. pp. 19–34. [PubMed] [Google Scholar]
- 24.Ichimura T, Isobe T, Okuyama T, Takahashi N, Araki K, Kuwano R, Takahashi Y. Molecular cloning of cDNA coding for brain-specific 14-3-3 protein, a protein kinase-dependent activator of tyrosine and tryptophan hydroxylases. Proc Natl Acad Sci USA. 1988;85:7084–7088. doi: 10.1073/pnas.85.19.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irizarry MC, Kim TW, McNamara M, Tanzi RE, George JM, Clayton DF, Hyman BT. Characterization of the precursor protein of the non-A beta component of senile plaques (NACP) in the human central nervous system. J Neuropathol Exp Neurol. 1996;55:889–895. doi: 10.1097/00005072-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Itagaki C, Isobe T, Taoka M, Natsume T, Nomura N, Horigome T, Omata S, Ichinose H, Nagatsu T, Greene LA, Ichimura T. Stimulus-coupled interaction of tyrosine hydroxylase with 14-3-3 proteins. Biochemistry. 1999;38:15673–15680. doi: 10.1021/bi9914255. [DOI] [PubMed] [Google Scholar]
- 27.Jackson D, Abercrombie ED, Zigmond MJ. Impact of l-dopa on striatal acetylcholine release: effects of 6-hydroxydopamine. J Pharmacol Exp Ther. 1993;267:912–918. [PubMed] [Google Scholar]
- 28.Jenco JM, Rawlingson A, Daniels B, Morris AJ. Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry. 1998;37:4901–4909. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- 29.Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998;44:S72–84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- 30.Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 31.Kim TD, Paik SR, Yang CH, Kim J. Structural changes in alpha-synuclein affect its chaperone-like activity in vitro. Protein Sci. 2000;9:2489–2496. doi: 10.1110/ps.9.12.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleppe R, Toska K, Haavik J. Interaction of phosphorylated tyrosine hydroxylase with 14-3-3 proteins: evidence for a phosphoserine 40-dependent association. J Neurochem. 2001;77:1097–1107. doi: 10.1046/j.1471-4159.2001.00318.x. [DOI] [PubMed] [Google Scholar]
- 33.Kowall NW, Hantraye P, Brouillet E, Beal MF, McKee AC, Ferrante RJ. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. NeuroReport. 2000;11:211–213. doi: 10.1097/00001756-200001170-00041. [DOI] [PubMed] [Google Scholar]
- 34.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 35.Kumer SC, Vrana KE. Intricate regulation of tyrosine hydroxylase activity and gene expression. J Neurochem. 1996;67:443–462. doi: 10.1046/j.1471-4159.1996.67020443.x. [DOI] [PubMed] [Google Scholar]
- 36.Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of α-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001;15:916–926. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, Brecha N, Edwards RH. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70:539–551. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- 38.Lotharius J, O'Malley KL. The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J Biol Chem. 2000;275:38581–38588. doi: 10.1074/jbc.M005385200. [DOI] [PubMed] [Google Scholar]
- 39.Markopoulou K, Wszolek ZK, Pfeiffer RF, Chase BA. Reduced expression of the G209A alpha-synuclein allele in familial Parkinsonism. Ann Neurol. 1999;46:374–381. doi: 10.1002/1531-8249(199909)46:3<374::aid-ana13>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 40.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 42.Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, DiMonte DA. Lack of nigral pathology in transgenic mice expressing human α-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- 43.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muslin AJ, Xing H. 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell Signal. 2000;12:703–709. doi: 10.1016/s0898-6568(00)00131-5. [DOI] [PubMed] [Google Scholar]
- 45.Nagatsu T, Levitt T, Udenfriend S. Tyrosine hydroxylase: the initial step in norepinephrine biosynthesis. J Biol Chem. 1964;239:2910–2917. [PubMed] [Google Scholar]
- 46.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M. Both familial Parkinson's disease mutations accelerate alpha-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 47.Neystat M, Lynch T, Przedborski S, Kholodilov N, Rzhetskaya M, Burke R. α-Synuclein expression in substantia nigra and cortex in Parkinson's disease. Mov Disord. 1999;14:417–422. doi: 10.1002/1531-8257(199905)14:3<417::aid-mds1005>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 48.Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J Neurosci. 1996;16:4135–4145. doi: 10.1523/JNEUROSCI.16-13-04135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O'Hara CM, Uhland-Smith A, O'Malley KL, Todd RD. Inhibition of dopamine synthesis by dopamine D2 and D3 but not D4 receptors. J Pharmacol Exp Ther. 1996;277:186–192. [PubMed] [Google Scholar]
- 50.Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, Wolozin B. α-Synuclein shares physical and functional homology with 14-3-3. J Neurosci. 1999;19:5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Payton JE, Perrin RJ, Wraight L, Clayton DF, George JM. Anomalous immunoreactivity of the Syn-1 antibody to α-synuclein. Soc Neurosci Abstr. 2001;27:93.4. [Google Scholar]
- 52.Perez RG, Squazzo SL, Koo EH. Enhanced release of amyloid β-protein from codon 670/671 “Swedish” mutant β-amyloid precursor protein occurs in both secretory and endocytic pathways. J Biol Chem. 1996;271:9100–9107. doi: 10.1074/jbc.271.15.9100. [DOI] [PubMed] [Google Scholar]
- 53.Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W-M, Selkoe DJ, Chen X, Stokin G, Koo EH. Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Aβ42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- 54.Pickel VM, Joh TH, Reis DJ. Ultrastructural localization of tyrosine hydroxylase in noradrenergic neurons of brain. Proc Natl Acad Sci USA. 1975;72:659–663. doi: 10.1073/pnas.72.2.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 56.Reinhard JF, Jr, Smith GK, Nichol CA. A rapid and sensitive assay for tyrosine-3-monooxygenase based upon the release of 3H2O and adsorption of [3H]-tyrosine by charcoal. Life Sci. 1986;39:2185–2189. doi: 10.1016/0024-3205(86)90395-4. [DOI] [PubMed] [Google Scholar]
- 57.Souza JM, Giasson BI, Lee VMY, Ischiropoulos H. Chaperone-like activity of synucleins. FEBS Lett. 2000;474:116–119. doi: 10.1016/s0014-5793(00)01563-5. [DOI] [PubMed] [Google Scholar]
- 58.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 59.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stokes AH, Hastings TG, Vrana KE. Cytotoxic and genotoxic potential of dopamine. J Neurosci Res. 1999;55:659–665. doi: 10.1002/(SICI)1097-4547(19990315)55:6<659::AID-JNR1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 62.Takeda A, Hashimoto M, Mallory M, Sundsumo M, Hansen L, Sisk A, Masliah E. Abnormal distribution of the non-Abeta component of Alzheimer's disease amyloid precursor/alpha-synuclein in Lewy body disease as revealed by proteinase K and formic acid pretreatment. Lab Invest. 1998;78:1169–1177. [PubMed] [Google Scholar]
- 63.Tanji M, Horwitz R, Rosenfeld G, Waymire JC. Activation of protein kinase C by purified bovine brain 14-3-3: comparison with tyrosine hydroxylase activation. J Neurochem. 1994;63:1908–1916. doi: 10.1046/j.1471-4159.1994.63051908.x. [DOI] [PubMed] [Google Scholar]
- 64.Tokuyasu KT. Use of poly(vinylpyrrolidone) and poly(vinylalcohol)for cryo-ultramicrotomy. Histochem J. 1989;21:163–171. doi: 10.1007/BF01007491. [DOI] [PubMed] [Google Scholar]
- 65.Ueda S, Aikawa M, Ishizuya-Oka A, Yamaoka S, Koibuchi N, Yoshimoto K. Age-related dopamine deficiency in the mesostriatal dopamine system of zitter mutant rats: regional fiber vulnerability in the striatum and the olfactory tubercle. Neuroscience. 2000;95:389–398. doi: 10.1016/s0306-4522(99)00451-0. [DOI] [PubMed] [Google Scholar]
- 66.Van Der Hoeven PC, Van Der Wal JC, Ruurs P, Van Blitterswijk WJ. Protein kinase C activation by acidic proteins including 14-3-3. Biochem J. 2000;347:781–785. doi: 10.1042/0264-6021:3470781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S, Kauffmann S, Hofele K, Spooren WP, Ruegg MA, Lin S, Caroni P, Sommer B, Tolnay M, Bilbe G. Neuropathology in mice expressing human alpha-synuclein. J Neurosci. 2000;20:6021–6029. doi: 10.1523/JNEUROSCI.20-16-06021.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wagner J, Palfreyman M, Zraika M. Determination of dopa, dopamine, dopac, epinephrine, norepinephrine, alpha-monofluoromethyldopa and alpha-difluoromethyldopa in various tissues of mice and rats using reversed-phase ion-pair liquid chromatography with electrochemical detection. J Chromatogr. 1979;164:41–54. doi: 10.1016/s0378-4347(00)81570-4. [DOI] [PubMed] [Google Scholar]
- 69.Waymire JC, Johnston JP, Hummer-Lickteig K, Lloyd A, Vigny A, Craviso GL. Phosphorylation of bovine adrenal chromaffin cell tyrosine hydroxylase. Temporal correlation of acetylcholine's effect on site phosphorylation, enzyme activation, and catecholamine synthesis. J Biol Chem. 1988;263:12439–12447. [PubMed] [Google Scholar]
- 70.Waymire JC, Craviso GL, Lichteig K, Johnston JP, Baldwin C, Zigmond RE. Vasoactive intestinal peptide stimulates catecholamine biosynthesis in isolated adrenal chromaffin cells: evidence for a cyclic AMP-dependent phosphorylation and activation of tyrosine hydroxylase. J Neurochem. 1991;57:1313–1324. doi: 10.1111/j.1471-4159.1991.tb08296.x. [DOI] [PubMed] [Google Scholar]
- 71.Withers GS, George JM, Banker GA, Clayton DF. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res Dev Brain Res. 1997;99:87–94. doi: 10.1016/s0165-3806(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 72.Zhang L, Wang H, Liu D, Liddington R, Fu H. Raf-1 kinase and exoenzyme S interact with 14-3-3 zeta through a common site involving lysine 49. J Biol Chem. 1997;272:13717–13724. doi: 10.1074/jbc.272.21.13717. [DOI] [PubMed] [Google Scholar]
- 73.Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR. Overexpression of human alpha-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 2000;866:33–43. doi: 10.1016/s0006-8993(00)02215-0. [DOI] [PubMed] [Google Scholar]