

Abstract
NMDA receptor antagonists produce region-specific neurodegeneration by an undetermined mechanism, but one proposed mechanism involves disinhibition. In certain areas of the brain, NMDA receptors mediate excitatory drive onto inhibitory interneurons. Thus, NMDA receptor/channel antagonists may reduce inhibition (i.e., produce “disinhibition”). If a sufficient level of disinhibition is produced, enhanced vulnerability to excitotoxicity may result. Furthermore, if there are region-specific differences in NMDA antagonist-induced disinhibition, this could underlie region-specific NMDA antagonist-induced neurotoxicity. In the present study, we tested this hypothesis by exposing rat brain slices to the NMDA receptor antagonist dizocilpine maleate (MK-801) and measuring MK-801-induced disinhibition in areas of higher and lower vulnerability to neurodegeneration [posterior cingulate/retrosplenial cortices (PCC/RSC) and parietal cortex, respectively]. Using whole-cell patch-clamp techniques, bicuculline-sensitive GABAAreceptor-mediated IPSCs were measured in biocytin-labeled pyramidal neurons in the PCC/RSC and parietal cortex. In the PCC/RSC, bath-applied MK-801 (10–40 μm) produced disinhibition, shown as a concentration-dependent decrease in spontaneous IPSC frequency and amplitude; MK-801 (40 μm) also reduced evoked IPSC amplitudes. In parietal cortex, MK-801 produced significantly less disinhibition. To determine whether disinhibition is caused by presynaptic or postsynaptic mechanisms, we tested the effects of MK-801 (40 μm) against miniature IPSC (mIPSC) frequency and amplitude in tetrodotoxin (TTX; 0.5 μm)-treated slices and found that MK-801 did not alter mIPSC frequency or amplitude. Taken together, these results suggest that NMDA receptors regulate activity of inhibitory interneurons and, consequently, GABA release in certain cortical areas. This region-specific reduction in inhibitory input to pyramidal cells could underlie the region-specific neurotoxicity of NMDA antagonists.
Keywords: MK-801, NMDA receptor, IPSCs, disinhibition, pyramidal cells, interneurons, cingulate cortex
Neurodegeneration can result from overactivation of NMDA receptors (Rothman and Olney 1986, 1987), causing excitotoxicity proposed to be responsible for certain neurological diseases. Consequently, NMDA antagonists were screened against animal models of epilepsy (Avoli and Oliver, 1987), ischemia (Aitken et al., 1988; Ford et al., 1989; Rod and Auer, 1989), and hypoglycemia (Wieloch, 1985).
The results from these studies were perplexing, however. NMDA antagonists were not always effective (Stasheff et al., 1989;Sveinbjornsdottir et al., 1993) nor were they always neuroprotective (MacDonald et al., 1990). In fact, some antagonists actually produced region-specific neurotoxicity (Olney et al., 1989,1991). In rats, the most affected areas were posterior cingulate cortex (PCC) and retrosplenial cortex (RSC); other areas were less sensitive (Allen and Iversen, 1990; Olney et al., 1991; Horvath et al., 1997). Pathomorphological changes varied with dose: low doses caused mitochondrial dilation, higher doses caused neuronal death (Olney et al., 1989, 1991; Fix et al., 1995; Horvath et al., 1997).
How do NMDA antagonists produce neurotoxicity? Olney and colleagues (Olney et al., 1991; Olney and Farber, 1995; Corso et al., 1997) proposed that NMDA antagonists produce neurodegeneration by disrupting inhibition. This disinhibition could be produced directly (by disrupting the local excitatory/inhibitory network) or indirectly (by acting through modulatory neurotransmitter systems). Results fromin vivo studies have supported Olney's hypothesis; however, no studies have directly tested this hypothesis in vitro in the PCC/RSC.
Using brain slices, we tested these hypotheses. We proposed that NMDA antagonists (1) reduce GABAergic inhibition in the PCC/RSC and (2) are more effective in the PCC/RSC than in less vulnerable regions, such as parietal cortex.
The first proposal is based on reports that NMDA receptors provide excitatory drive onto inhibitory interneurons in several brain areas, including the hippocampus (Grunze et al., 1996) and olfactory cortex (Schoppa et al., 1998). Thus, NMDA antagonists produce disinhibition in these areas. We extend these findings to the PCC/RSC. The second proposal is important regarding NMDA antagonist-induced neurodegeneration, because if NMDA antagonists produce more disinhibition in PCC/RSC than elsewhere, this may underlie their region-specific neurotoxicity. Finally, the use of brain slices may help to determine the level at which NMDA antagonists disrupt inhibition (i.e., in the local excitatory/inhibitory circuits, versus at the level of extrinsic modulatory inputs).
To test these proposals, we used the NMDA antagonist dizocilpine maleate (MK-801) to produce disinhibition in slices of PCC/RSC and parietal cortex. Disinhibition was assessed using whole-cell patch-clamp recordings in pyramidal cells to measure GABAA-mediated IPSC frequencies and amplitudes. Here we provide the first direct electrophysiological evidence that NMDA antagonists reduced inhibitory synaptic drive onto pyramidal cells in the PCC/RSC. Also, the mechanism for region-specific vulnerabilities may be explained by our results, because MK-801 produces more disinhibition in cortical areas most vulnerable to NMDA antagonist-induced neurotoxicity.
Preliminary results have been published previously in abstract form (Li et al., 2000).
MATERIALS AND METHODS
Cortical slices. Cortical slices were prepared from young, male Sprague Dawley rats (postnatal day 15–25). Rats were isoflurane-anesthetized and decapitated. The brains were quickly removed from the skulls and placed in cold (4°C) artificial CSF (aCSF) containing (in mm): 120 NaCl, 3.3 KCl, 1.23 NaH2PO4, 25 NaHCO3, 1.2 MgSO4, 1.8 CaCl2, and 10 d-glucose at pH 7.3, previously saturated with 95%O2/5%CO2. Coronal cortical slices (300 μm thickness) containing the PCC/RSC or the parietal cortex (Paxinos and Watson, 1986) were cut with a Vibratome(Model 752; Campden, Berlin, Germany) and incubated in a holding chamber continuously bubbled with 95% O2 and 5% CO2 at room temperature (22–24°C).
Whole-cell voltage-clamp recording. Our whole-cell patch-clamp techniques have been described in our previous publication (Mott et al., 1999). For recording, patch pipettes were pulled from borosilicate glass capillary tubing (1.5 mm outer diameter, 1.05 mm inner diameter; World Precision Instruments, Sarasota, FL) on a Flaming-Brown horizontal microelectrode puller (Model P-97; Sutter Instrument Co., Novato, CA). Pipettes were filled with an intracellular solution containing (in mm): 130 Cs-gluconate, 7 CsCl, 10 HEPES, 4 Mg-ATP, pH = 7.25. The quaternary lidocaine derivative QX-314 (4 mm) (Sigma, St. Louis, MO) was also included to suppress fast sodium currents. Osmolarity was adjusted to 280 mOsm. Pipette resistances generally were in the range of 4–7MΩ. Biocytin (0.3–0.4%) (Sigma) was also added to the intracellular solution for later visualization of the morphology of the recorded cells.
After >1 hr of incubation in the holding chamber, a slice was transferred to a small submersion chamber maintained at room temperature (22–24°C) and secured in place with a bent piece of platinum wire resting on the top of the slice. Individual cells were visualized using an infrared differential interference contrast ZeissAxioskop microscope and a 40× water immersion objective. Tight seals (>1 GΩ) were obtained on pyramidal-shaped cells, and whole-cell recordings were made after rupturing the cell membrane with gentle suction. After establishment of the whole-cell recording configuration, stable long-lasting tight-seal recordings were achieved in most cases. Spontaneous and evoked IPSCs (eIPSCs) were recorded continuously using an Axopatch 1-D amplifier (Axon Instruments, Foster City, CA). Output current signals were DC-coupled to a digital oscilloscope (Nicolet Model 410). Series resistance was monitored throughout the recordings; a cell was discarded if it changed significantly (>20% of the control). In addition, a PCM/VCR recorder (Model 400; A. R. Vetter Co, Rebersburg, PA) was used to capture all tracings of synaptic events for off-line analysis and archiving. The stored signal was further analyzed using Strathclyde Electrophysiology Software Whole Cell Program (courtesy of Dr. John Dempster, University of Strathclyde, Glasgow, UK) with an interface (BNC-2090; National Instruments, Austin, TX) to a PC-based computer.
Cortical interneurons were selected on the basis of the shape of the soma (small and round under DIC microscopy) and their characteristic firing pattern. To determine the firing pattern of a cortical interneuron while achieving the tight seal offered by Cs-gluconate solution under the voltage clamp, the tip of a patch pipette was filled with a solution containing K-gluconate (in mm: 130 K-gluconate, 7 KCl, 10 HEPES, 4 Mg-ATP, and 0.3 Tris-GTP, pH 7.25), then backfilled with a solution containing Cs-gluconate (see above). The dialysis of the recorded cell with the Cs-gluconate solution could be observed from the distortions of shapes of the action potentials ∼10 min after whole configuration was established. NMDA receptor-mediated EPSCs were then recorded from interneurons.
Electrical stimulation. A monopolar tungsten electrode (A-M System, Carlsborg, WA) was placed ∼50–70 μm lateral to the soma of the recorded pyramidal cells in the same layer. The stimulus threshold was first determined by increasing the intensity of the rectangular wave pulse until detectable responses occurred. Then constant current rectangular stimulus pulses 50% higher than threshold intensity with a duration of 0.1 msec and interval of 0.0166 Hz were delivered through the electrode by an isolated stimulator (Grass S88; Grass Instruments, Quincy, MA).
Histological identification of pyramidal cells. During recording, pyramidal cells were filled with biocytin. After the end of the recording, the slice was allowed to stay in the recording chamber for an additional 10–20 min for further biocytin transport within the axon. The slices were then placed overnight in 4% paraformaldehyde and 0.05% glutaraldehyde in 0.1 m PBS. The slices were washed thoroughly in PBS and incubated in 0.1m Tris-buffered saline (TBS) containing 1% H2O2 for 30 min. The slices were then incubated overnight at 4°C with avidin–biotin–peroxidase complex (ABC kit; Vector Labs, Burlingame, CA) in TBS containing 0.05% Triton X-100. The slices were then rinsed three times in PBS, reacted in a solution containing 3,3′-diaminobenzidine (DAB kit, Vector Labs), then cleared and mounted. The morphology of the biocytin-filled pyramidal cells was examined with a light microscope, and cells were drawn using a camera lucida.
Statistical analysis of data and drug application. Data were analyzed off-line using Strathclyde Electrophysiological Software. The Kolmogorov–Smirnov (K-S) statistical test was used to compare two different cumulative distributions using Origin 5.0 for Windows (MicroCal Software, Norththampton, MA). Paired and unpairedt tests and one-way ANOVA tests were also used, when appropriate. All group data are presented as mean ± SEM.
MK-801 and TTX were purchased from RBI (Natick, MA).d-(-)2-amino-5-phosphonovaleric acid (d-AP5) and bicuculline methiodine (BMI) were purchased from Sigma. All drugs were dissolved directly in the aCSF and bath-applied in the perfusion medium for ∼20 min, unless noted otherwise.
RESULTS
Pyramidal cells were recorded in layers II–VI of the neocortex. We investigated these cortical strata in the PCC/RSC and the parietal cortex. The location of the PCC/RSC and the parietal cortex are illustrated in Figure1A. Data were acquired from 54 pyramidal cells, 6 interneurons in the PCC/RSC, and 49 pyramidal cells from the parietal cortex. According to the location of their somata, pyramidal cells are divided into three subgroups: the superficial layer (II–III), layer IV, and the deep layer (V–VI). In the PCC/RSC, 25 (46%) pyramidal cells are in the superficial layer, 13 (24%) in layer IV, and 16 (30%) in the deep layer. In parietal cortex, 20 (40%) pyramidal cells are in the superficial layer, 14 (29%) in layer IV, and 15 (31%) in the deep layer. All six interneurons are recorded from PCC/RSC layers II and III. Because of the irreversible blockade of NMDA receptors by MK-801, only a single experiment was done from each cortical slice treated with MK-801.
Fig. 1.
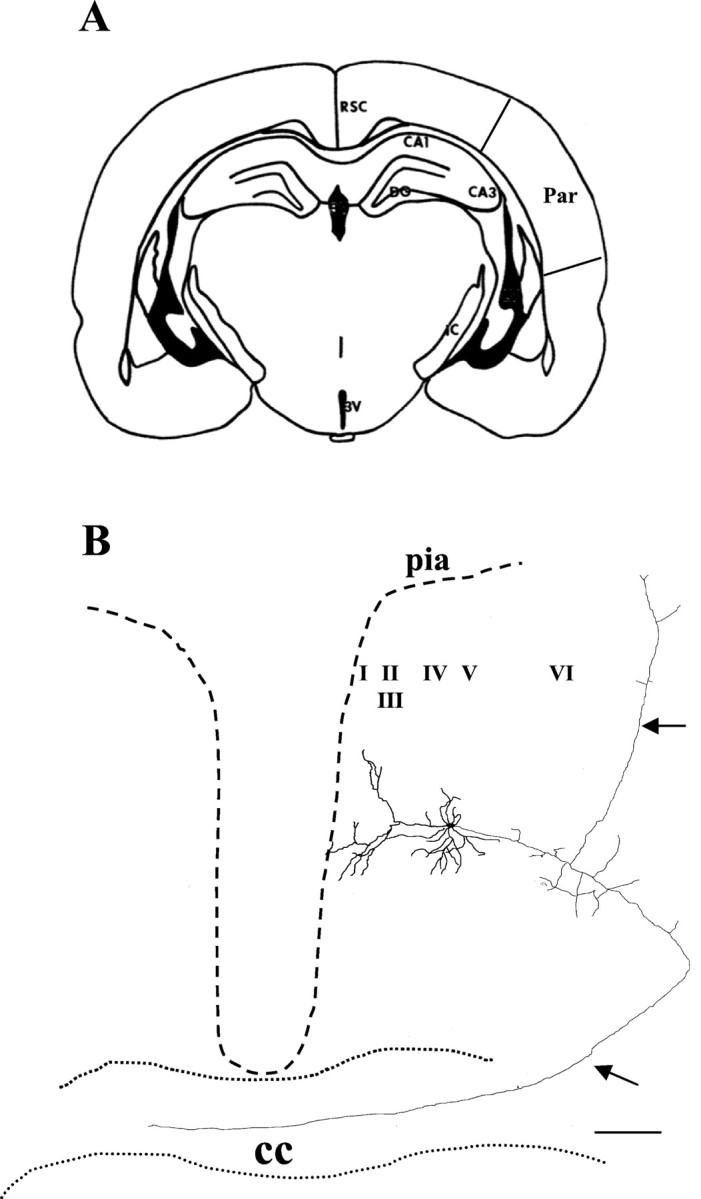
Camera lucida reconstruction of a PCC/RSC pyramidal cell filled with biocytin. A, Coronal section of a cortical slice containing PCC/RSC and parietal cortex.B, Camera lucida reconstruction of a biocytin-filled, layer V pyramidal cell recorded in the PCC/RSC. This pyramidal cell was filled with biocytin during the experiment. An apical dendrite arising from the soma extends toward the pial surface and branches off to form multiple tufts. In addition to a typical apical dendrite tree, a long axon originating from the soma projects to the contralateral side of the hemisphere through the corpus callosum. Another branch of the axon projects ipsilaterally to the lateral cortex. The pia is near the end of the apical dendrite branches. Dashed lines represent the approximate pial surface of the cortex. RSC, Retrosplenial cortices; Par, parietal cortex;CC, corpus callosum;I–VI, cortical lamina;DG, dentate gyrus; 3V, third ventricle;CA, cornus ammonis. Arrows denote axon. Scale bar, 200 μm.
Morphology of dendritic and axonal arbors of pyramidal cells in the PCC/RSC
The morphology of each recorded cell was assessed with biocytin staining to unambiguously distinguish pyramidal cells from other cell types. We were able to recover histologically ∼90% of all recorded cells in the PCC/RSC areas. The dendritic arbor of pyramidal cells in the PCC/RSC was characterized by a long apical dendrite that usually extended toward the pial surface, where typically it branched extensively to form multiple small terminal tufts just under the pial surface. Along the length of the apical dendrites are numerous obliquely branched dendritic collaterals. Basal dendrites extended outward from the lower portion of the soma; these basal dendrites ascended or descended with gradual tapering. The pyramidal cell axonal arbor was densely distributed around the soma and also extended widely, both horizontally and vertically. These morphological characteristics are consistent with cortical pyramidal cells described elsewhere (Kim and Connors, 1993; Lubke et al., 1996; Reyes and Sakmann, 1999; Feldmeyer and Sakmann, 2000). Figure1B shows a camera lucida reconstruction of a PCC/RSC pyramidal cell. The soma of this pyramidal cell is located in layer V, and it sends a long axon projecting to the contralateral side across the corpus callosum (Sripanidkulchai and Wyss, 1987).
MK-801 decreases the spontaneous IPSC frequency in pyramidal cells of the PCC/RSC
Spontaneous IPSCs (sIPSCs) were recorded from pyramidal cells in the PCC/RSC using whole-cell voltage-clamp techniques. The holding potentials were +10 or +30 mV, and the calculatedECL was approximately −40 mV for the internal solution used. Under these experimental conditions, the sIPSCs were inward currents at a holding potential of −70 mV (Fig.2A) and were robust outward currents at a holding potential of +30 mV (Fig.2B). At the end of the experiment, the recorded sIPSCs were abolished by bath application of a selective GABAA receptor antagonist, BMI (20 μm) (Fig. 2C). NMDA receptor-mediated EPSCs might be present when a cell was depolarized to +40 mV (Hestrin, 1992). Accordingly, we also assessed whether NMDA receptor-mediated EPSCs were also present when a cell was held at +30 mV without blocking excitatory transmissions. We examined eight PCC/RSC pyramidal cells in the presence of BMI (20 μm). When the holding potential was held at −70 mV, fast inward currents mediated by AMPA receptors dominated, and no NMDA receptor-mediated EPSCs were observed (Fig.2D). NMDA receptor-mediated EPSCs only become detectable after the holding potential was changed to +30 mV (data not shown). However, long slow outward currents were recorded in only four of the nine cells tested, and the frequency of recorded NMDA–EPSCs is very low. These data indicated that the contribution of NMDA receptor-mediated EPSCs was insignificant and negligible in cells held at +30 mV. Figure 2, E and F, shows evoked IPSCs recorded in a PCC/RSC pyramidal cell in the presence of DNQX (20 μm) and d-AP5 (50 μm). A plot of the amplitude of evoked IPSCs against holding potentials (Fig. 2E) indicated an x-intercept of −39 mV, closely approximating the calculated reversal potential of ECL (Fig.2F). These results indicated that the recorded IPSCs were mediated by the activation of GABAAreceptors.
Fig. 2.
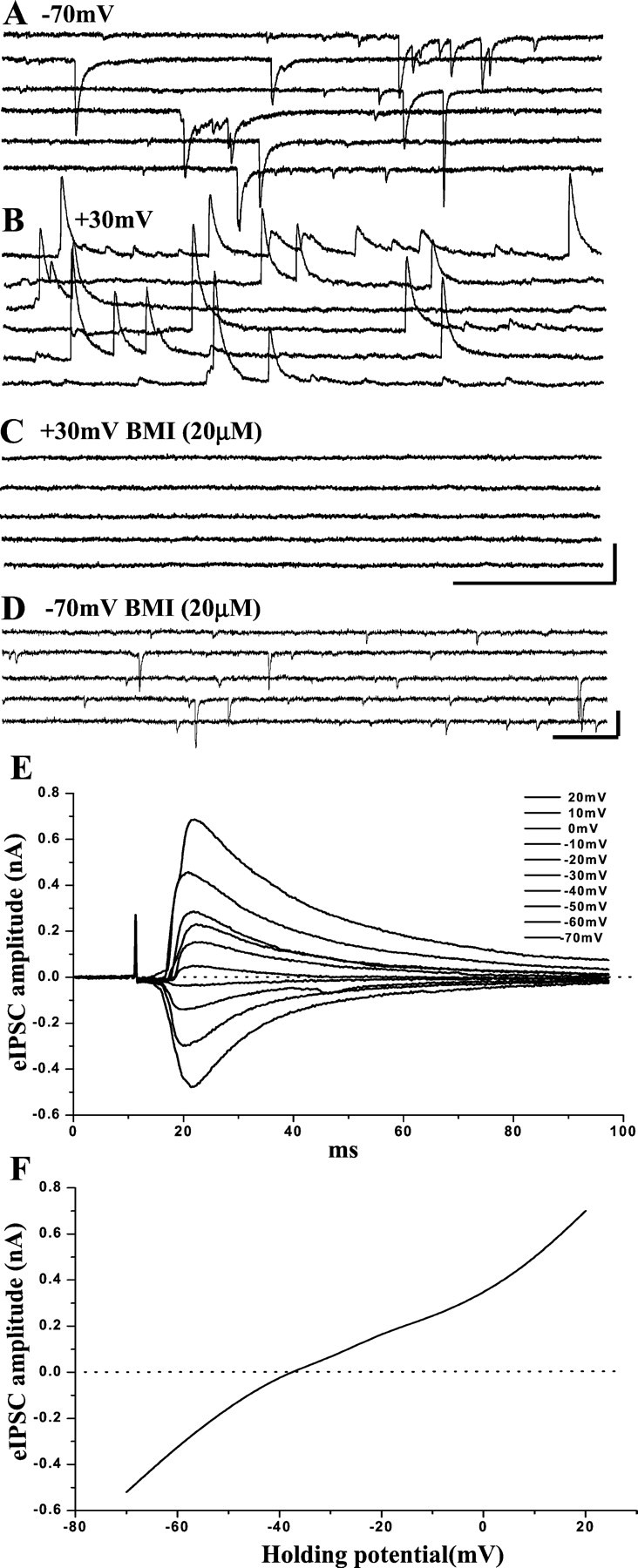
GABAA receptor-mediated IPSCs recorded in PCC/RSC pyramidal cells. Whole-cell recordings were performed using a CsCl-based internal solution at holding potentials of −70 mV and +30 mV. When the holding potential was −70 mV (A), inward currents were recorded. When the cell was held at +30 mV, the currents became outward (B). The GABAA receptor antagonist BMI (20 μm) abolishes the currents (C). In the presence of BMI (20 μm) and at the holding potential of −70 mV, fast inward currents are recorded from another pyramidal cell and can be blocked by DNQX (20 μm) (D). In the presence of d-AP5 (50 μm) and DNQX (20 μm), amplitudes of evoked IPSCs recorded from another pyramidal cell held at potentials ranged from −70 to +20 mV (E). An I–V curve was constructed (F) based on the evoked synaptic responses shown in E. The apparent reversal potential was approximately −39 mV. These findings indicate that the recorded IPSCs were mediated by GABAA receptors. Calibration:A–C, 500 msec, 0.1 nA; D, 100 msec, 50 pA.
Effects of MK-801 on sIPSCs in PCC/RSC pyramidal cells are demonstrated in Figures 3 and4. MK-801 suppressed two IPSC properties: amplitude and frequency (the latter reflected as an increase in the interval between sIPSCs). Figure 3 shows recordings from a layer V pyramidal cell. At a holding potential of +30 mV, the recorded IPSCs were outward currents (Fig. 3A, top panel). Bath application of the NMDA antagonist MK-801 (40 μm) caused a significant decrease in the frequency and amplitude of sIPSCs in this pyramidal cell (Fig.3A, second panel from top). The IPSCs were abolished by bath-applied BMI (20 μm) and recovered partially on washout (Fig. 3A, bottom two panels). The cumulative probability distributions for these changes are shown in Figure 3B. As shown, bath-applied MK-801 produced a rightward shift in the distribution of sIPSC intervals (Fig. 3B, left panel), indicating a decrease in sIPSC frequency (K-S test; p< 0.01). In addition, sIPSC amplitudes were also reduced significantly (K-S test; p < 0.001), as shown in Figure3B (right panel). The morphology of this pyramidal cell is shown in Figure 3C.
Fig. 3.
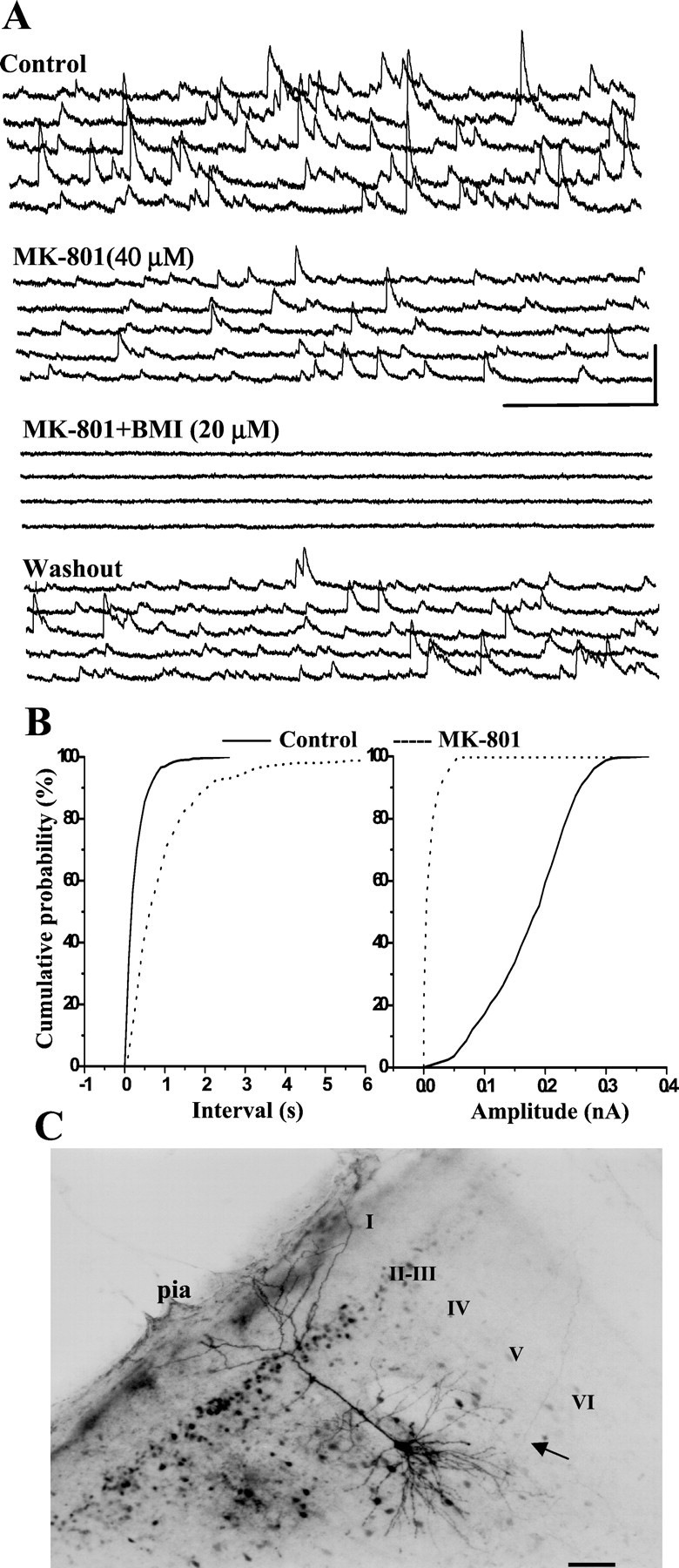
MK-801 decreases GABAAreceptor-mediated sIPSCs. A, In a layer V PCC/RSC pyramidal cell held at +30 mV, bath-applied MK-801 (40 μm) decreased the frequency and amplitude of sIPSCs. When BMI (20 μm) was added, sIPSCs were blocked reversibly. Calibration: 500 msec, 0.1 nA. B, In the same cell, the cumulative inter-event interval distribution shows a significant increase in the inter-event interval caused by MK-801 (p < 0.01; K-S test). The cumulative amplitude distribution also shows a significant decrease in amplitude (p < 0.001; K-S test). C, Photomicrograph of the same cell filled with biocytin.I–VI, Cortical lamina.Arrow denotes axon. Scale bar, 100 μm.
Fig. 4.
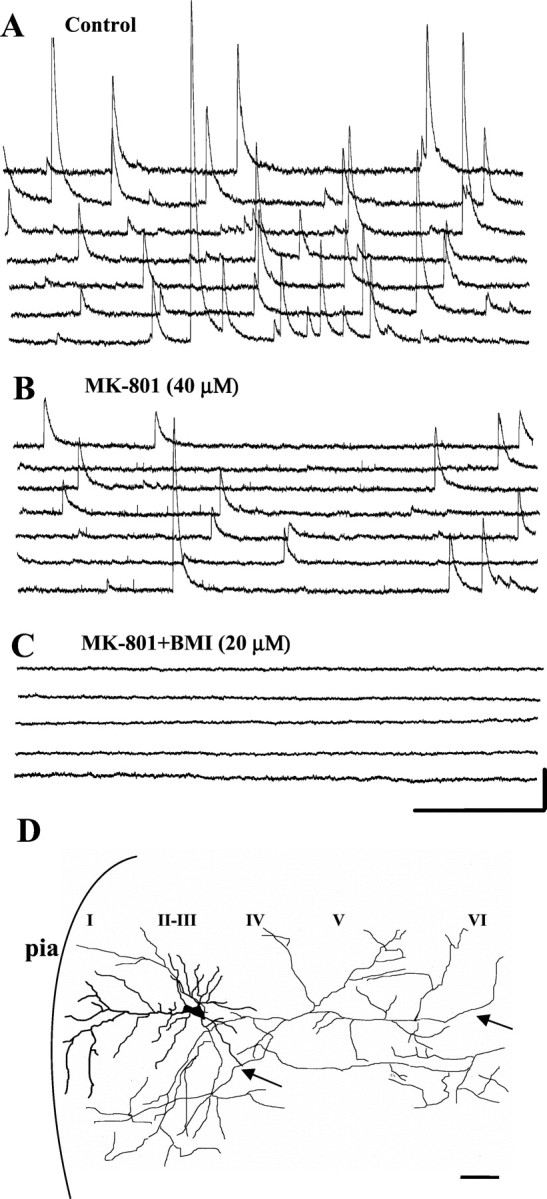
Effect of MK-801 on sIPSCs from a layer III cell. In a layer III PCC/RSC pyramidal cell held at +30 mV, bath-applied MK-801 (40 μm) (B) decreased sIPSCs compared with control (A), an effect similar to that seen in layer V (Fig. 3). Recorded sIPSCs were abolished by BMI (20 μm) (C). Calibration: 500 msec, 0.1 nA. D, Camera lucida reconstruction of the same cell. As shown, this is also a pyramidal cell, with processes slightly different from the cell shown in Figure 3, although the pharmacological responses to MK-801 were similar. I–VI, Cortical lamina. Arrows denote axon. Scale bar, 100 μm.
This effect was not limited to layer V pyramidal cells. A similar effect was also seen in a layer III pyramidal cell of the PCC/RSC, as shown in Figure 4. This pyramidal cell responded to bath-applied MK-801 (40 μm) with a significant decrease in sIPSC frequency (Figs. 4A,5B). In addition, amplitudes of sIPSCs were also attenuated by MK-801. Although large-amplitude sIPSCs apparently dominated in this layer III pyramidal cell, the morphology of this pyramidal cell, as shown in Figure4D, is similar to that of the pyramidal cell illustrated in Figure 4C. We found similar MK-801-mediated effects in layers II–VI as well.
Fig. 5.
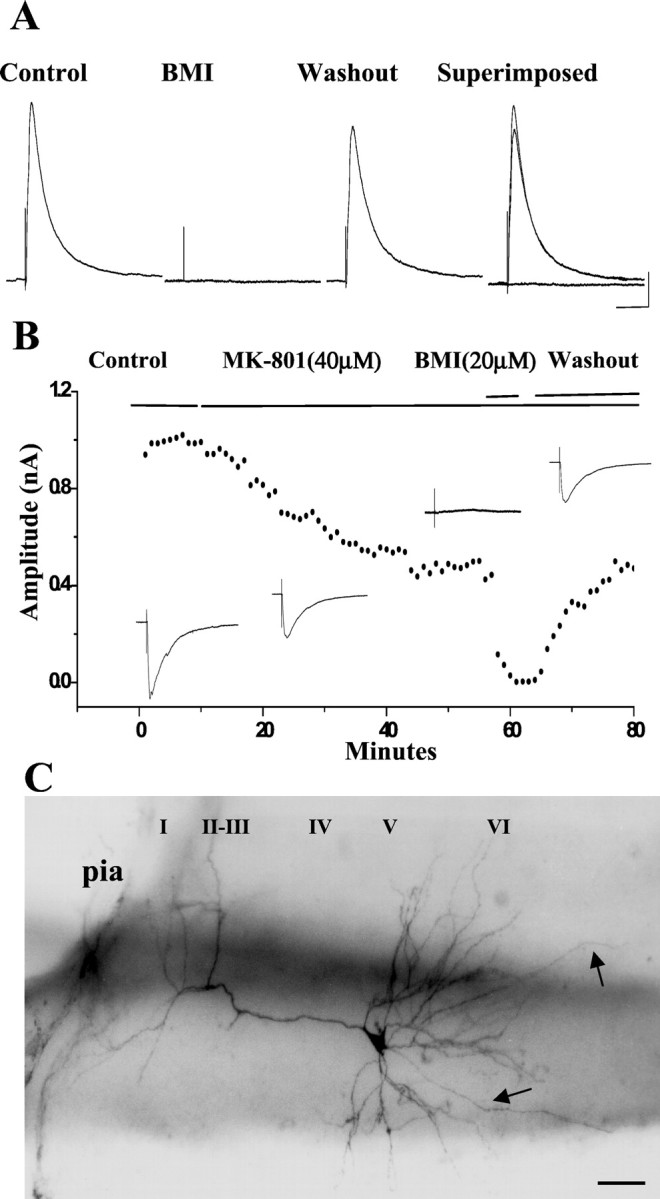
Effect of MK-801 on eIPSCs. A, In a layer IV PCC/RSC pyramidal cell held at +5mV, outward eIPSCs were recorded. These eIPSCs were completely and reversibly blocked by BMI (20 μm), as was seen with sIPSCs (Fig.2A–C), indicating that eIPSCs were mediated by GABAA receptors. Calibration: 50 msec, 0.1 nA. B, In a layer V PCC/RSC pyramidal cell held at +5mV, the eIPSC time course after bath-applied MK-801 (40 μm), BMI (20 μm), and washout is shown. Insetsillustrate the averaged peak IPSC traces corresponding to each treatment. C, Photomicrograph of the pyramidal cell shown in B filled with biocytin.I–VI, Cortical lamina.Arrow denotes axon. Scale bar, 100 μm.
Inhibitory effects of MK-801 on sIPSCs of pyramidal cells were compared among the three groups. Bath-applied MK-801 (40 μm) reduced sIPSC frequency by 43.3 ± 3.8% (n = 16) compared with the control in the superficial group (layers II and III), 44.4 ± 4.6% (n = 11) in layer IV, and 44 ± 3.0% (n = 13) in the deep layer (layers V and VI). There are no significant differences in mean IPSC frequency among three groups (p > 0.05; one-way ANOVA), so we pooled data obtained from pyramidal cells recorded in PCC/RSC layers II–VI. Overall, bath application of MK-801 (40 μm) reduced the frequency of sIPSCs by 43.8 ± 2.2% (n = 40) relative to control (p< 0.05; paired t test).
We also examined the concentration dependence of MK-801-induced decreases in the frequency of sIPSCs using three concentrations of MK-801 bath-applied in sequence. We monitored sIPSCs for ∼20 min after each concentration was applied. Overall, reductions of mean inter-event intervals were 15.4 ± 1.4, 28.6 ± 2.3, and 44.3 ± 3.1% after bath application of MK-801 at the doses of 10, 20, and 40 μm, respectively (p < 0.05; one-way ANOVA; n = 4).
We also tested the effect of the widely used competitive NMDA receptor antagonist d-AP5. Similar to MK-801, d-AP5 (50 μm) significantly (K-S test; p < 0.05) decreased the frequency of sIPSCs in two PCC/RSC pyramidal cells tested (data not shown).
MK-801 reduced the amplitude of evoked IPSCs in pyramidal cells in the PCC/RSC
In these experiments, MK-801 was also tested against evoked IPSCs of PCC/RSC pyramidal cells. The pyramidal cells were held at −5 or +5mV, and an eIPSC was elicited by a single pulse delivered lateral to the recording electrode (see Materials and Methods). These eIPSCs were abolished by bath application of GABAA receptor antagonist BMI (20 μm), indicating that eIPSCs are mediated by GABAA receptors (Fig.5A).
The time course of the MK-801-induced effect on eIPSC of a layer V PCC/RSC is shown in Figure 5B. As shown, the evoked IPSCs were gradually, but significantly, reduced by bath-applied MK-801. At a holding potential of +5mV, recordings were made in control solution for 10 min, then MK-801 (40 μm) was bath-applied. Five minutes after addition of MK-801, the eIPSCs began to decline. By 30 min after treatment, eIPSCs were reduced from 1.0 to 0.5 nA (i.e., 50% of control). Again, the eIPSCs were blocked by bath-applied BMI (20 μm) and recovered during BMI washout. Overall, MK-801 (40 μm) significantly reduced evoked responses to 51.3 ± 2.3% of control (n = 13; p < 0.05; paired t test).
Comparison of effects of MK-801 on sIPSCs and eIPSCs in the PCC/RSC and the parietal cortex
Several in vivo animal studies have demonstrated that pathomorphological changes induced by NMDA antagonists occur in a number of different areas of the brain, but with different sensitivities among the affected areas (Olney et al., 1991; Fix et al., 1993; Horvath et al., 1997). For example, MK-801 causes more severe damage to neurons in the PCC/RSC, whereas some other cortical areas were less affected.
We hypothesize that one mechanism by which MK-801 produces region-specific neurodegeneration is that MK-801 more effectively produces disinhibition in a vulnerable area (e.g., PCC/RSC) than a less vulnerable area (e.g., parietal cortex). To address this, we recorded sIPSCs and eIPSCs from 49 pyramidal cells in the parietal cortex.
Figure 6 shows the effect of MK-801 on sIPSCs recorded in a layer V parietal pyramidal cell. The sIPSCs were only slightly reduced by MK-801 (40 μm) (Fig.6A). Although MK-801 significantly increased the inter-event intervals (K-S test; p < 0.05) (Fig.6B, left panel), MK-801 did not have a significant effect on the cumulative amplitude distribution (K-S test; p > 0.05) (Fig. 6B,right panel).
Fig. 6.
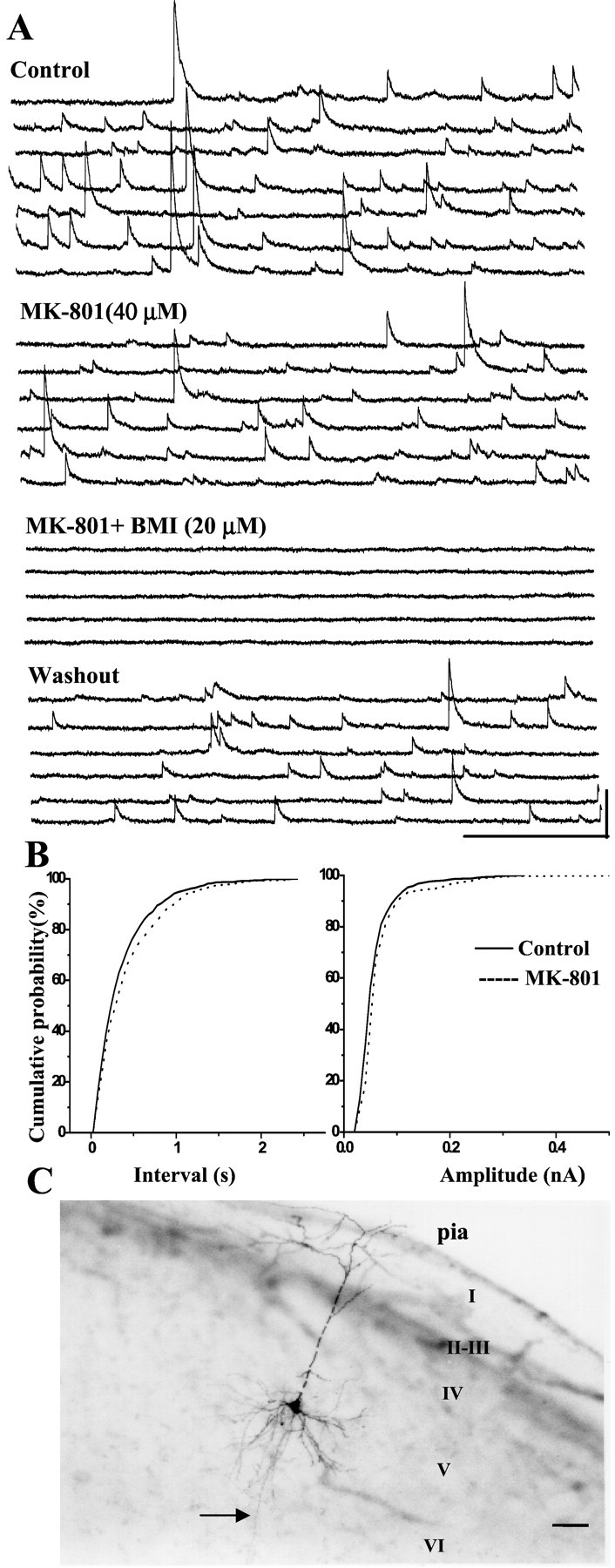
MK-801 is less effective on sIPSCs of pyramidal cells in the parietal cortex. A, At the holding potential of +30 mV, bath-applied MK-801 (40 μm) decreased sIPSCs in a layer V pyramidal cell in the parietal cortex. When BMI (20 μm) was added, sIPSCs were abolished, then recovered after washout. Calibration: 500 msec, 0.1 nA.B, In the same cell, the cumulative inter-event interval distribution shows a slight but significant increase in the inter-event interval (p < 0.05; K-S test). In contrast, MK-801 did not significantly decrease the sIPSC amplitude in the parietal cortex, as shown by the cumulative amplitude distribution (p > 0.05; K-S test). C, Photomicrograph of the same cell filled with biocytin.I–VI, Cortical lamina.Arrow denotes axon. Scale bar, 100 μm. (In the main dendrite, the small apparent gaps are artifacts: they arose after coverslipping. During the experiment, the dendrite was intact, as in all other experiments.)
Depressive effects of MK-801 on pyramidal cell sIPSCs were also compared among the three groups. Bath-applied MK-801 (40 μm) reduced frequency of sIPSCs by 21.9 ± 4.8% (n = 14) compared with control in the superficial group (layers II and III), 23.9 ± 5.4% (n = 12) in layer IV, and 23.4 ± 3.5% (n = 13) in the deep layers (layers V and VI). No significant differences in mean IPSC frequency among three groups were observed (p > 0.05; one-way ANOVA). Accordingly, we also pooled data obtained from pyramidal cells recorded in layers II–VI of parietal cortex. Bath application of MK-801 (40 μm) reduced the frequency of sIPSCs by 22.9 ± 2.6% (n = 39) relative to control (p < 0.05; pairedt test). In contrast, in the PCC/RSC, mean sIPSC frequency was reduced by 43.8 ± 2.2% (n = 40). In addition, we found that in ∼19% pyramidal cells recorded in parietal cortex, mean sIPSC frequency was reduced by MK-801 (40 μm) <10% (ranging from 2 to 10%), compared with 5% of those in the PCC/RSC.
Overall inhibitory effect of MK-801 on sIPSC frequency of each group recorded from two brain regions is summarized in Figure7. There is a significantly greater depression of sIPSC frequency by MK-801 in the PCC/RSC versus the parietal cortex (unpaired t test; p < 0.05) (Fig. 7A). The amplitudes of IPSCs are also suppressed by MK-801 in the PCC/RSC. Taken together, these results indicate that, at a dose of 40 μm, MK-801 caused greater disinhibition of pyramidal cells in the RSC than in the parietal cortex.
Fig. 7.
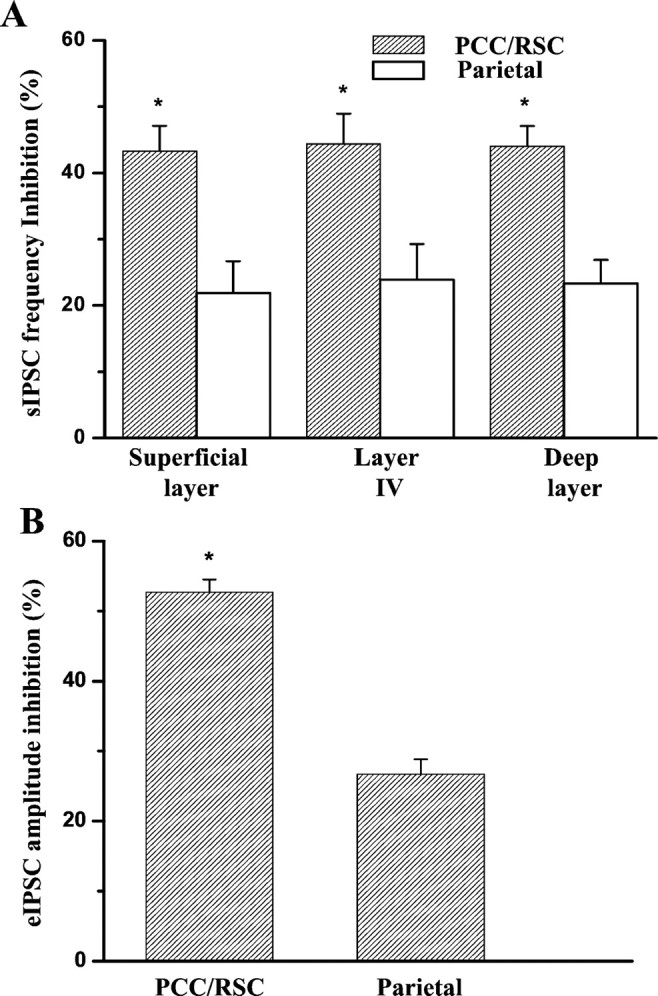
Comparison of MK-801 on sIPSCs and eIPSCs of pyramidal cells in the PCC/RSC and the parietal cortex.A, Graph shows data indicating the reduction in sIPSC frequency in PCC/RSC pyramidal cells [superficial layers (n = 16), layer IV (n = 11), and the deep layers (n = 13)] and in the parietal cortex [superficial layer (n = 14), layer IV (n = 12), and the deep layers (n = 13)]. MK-801 (40 μm) significantly decreased the average sIPSC frequency of pyramidal cells in the PCC/RSC compared with those in the parietal cortex (*p < 0.05; unpaired t test).B, Graph shows that MK-801 (40 μm) significantly inhibited the average peak amplitude of eIPSCs in pyramidal cells (n = 10) in the PCC/RSC compared with the parietal cortex (n = 9) (*p < 0.05; unpaired t test).
Similar differences were also observed on eIPSCs of pyramidal cells in the parietal cortex as shown in Figure7B. The mean amplitude of eIPSCs of pyramidal cells recorded from the PCC/RSC was decreased by 52 ± 1.8% (n = 10) of control. However, the average eIPSCs amplitude of pyramidal cells recorded from the parietal cortex was decreased by only 27 ± 2.1% (n = 9) of control after bath application of 40 μm MK-801. There is a significant difference between two groups (p < 0.05; unpairedt test).
MK-801 inhibits NMDA receptor-mediated EPSCs in PCC/RSC interneurons
It has been demonstrated that excitability of interneurons in the entorhinal cortex (Jones and Buhl, 1993) and auditory cortex (Bandrowski et al., 2001), the olfactory bulb (Schoppa et al., 1998) of rats can be modulated via excitatory synaptic input mediated by NMDA receptors. To assess whether MK-801 has a direct effect on NMDA receptor-mediated EPSCs in GABAergic interneurons, we isolated NMDA receptor-mediated EPSCs from six PCC/RSC layer II–III interneurons. Figure 8B shows a typical firing pattern of a PCC/RSC layer III interneuron (Kawaguchi, 1995; Zhou and Hablitz, 1998) compared with that of a PCC/RSC layer IV pyramidal cell (Fig. 8A). When the interneuron was held at −70 mV, DNQX-sensitive or AMPA/kainate receptor-mediated fast EPSCs were dominant (Fig. 8C). When the holding potential was changed to +50 mV, there were few, very long slow outward currents (Fig. 8D). These could be blocked byd-APV (n = 2; data not shown), suggesting they are NMDA receptor mediated.
Fig. 8.
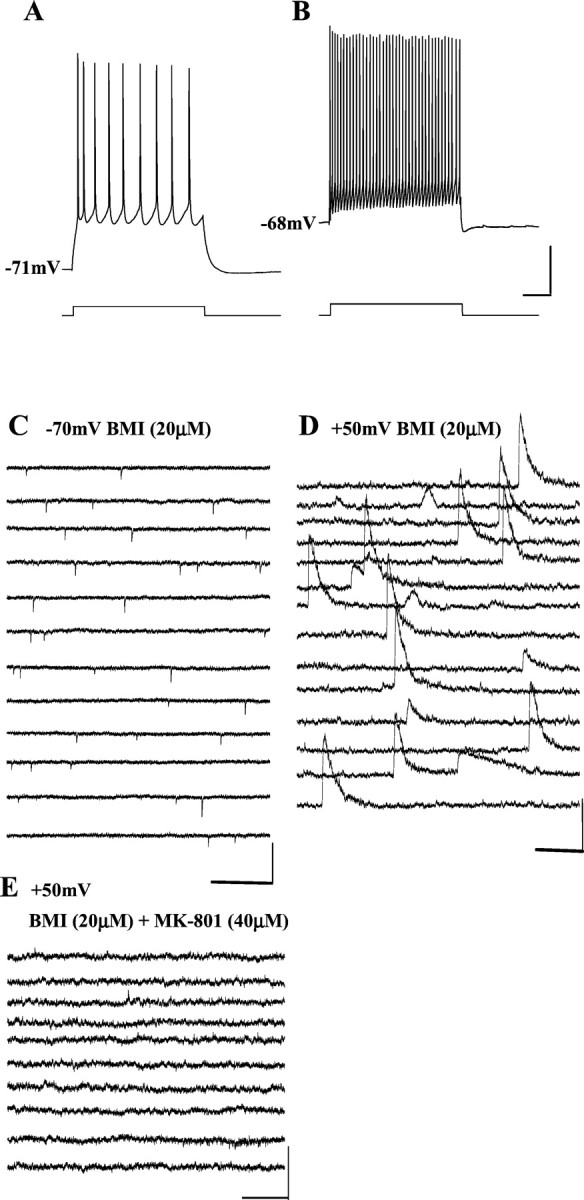
MK-801 blocks NMDA receptor-mediated EPSCs in PCC/RSC interneurons. A, Current-clamp recording of a PCC/RSC layer IV pyramidal cell in response to depolarizing current injection. The firing pattern of this pyramidal cell shows slow action potentials and frequency adaptation, characteristics typical for pyramidal cells. B, Current-clamp recording of a PCC/RSC layer III interneuron in response to depolarizing current injection. This interneuron fires rapidly and lacks frequency adaptation, characteristics typical for interneurons. Calibration:A, B, 200 msec, 20 mV, 80 pA.C, At the holding potential of −70 mV and in the presence of BMI (20 μm), DNQX-sensitive fast inward EPSCs are dominant in the interneuron. Calibration: 500 msec, 50 pA.D, When this interneuron was voltage-clamped at +50 mV and in the presence of BMI (20 μm), the long slow EPSCs were dominant. Calibration: 1000 msec, 100 pA. E, The slow EPSCs were blocked after administration of MK-801 (40 μm). Calibration: 1000 msec, 50 pA.
Twenty minutes after bath-applied MK-801 (40 μm), these NMDA receptor-mediated EPSCs were abolished (Fig.8E). MK-801 completely blocked NMDA receptor-mediated EPSCs in all six PCC/RSC interneurons tested. These results suggested that PCC/RSC interneurons receive NMDA receptor-mediated input; the absence of excitatory afferent drive onto the PCC/RSC interneurons could result in a decrease of inhibitory transmission from interneurons onto pyramidal cells.
MK-801 did not inhibit mIPSCs of pyramidal cells in the PCC/RSC
The results described above demonstrated that a block of NMDA receptors by the NMDA antagonist MK-801 reduced GABAA receptor-mediated IPSCs in PCC/RSC pyramidal cells. To determine whether the decrease of sIPSCs and eIPSCs in pyramidal cells was caused by a reduction of action potential-dependent or action potential-independent mechanisms, we used TTX, a Na+ channel blocker, to block synaptic events that are dependent on action potentials. Under such conditions, synaptic responses obtained in the presence of TTX are caused by quantal release and referred to as miniature IPSCS (mIPSCs).
We investigated the effect of MK-801 on mIPSCs in pyramidal cells in the PCC/RSC and found that MK-801 did not affect mIPSC frequency or amplitude. Figure 9A shows recordings of sIPSCs from a PCC/RSC pyramidal cell in layer III before and after application of TTX (0.5 μm). TTX caused a decrease in the mean sIPSC amplitude, indicating that a significant portion of the sIPSCs was caused by action potential-dependent GABA release from inhibitory interneurons. In the presence of TTX and at a holding potential of +30 mV, the average mIPSC frequency in the pyramidal cells was 4.5 ± 0.26/sec and the mean amplitude was 35 ± 6.2 pA (n = 5). After pretreatment of the slice with TTX for >30 min, bath application of MK-801 (40 μm) had no effect on the frequency (4.2 ± 0.23/sec) or amplitude (33 ± 5.8 pA) of mIPSCs (Fig.9A). In contrast to the effects of MK-801 on spontaneous IPSCs, there were no significant changes in the cumulative probability distribution of either mIPSC frequency (Fig. 9B,left panel) or amplitude (Fig. 9B,right panel) after bath application of MK-801 (K-S;p > 0.05). These results indicated that MK-801 exerts its inhibitory effect on IPSCs of pyramidal cells through an action potential-dependent mechanism.
Fig. 9.
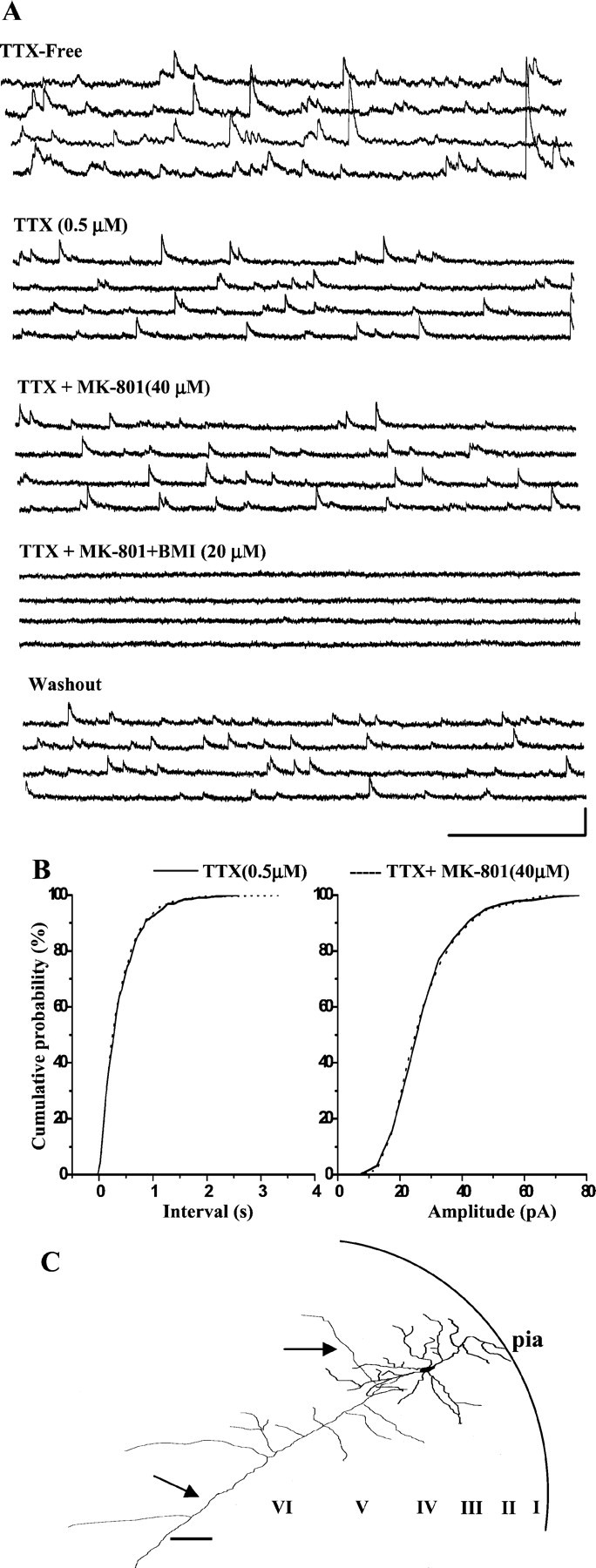
MK-801 does not suppress mIPSCs. A, In a PCC/RSC layer III pyramidal cell held at +30 mV, bath-applied TTX (0.5 μm) abolished all action potential-dependent IPSCs, leaving only mIPSCs. Bath-applied MK-801 (40 μm) had no effect on mIPSCs, whereas BMI (20 μm) reversibly blocked mIPSCs. Calibration: 500 msec, 0.05 nA.B, Cumulative mIPSC inter-event and amplitude distributions were not significantly changed (K-S test;p > 0.05), indicating that MK-801 produces disinhibition through an action potential-dependent mechanism.C, Camera lucida reconstruction of the same cell filled with biocytin. I–VI, Cortical lamina.Arrows denote axon. Scale bar, 100 μm.
DISCUSSION
Using whole-cell patch-clamp techniques combined with morphological identification of neurons in rat brain slices, we have investigated the effect of the noncompetitive NMDA receptor antagonist MK-801 on sIPSCs and eIPSCs in pyramidal cells of PCC/RSC and parietal cortex. Our results demonstrate that in PCC/RSC pyramidal cells, GABAA receptor-mediated synaptic transmission is reduced by NMDA receptor antagonists. These observations suggest that NMDA receptor-mediated excitation of interneurons facilitates GABAergic inhibition of PCC/RSC pyramidal cells.
The modulation of GABAergic inhibition by MK-801 in two regions with different vulnerabilities to NMDA antagonist-induced neurotoxicity
MK-801 has neuroprotective effects in certain CNS disorders (Foster et al., 1988; Dirnagl et al., 1990). However, MK-801 also has negative effects in vivo, including altered behavior (Sams-Dodd, 1997) and temperature regulation (Colbourne et al., 1999). Several in vivo studies reported that chronic injection of low-dose MK-801 [or other NMDA receptor antagonists, e.g., phencyclidine (PCP)] causes neuronal vacuolization in the PCC/RSC (Olney et al., 1989; Fix et al., 1993; Corso et al., 1997). This vacuolization can be reversible at low doses; however, irreversible neuronal damage occurs after high doses or repeated low doses (Fix et al., 1993).
Given these results, a fundamental question arose: how could an agent that had been expected to be neuroprotective turn out to be neurotoxic? On the basis of their whole animal studies, Olney and others proposed that these drugs might impair neuronal inhibition, leading to hyperactivity and neuronal death (Olney et al., 1991; Corso et al., 1997; Horvath et al., 1997). However, neurotoxic mechanisms underlying this phenomenon may be complex and could include those mediated by direct disruption of GABAergic function (Olney et al., 1991) or through a more indirect mechanism via modulatory neurotransmitters acting in vulnerable regions [such as cholinergic (Olney et al., 1991;), serotoninergic (Loscher and Honack, 1991), adrenergic (Loscher and Honack, 1991; Farber et al., 1995a), and dopaminergic (Sharp et al., 1994; Farber et al., 1996). Either way, a significant loss of inhibition could result in hyperexcitability, and this could lead to excitotoxicity. Consistent with these proposals are studies in which MK-801 increased glucose utilization in regions vulnerable to neurotoxicity, including the cingulate and entorhinal cortices (Kurumaji and McCulloch, 1990; Nehls et al., 1990; Patel and McCulloch, 1995). These results suggest that the vulnerable areas were highly activated by MK-801. The use of brain slices allowed us to assess disinhibition with less interference from modulating inputs and to determine the extent of disinhibition produced within localized networks. In our study, we have found that NMDA antagonists greatly reduce IPSCs in the PCC/RSC, an area of high susceptibility to NMDA antagonist-induced neurotoxicity.
With respect to the regional vulnerability to NMDA antagonist-induced neurotoxicity in vivo, the PCC/RSC is more susceptible than other cortical areas detected with a range of methods, including hematoxylin and eosin, silver stains, and electron microscopy (Olney et al., 1989; Fix et al., 1993; Corso et al., 1997;Horvath et al., 1997). We have found regional differences in the disinhibitory effects of MK-801 in vitro that parallelin vivo histopathological studies. Specifically, PCC/RSC pyramidal sIPSCs were significantly decreased by MK-801, whereas parietal pyramidal sIPSCs were less affected. Similar to this weak effect of MK-801 on parietal sIPSCs, Salin and Prince (1996)demonstrated that d-APV, a competitive NMDA receptor antagonist, only minimally inhibited sIPSC frequency in rat somatosensory cortex, another area less vulnerable to MK-801-induced neurotoxicity (Olney et al., 1989, 1991). We therefore postulate that these regional differences in disinhibition may explain, at least in part, the regional difference in the vulnerability to NMDA antagonists-induced neurotoxicity.
Our data indicate that MK-801-induced reduction in excitatory synaptic input onto GABAergic interneurons could be responsible for reducing pyramidal cell sIPSC frequency, although this is indirect evidence. However, we also isolated NMDA receptor-mediated EPSCs in interneurons and found that MK-801 blocked these EPSCs, providing more direct evidence for NMDA antagonist-induced disinhibition. Entorhinal interneurons receive NMDA receptor-mediated input (Jones and Buhl, 1993); however, some hippocampal interneurons may not have NMDA receptors (McBain and Dingledine, 1993). Similar to the hippocampal interneurons (McBain and Dingledine, 1993; Mott et al., 1997), cortical interneurons have heterogeneous anatomic morphologies and physiological functions (Kawaguchi 1995; Kawaguchi and Kubota, 1996). These differences may contribute to regional vulnerabilities in MK-801-induced neurotoxicity.
Systemic administration of MK-801 results in altered field potentials, specifically limited to layer III of rat medial entorhinal cortex (Gloveli et al., 1997). A marked increase in immediate early gene c-fos expression in layer III was also noted in rats pretreated with MK-801 (Vaisanen et al., 1999). These results suggested that cells in layer III are more sensitive to NMDA receptor antagonists. However, our experiments demonstrated that reduction in inhibitory transmission or disinhibition induced by MK-801 is not layer specific, although there are interlaminar differences in sIPSC frequency in rat somatosensory cortex (Salin and Prince, 1996).
MK-801 modulates pyramidal cell IPSCs via an action potential-dependent mechanism
Reduction of inhibitory transmission by NMDA antagonists has been observed in hippocampus (Hablitz and Langmoen, 1986; Grunze et al., 1996) and basolateral amygdala (Rainnie et al., 1991). In rat olfactory bulb, NMDA receptor activation is required for dendrodendritic inhibition (Schoppa et al., 1998). Direct infusion of d-APV bilaterally into the accessory olfactory bulb produced epileptiform seizures in adult female mice (Brennan and Keverne, 1989), an effect consistent with disinhibition. These investigators suggested that reduced inhibitory transmission could result from a loss of excitatory drive onto interneurons in the local circuit.
Our findings are compatible with this mechanism. MK-801 dramatically reduced the frequency of sIPSCs and the amplitude of eIPSCs but did not affect the frequency or amplitude of TTX-insensitive mIPSCs. This argues that MK-801 did not affect the postsynaptic efficacy of released GABA and did not affect quantal size or probability of spontaneous release. [Disinhibition produced by presynaptic mechanisms has been observed elsewhere, such as in studies of the effects of opioids and cannabinoids (Cohen et al., 1992; Hoffman and Lupica 2000)]. MK-801 seemed to affect action potential-dependent GABA release, compatible with an effect in inhibitory interneurons on either action potential-induced calcium influx into the presynaptic terminal or an effect on action potential initiation or propagation. Consequently, future experiments will focus on the effects of MK-801 on synaptic currents and resting spontaneous activity in interneurons.
Relation to proposed models
These results support the hypothesis that NMDA antagonists produce disinhibition within a PCC/RSC excitatory/inhibitory network. This disinhibition may be attributable partially to a local effect, because these findings were obtained in brain slices in which many of the modulatory input fibers have been severed.
These results, however, do not rule out the importance of inputs from other brain areas shown to play a role in NMDA antagonist-induced neurotoxicity [e.g., inputs from the anterior thalamus (Tomitaka et al., 2000) or cholinergic nuclei (Corso et al., 1997)]. This may be important, because it is not clear whether disinhibition alone is sufficient to produce neurodegeneration in the PCC/RSC. For example, if disinhibition is sufficient, then local injection of NMDA antagonists into the PCC or RSC might be expected to produce localized neurodegeneration. However, the results vary regarding the in vivo effects of direct injections of NMDA antagonists:d-APV injected directly into the cingulate caused neuronal vacuolization (Olney et al., 1989), whereas MK-801 injected into the RSC did not induce HSP70 heat shock protein, but instead, HSP70 was induced after bilateral (but not unilateral) MK-801 injections into the anterior thalamus (Tomitaka et al., 2000). Based on the latter findings, a model was constructed in which excitatory connections from the anterior thalamus to the RSC mediate MK-801-induced neurotoxicity, and it is within the thalamus that local modulation of GABAergic inhibition occurs (Tomitaka et al., 2000). That model is entirely consistent with their findings. However, our results suggest that there also is modulation of GABAergic function at the local cortical level. It is possible that locally mediated disinhibition (as seen in our slices) may not be sufficient to produce neurodegeneration. However, local disinhibition could become critical when modulated by other neurotransmitters or external excitatory inputs, then leading to full-blown neurodegeneration. Given that region-specific disinhibition seen in vitro does parallel region-specific neurodegeneration seen in vivo, it is plausible that disinhibition may be a candidate mechanism underlying neurotoxicity.
Clinical significance
These findings may have wide-ranging clinical significance because drugs with NMDA antagonist activity are being used clinically, and many more are under development. These include drugs for pain, epilepsy, and Parkinson's disease (Subramaniam et al., 1995; Boyce et al., 1999;Parsons et al., 1999). Also, certain abused drugs, such as PCP and ketamine, are NMDA antagonists (Olney et al., 1987). Potent NMDA antagonists such as PCP produce bizarre and disturbing behavioral effects in humans, but it has not been determined whether this reflects neurotoxicity. If so, it will be important to better understand the neurotoxic mechanisms and to reduce the risk of damage. One strategy involves using NMDAR-glycine site antagonists, which lack neurotoxicity (Berger et al., 1994; Auer, 1997). However, noncompetitive channel antagonists may still prove useful, so it will be important to better understand their neurotoxic mechanisms and the characteristics that increase vulnerability to neurotoxicity [e.g., age or gender (Auer, 1996; Wozniak et al., 1996)].
Also, NMDA antagonists are proposed to model psychiatric conditions such as schizophrenia (Ellison, 1995; Farber et al., 1995b). If there are shared mechanisms between drug-induced neurotoxicity and the etiologies of schizophrenia, these findings may have broad implications for psychiatry.
Footnotes
This work was supported by the Department of Defense (Neurotoxin Exposure Treatment Research Program, DAMA17-99-1-9541; Q.L, S.C., D.V.L., W.A.W.), The Department of Veteran Affairs (W.A.W., S.C), and the National Institutes of Health (DA-06735; D.V.L).
Correspondence should be addressed to Dr. Qiang Li, Neurology Research, Veterans Administration Medical Center, Room 24, Building 16, 508 Fulton Street, Durham, NC 27705. E-mail:Liq@duke.edu.
REFERENCES
- 1.Aitken PG, Balestrino M, Somjen GG. NMDA antagonists: lack of protective effect against hypoxic damage in CA1 region of hippocampal slice. Neurosci Lett. 1988;89:187–192. doi: 10.1016/0304-3940(88)90379-5. [DOI] [PubMed] [Google Scholar]
- 2.Allen HL, Iversen LL. Phencyclidine, dizocilpine and cerebrocortical neurons. Science. 1990;247:221. doi: 10.1126/science.2403696. [DOI] [PubMed] [Google Scholar]
- 3.Auer RN. Effect of age and sex on N-methyl-d-aspartate antagonist-induced neuronal necrosis in rats. Stroke. 1996;27:743–746. doi: 10.1161/01.str.27.4.743. [DOI] [PubMed] [Google Scholar]
- 4.Auer RN. Structural neurotoxicologic investigation of the glycine antagonist 5-nitro-6,7-dichloroquinoxalinedione (ACEA-1021). Neurotoxicology. 1997;18:53–62. [PubMed] [Google Scholar]
- 5.Avoli M, Oliver A. Bursting in human epileptogenic neocortex is depressed by an N-methyl-d-aspartate antagonist. Neurosci Lett. 1987;76:249–258. doi: 10.1016/0304-3940(87)90724-5. [DOI] [PubMed] [Google Scholar]
- 6.Bandrowski AE, Aramakis VB, Moore SL. Metabotropic glutamate receptors modify ionotropic glutamate responses in neocortical pyramidal cells and interneurons. Exp Brain Res. 2001;136:25–40. doi: 10.1007/s002210000556. [DOI] [PubMed] [Google Scholar]
- 7.Berger P, Farrel K, Sharp F, Skolnick P. Drugs acting at the strychnine-insensitive glycine receptor do not induce HSP-70 protein in the cingulate cortex. Neurosci Lett. 1994;168:147–150. doi: 10.1016/0304-3940(94)90437-5. [DOI] [PubMed] [Google Scholar]
- 8.Boyce S, Wyatt A, Webb JK, O'Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NM. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology. 1999;38:611–623. doi: 10.1016/s0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- 9.Brennan PA, Keverne EB. Impairment of olfactory memory by local infusions of non-selective excitatory amino acid receptor antagonists into the accessory olfactory bulb. Neuroscience. 1989;33:463–468. doi: 10.1016/0306-4522(89)90398-9. [DOI] [PubMed] [Google Scholar]
- 10.Cohen GA, Doze VA, Madison DV. Opioid inhibition of GABA release from presynaptic terminals of rat hippocampal interneurons. Neuron. 1992;9:325–335. doi: 10.1016/0896-6273(92)90171-9. [DOI] [PubMed] [Google Scholar]
- 11.Colbourne F, Rakic D, Auer RN. the effects of temperature and scopolamine on N-methyl-d-aspartate antagonist-induced neuronal necrosis in the rat. Neuroscience. 1999;90:87–94. doi: 10.1016/s0306-4522(98)00375-3. [DOI] [PubMed] [Google Scholar]
- 12.Corso TD, Sesma MA, Tenkova TI, Der TC, Wozniak DF, Farber NB, Olney JW. Multifocal brain damage induced by phencyclidine is augmented by pilocarpine. Brain Res. 1997;752:1–14. doi: 10.1016/s0006-8993(96)01347-9. [DOI] [PubMed] [Google Scholar]
- 13.Dirnagl U, Tanabe J, Pulsinelli W. Pre- and post-treatment with MK-801 but not pretreatment alone reduces neocortical damage after focal cerebral ischemia in the rat. Brain Res. 1990;527:62–68. doi: 10.1016/0006-8993(90)91060-t. [DOI] [PubMed] [Google Scholar]
- 14.Ellison G. The N-methyl-d-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias. Brain Res Rev. 1995;20:250–267. doi: 10.1016/0165-0173(94)00014-g. [DOI] [PubMed] [Google Scholar]
- 15.Farber NB, Foster J, Duhan NL, Olney JW. Alpha 2 adrenergic agonists prevent MK-801 neurotoxicity. Neuropsychopharmacology. 1995a;12:347–349. doi: 10.1016/0893-133X(95)00048-I. [DOI] [PubMed] [Google Scholar]
- 16.Farber NB, Wozniak DF, Price MT, Labruyere J, Huss J, St. Peter H, Olney JW. Age-specific neurotoxicity in the rat associated with NMDA receptor blockade: potential relevance to schizophrenia? Biol Psychiatry. 1995b;38:788–796. doi: 10.1016/0006-3223(95)00046-1. [DOI] [PubMed] [Google Scholar]
- 17.Farber NB, Foster J, Duhan NL, Olney JW. Olanzapine and fluperlapine mimic clozapine in preventing MK-801 neurotoxicity. Schizophrenia Res. 1996;21:33–37. doi: 10.1016/0920-9964(96)00024-2. [DOI] [PubMed] [Google Scholar]
- 18.Feldmeyer D, Sakmann B. Synaptic efficacy and reliability of excitatory connections between the principal neurons of the input (layer 4) and output layer (layer 5) of the neocortex. J Physiol (Lond) 2000;525:31–39. doi: 10.1111/j.1469-7793.2000.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fix AS, Horn JW, Writhman KA, Johnson CA, Long GG, Storts RW, Farber N, Wozniak DF, Olney JW. Neuronal vacuolization and necrosis induced by the noncompetitive N-methyl-d-aspartate (NMDA) antagonist MK(+)801 (dizocilpine maleate): a light and electron microscopic evaluation of the rat retrosplenial cortex. Exp Neurol. 1993;123:204–215. doi: 10.1006/exnr.1993.1153. [DOI] [PubMed] [Google Scholar]
- 20.Fix AS, Wozniak DF, Truex LL, McEwen M, Miller JP, Olney JW. Quantitative analysis of factors influencing neuronal necrosis induced by MK-801 in the rat posterior cingulate/retrosplenial cortex. Brain Res. 1995;696:194–204. doi: 10.1016/0006-8993(95)00842-e. [DOI] [PubMed] [Google Scholar]
- 21.Ford LM, Sanberg PR, Norman AB, Fogelson MH. MK-801 prevents hippocampal neurodegeneration in neonatal hypoxic-ischemic rats. Arch Neurol. 1989;46:1090–1096. doi: 10.1001/archneur.1989.00520460072016. [DOI] [PubMed] [Google Scholar]
- 22.Foster AC, Gill R, Woodruff GN. Neuroprotective effects of MK-801 in vivo: selectivity and evidence for delayed degeneration mediated by NMDA receptor activation. J Neurosci. 1988;8:4745–4754. doi: 10.1523/JNEUROSCI.08-12-04745.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gloveli T, Iserhot C, Schmitz D, Castren E, Behr J, Heinemann U. Systemic administration of the phencyclidine compound MK-801 affects stimulus-induced field potentials selectively in layer III of rat medial entorhinal cortex. Neurosci Lett. 1997;221:93–96. doi: 10.1016/s0304-3940(96)13307-3. [DOI] [PubMed] [Google Scholar]
- 24.Grunze HCR, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, Greene RW. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16:2034–2043. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hablitz JJ, Langmoen IA. N-methyl-d-aspartate receptor antagonist reduce synaptic excitation in the hippocampus. J Neurosci. 1986;6:102–106. doi: 10.1523/JNEUROSCI.06-01-00102.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature. 1992;357:686–689. doi: 10.1038/357686a0. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman AF, Lupica CR. Mechanism of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horvath ZC, Czopf J, Buzsaki G. MK-801-induced neuronal damage in rats. Brain Res. 1997;753:181–195. doi: 10.1016/s0006-8993(96)01290-5. [DOI] [PubMed] [Google Scholar]
- 29.Jones RSG, Buhl EH. Basket-like interneurons in layer II of the entorhinal cortex exhibit a powerful NMDA-mediated synaptic excitation. Neurosci Lett. 1993;149:35–39. doi: 10.1016/0304-3940(93)90341-h. [DOI] [PubMed] [Google Scholar]
- 30.Kawaguchi Y. Physiological subgroups of nonpyramidal cells with specific morphological characteristics in layer II/III of rat frontal cortex. J Neurosci. 1995;15:2638–2655. doi: 10.1523/JNEUROSCI.15-04-02638.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawaguchi Y, Kubota Y. Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J Neurosci. 1996;16:2701–2715. doi: 10.1523/JNEUROSCI.16-08-02701.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim HG, Connors BW. Apical dendrites of the neocortex: correlation between sodium- and calcium-dependent spiking and pyramidal cell morphology. J Neurosci. 1993;13:5301–5311. doi: 10.1523/JNEUROSCI.13-12-05301.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurumaji A, McCulloch J. Effects of unilateral intrahippocampal injection of MK-801 upon local cerebral glucose utilisation in conscious rats. Brain Res. 1990;518:342–346. doi: 10.1016/0006-8993(90)90995-n. [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Clark S, Wilson WA, Lewis DV. MK-801, an NMDA receptor antagonist, modulates the inhibitory postsynaptic currents (IPSCs) in pyramidal neurons in the rat cingulate cortex. Soc Neurosci Abstr. 2000;29:431. [Google Scholar]
- 35.Loscher W, Honack D. The novel, competitive N-methyl-d-aspartate (NMDA) antagonist CGP 37849 preferentially induces phencyclidine-like behavioral effects in kindled rats: attenuation by manipulation of dopamine, alpha-1 and serotonin 1A receptor. J Pharmacol Exp Ther. 1991;257:1146–1153. [PubMed] [Google Scholar]
- 36.Lubke J, Markram H, Frotscher M, Sakmann B. Frequency and dendrite distribution of autopses established by layer 5 pyramidal neurons in the developing rat neocortex: comparison with synaptic innervation of adjacent neurons of the same class. J Neurosci. 1996;16:3209–3218. doi: 10.1523/JNEUROSCI.16-10-03209.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacDonald JW, Silverstein FS, Johnston MV. MK-801 pretreatment enhances N-methyl-d-aspartate-mediated brain injury and increases brain N-methyl-d-aspartate antagonist recognition site binding in rats. Neuroscience. 1990;38:103–113. doi: 10.1016/0306-4522(90)90377-g. [DOI] [PubMed] [Google Scholar]
- 38.McBain CJ, Dingledine R. Heterogeneity of synaptic glutamate receptors on CA3 stratum radiatum interneurones of rat hippocampus. J Physiol (Lond) 1993;462:373–392. doi: 10.1113/jphysiol.1993.sp019560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mott DD, Turner DA, Okazaki MM, Lewis DV. Interneurons of the rat dentate gyrus: morphological and electrophysiological heterogeneity. J Neurosci. 1997;17:3990–4005. doi: 10.1523/JNEUROSCI.17-11-03990.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mott DD, Li Q, Okazaki MM, Turner DA, Lewis DV. GABAB-receptor-mediated currents in interneurons of the dentate-hilus border. J Neurophysiol. 1999;82:1438–1450. doi: 10.1152/jn.1999.82.3.1438. [DOI] [PubMed] [Google Scholar]
- 41.Nehls DG, Park CK, MacCormack AG, McCulloch J. The effects of N-methyl-d-aspartate receptor blockade with MK-801 upon the relationship between cerebral blood flow and glucose utilisation. Brain Res. 1990;511:271–279. doi: 10.1016/0006-8993(90)90172-8. [DOI] [PubMed] [Google Scholar]
- 42.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 43.Olney JW, Price MT, Labruyere J, Salles KS, Frierdich G, Mueller M, Silverman E. Anti-parkinsonian agents are phencyclidine agonists and N-methyl-aspartate antagonists. Eur J Pharmacol. 1987;142:319–320. doi: 10.1016/0014-2999(87)90123-3. [DOI] [PubMed] [Google Scholar]
- 44.Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- 45.Olney JW, Labruyere J, Wang G, Wozniak DF, Price MT, Sesma MA. NMDA antagonist neurotoxicity: mechanism and prevention. Science. 1991;254:1515–1518. doi: 10.1126/science.1835799. [DOI] [PubMed] [Google Scholar]
- 46.Patel TR, McCulloch J. AMPA receptor antagonism attenuates MK-801-induced hypermetabolism in the posterior cingulate cortex. Brain Res. 1995;686:254–258. doi: 10.1016/0006-8993(95)00483-7. [DOI] [PubMed] [Google Scholar]
- 47.Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist: a review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 48.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 49.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Excitatory transmission in the basolateral amygdala. J Neurophysiol. 1991;66:986–998. doi: 10.1152/jn.1991.66.3.986. [DOI] [PubMed] [Google Scholar]
- 50.Reyes A, Sakmann B. Developmental switch in the short-term modification of unitary EPSPs evoked in layer 2/3 and layer 5 pyramidal neurons of rat neocortex. J Neurosci. 1999;19:3827–3835. doi: 10.1523/JNEUROSCI.19-10-03827.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rod MR, Auer RN. Pre- and post-ischemic administration of dizocilpine (MK-801) reduces cerebral necrosis in the rat. Can J Neurol Sci. 1989;16:340–344. doi: 10.1017/s031716710002919x. [DOI] [PubMed] [Google Scholar]
- 52.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 53.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. Trends Neurosci. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 54.Salin PA, Prince DA. Spontaneous GABAA receptor-mediated inhibitory currents in adult rat somatosensory cortex. J Neurophysiol. 1996;75:1573–1588. doi: 10.1152/jn.1996.75.4.1573. [DOI] [PubMed] [Google Scholar]
- 55.Sams-Dodd F. Effects of novel antipsychotic drugs on phencyclidine-induced stereotyped behavior and social isolation in the rat social interaction test. Behav Pharmacol. 1997;8:196–215. [PubMed] [Google Scholar]
- 56.Schoppa NE, Kinzie M, Sahara Y, Segerson TP, Westbrook GL. Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci. 1998;18:6790–6802. doi: 10.1523/JNEUROSCI.18-17-06790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharp FR, Butman M, Aardalen K, Nickolenko J, Nakki R, Massa SM, Swanson A, Sagar SM. Neuronal injury produced by NMDA antagonists can be detected using heat shock proteins and can be blocked with antipsychotics. Psychopharmacol Bull. 1994;30:555–560. [PubMed] [Google Scholar]
- 58.Sripanidkulchai K, Wyss JM. The laminar organization of efferent neuronal cell bodies in the retrosplenial granular cortex. Brain Res. 1987;406:255–269. doi: 10.1016/0006-8993(87)90790-6. [DOI] [PubMed] [Google Scholar]
- 59.Stasheff SF, Anderson WW, Clark S, Wilson WA. NMDA antagonists differentiate epileptogenesis from seizure expression in an in vitro model. Science. 1989;245:648–651. doi: 10.1126/science.2569762. [DOI] [PubMed] [Google Scholar]
- 60.Subramaniam S, Rho JM, Penix L, Donevan SD, Fielding RP, Rogawski MA. Felbamate block of the N-methyl-d-aspartate receptor. J Pharmacol Exp Ther. 1995;273:878–886. [PubMed] [Google Scholar]
- 61.Sveinbjornsdottir S, Sander JW, Upton D, Thompson PJ, Patsalos PN, Hirt D, Emre M, Lowe D, Duncan JS. The excitatory amino acid antagonist d-CPP-ene (SDZ EAA-494) in patients with epilepsy. Epilepsy Res. 1993;16:165–174. doi: 10.1016/0920-1211(93)90031-2. [DOI] [PubMed] [Google Scholar]
- 62.Tomitaka S, Tomitaka M, Tolliver BK, Sharp FR. Bilateral blockade of NMDA receptors in anterior thalamus by dizocilpine (MK-801) injures pyramidal neurons in rat retrosplenial cortex. Eur J Neurosci. 2000;12:1420–1430. doi: 10.1046/j.1460-9568.2000.00018.x. [DOI] [PubMed] [Google Scholar]
- 63.Vaisanen J, Linden AM, Lakso M, Wong G, Heinemann U, Castren E. Excitatory actions of NMDA receptor antagonists in rat entorhinal cortex and cultured entorhinal cortical neurons. Neuropsychopharmacology. 1999;21:137–146. doi: 10.1016/S0893-133X(99)00006-8. [DOI] [PubMed] [Google Scholar]
- 64.Wieloch T. Hypoglycemia-induced damage prevented by an N-methyl-d-aspartate antagonist. Science. 1985;230:681–683. doi: 10.1126/science.2996146. [DOI] [PubMed] [Google Scholar]
- 65.Wozniak DF, Brosnon-Watters G, Nardi A, McEwen M, Corso TD, Olney JW, Fix AS. MK-801 neurotoxicity in male mice: histologic effects and chronic impairment in spatial learning. Brain Res. 1996;707:165–179. doi: 10.1016/0006-8993(95)01230-3. [DOI] [PubMed] [Google Scholar]
- 66.Zhou FM, Hablitz JJ. AMPA receptor-mediated EPSCs in rat neocortical layer II/III interneurons have rapid kinetics. Brain Res. 1998;780:166–169. doi: 10.1016/s0006-8993(97)01311-5. [DOI] [PubMed] [Google Scholar]