

Abstract
Cerebral ischemia is a major cause of brain dysfunction. Using a model of delayed death induced by a brief, transient oxygen and glucose deprivation, we studied here how this affected the structural organization of hippocampal synaptic networks. We report that brief anoxic–hypoglycemic episodes rapidly modified the structure of synapses. This was characterized, at the electron microscopic level, by a transient increase in the proportion of perforated synapses, followed after 2 hr by an increase in images of multiple synapse boutons. These changes were considerable because 10–20% of all synapses were affected. This structural remodeling was correlated by three kinds of modifications observed using two-photon confocal microscopy: the growth of filopodia, occurring shortly (5–20 min) after anoxia–hypoglycemia, enlargements of existing spines, and formation of new spines, both seen mainly 20–60 min after the insult. All of these structural changes were calcium and NMDA receptor dependent and thus reproduced, to a larger scale, those associated with synaptic plasticity. Concomitantly and related to the severity of anoxia–hypoglycemia, we could also observe spine loss and images of spine, dendrite, or presynaptic terminal swellings that evolved up to membrane disruption. These changes were also calcium dependent and reduced by NMDA receptor antagonists. Thus, short anoxic–hypoglycemic episodes, through NMDA receptor activation and calcium influx, resulted in a profound structural remodeling of synaptic networks, through growth, formation, and elimination of spines and synapses.
Keywords: ischemia, synaptic plasticity, spines, morphology, synaptogenesis, anoxic LTP
Reduced supply of oxygen and glucose such as occurs during failure of blood circulation is a primary cause of brain damage. A central mechanism responsible for the neuronal death that occurs under these conditions is a massive release of glutamate (Lee et al., 1999) by excitatory terminals and glial cells. This process appears, at least partly, to be mediated by a reverse functioning of cellular uptake systems (Rossi et al., 2000; Jabaudon et al., 2001). Glutamate then activates excitatory amino acid receptors and probably leads to toxicity through excessive calcium influx and calcium overload of neurons (Lee et al., 1999). When ischemia is important, excitotoxicity results in immediate cell death. However, if the insult is less severe, cell death is considerably reduced and is usually delayed. Because recovery of function critically depends on protection of these less affected regions, it is important to understand the regulatory mechanisms activated in these areas by ischemia.
To this end, we examined here how production of a short, transient period of anoxia–hypoglycemia affected synaptic connectivity within the network. Much recent evidence has revealed that synapses express a high degree of functional and structural plasticity (Luscher et al., 2000). Modifications of synaptic efficacy, such as long-term potentiation (LTP), are believed to underlie important functions of the brain and have been reported in association with anoxia (anoxic LTP;Crepel et al., 1993; Hammond et al., 1994; Hsu and Huang, 1997). Also, recent studies suggest that this functional plasticity is associated with a structural remodeling of synapses (Luscher et al., 2000). Formation of filopodia (Maletic-Savatic et al., 1999), possible precursors of spines (Ziv and Smith, 1996; Fiala et al., 1998), formation of new spines, or new types of synapses (Engert and Bonhoeffer, 1999; Toni et al., 1999) have been observed in association with induction of LTP. Also, modulation of the intracellular calcium concentration in spines appears to generate changes in the shape of spines (Segal et al., 2000). Finally, some evidence suggests that lasting interference with the function of excitatory amino acid receptors affects spine number (Kirov and Harris, 1999; McKinney et al., 1999). Thus, anoxia could represent a condition in which the structural organization of synapses is modified. Previous work has indeed shown that interruption of cerebral blood flow or anoxia results in appearance of focal dendritic swellings (Hsu and Buzsaki, 1993; Park et al., 1996; Werth et al., 1998) or disappearance of dendritic spines (Hori and Carpenter, 1994; Park et al., 1996; Hasbani et al., 2001). There is also strong evidence that swelling and spine loss are caused by activation of excitatory amino acid receptors (Park et al., 1996;Smart and Halpain, 2000). Transient ischemic episodes were therefore likely to affect synaptic networks. We examined here this issue using electron and confocal microscopy and provide evidence that an intense process of synaptic growth and remodeling, similar to that associated with plasticity, occurs concomitantly with swelling, bursting, and elimination of spines, thereby markedly affecting connectivity within the network.
MATERIALS AND METHODS
Preparation of cultures and anoxia–hypoglycemia experiments. Hippocampal organotypic slice cultures were prepared from 7-d-old rats and maintained 10–15 d in culture as described previously (Stoppini et al., 1991). Electrophysiology was performed in an interface-type chamber, with slice cultures continuously perfused with a medium containing (in mm): 124 NaCl, 1.6 KCl, 2.5 CaCl2, 1.5 MgCl2, 24 NaHCO3, 1.2 KH2PO4, 10 glucose, and 2 ascorbic acid, pH 7.4 (temperature of 33°C). EPSPs were recorded in the stratum radiatum of the CA1 area and evoked by stimulation of a group of CA3 neurons. For two-photon confocal microscopy experiments, slice cultures were placed on an infrapatch setup (Luigs and Neuman, Ratingen, Germany) under continuous perfusion (4 ml/min). In both cases, anoxia–hypoglycemia was produced by replacing oxygen with N2 and switching to a medium containing sucrose instead of glucose.
Two-photon confocal microscopy. CA1 pyramidal neurons were patched using electrodes filled with a medium containing (in mm): 130 K-gluconate, 4 NaCl, 5 EGTA, 20 HEPES, 1 CaCl2, 1 MgCl2, 0.2 Na3GTP, and 2 Na2ATP, pH 7.2–7.4 (290 mOsm, 0.04–0.06% sulforhodamine). Imaging of dendritic spines was performed through a 40× water immersion objective using a Bio-Rad (Hercules, CA) MRC1024 scan head and a Mira 900 laser (Coherent Inc., Santa Clara, CA) set at 830 nm wavelength. Analyses were performed by focusing on small dendritic segments (100–200 μm in length), located on secondary and tertiary dendrites, and taking images every 2–5 min over a 1–2 hr period. The size of the dendritic segments under analysis remained constant throughout the experiments, as indicated by measurements made between specific markers (spines and branch points) easily identifiable on those segments. Filopodia were defined as thin, dynamic dendritic protrusions of over 2 μm in length. Spine enlargements were considered when the spine head diameter increased by a factor of two.
Electron microscopy processing. At different times (15 min, 30 min, and 2 hr) after anoxia, slice cultures were fixed and processed for electron microscopy as described previously (Toni et al., 2001). Briefly, cultures were fixed overnight at 4°C in 3% glutaraldehyde, rinsed in 0.1 m phosphate buffer (pH 7.4), and post-fixed in a fresh solution of 1% osmium tetroxide (OsO4) with 1.5% potassium chromium trisoxalate (K3Cr(C2O4))3(Aldrich, Milwaukee, WI), pH 9.5, for 2 hr. After a 5 min rinse in distilled water adjusted to pH 9.5 with KOH, the samples were dehydrated in ethanol and propylene oxide and embedded in Epon (Fluka, Buchs, Switzerland). For light microscopy, sections were stained with methylene blue. For serial electron microscopy, ribbons of up to 60 sections were cut in the middle portion of the apical arborization of CA1 pyramidal neurons (ultratome Ultracut-E; Leica, Deerfield, IL) and collected on single-slotted Formvar-coated grids. Sections were stained for 20 min in 5% uranyl acetate and 30 sec in lead citrate and analyzed on a Philips CM10 electron microscope at a magnification of 8900× to 28,500×.
Morphological analyses. Synapses were defined by a clear postsynaptic density facing at least three presynaptic vesicles, perforated synapses by the presence of a discontinuity in the postsynaptic density (Geinisman et al., 1987), and multiple synapse boutons (MSBs) by the presence of two independent dendritic spines contacting the same axon terminal (Sorra and Harris, 1993). The proportion of perforated synapses and MSBs was determined by taking random pictures of synapses in the middle one-third of the stratum radiatum and defining those corresponding to the above criteria.
For stereological analyses, five to six serial sections per culture were examined, and synapses present in a volume of 31.3–71.2 μm3 were analyzed. The dissector procedure was performed as described by Geinisman et al. (1996). For three-dimensional analyses, 45 MSBs were reconstructed out of seven hippocampal slice cultures. They were selected on the test section based on the presence of two clearly recognizable spines contacting the same terminal. The profiles were then photographed serially at a magnification of at least 15,500× and three dimensionally reconstructed using a software developed by J. C. Fiala and K. M. Harris (Boston University, Boston, MA).
Data are presented as a mean ± SEM, with n indicating the number of synapses or slice cultures analyzed, as indicated. Statistical analyses were performed using the Student's ttest.
RESULTS
Using a model of hippocampal organotypic slice cultures, we investigated the effects of brief, transient episodes (2–10 min) of anoxia–hypoglycemia on spine morphology. These short episodes produced only minimal damage to the tissue and enhanced synaptic transmission. In accordance with previous reports (Crepel et al., 1993; Hsu and Huang, 1997), a lasting increase in the size of EPSPs was recorded in the CA1 area of hippocampal slice cultures (Fig.1A) by stimulation of a group of CA3 neurons. This effect was NMDA receptor dependent, because it could be prevented by treatment of slice cultures with 100 μmd-AP-5 (Fig.1A). These short anoxic–hypoglycemic episodes did not produce acute cell death. As shown on semithin sections, the morphology of pyramidal neurons in the CA1 area was still preserved 2–6 hr after the anoxia–hypoglycemia (Fig. 1B). Electron microscopic analyses further confirmed that there were no obvious signs of cell death or gross morphological alterations at those time points (Fig. 1C). Propidium iodide experiments revealed only rare cases of labeled neurons up to 6 hr after the anoxia (Fig.1D). However, staining increased significantly after 24–48 hr, thereby indicating a process of delayed cell death (Laake et al., 1999) (Fig. 1E).
Fig. 1.
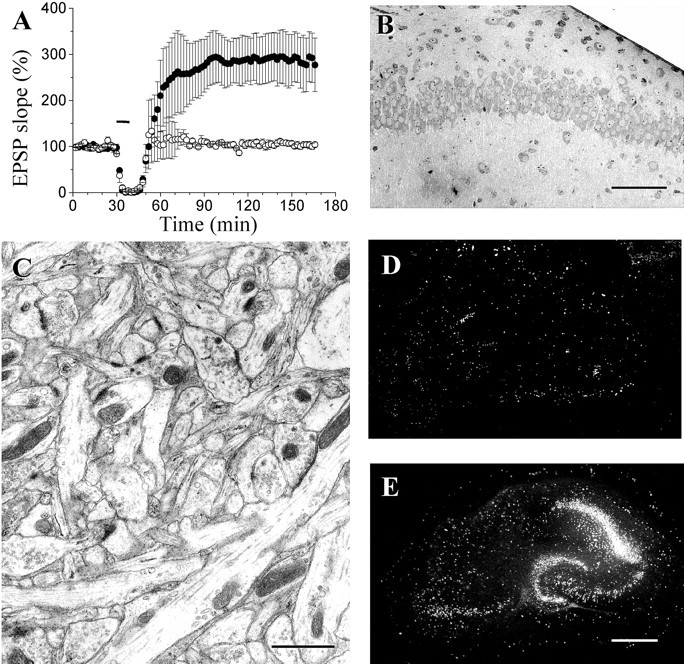
Enhancement of excitatory transmission and delayed cell death induced by short anoxic–hypoglycemic episodes.A, Changes in EPSP slope produced by a short anoxia–hypoglycemia (blackbar indicates 10 min) in hippocampal organotypic slice cultures in the absence (filled circles; n = 5) or presence (open circles; n = 4) of the NMDA receptor antagonist d-AP-5 (100 μm). Data are mean ± SEM. B, Semithin section of the CA1 area of a slice culture fixed 6 hr after a short (10 min) anoxia—hypoglycemia. Scale bar, 100 μm. C, Electron microscopic image of the stratum radiatum of a slice culture fixed 6 hr after a short (10 min) anoxia–hypoglycemia. Scale bar, 1 μm.D, E, Propidium iodide staining of slice cultures showing only a limited, nonspecific cell death 6 hr (D) after a short anoxia–hypoglycemia but a more intense delayed cell death 48 hr after the injury (E), Scale bar, 500 μm.
A more careful analysis of the morphology of excitatory synapses in these challenged cultures revealed, however, several changes taking place during the first 2 hr after the anoxia–hypoglycemia. A first observation was a marked increase in the proportion of synapses with perforated postsynaptic densities in slice cultures fixed 15–30 min after the anoxic episode (Fig.2A,C). This change was detectable by analyzing the entire population of spine profiles. It was significant using both single-section analysis applied to a large number of spine profiles (30 ± 2.0 vs 14 ± 3.5%; 2557 synapses analyzed; 11 slice cultures; p < 0.01) and unbiased stereological methods applied to a smaller group of serial sections (35.0 ± 3.7 vs 16.3 ± 0.7; 667 synapses analyzed; 10 slice cultures; p < 0.01) (Fig.2C). The increase in perforated synapses could be prevented by treatment of slice cultures with 100 μmd-AP-5 applied during the anoxic episode (Fig.2C). Also, the change was only transient, because the proportion of perforated synapses was back to control values after 2 hr. At that time, however, another modification could be observed, consisting of an increase in the proportion of MSBs (Fig.2B). The increase in MSBs was also highly significant and confirmed using both single-section analysis (15.0 ± 0.4 vs 5.0 ± 0.4%; 2383 synapses analyzed; 11 slice cultures;p < 0.01) and unbiased stereological methods (21.4 ± 1.4 vs 6.4 ± 0.7; 444 synapses analyzed; 11 slice cultures; p < 0.01) (Fig. 2D). Application of d-AP-5 (100 μm) during the anoxic episode also completely prevented the increase in MSB proportion (Fig. 2D). These changes thus reproduced those reported after LTP induction (Toni et al., 1999).
Fig. 2.
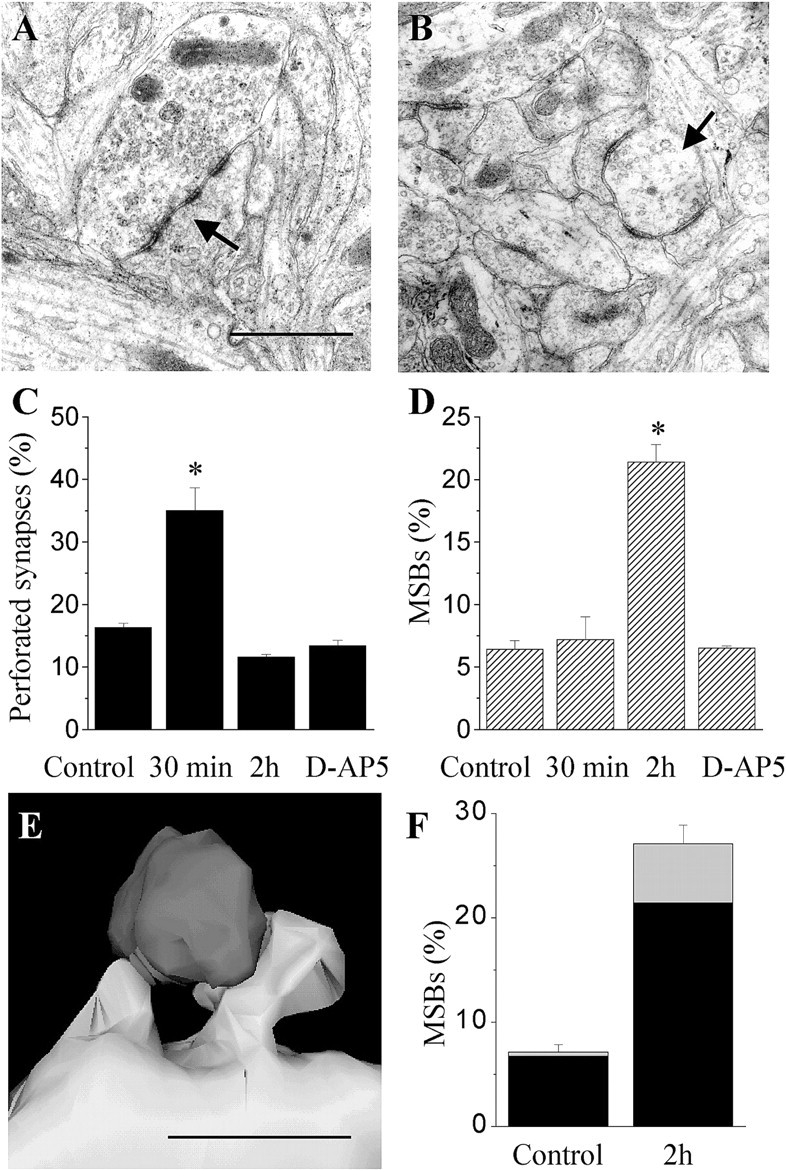
Anoxia–hypoglycemia induced morphological remodeling of spine synapses. A, Illustration of a synapse with a perforated postsynaptic density (arrow). Scale bar, 1 μm. B, Illustration of an MSB in which three individual spines contact the same terminal.C, Increase in the proportion of synapses with a perforated PSD observed 15–30 min after a short (10 min) anoxia–hypoglycemia. Data are mean ± SEM, obtained through analysis of 10 slice cultures using unbiased stereological methods (667 synapses analyzed). Note that the change in perforated synapses is prevented by treatment with the NMDA receptor antagonistd-AP-5. D, Increase in the proportion of MSBs observed 2 hr after a short anoxia–hypoglycemia. Data are mean ± SEM, obtained through analysis of 11 slice cultures using unbiased stereological methods (444 synapses analyzed).E, Illustration of an MSB in which the two spines arise from the same dendrite in a slice culture fixed 2 hr after anoxia–hypoglycemia. Scale bar, 1 μm. F, Proportion of MSBs with two spines arising from the same dendrite (gray columns) or different dendrites (black column) under control conditions and 2 hr after anoxia–hypoglycemia.
To investigate the origin of the spines in MSBs, we proceeded to three-dimensional reconstruction of cases observed under control conditions or in slice cultures fixed 2 hr after anoxia–hypoglycemia (Fig. 2E). Two of 30 MSBs reconstructed under control conditions had spines originating from the same dendrite (6.7%) (Fig.2F). For comparison, 4 of 15 MSBs reconstructed 2 hr after anoxia exhibited adjacent spines contacting the same terminal (26.7%). When expressed as a ratio of the proportion of MSBs observed under the two conditions (Fig. 2F), the results suggested that both MSBs with spines originating from the same dendrite and MSBs with spines from different dendrites increased after anoxia–hypoglycemia.
Because these observations suggested a process of synapse formation similar to that reported after LTP induction, we investigated the changes in spine morphology that could be detected using two-photon confocal microscopy. To visualize dendrites and spines, CA1 pyramidal neurons were patched under visual control using an electrode filled with sulforhodamine (0.04–0.06%). Within 10–15 min of whole-cell access, the entire dendritic arborization could be visualized, and the changes in the morphology of spines could be analyzed repetitively (every 2–5 min) before and after a short (2–10 min) anoxic–hypoglycemic episode. Several modifications of spines were identified. As illustrated in Figure 3, a first change triggered by anoxia was the appearance of thin filopodia. Under control conditions, filopodia were occasionally observed (1 of 11 experiments with 60–80 min of repetitive analysis). After an anoxic–hypoglycemic episode, their occurrence markedly increased (17 of 24 experiments). When expressed as number of filopodia observed per 100 μm of dendritic length, the increase was statistically significant (p < 0.05) (Fig. 3B). Most filopodia initially appeared 5–20 min after the onset of anoxia (Fig. 3C). Then, they grew in size and retracted, although usually not completely, over the time course of the next hour. The triggering of filopodia by anoxia–hypoglycemia was calcium dependent, because both intracellular injection of 10 mmBAPTA, a calcium chelator (n = 11), or reduction of the extracellular calcium concentration (0.1 mmCa2+ and 10 mmMg2+; n = 10) prevented their occurrence. Furthermore, the induction of filopodia was blocked by treatment of slice cultures with the NMDA receptor antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) (40 μm;n = 12).
Fig. 3.

Anoxia–hypoglycemia induced formation of filopodia. A, Time sequence illustrating the formation and modifications of a filopodia induced after a short (10 min) anoxia–hypoglycemia. Scale bar, 5 μm. B, Quantitative analysis of the number of filopodia per 100 μm dendritic length observed over periods of 80 min of observation under control conditions (white column), after a short anoxia–hypoglycemia (black column), after an anoxia–hypoglycemia but with 10 mm BAPTA in the pipette solution (gray column), or a medium containing low calcium (0.1 mm), high magnesium (10 mm), or MK-801 (40 μm; dashed column). Data are mean ± SEM of 11–24 experiments. *p < 0.05.C, Time interval at which filopodia were seen to appear after anoxia–hypoglycemia. Data are expressed as percentage of the total number of events.
A second type of changes induced by anoxia were modifications of the morphology of dendritic spines. Figure4A illustrates three examples of enlargements of the spine head observed after an anoxic–hypoglycemic episode. Similar cases were seen in 13 of 24 experiments, whereas only 1 of 11 experiments showed such changes under control conditions. When expressed per unit length of dendrite, the occurrence of these spine enlargements significantly increased (p < 0.05) (Fig. 4C, black columns). These changes appeared to be spine specific, because neighboring spines were not affected. As illustrated in Figure 4,C and D, they mainly occurred between 20 and 40 min after the anoxia, and they were prevented by intracellular injection of 10 mm BAPTA by reducing the extracellular calcium concentration (0.1 mmCa2+ and 10 mmMg2+), as well as by blocking NMDA receptors with 40 μm MK-801.
Fig. 4.

Anoxia–hypoglycemia induced enlargement of spines and formation of new spines. A, Representative examples of shape changes and enlargement of spine heads observed after an anoxic–hypoglycemic episode (asterisks; 2 min anoxia). Scale bars, 5 μm. B, Representative examples of formation of a new spine after anoxia–hypoglycemia (asterisks; 2 and 10 min anoxia). Scale bars, 5 μm.C, Quantitative analysis of the number of spine enlargements (black columns) and new spines (dashed columns) observed per 100 μm dendritic length under control conditions, after a short (2–10 min) anoxia–hypoglycemia, after an anoxia–hypoglycemia but with either 10 mm BAPTA in the pipette solution or a medium containing low calcium (0.1 mm), high magnesium (10 mm), or with MK-801 (40 μm). Data are mean ± SEM of 10–24 experiments. *p < 0.05. C, Time interval at which spine enlargements and new spines were seen to appear after anoxia–hypoglycemia. Data are expressed as percentage of the total number of events.
Associated with the spine enlargements, we also observed frequently a process of new spine formation. In 13 of 24 experiments, new spines were seen to appear on the dendritic segment under analysis. Two typical examples are illustrated in Figure 4B. Images from sections below and above the labeled dendrite were systematically recorded, allowing in this way to exclude that the new spines could have been mistaken because initially present on a different plane of section. Formation of new spines rarely occurred under control conditions (1 of 11 experiments). When expressed per unit length of dendrite, this mechanism was markedly enhanced by anoxia–hypoglycemia (p < 0.05) (Fig. 4C, dashed columns). This effect was calcium dependent, as well as NMDA receptor dependent. Curiously, BAPTA was less efficient in preventing the phenomenon. These new spines usually started to be detectable between 30 and 60 min after the anoxic episode and persisted until the end of the experiment, up to 90 min in some cases.
Although all of the changes reported above suggested mechanisms of synaptic growth, there were also modifications that lead to a decrease of synaptic connectivity. Figure 5illustrates a process referred to as dendritic swelling (Hsu and Buzsaki, 1993; Park et al., 1996). This phenomenon was observed in 17 of 28 experiments, whereas it occurred only once in 11 experiments under control conditions. Interestingly, Figure 5A reveals that these dendritic swellings could evolve within 10–30 min into a complete disruption of the dendritic structure through a bursting phenomenon. Of 31 cases, 17 (55%) evolved to bursting within 30 min. This phenomenon was seen with both single- and two-photon confocal microscopy and occurred even without repetitive illumination of the labeled cell. They were thus not produced by laser illumination. The frequency of these dendritic swellings correlated well with the severity (duration) of the anoxic–hypoglycemic episode (Fig.5B). They were calcium dependent and could be prevented by intracellular injection of BAPTA or by application of MK-801 (Fig.5B). The swellings were seen mainly between 20 and 40 min after the anoxia and tended to occur faster when the anoxia was more prolonged. Associated with the swellings, there was usually a process of spine loss. Figure 5A shows an example in which swellings were accompanied by a smoothing of the dendrite and disappearance of spines. In 11 of 17 experiments in which swellings occurred, spine loss was also observed, whereas spine loss without swellings was observed only twice. Also, swellings were usually preceded by a few minutes of spine loss (5.1 ± 1.3 min; n = 11). As for swellings, spine loss was calcium dependent and reduced by blockade of NMDA receptors. Interestingly, the structures from which these swellings originated could be spines and dendrites but also axonal varicosities. Figure 6 illustrates confocal and electron microscopic images showing swellings at the level of a postsynaptic spine (Fig.6A,B), as well as at the level of a DiI-labeled axonal varicosity (Fig. 6C) and a presynaptic terminal (Fig. 6D). Thus, swelling is a mechanism that concerned both presynaptic and postsynaptic structures.
Fig. 5.

Anoxia–hypoglycemia-induced swelling and spine loss. A, Time sequence illustrating the formation of dendritic and spine swellings after a short (10 min) anoxia. Note that the formation of dendritic swellings is associated with a smoothing of the dendrite and the disappearance of spines (asterisks). These swellings progressively increased in size over a time course of 10–30 min and finally burst out, disrupting the cellular membrane (arrows). Scale bar, 10 μm.B, Quantitative analysis of the number of swellings (black columns) and spine loss (dashed columns) observed per 100 μm dendritic length under control conditions, after a short anoxia–hypoglycemia (2–5 min,n = 12; 10 min, n = 16), or after an anoxia–hypoglycemia but with either 10 mm BAPTA in the pipette solution (n = 11) or a medium containing low calcium (0.1 mm), high magnesium (10 mm; n = 10), or MK-801 (40 μm; n = 12). Data are mean ± SEM. *p < 0.05; **p < 0.01.C, Time interval at which swellings and spine loss were seen to occur after anoxia–hypoglycemia. Data are expressed as percentage of the total number of events.
Fig. 6.
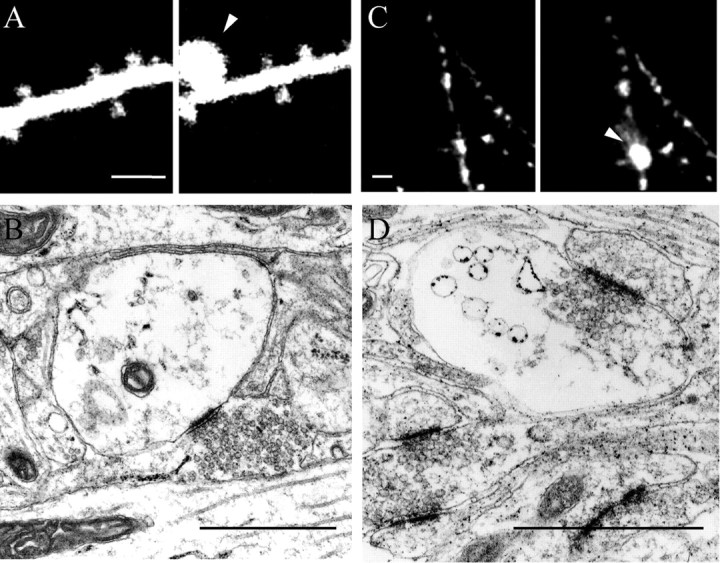
Anoxia–hypoglycemia-induced swelling may occur at the level of postsynaptic spines, as well as presynaptic terminals.A, Illustration of a case of swelling initiated at the level of a spine head. Scale bar, 5 μm. B, Electron microscopic image showing a similar case in which the swollen structure still receives a synaptic contact. C, Confocal microscopy of a DiI-labeled axonal fiber illustrating swelling at the level of one of the varicosities. Scale bar, 5 μm. D, Electron microscopic image showing swelling at the level of a presynaptic terminal. Scale bar, 1 μm.
DISCUSSION
The present study shows that brief periods of anoxia–hypoglycemia, which produce minimal damage to the tissue and do not result in acute cell death, are associated within the first 2 hr with an intense remodeling of synaptic connections, both through a process of growth and formation of new spines and new synapses and through spine loss, swelling, and destruction of existing synapses, axon terminals, and dendrites. The two processes appeared to occur independently because they were sometimes seen concomitantly on the same dendritic branches. They were both calcium and NMDA receptor dependent and could contribute to the early pathological events leading to delayed death.
A particularly interesting aspect of the present results is that the growth mechanisms reported here after anoxia show a strong analogy with the changes that have been described in association with synaptic plasticity and LTP induction (Luscher et al., 2000). In both cases, changes in synaptic efficacy take place. This is well documented for LTP, but this has also been reported for anoxia (Crepel et al., 1993;Hammond et al., 1994; Hsu and Huang, 1997). As illustrated here, this anoxic potentiation, similar to LTP, involves a long-lasting, NMDA receptor-dependent increase of synaptic responses. Furthermore, evidence suggests that, under both conditions, changes in the expression of glutamate receptor subunits may take place (Shi et al., 1999, 2001; Kobayashi and Millhorn, 2001; Optiz et al., 2001). The present study shows in addition that there is also a remarkable analogy in terms of the structural modifications of spines. Growth of filopodia, changes of the ultrastructure of spines, and formation of new spines and synapses occur under both conditions. Most interesting, these modifications take place with a very similar time sequence. As reported by Maletic-Savatic et al. (1999) after LTP, the first changes that we observed here after anoxia–hypoglycemia were the growth of filopodia. They occurred 5–20 min after the anoxic episode, and they were calcium and NMDA receptor dependent. Furthermore, we could detect formation of new spines 20–60 min after anoxia, similar to what has been reported by Engert and Bonhoeffer (1999) after LTP. Associated with this process of spine formation, we could also observe many cases of spine enlargements that were not observed previously in confocal studies of LTP. Similar images, however, were seen recently using confocal microscopic analyses of spines labeled with GFP-tagged postsynaptic density 95 (PSD-95) (Marrs et al., 2001). This study clearly indicated a highly dynamic nature of the synaptic membrane and PSD that is likely to reflect some of the changes reported using electron microscopy (Toni et al., 2001). Both here after anoxia and after LTP induction, electron microscopic analyses reveal major differences in the ultrastructural characteristics of spines. In particular, we found a marked but transient increase in the proportion of spines with perforated PSDs ∼30 min after anoxia, a phenomenon that is followed 2 hr by an increase in the proportion of MSBs. This sequence of changes thus fully corresponds to that reported after LTP or in association with associative learning (Geinisman, 1993; Toni et al., 1999; Geinisman et al., 2001). It is also interesting to note that the synapses with perforated PSDs seen after LTP are usually particularly large and exhibit features that suggest an important remodeling of synaptic membranes (Toni et al., 2001). They could thus correspond to the spine enlargements seen here with confocal microscopy and the dynamic changes reported recently at the level of the PSD (Marrs et al., 2001). Although the physiological and functional significance of these morphological changes remains unclear, an obvious interpretation is that they reflect a calcium- and NMDA receptor-dependent process of growth and synapse formation. The evidence suggesting an activation of proteases and the involvement of actin filament or other elements of the cytoskeleton during anoxia supports this interpretation (Friedman et al., 1998; Brana et al., 1999; Smart and Halpain, 2000).
One important characteristic of this growth process in the case of anoxia is that it is widespread and takes place at a very large scale. The number of filopodia, of newly formed spines or of spines that became enlarged, increased by almost an order of magnitude. Interestingly, the frequency of these events did not seem to vary between 2 and 10 min after anoxia–hypoglycemia, in contrast to the number of swellings (Fig. 5). Overall, the number of spines that significantly changed shape or were newly formed after anoxia represented between 2 and 5% of all visible spines. At the ultrastructural level, the number of synapses that showed changes at the level of their PSD was even larger and represented 15–20% of all spine profiles. Accordingly, the magnitude of these changes may be expected to significantly alter excitability properties and information processing within the network. In this respect, these data are consistent with the observation of marked differences in the density of spines on CA1 neurons after preparing acute slices, a process that involves exposure to a short ischemia (Kirov et al., 1999). Thus, through the release of glutamate, ischemia could activate mechanisms analogous to those implicated in synaptic plasticity but to a larger scale. Consistent with this, the changes reported here, like those described after LTP, are calcium and NMDA receptor dependent. This phenomenon could very well have deleterious effects by negatively affecting the functioning of the network and, through increased excitability, initiate intracellular cascades responsible for delayed cell death (Obrenovitch and Urenjak, 1997).
An additional mechanism susceptible to lead to tissue damage is that responsible for swelling and bursting of neuronal structures. The two mechanisms are probably distinct and unrelated, although they sometimes occurred concomitantly. Swelling and bursting were more pronounced with more severe anoxia and they were calcium dependent (Abdel-Hamid et al., 1997) but less NMDA receptor sensitive than growth mechanisms (Park et al., 1996; Smart and Halpain, 2000). Also, swelling was probably causally related to spine loss, because the two phenomena were usually associated with swelling slightly preceding spine loss. Whereas spine loss has been reported recently to be only transient (Hasbani et al., 2001), swelling, and particularly bursting, appears to be a potentially very damaging process, disrupting the membrane continuity. It is very interesting in this respect that the phenomenon may in fact occur on dendrites, spines, as well as presynaptic terminals, being therefore not structure specific.
In conclusion, the present study shows that a short episode of anoxia–hypoglycemia markedly affects the organization of synaptic networks: it modifies the structure and function of synapses and triggers the growth of new spines, while, at the same time, other spines, dendrites and axonal varicosities are being destroyed. These changes are thus likely to contribute to the functional impairment that characterizes tissue injured by a transient ischemia.
P.J. and I.N. contributed equally to this work.
This work was supported by Swiss National Science Foundation Grant 31-56852.99 and Program Scientific Co-operation between Eastern Europe and Switzerland Grant 7UKPJ062401. We thank M. Moosmayer and L. Parisi for excellent technical support and Fred Pillonel for photographic work.
Correspondence should be addressed to Prof. Dominique Muller, Neuropharmacology, University Medical Center, 1211 Geneva 4, Switzerland. E-mail: dominique.muller@medecine.unige.ch.
REFERENCES
- 1.Abdel-Hamid KM, Tymianski M. Mechanisms and effects of intracellular calcium buffering on neuronal survival in organotypic hippocampal cultures exposed to anoxia/aglycemia or to excitotoxins. J Neurosci. 1997;17:3538–3553. doi: 10.1523/JNEUROSCI.17-10-03538.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brana C, Benham CD, Sundstrom LE. Calpain activation and inhibition in organotypic rat hippocampal slice cultures deprived of oxygen and glucose. Eur J Neurosci. 1999;11:2375–2384. doi: 10.1046/j.1460-9568.1999.00653.x. [DOI] [PubMed] [Google Scholar]
- 3.Crepel V, Hammond C, Chinestra P, Diabira D, Ben-Ari Y. A selective LTP of NMDA receptor-mediated currents induced by anoxia in CA1 hippocampal neurons. J Neurophysiol. 1993;70:2045–2055. doi: 10.1152/jn.1993.70.5.2045. [DOI] [PubMed] [Google Scholar]
- 4.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 5.Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman JE, Chow EJ, Haddad GG. State of actin filaments is changed by anoxia in cultured rat neocortical neurons. Neuroscience. 1998;82:421–427. doi: 10.1016/s0306-4522(97)00217-0. [DOI] [PubMed] [Google Scholar]
- 7.Geinisman Y. Perforated axospinous synapses with multiple, completely partitioned transmission zones: probable structural intermediates in synaptic plasticity. Hippocampus. 1993;3:417–434. doi: 10.1002/hipo.450030404. [DOI] [PubMed] [Google Scholar]
- 8.Geinisman Y, Morrell F, de Toledo-Morrell L. Axospinous synapses with segmented postsynaptic densities: a morphologically distinct synaptic subtype contributing to the number of profiles of “perforated” synapses visualized in random sections. Brain Res. 1987;423:179–188. doi: 10.1016/0006-8993(87)90838-9. [DOI] [PubMed] [Google Scholar]
- 9.Geinisman Y, Gundersen HJ, van der Zee E, West MJ. Unbiased stereological estimation of the total number of synapses in a brain region. J Neurocytol. 1996;25:805–819. doi: 10.1007/BF02284843. [DOI] [PubMed] [Google Scholar]
- 10.Geinisman Y, Berry RW, Disterhoft JF, Power JM, van der Zee EA. Associative learning elicits the formation of multiple-synapse boutons. J Neurosci. 2001;21:5568–5573. doi: 10.1523/JNEUROSCI.21-15-05568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammond C, Crepel V, Gozlan H, Ben-Ari Y. Anoxic LTP sheds light on the multiple facets of NMDA receptors. Trends Neurosci. 1994;17:497–503. doi: 10.1016/0166-2236(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 12.Hasbani MJ, Schlief ML, Fisher DA, Goldberg MP. Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci. 2001;21:2393–2403. doi: 10.1523/JNEUROSCI.21-07-02393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hori N, Carpenter DO. Functional and morphological changes induced by transient in vivo ischemia. Exp Neurol. 1994;129:279–289. doi: 10.1006/exnr.1994.1170. [DOI] [PubMed] [Google Scholar]
- 14.Hsu KS, Huang CC. Characterization of the anoxia-induced long-term synaptic potentiation in area CA1 of the rat hippocampus. Br J Pharmacol. 1997;122:671–681. doi: 10.1038/sj.bjp.0701409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu M, Buzsaki G. Vulnerability of mossy fiber targets in the rat hippocampus to forebrain ischemia. J Neurosci. 1993;13:3964–3979. doi: 10.1523/JNEUROSCI.13-09-03964.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jabaudon D, Scanziani M, Gahwiler BH, Gerber U. Acute decrease in net glutamate uptake during energy deprivation. Proc Natl Acad Sci USA. 2001;97:5610–5615. doi: 10.1073/pnas.97.10.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirov SA, Harris KM. Dendrites are more spiny on mature hippocampal neurons when synapses are inactivated. Nat Neurosci. 1999;2:878–883. doi: 10.1038/13178. [DOI] [PubMed] [Google Scholar]
- 18.Kirov SA, Sorra KE, Harris KM. Slices have more synapses than perfusion-fixed hippocampus from both young and mature rats. J Neurosci. 1999;19:2876–2886. doi: 10.1523/JNEUROSCI.19-08-02876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi S, Millhorn DE. Regulation of N-methyl-d-aspartate receptor expression and N-methyl-d-aspartate-induced cellular response during chronic hypoxia in differentiated rat PC12 cells. Neuroscience. 2001;101:1153–1162. doi: 10.1016/s0306-4522(00)00435-8. [DOI] [PubMed] [Google Scholar]
- 20.Laake JH, Haug FM, Wieloch T, Ottersen OP. A simple in vitro model of ischemia based on hippocampal slice cultures and propidium iodide fluorescence. Brain Res Brain Res Protoc. 1999;4:173–184. doi: 10.1016/s1385-299x(99)00021-5. [DOI] [PubMed] [Google Scholar]
- 21.Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 22.Luscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- 23.Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- 24.Marrs GS, Green SH, Dailey ME. Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat Neurosci. 2001;4:1006–1013. doi: 10.1038/nn717. [DOI] [PubMed] [Google Scholar]
- 25.McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- 26.Obrenovitch TP, Urenjak J. Altered glutamatergic transmission in neurological disorders: from high extracellular glutamate to excessive synaptic efficacy. Prog Neurobiol. 1997;51:39–87. doi: 10.1016/s0301-0082(96)00049-4. [DOI] [PubMed] [Google Scholar]
- 27.Optiz T, Grooms SY, Bennett MV, Zukin RS. Remodeling of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit composition in hippocampal neurons after global ischemia. Proc Natl Acad Sci USA. 2001;97:13360–13365. doi: 10.1073/pnas.97.24.13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JS, Bateman MC, Goldberg MP. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis. 1996;3:215–227. doi: 10.1006/nbdi.1996.0022. [DOI] [PubMed] [Google Scholar]
- 29.Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- 30.Segal I, Korkotian I, Murphy DD. Dendritic spine formation and pruning: common cellular mechanisms? Trends Neurosci. 2000;23:53–57. doi: 10.1016/s0166-2236(99)01499-x. [DOI] [PubMed] [Google Scholar]
- 31.Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- 32.Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–1816. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 33.Smart FM, Halpain S. Regulation of dendritic spine stability. Hippocampus. 2000;10:542–554. doi: 10.1002/1098-1063(2000)10:5<542::AID-HIPO4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 34.Sorra KE, Harris KM. Occurrence and three-dimensional structure of multiple synapses between individual radiatum axons and their target pyramidal cells in hippocampal area CA1. J Neurosci. 1993;13:3736–3748. doi: 10.1523/JNEUROSCI.13-09-03736.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stoppini L, Buchs P-A, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 36.Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D. LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature. 1999;402:421–425. doi: 10.1038/46574. [DOI] [PubMed] [Google Scholar]
- 37.Toni N, Buchs PA, Nikonenko I, Povilaitite P, Parisi L, Muller D. Remodeling of synaptic membranes after induction of long-term potentiation. J Neurosci. 2001;21:6245–6251. doi: 10.1523/JNEUROSCI.21-16-06245.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Werth JL, Park TS, Silbergeld DL, Rothman SM. Excitotoxic swelling occurs in oxygen and glucose deprived human cortical slices. Brain Res. 1998;782:248–254. doi: 10.1016/s0006-8993(97)01286-9. [DOI] [PubMed] [Google Scholar]
- 39.Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/s0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]