

Abstract
Amyloid β-peptide (Aβ) fibril deposition on cerebral vessels produces cerebral amyloid angiopathy that appears in the majority of Alzheimer's disease patients. An early onset of a cerebral amyloid angiopathy variant called hereditary cerebral hemorrhage with amyloidosis of the Dutch type is caused by a point mutation in Aβ yielding AβGlu22→Gln. The present study addresses the effect of amyloid fibrils from both wild-type and mutated Aβ on vascular cells, as well as the putative protective role of antioxidants on amyloid angiopathy. For this purpose, we studied the cytotoxicity induced by Aβ1–40 Glu22→Gln and Aβ1–40 wild-type fibrils on human venule endothelial cells and rat aorta smooth muscle cells. We observed that AβGlu22→Gln fibrils are more toxic for vascular cells than the wild-type fibrils. We also evaluated the cytotoxicity of Aβ fibrils bound with acetylcholinesterase (AChE), a common component of amyloid deposits. Aβ1–40 wild-type–AChE fibrillar complexes, similar to neuronal cells, resulted in an increased toxicity on vascular cells. Previous reports showing that antioxidants are able to reduce the toxicity of Aβ fibrils on neuronal cells prompted us to test the effect of vitamin E, vitamin C, and 17β-estradiol on vascular damage induced by Aβwild-type and AβGlu22→Gln. Our data indicate that vitamin E attenuated significantly the Aβ-mediated cytotoxicity on vascular cells, although 17β-estradiol and vitamin C failed to inhibit the cytotoxicity induced by Aβ fibrils.
Keywords: Alzheimer's disease, CAA, HCHWA-D, amyloid, vitamin E, 17β-estradiol, vitamin C, oxidative stress, acetylcholinesterase, endothelial cells, vascular smooth muscle cells
Cerebral amyloid angiopathy (CAA) is linked to most cases of Alzheimer's disease (AD). CAA is characterized by the deposition of amyloid β-peptide (Aβ) mainly in the media and adventitia of both leptomeningeal and intracortical vessels (Vinters et al., 1988). An early onset of CAA occurs in the hereditary cerebral hemorrhage with amyloidosis of the Dutch type (HCHWA-D) (van Duinen et al., 1987). HCHWA-D is caused by a point mutation in the Aβ-encoding gene, which produces the substitution of Glu→Gln at position 22 (Levy et al., 1990), which renders Aβ more fibrillogenic (Wisniewski et al., 1991; Alvarez et al., 1997).
Vascular amyloid deposits of both CAA and HCHWA-D, similar to senile plaques from the brain parenchyma of AD patients, contain molecules other than Aβ (Snow et al., 1988; van Duinen et al., 1995; Verbeek et al., 1998). The enzyme acetylcholinesterase (AChE) has been reported in CAA deposits (Mesulam et al., 1992), which could be relevant for CAA pathology because AChE is able to induce Aβ aggregation into fibrils (Inestrosa et al., 1996; Alvarez et al., 1998). Moreover, those Aβ–AChE fibrillar complexes are more toxic for neuronal cells than Aβ fibrils alone (Alvarez et al., 1998; Muñoz and Inestrosa, 1999).
Amyloid-associated pathophysiology has been reported to involve oxidative stress (Behl, 1997; Miranda et al., 2000). The role of oxidative stress in AD is strengthened by in vitro findings showing that Aβ increases H2O2 in cells (Behl et al., 1994), whereas catalase, an enzyme that converts H2O2 to O2 and H2O, blocks Aβ toxicity (Behl et al., 1994), although there is controversy on the role of H2O2 in Aβ-mediated cell damage (Zhang et al., 1996). Moreover, neuroblastoma cells (Neuro 2a), which are resistant to Aβ, glutamate, and H2O2, contain high levels of the antioxidant glutathione (Calderón et al., 1999). In accordance with the oxidative pathophysiological hypothesis, vitamin E (vit E) and other antioxidants have demonstrated protective properties on neuronal cells against the Aβ-mediated neurotoxicity (Behl et al., 1992; Pappolla et al., 1997). The hormone 17β-Estradiol (E2), with well known antioxidant properties, also protects neuronal cells against Aβ-mediated neurotoxicity (Goodman et al., 1996; Behl et al., 1997a; Bonnefont et al., 1998). Moreover, vit E and E2 have been associated with the retardation of the onset and progression of AD (Tang et al., 1996;Kawas et al., 1997; Sano et al., 1997; Morris et al., 1998).
In the present work, we studied the vascular toxicity induced by different types of Aβ fibrils involved in amyloid angiopathy. First, we investigated the toxicity induced by Aβ1–40wild type (Aβwt) and AβGlu22→Gln on endothelial and vascular smooth muscle cells (VSMC). Second, the effect of the presence of AChE in the Aβ fibrillar complexes on these target cells was assessed. Finally, we evaluated the protective effect of vit E, vitamin C (vit C), and E2 on vascular cells challenged by Aβ fibrils.
MATERIALS AND METHODS
Materials. Synthetic Aβ peptides corresponding to the human Aβwt sequence and the Aβ1–40 Dutch variant that contains a glutamic acid to glutamine substitution (AβGlu22→Gln) were used in the present work. All of the peptides were obtained from Chiron (Emeryville, CA), Sigma (St. Louis, MO), and Calbiochem (Postfach, Germany). Chemicals, culture media, and sera were obtained from Sigma, Roche (Alameda, CA), Merck (Darmstadt, Germany), Invitrogen (Paisley, UK), and Molecular Probes (Leiden, The Netherlands).
Cell lines. Rat aorta smooth muscle cells (A7r5) were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). Human venule endothelial cells (HUVEC) were grown in M-199 medium supplemented with 10% FBS, 3.2 mm glutamine, and antibiotics.
AChE purification. The tetrameric G4AChE form (sedimentation coefficient of 10.7 S) was purified from bovine caudate nucleus, using acridine-affinity chromatography as described previously (Inestrosa et al., 1987). The specific activity (6000 U/mg protein) was determined by the method of Ellman et al. (1961). The staining intensity after SDS-PAGE (a single band of 66 kDa) was used to verify its purity.
Aggregation assay (turbidity). The aggregation assay was performed as described previously (Muñoz and Inestrosa, 1999). Briefly, peptide stock solutions were prepared by dissolving freeze-dried aliquots of AβGlu22→Gln and Aβwt in dimethylsulfoxide. Peptide stock aliquots were diluted in 0.1 m Tris-HCl, pH 7.4, to a final concentration of 96.6 μm Aβ. For the aggregation assays with AChE, peptide stock aliquots were added to 0.1 m Tris-HCl containing AChE (100 nm). The solutions were stirred continuously (210 rpm) at room temperature for 48 hr. Aggregation was measured by turbidity at 405 nm versus buffer blank.
Amyloid fibril isolation. Preformed fibrils were washed four times with PBS by centrifugation at 14,000 rpm for 30 min to remove the soluble Aβ and AChE. Pellets were homogenized in PBS. Aliquots of the homogenate were transferred to a denaturing buffer and subjected to Tris-tricine SDS-PAGE (Schagger et al., 1988) to quantify the concentrations of Aβ peptide contained in the fibrils. This was achieved by densitometric scanning using Aβ and AChE with known concentrations as standards. Data were processed by the GS365W program from Hoefer Scientific Instruments (San Francisco, CA).
Congo red staining. The Aβ fibrils and Aβ–AChE complexes were mounted on slides coated with 0.5% gelatin. Samples were dried overnight at room temperature and stained by using the alkaline Congo red (CR) method as described previously (Puchtler et al., 1961). Briefly, a saturated CR solution was prepared with 80% ethanol and 0.005% NaOH and NaCl saturated and was then filtered. Samples were incubated with CR solution for 20 min, dehydrated with increasing alcohol grades, cleared with xylene, and mounted in Canadian balsam. Slides were observed with a Nikon (Tokyo, Japan) Optiphot microscope configured for polarized light.
Electron microscopy. The amyloid fibrils were placed on Formvar carbon-coated 300 mesh nickel grids and negatively stained with 3% phosphotungstic acid solution for 1 min (Inestrosa et al., 1996). Grids were examined under a Philips EM-300 electron microscope at 60 kV.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide reduction and lactic dehydrogenase release assays. Cells were seeded in 96-well plates in serum- and phenol red-free medium with 2 μm insulin at a density of 4 × 103 cells/100 μl per well. PBS (10 μl) (control), Aβ fibrils, and AChE–Aβ complexes were added at different concentrations to wells. Experiments to study the effect of AChE alone on vascular cells were performed by adding the enzyme to cell cultures at a range of concentrations (0.1–25 nm). In the experiments with antioxidants, 1 mm vit E, 100 μm vit C, or E2 (0, 1, or 10 μm) was added 2 hr before the amyloid fibrils. The hydrophobic vit E and E2 were dissolved in ethanol to a final concentration of 5 nm ethanol per well. Cells were incubated for 48 hr at 37°C, after which cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction method (Mosmann, 1983) and lactic dehydrogenase (LDH) release as described previously (Muñoz and Inestrosa, 1999). Briefly, after the addition of 11 μl of MTT stock solution (5 mg/ml), the reaction was terminated 4 hr later with 110 μl of stop solution (50% dimethylformamide and 20% SDS at pH 4.7). MTT reduction was determined in a Labsystems (Espoo, Finland) Uniskam I spectrophotometer at 540 and 650 nm. LDH release was measured in the medium of cytotoxicity assays. Fifty microliters of culture supernatants were collected from each well, and LDH activities were determined with a colorimetric LDH assay kit (Promega, Madison, WI). Total cellular LDH activity was determined by lysing the cells with the kit lysis buffer.
Phosphatydylserine translocation and O2·− production measurements. HUVEC and A7r5 cells were seeded in 24-well plates in serum- and phenol red-free medium with 2 μminsulin at a density of 50 × 103cells/300 μl per well. Cells were preincubated with 1 mm vit E or 10 μmE2, after which PBS (control), Aβ (10 μm final concentration), or H2O2 (50 mm) were added. Cells were incubated for 4 or 48 hr at 37°C, and translocation of plasma membrane phosphatydylserine (a marker of apoptosis) was detected by annexin-V-Fluos binding according to the protocol of the manufacturer. O2·− production was measured by the intracellular oxidation of dihydroethydium (100 nm). Annexin-V binding and ethydium production were quantified on >4000 cells per experimental condition with a Becton-Dickinson (Franklin Lakes, NJ) FACScan cytometer. Debris was excluded on the basis of forward and side light-scattering properties. Experiments run in parallel also showed that cells analyzed presented intact membranes, demonstrated by the exclusion of propidium iodide. The putative effect of lot-to-lot variation of Aβ in aggregation and toxicity was discarded by using different lots purchased from different companies. In all cases, the Dutch variant was more toxic than the wild-type Aβ, because it has been demonstrated previously in PC12 cells, a well known cell target model for Aβ-mediated cytotoxicity (our unpublished observations).
Determination of superoxide dismutase activity. Superoxide dismutase (SOD) activity was evaluated by measuring the inhibition of cytochrome c reduction by SOD from homogenized vascular cells. Cytochrome c reduction was induced by O2·− (superoxide radical) generated by the xanthine–xanthine oxidase system (Fridovich, 1985). The reaction was initiated by adding xanthine oxidase (10 μl of a 0.7 U/ml solution) and 5–150 μl of SOD standard (44.8 U/ml) or 20–200 μl of homogenized vascular cell samples to 1 ml of reaction buffer (50 mm potassium phosphate at pH 7.8, 0.1 mm EDTA, 50 μm xanthine, and 10 μm cytochrome c). Reaction was followed at 550 nm for 2–6 min at 25°C with a Lambda 2 spectrophotometer (PerkinElmer Life Sciences, Ueberlingen, Germany). Results are expressed as units of SOD per milligram of protein. Proteins were quantified in homogenized vascular cells as described previously (Lowry et al., 1951).
Determination of nitric oxide synthase activity. Nitric oxide synthase (NOS) was evaluated by measuring the oxidation of oxyhemoglobin to methemoglobin by NO (Knowles et al., 1990). Homogenized vascular cell samples (100 μl) were preincubated for 5 min with the reaction buffer (40 mm potassium phosphate at pH 7.2, 1.6 μm oxyhemoglobin, and 1 mm MgCl2) at 37°C. The reaction was initiated by the addition of 1 mml-arginine and 1 mm NADPH. Oxidation of oxyhemoglobin was measured for 2 min at 37°C by the change in absorbance at 401 versus 411 nm registered with a Lambda 2 spectrophotometer. The results are expressed as nanomoles of NO per milligram of protein per minute. Proteins were quantified in homogenized vascular cells as described previously (Lowry et al., 1951).
Statistical analysis. Data were expressed as the mean ± SEM of the values from the number of experiments performed in triplicate as indicated in the corresponding figures. Data were evaluated statistically by using the Student's t test. For multiple comparisons, a one-way ANOVA test by the addition of the Kruskal–Wallis test was used. p < 0.05 was the minimum significance level.
RESULTS
Characterization of Aβ fibrils and Aβ–AChE complexes
In the present work, we used Aβ1–40instead of Aβ1–42 because both CAA and HCHWA-D vascular deposits are mostly composed of the Aβ1–40 type (Castaño et al., 1996;Hamano et al., 1997). To avoid anomalous results is essential to characterize the amyloid nature of the AβGlu22→Gln fibrils and AβGlu22→Gln–AChE complexes, because it was performed when AChE was described as a fibrillogenic agent for Aβwt1–40 (Inestrosa et al., 1996; Alvarez et al., 1998) and Aβwt1–42 (Muñoz and Inestrosa, 1999). The following set of criteria were applied: turbidometry, electron microscopy, and Congo red staining. Turbidometric assays showed an increase of the maximal amount of aggregates formed in the presence of AChE on the AβGlu22→Gln fibrillation (Fig.1). The morphology of AβGlu22→Gln fibrils was examined by electron microscopy, showing typical unbranched fibrils with no morphological differences regardless of the presence or absence of AChE (Fig.2), as has been reported previously for Aβwt and Aβwt–AChE complexes (Inestrosa et al., 1996). The amyloid quality of the fibrils was further assessed by staining with Congo red, which produced birefringent Maltese crosses in both types of fibrils observed under polarized light (Fig. 2, insets). The presence of AChE in the fibrillar complexes was demonstrated by the hydrolysis of the specific substrate acetylthiocholine (Fig.3A) and by SDS-PAGE in which AChE bands appeared in both types of complexes with Aβwt and AβGlu22→Gln(Fig. 3B). The amount of AChE bound to the fibrils differed depending on the type of peptides used, with Aβwt binding more AChE than AβGlu22→Gln (Fig. 3A,inset).
Fig. 1.

Aggregation assay of AβGlu22→Glnand AβGlu22→Gln with AChE followed by turbidometry at 405 nm. Data are from one representative experiment from the initial time at 0 up to 360 min.
Fig. 2.
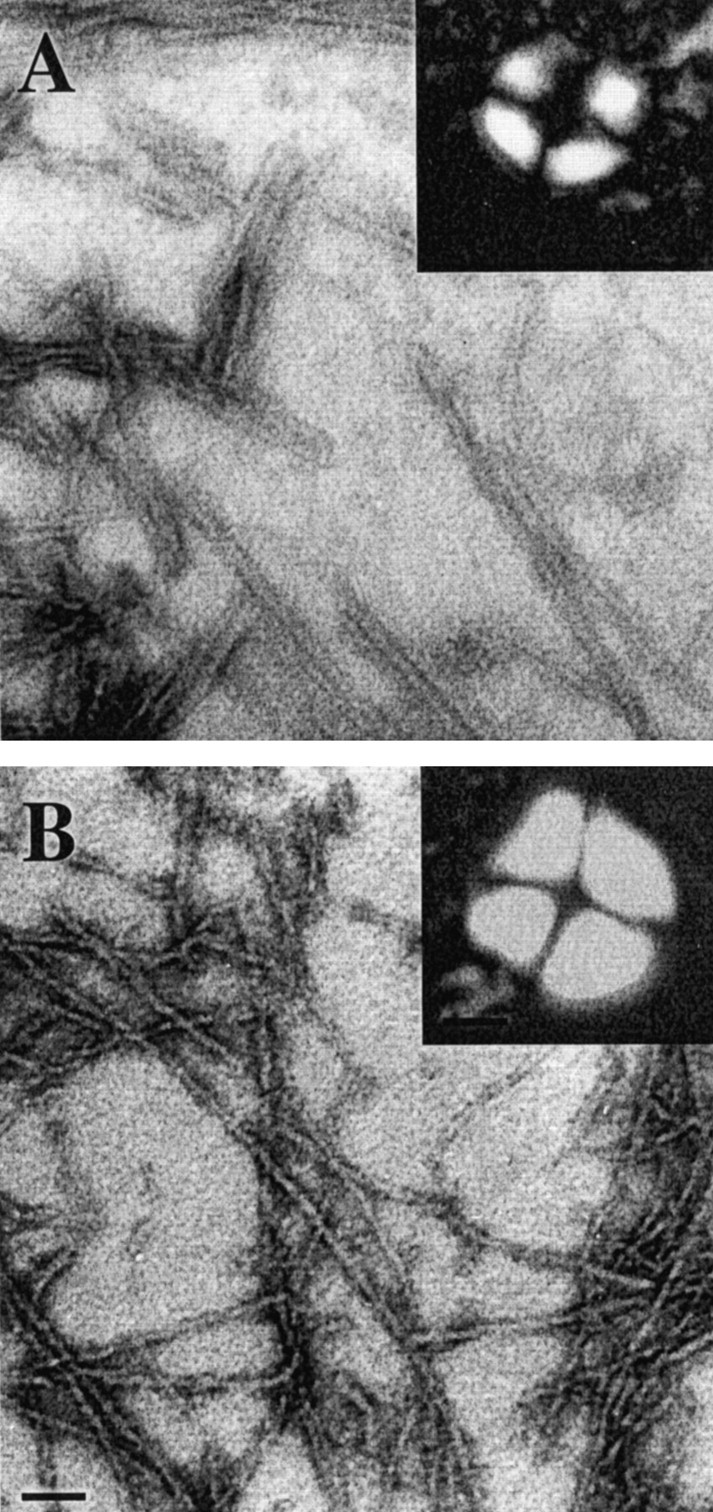
Electronic micrographs of negatively stained AβGlu22→Gln fibrils (A) and AβGlu22→Gln–AChE complexes (B). Scale bar, 0.15 μm. Insets are pictures of Maltese crosses obtained for both AβGlu22→Gln fibrils (A) and AβGlu22→Gln–AChE complexes (B) by staining with Congo red. Pictures were taken with a Nikon Optiphot microscope configured for polarized light. Scale bar, 15 μm.
Fig. 3.

Presence of AChE in AβGlu22→Gln–AChE complexes. It was demonstrated by the hydrolysis of the substrate acetylthiocholine (A) following the method of Ellman et al. (1961). Briefly, aliquots of 3 μl of AβGlu22→Gln fibrils and AβGlu22→Gln–AChE complexes were incubated in triplicate with acetylthiocoline, and the reaction was stopped with tacrine. Absorbances were determined at 412 nm. In theinset in A, the amount of AChE bound to 10 μg of both amyloid fibril types is shown. The AChE bound to the complexes was calculated by densitometric scanning of the SDS-PAGE bands of AChE compared with known concentrations of AChE. Data are mean ± SEM (error bars) values of five to nine separate experiments. *p < 0.05 by nonpaired Student'st tests versus the respective values of Aβwt–AChE complexes. B shows an SDS-PAGE with samples from both types of fibrils and complexes used in the present work. The different lanes were occupied as follows: lane 1, purified Aβwt fibrils;lane 2, Aβwt–AChE complexes; lane 3, AβGlu22→Gln fibrils; and lane 4, AβGlu22→Gln–AChE complexes.
Cytotoxicity of Aβ fibrils and Aβ–AChE complexes on vascular cells
We used endothelial cells and VSMC because they degenerate in both CAA and HCHWA-D (Wisniewski et al., 1992; Wisniewski and Wegiel, 1994;Kalaria, 1997; Zhang et al., 1998). Cytotoxicity on vascular cells was evaluated at 6, 48, and 120 hr in the presence of 10 μmAβGlu22→Gln fibrils (Fig.4A). A series of experiments was also performed using different concentrations of Aβ (Fig. 4B). The incubation time and fibril concentration for subsequent experiments were set at 48 hr and 10 μm Aβ fibrils, because there were no significant differences between 48 and 120 hr of incubation and between 10 and 25 μm Aβ fibrils. HUVEC cells (Fig.4C) showed a higher sensitivity to the toxic action of the Aβwt and AβGlu22→Glnfibrils than A7r5 cells (Fig. 4E), and the highest cytotoxicity was produced by the Dutch Aβ variant on both types of cells. The effect of Aβ–AChE complexes depended on the type of Aβ; thus, with Aβwt, the complexes showed higher toxicity than with Aβwt alone on both types of vascular cells, measured by MTT reduction (Fig.4C,E). No significant differences were obtained in A7r5 cells with AβGlu22→Gln fibrils and AβGlu22→Gln–AChE complexes (Fig.4E), whereas on HUVEC cells, AβGlu22→Gln–AChE complexes resulted less toxic than fibrils alone (Fig. 4C). Controls run with AChE alone at a range of concentrations, including those bound to the fibrils, did not induce a loss of vitality in either type of cell (Fig.4D).
Fig. 4.
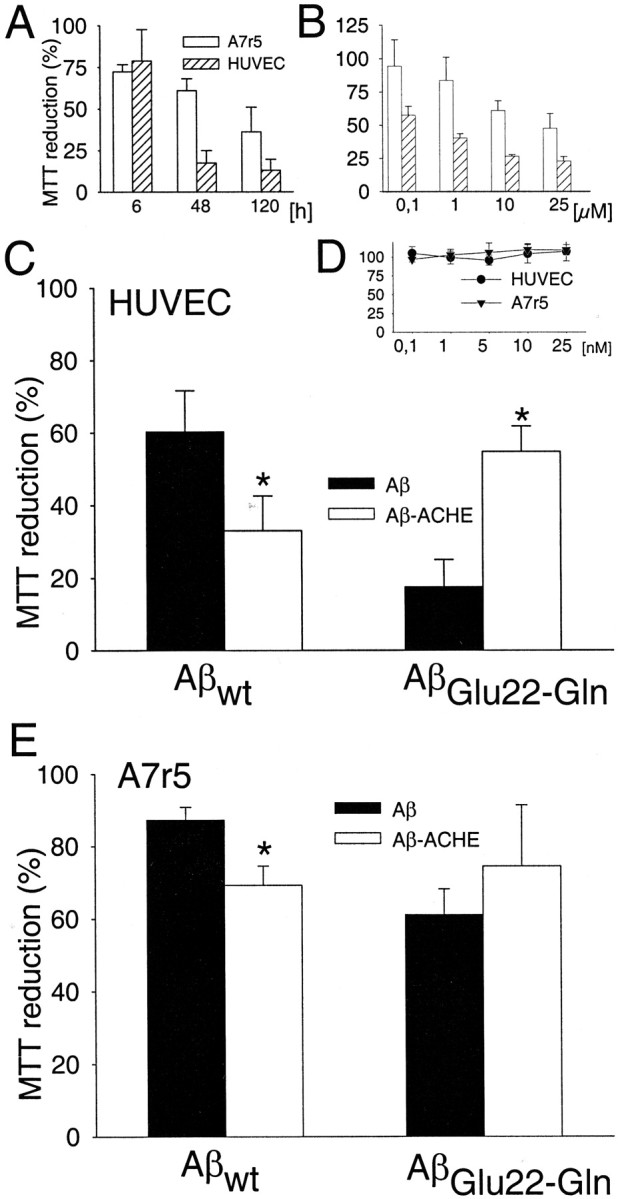
Cytotoxicity of Aβ fibrils and Aβ–AChE complexes on HUVEC (C) and A7r5 (E) cells are expressed as MTT reduction percentages obtained from the incubation with 10 μmfibrils alone (black bars) and complexes with AChE (white bars) for 48 hr. Data are mean ± SEM (error bars) values of five experiments performed in triplicate. *p < 0.05 by nonpaired Student's ttest versus the respective experiments performed with fibrils without AChE. A, MTT reduction percentages obtained with 10 μm AβGlu22→Gln fibrils at different incubation times. Data are mean ± SEM (error bars) values of three experiments performed in triplicate. B, MTT reduction percentages obtained after 48 hr of treatment with increasing concentrations of AβGlu22→Gln fibrils. Data are mean ± SEM (error bars) values of five to eight experiments performed in triplicate. D, MTT reduction percentages obtained by incubating A7r5 and HUVEC cells for 48 hr with increasing concentrations of AChE alone in the range that is bound to the fibrils. Data are mean ± SEM (error bars) values of three experiments performed in triplicate.
Aβ and oxidative stress
Oxidative stress has been proposed as one of the possible mechanisms of Aβ-mediated toxicity in both neuronal (Calderón et al., 1999) and endothelial (Thomas et al., 1996) cells. The different sensitivity of A7r5 and HUVEC cells to the amyloid peptides might indicate differences in the antioxidant cellular protection, a hypothesis also proposed for several neuronal cell lines (Calderón et al., 1999). To investigate whether Aβ-mediated toxicity involves the production of free radicals and subsequent apoptosis in vascular cells, we performed experiments to determine the O2·− production and the presence of phosphatydylserine in the outer leaflet of the plasma membrane, a specific marker for apoptosis, on HUVEC and A7r5 cells challenged with Aβ. The annexin-V (apoptosis marker) versus dihidroethydium (O2·− production) dot plots of Figure 5 show that Aβ incubation induced a marked increase in O2·−-positive HUVEC cells (Fig. 5A) but not in A7r5 cells (Fig.5B). However, both cell types showed a significant increase in the number of cells positive to annexin-V, demonstrating that the Aβ-induced cytotoxicity on vascular cells is mediated by apoptosis. The effect of H2O2, a well known prooxidant, was also evaluated on HUVEC cells (Fig.5C). The dot plots obtained with H2O2 were similar to those obtained with Aβ (Fig. 5A), reinforcing the hypothesis that oxidative stress is directly involved in Aβ-mediated vascular cytotoxicity.
Fig. 5.
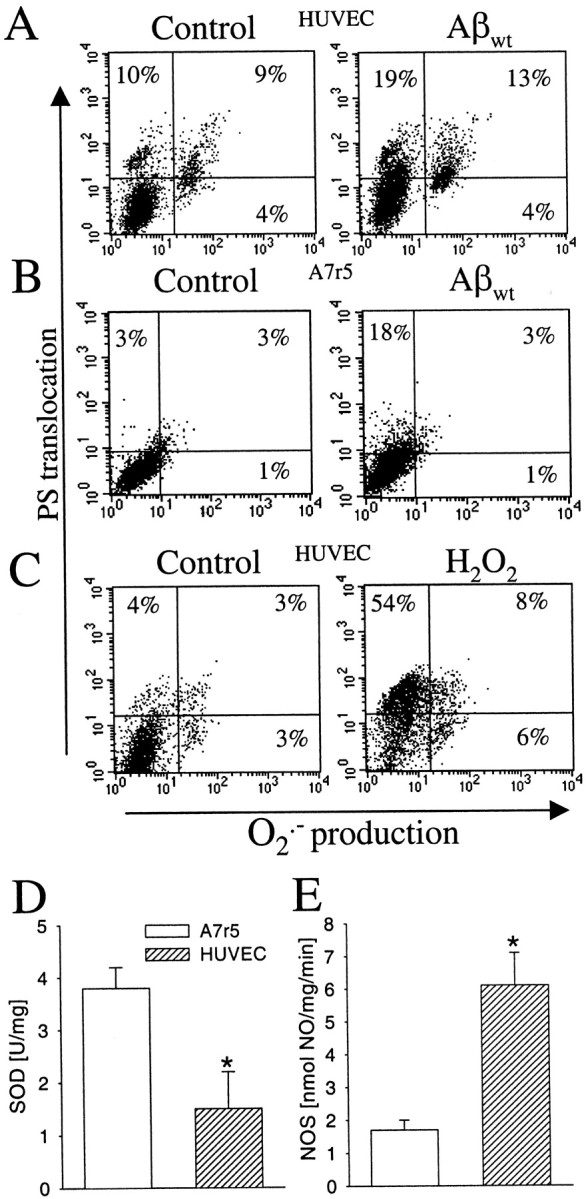
Oxidative stress and Aβ-mediated vascular cytotoxicity. Phosphatydylserine translocation to the outer membrane and O2·− production on HUVEC cells (A) and A7r5 cells (B) exposed to 10 μm Aβwt fibrils and HUVEC cells (C) with 50 μmH2O2 at 48 hr. Percentages indicate cells positive for annexin-V binding (top quadrants) and O2·− production (right quadrants) in regard to the controls (bottom left quadrant). Activity of SOD (D) and NOS (E) in A7r5 and HUVEC cells. Results are expressed in relation to milligrams of protein. Data are mean ± SEM (error bars) values of seven to eight cell samples analyzed in triplicate. *p < 0.05 by nonpaired Student'st test versus the respective results on A7r5 cells.
The different O2·− production between HUVEC and A7r5 cells in response to Aβ might be related to a differential balance of the prooxidant and antioxidant systems on each cell type. Therefore, we studied the SOD activity, the enzyme responsible to eliminate O2·− on vascular cells, and NOS activity. The interest in studying NOS activity lies on the observation that superoxide radicals can interact with NO to produce the highly prooxidant peroxynitrite radical (Beckman, 1996). HUVEC cells showed a lower SOD activity (twofold lower than that of A7r5 cells) (Fig. 5D). On the other hand, NO generation was fourfold higher in HUVEC cells than in A7r5 cells (Fig. 5E), as expected for endothelial cells (Moncada et al., 1988). Both results are consistent with a lower antioxidant protection of HUVEC cells compared with A7r5 cells.
The role of vitamin E, vitamin C, and E2 on Aβ-mediated vascular cytotoxicity
The protective role of different antioxidants on HUVEC cells challenged with Aβwt fibrils was studied by measuring apoptosis by the annexin-V method (Fig.6A). The percentage of annexin-V-positive cells doubled in the presence of Aβ. Preincubation of HUVEC cells with vit E reduced significantly the percentage of annexin-V-positive cells, whereas preincubation with E2 showed no statistically significant reduction of annexin-V staining. Similarly, vit E, but not E2, attenuated significantly the Aβ-mediated cytotoxicity for both wild type and Dutch variant on HUVEC cells measured by MTT reduction (Figs. 6B,7A). The lack of protection by E2 against Aβ-mediated cytotoxicity was also confirmed by measuring the LDH release on both types of cells (Fig.7B). LDH release in response to Aβ was similar in the presence or absence of E2. The other classic vitamin with antioxidant activity, vit C, at 100 μm did not show any protective effect on vascular cells in response to the insult with Aβ fibrils (Fig.6B). Higher concentrations of vit C were discarded because preliminary experiments at 1 mm produced a significant decrease in the viability of the vascular cells (data not shown). The protective effect of the three antioxidants against Aβ was also tested on A7r5 cells (Figs. 6B, 7). As described previously, these cells were more resistant to Aβ toxicity than HUVEC cells. Consequently, no major differences in the Aβ toxicity were identified in the presence of the antioxidants.
Fig. 6.
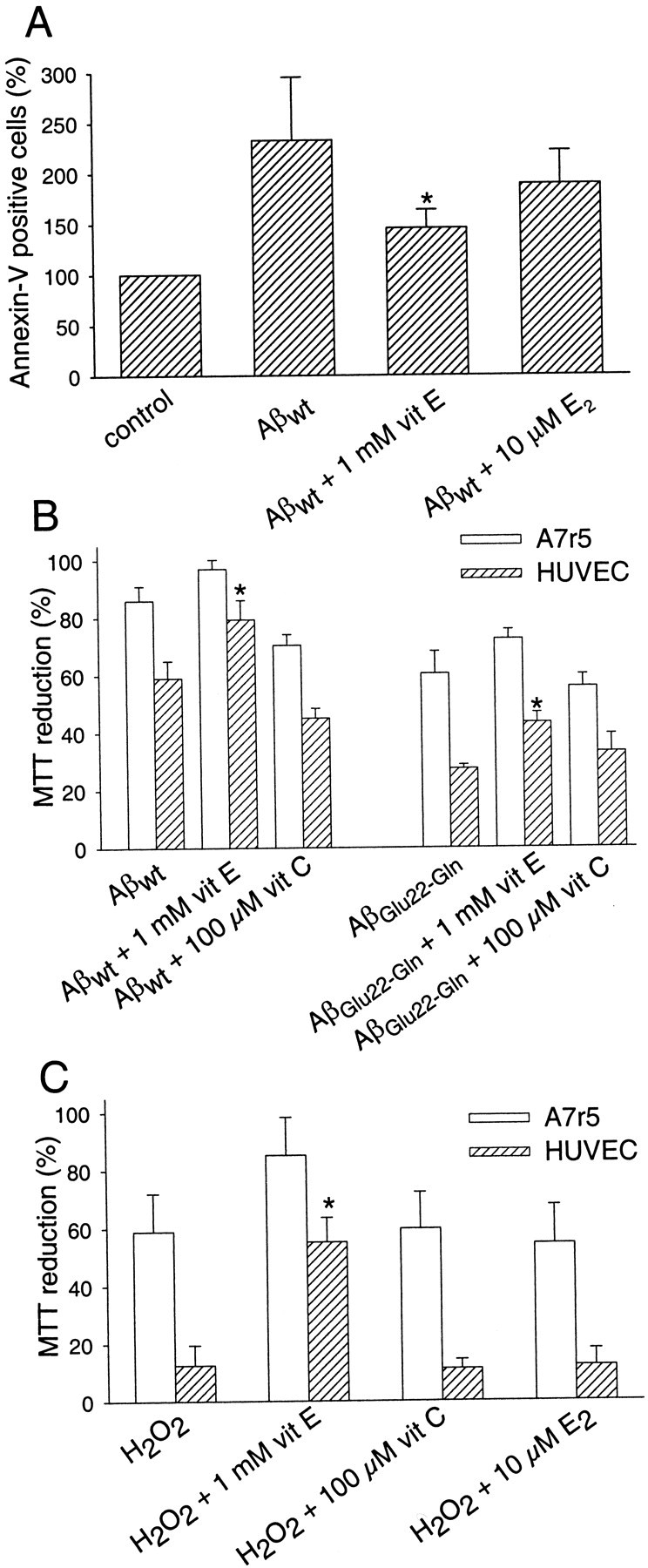
Aβ-mediated vascular cytotoxicity and protection by vit E. A, Annexin-V-positive HUVEC cells obtained after preincubating 1 mm vit E and 10 μmE2 for 2 hr before adding 10 μmAβwt. Cells were incubated with the fibrils for 48 hr. Cell controls (18 ± 4% annexin-V-positive cells) were assumed as 100%. Data are mean ± SEM (error bars) values of four experiments performed in duplicate. *p < 0.05 by a one-way ANOVA test by the addition of the Kruskal–Wallis test to compare the respective treatments versus fibrils alone.B, MTT reduction percentages obtained by preincubating HUVEC and A7r5 cells with 1 mm vit E and 100 μm vit C for 2 hr before adding 10 μmAβwt or AβGlu22→Gln fibrils. Cells were incubated with the fibrils for 48 hr. Data are mean ± SEM (error bars) values of four to six experiments performed in triplicate. *p < 0.05 by a one-way ANOVA test by the addition of the Kruskal–Wallis test to compare the respective treatments versus fibrils alone. C, H2O2 MTT reduction percentages obtained by preincubating vascular cells for 2 hr with 1 mm vit E, 100 μm vit C, and 10 μm E2. Cells were challenged for 48 hr with 100 μm (A7r5 cells) and 50 μm (HUVEC cells) H2O2. Data are mean ± SEM (error bars) values of three to five experiments performed in triplicate. *p < 0.05 by a one-way ANOVA test by the addition of the Kruskal–Wallis test to compare the respective treatments versus H2O2 alone.
Fig. 7.
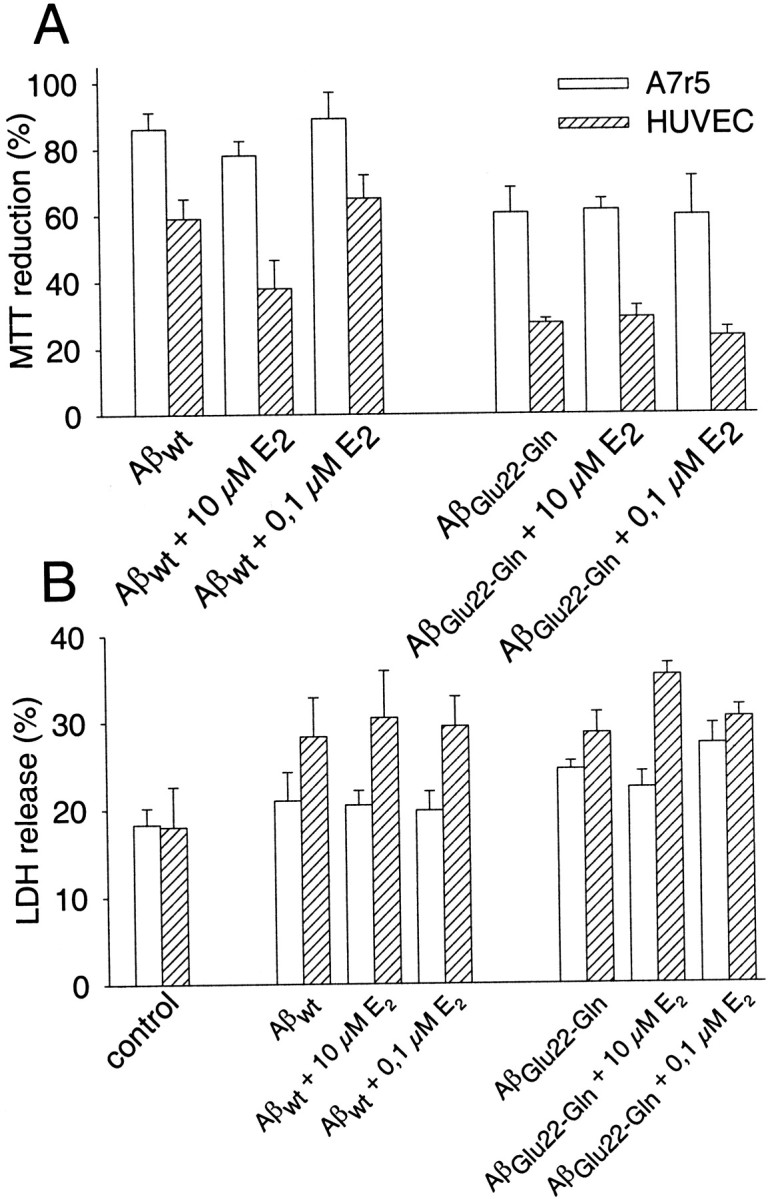
Effect of E2 on the cytotoxicity induced by Aβwt and AβGlu22→Gln fibrils on vascular cells. A, MTT reduction percentages obtained by preincubating HUVEC and A7r5 cells with 0.1 and 10 μmE2 for 2 hr before adding 10 μmAβwt or AβGlu22→Gln fibrils. Cells were incubated with fibrils for 48 hr. Data are mean ± SEM (error bars) values of 7–14 experiments performed in triplicate.B, LDH release obtained by preincubating HUVEC and A7r5 cells with 0.1 and 10 μm E2 for 2 hr before adding 10 μm Aβwt or AβGlu22→Gln fibrils. Cells were incubated with fibrils for 48 hr. Results are expressed as percentages with respect to the total LDH assumed as 100%. Data are mean ± SEM (error bars) values of three to five experiments performed in triplicate.
A similar pattern of protection with the three antioxidants was obtained when both cell types were challenged with H2O2 instead of Aβ (Fig.6C). First, HUVEC cells were more sensitive to the oxidative stress than A7r5 cells (Fig. 6C), even when the concentration of H2O2 used for the experiments performed on HUVEC cells (50 μm) was lower than that used for the experiments performed on A7r5 cells (100 μm). Second, as shown with the Aβ challenge, only vit E was able to attenuate the H2O2-mediated oxidative insult by increasing the cell viability on both types of cells.
DISCUSSION
CAA is a pathology frequently linked to Alzheimer's disease (Vinters et al., 1988). The amyloid vascular deposits from CAA are similar to the brain senile plaques, including the presence of several molecules such as AChE (Mesulam et al., 1992), which has been reported to increase the neurotoxicity of Aβwt fibrils (Muñoz and Inestrosa, 1999). In addition, CAA is the key pathological feature of patients suffering from HCHWA-D, although in this case, the angiopathy is more extensive and develops at early ages, without the presence of mature senile plaques in the cerebral parenchyma (van Duinen et al., 1987; Maat-Schieman et al., 1992). The pathophysiology underlying the Aβ-mediated vascular damage is not fully understood, but oxidative stress could play a key role as in the case of the Aβ-mediated neuronal degeneration (Behl, 1997; Miranda et al., 2000).
In the present study, we determined the cytotoxic effect of Aβwt and AβGlu22→Glnfibrils on vascular cells and the modulation of the cytotoxicity by AChE. Furthermore, we evaluated whether well known antioxidants such as vit E, vit C, and E2 might reverse the vascular toxicity conferred by Aβ fibrils.
Amyloid fibrils and cytotoxicity
Our results show that Aβwt and AβGlu22→Gln fibrils induce toxicity on vascular cells with characteristics similar to that reported on neuronal cells (Bonnefont et al., 1998). AβGlu22→Gln fibrils were more toxic than those of Aβwt on vascular cells. These findings suggest a key role for fibril stability on the cytotoxicity, because previous studies have shown that the Gln mutation stabilizes the fibrils and hence increases the fibrillogenic nature of this Aβ type (Wisniewski et al., 1991; Fraser et al., 1992), despite the absence of any major folding difference between the wild-type and the mutated peptide (George and Howlett, 1999). Therefore, a plausible explanation could be related to an increased attachment of AβGlu22→Gln to the extracellular matrix of vascular cells, which has demonstrated the ability to bind and assemble amyloid, hence enhancing its toxicity (Watson et al., 1997; van Nostrand et al., 1998).
Aβ–AChE complexes
We found that the AChE–Aβ complexes formed with Aβwt are significantly more toxic than Aβwt fibrils alone for both target cells, consistent with previous reports on neuronal cells (Alvarez et al., 1998; Muñoz and Inestrosa, 1999). These results might be related to the enhancement of the Aβwtfibril stability by AChE (Inestrosa et al., 1996). Those findings suggest that the AChE bound to the amyloid vascular deposits could act as an enhancer of the vascular degeneration observed in CAA, because AChE has been reported to be present in amyloid vascular deposits (Mesulam et al., 1992).
On the other hand, AβGlu22→Gln–AChE complexes showed no differences on VSMC compared with the AβGlu22→Gln fibrils alone, and endothelial cells showed a significant increase in cell survival. The lack of morphological differences observed for either kind of fibril by electron microscopy analysis suggests that the differences could be in the fibril stability, AβGlu22→Gln–AChE complexes being less stable than AβGlu22→Glnfibrils. This might be the result of an anomalous pattern in the association between AβGlu22→Gln and AChE compared with other amyloid peptides (Inestrosa et al., 1996; Alvarez et al., 1997; Muñoz et al., 1999). Our data also showed a lower binding of AChE to AβGlu22→Gln, an observation that it might be related to the fast fibrillation of AβGlu22→Gln, resulting in fewer molecules of AChE being bound or to a decrease in the binding affinity attributable to the changes in the Aβ sequence. The pathophysiological relevance of AβGlu22→Gln–AChE complexes is unknown at present because there is no evidence for the presence of AChE in HCHWA-D vascular deposits.
Free radicals and Aβ-mediated cytotoxicity
Several studies have reported a release of O2·− in response to Aβ in vascular cells (Thomas et al., 1996; Suo et al., 1997), linking the toxicity of Aβ to oxidative stress, as was observed on HUVEC cells. We found that endothelial cells were more sensitive to H2O2 and Aβ fibrils than VSMC, an observation that correlates with the higher rate of O2·− production on HUVEC cells. Furthermore, endothelial cells showed low levels of SOD activity, which could render these cells more vulnerable to oxidative insults and, therefore, to the action of Aβ. This observation is in agreement with previous studies reporting that exogenous addition of SOD reverses the effect of Aβ in endothelial cells (Crawford et al., 1997; Thomas et al., 1997; Price et al., 1997). NO production was also higher in endothelial cells than in VSMC. NO can react with the prooxidant O2·−to form peroxynitrite (Beckman, 1996), which is considered a powerful oxidant in AD-associated brain damage (Smith et al., 1997). All of these results point to a correlation between oxidative stress and the cytotoxicity ratio. We hypothesize that endothelium is more sensitive to Aβ-mediated apoptosis attributable to the lower intracellular antioxidant activity and the high production of O2·− and NO.
Protection with antioxidants
Both vit E and vit C have antioxidant properties. In the case of the former, it seems to be associated with the prevention of lipid peroxidation (Halliwell and Gutteridge, 1984) by trapping the peroxyl radicals (Naiki et al., 1998). In the case of vit C, its involvement as a protective antioxidant agent is controversial. On one hand, its physiological role appears to be mainly directed to restore the antioxidant properties of vit E (Tappel, 1968), whereas at high concentrations it acts as a prooxidant (Halliwell, 1999).
As discussed above, the oxidative stress hypothesis is well suited to explain the toxic effect of β-amyloid in both neuronal cells (Miranda et al., 2000) and vascular cells (this study). Additional support for the oxidative stress hypothesis was provided by several studies showing the neuroprotective effects of different antioxidants, such as vit E (Behl et al., 1992), vit C (Yallampalli et al., 1998), E2 (Goodman et al., 1996), melatonin (Pappolla et al., 1997), and lazaroids (Behl et al., 1997b), against β-amyloid-induced toxicity. Biological effects found for many of these antioxidants in laboratory experiments have also been matched by epidemiological studies reporting the beneficial effects of antioxidants (Tang et al., 1996; Sano et al., 1997).
Our data show that vit E protects endothelial cells against Aβ challenge, whereas vit C failed to inhibit the Aβ- and H2O2-mediated cytotoxicity. In the case of vit C, reports showing a lack of neuroprotection have also appeared (Lockhart et al., 1994).
The protective effect of E2 against Aβ has been shown previously on neuronal cells (Goodman et al., 1996; Behl et al., 1997a; Bonnefont et al., 1998). However, E2 was unable to reverse the toxicity of Aβ or H2O2 in vascular cells (this study), despite its antioxidant properties (Sugioka et al., 1987). These findings do not rule out that E2, via one or more of its different mechanisms of action (Nadal et al., 2001), plays a protective role in AD. E2 exerts a wide range of positive effects on both the CNS (Honjo et al., 1992; Bonnefont et al., 1998; Inestrosa et al., 1998; Toran-Allerand, 2000) and the vascular system (Ruehlmann et al., 1998; Mendelsohn and Karas 1999; Valverde et al., 1999). All or part of these effects of E2 may be related to the protective role on neuronal cells challenged with Aβ (Goodman et al., 1996; Behl et al., 1997a; Bonnefont et al., 1998). However, they appear to be ineffective at the vascular level for amyloid-associated pathology.
In summary, AβGlu22→Gln fibrils are more toxic for vascular cells than Aβwt wild-type fibrils, suggesting that the early onset of HCWA-D is related to the high toxicity rate induced by AβGlu22→Glnfibrils. Additional work is needed to verify that AβGlu22→Gln toxicity is related to the stability of the fibrils. The presence of AChE in the Aβwt fibrils could also be a risk factor for CAA attributable to the enhancer effect of the amyloid toxicity by the enzyme. Finally, we postulate that one of the mechanisms of action of Aβ fibrils is the generation of oxidative stress on vascular cells and that the high sensitivity of endothelial cells to Aβ fibrils is related to their lower protection against oxidative stress (low levels of SOD and high levels of NOS activity), a hypothesis that receives additional support from the observation that the antioxidant vit E reverses the Aβ-mediated cytotoxicity.
Footnotes
This work was supported by grants from Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) (Ministerio de Educación, Chile Grant 3980024) and Fondo de Investigación Sanitaria (Ministerio de Sanidad, Spain Grant 01-1029) to F.J.M.; Dirección General de Investigación Científica y Técnica (Ministerio de Ciencia y Tecnología, Spain Grant BIO99-0508) to G.G.-G.; Human Frontier Science Program and Distinció de la Generalitat de Cataluña (Spain) to M.A.V.; FONDECYT Grant 2990087 to C.O.; and Fondo de Investigación Avanzada en Áreas Prioritarias-Biomedicina Grant 1389001, Millenium Institute for Fundamental and Applied Biology Grant P-99-007-F, and the Presidential Chair in Science from the Chilean Government to N.C.I. We acknowledge Aoife Currid for proofreading this manuscript.
Correspondence should be addressed to Dr. Francisco J. Muñoz, Unitat de Senyalització Cel·lular, Universitat Pompeu Fabra, Calle Dr. Aiguader, 80, Barcelona 08003, Spain. E-mail:paco.munoz@cexs.upf.es.
REFERENCES
- 1.Alvarez A, Opazo C, Alarcón R, Garrido J, Inestrosa NC. Acetylcholinesterase promotes the aggregation of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J Mol Biol. 1997;272:348–361. doi: 10.1006/jmbi.1997.1245. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez A, Alarcón R, Opazo C, Campos EO, Muñoz FJ, Calderón FH, Dajas F, Gentry MK, Doctor BP, De Mello FG, Inestrosa NC. Stable complexes involving acetylcholinesterase and amyloid-beta peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer's fibrils. J Neurosci. 1998;18:3213–3223. doi: 10.1523/JNEUROSCI.18-09-03213.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 4.Behl C. Amyloid beta-protein toxicity and oxidative stress in Alzheimer's disease. Cell Tissue Res. 1997;290:471–480. doi: 10.1007/s004410050955. [DOI] [PubMed] [Google Scholar]
- 5.Behl C, Davis JB, Cole GM, Schubert D. Vitamin E protects nerve cells from amyloid β protein toxicity. Biochem Biophys Res Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 6.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 7.Behl C, Skutella T, Lezoualc'h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997a;51:535–541. [PubMed] [Google Scholar]
- 8.Behl C, Trapp T, Skutella T, Holsboer F. Protection against oxidative stress-induced neuronal cell death-a novel role for RU486. Eur J Neurosci. 1997b;9:912–920. doi: 10.1111/j.1460-9568.1997.tb01442.x. [DOI] [PubMed] [Google Scholar]
- 9.Bonnefont AB, Muñoz FJ, Inestrosa NC. Estrogen protects neuronal cells from the cytotoxicity induced by acetylcholinesterase-amyloid complexes. FEBS Lett. 1998;441:220–224. doi: 10.1016/s0014-5793(98)01552-x. [DOI] [PubMed] [Google Scholar]
- 10.Calderón FH, Bonnefont A, Muñoz FJ, Fernández V, Videla LA, Inestrosa NC. PC12 and Neuro 2a cells have different susceptibilities to acetylcholinesterase-amyloid complexes amyloid25–35 fragment glutamate hydrogen peroxide. J Neurosci Res. 1999;56:620–631. doi: 10.1002/(SICI)1097-4547(19990615)56:6<620::AID-JNR8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 11.Castaño EM, Prelli F, Soto C, Beavis R, Matsubara E, Shoji M, Frangione B. The length of amyloid-beta in hereditary cerebral hemorrhage with amyloidosis Dutch type. Implications for the role of amyloid-beta 1–42 in Alzheimer's disease. J Biol Chem. 1996;271:32185–32191. doi: 10.1074/jbc.271.50.32185. [DOI] [PubMed] [Google Scholar]
- 12.Crawford F, Suo Z, Fang C, Sawar A, Su G, Arendash G, Mullan M. The vasoactivity of A beta peptides. Ann NY Acad Sci. 1997;826:35–46. doi: 10.1111/j.1749-6632.1997.tb48459.x. [DOI] [PubMed] [Google Scholar]
- 13.Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 14.Fraser PE, Nguyen JT, Inouye H, Surewicz WK, Selkoe DJ, Podlisny MB, Kirschner DA. Fibril formation by primate rodent Dutch-haemorrhagic analogues of Alzheimer amyloid beta-protein. Biochemistry. 1992;31:10716–10723. doi: 10.1021/bi00159a011. [DOI] [PubMed] [Google Scholar]
- 15.Fridovich I. CRC Handbook of methods for oxygen radical research (Greenwald RA, ed), pp 213–220. CRC; Boca Raton, FL: 1985. [Google Scholar]
- 16.George AR, Howlett DR. Computationally derived structural models of the beta-amyloid found in Alzheimer's disease plaques and the interaction with possible aggregation inhibitors. Biopolymers. 1999;50:733–741. doi: 10.1002/(SICI)1097-0282(199912)50:7<733::AID-BIP6>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity oxidative injury amyloid β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 18.Halliwell B. Vitamin C: poison, prophylactic or panacea? Trends Biochem Sci. 1999;24:255–259. doi: 10.1016/s0968-0004(99)01418-8. [DOI] [PubMed] [Google Scholar]
- 19.Halliwell B, Gutteridge JM. Lipid peroxidation, oxygen radicals, cell damage, and antioxidant therapy. Lancet. 1984;1:1396–1397. doi: 10.1016/s0140-6736(84)91886-5. [DOI] [PubMed] [Google Scholar]
- 20.Hamano T, Yoshimura M, Yamazaki T, Shinkai Y, Yanagisawa K, Kuriyama M, Ihara Y. Amyloid β-protein (Aβ) accumulation in the leptomeninges during aging and in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:922–932. doi: 10.1097/00005072-199708000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Honjo H, Tamura T, Matsumoto Y, Kawata M, Ogino Y, Tanaka K, Yamamoto T, Ueda S, Okada H. Estrogen as a growth factor to central nervous cells. Estrogen treatment promotes development of acetylcholinesterase-positive basal forebrain neurons transplanted in the anterior eye chamber. J Steroid Biochem Mol Biol. 1992;41:633–635. doi: 10.1016/0960-0760(92)90395-y. [DOI] [PubMed] [Google Scholar]
- 22.Inestrosa NC, Roberts WL, Marshall TL, Rosenberry TL. Acetylcholinesterase from bovine caudate nucleus is attached to membranes by a novel subunit distinct from those of acetylcholinesterases in other tissues. J Biol Chem. 1987;262:4441–4444. [PubMed] [Google Scholar]
- 23.Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, Linker C, Casanueva O, Soto C, Garrido J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 24.Inestrosa NC, Marzolo MP, Bonnefont AB. Cellular and molecular basis of estrogen's neuroprotection. Potential relevance for Alzheimer's disease. Mol Neurobiol. 1998;17:73–86. doi: 10.1007/BF02802025. [DOI] [PubMed] [Google Scholar]
- 25.Kalaria RN. Cerebrovascular degeneration is related to amyloid-beta protein deposition in Alzheimer's disease. Ann NY Acad Sci. 1997;826:263–271. doi: 10.1111/j.1749-6632.1997.tb48478.x. [DOI] [PubMed] [Google Scholar]
- 26.Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology. 1997;48:517–521. doi: 10.1212/wnl.48.6.1517. [DOI] [PubMed] [Google Scholar]
- 27.Knowles RG, Merrett M, Salter M, Moncada S. Differential induction of brain lung and liver nitric oxide synthase by endotoxin in the rat. Biochem J. 1990;270:833–836. doi: 10.1042/bj2700833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 29.Lockhart BP, Benicourt C, Junien JL, Privat A. Inhibitors of free radical formation fail to attenuate direct beta-amyloid25–35 peptide-mediated neurotoxicity in rat hippocampal cultures. J Neurosci Res. 1994;39:494–505. doi: 10.1002/jnr.490390416. [DOI] [PubMed] [Google Scholar]
- 30.Lowry OH, Rosebrough NJ, Farr L, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:267–275. [PubMed] [Google Scholar]
- 31.Maat-Schieman ML, van Duinen SG, Haan J, Roos RA. Morphology of cerebral plaque-like lesions in hereditary cerebral hemorrhage with amyloidosis (Dutch). Acta Neuropathol (Berl) 1992;84:674–679. doi: 10.1007/BF00227745. [DOI] [PubMed] [Google Scholar]
- 32.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 33.Mesulam M-M, Carson K, Price B, Geula C. Cholinesterases in the amyloid angiopathy of Alzheimer's disease. Ann Neurol. 1992;31:565–569. doi: 10.1002/ana.410310517. [DOI] [PubMed] [Google Scholar]
- 34.Miranda S, Opazo C, Larrondo LF, Muñoz FJ, Ruiz F, Leighton F, Inestrosa NC. The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer's disease. Prog Neurobiol. 2000;62:633–648. doi: 10.1016/s0301-0082(00)00015-0. [DOI] [PubMed] [Google Scholar]
- 35.Moncada S, Radomski MW, Palmer RM. Endothelium-derived relaxing factor. Identification as nitric oxide and role in the control of vascular tone and platelet function. Biochem Pharmacol. 1988;37:2495–2501. doi: 10.1016/0006-2952(88)90236-5. [DOI] [PubMed] [Google Scholar]
- 36.Morris MC, Beckett LA, Scherr PA, Hebert LE, Bennett DA, Field TS, Evans DA. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12:121–126. doi: 10.1097/00002093-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 38.Muñoz FJ, Inestrosa NC. Neurotoxicity of acetylcholinesterase-amyloid-β-peptide aggregates is dependent on the type of Aβ-peptide and the AChE concentration present in the complexes. FEBS Lett. 1999;450:205–209. doi: 10.1016/s0014-5793(99)00468-8. [DOI] [PubMed] [Google Scholar]
- 39.Nadal A, Díaz M, Valverde MA. The estrogen trinity: membrane, cytosolic and nuclear effects. News Physiol Sci. 2001;16:251–255. doi: 10.1152/physiologyonline.2001.16.6.251. [DOI] [PubMed] [Google Scholar]
- 40.Naiki H, Hasegawa K, Yamaguchi I, Nakamura H, Gejyo F, Nakakuki K. Apolipoprotein E and antioxidants have different mechanism of inhibiting Alzheimer's β-amyloid fibril formation in vitro. Biochemistry. 1998;37:17882–17889. doi: 10.1021/bi980550y. [DOI] [PubMed] [Google Scholar]
- 41.Pappolla MA, Sos M, Omar RA, Bick RJ, Hickson-Bick DL, Reiter RJ, Efthimiopoulos S, Robakis NK. Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J Neurosci. 1997;17:1683–1690. doi: 10.1523/JNEUROSCI.17-05-01683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price JM, Sutton ET, Hellermann A, Thomas T. Beta-amyloid induces cerebrovascular endothelial dysfunction in the rat brain. Neurol Res. 1997;19:534–538. doi: 10.1080/01616412.1997.11740853. [DOI] [PubMed] [Google Scholar]
- 43.Puchtler H, Sweat F, Levine M. On the binding of Congo red by amyloid. J Histochem Cytochem. 1961;10:355–364. [Google Scholar]
- 44.Ruehlmann DO, Steinert JR, Valverde MA, Jacob R, Mann GE. Environmental estrogenic pollutants induce acute vascular relaxation by inhibiting L-type Ca2+ channels in smooth muscle cells. FASEB J. 1998;12:613–619. doi: 10.1096/fasebj.12.7.613. [DOI] [PubMed] [Google Scholar]
- 45.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 46.Schagger H, Aquila H, Von Jagow G. Coomassie blue-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for direct visualization of polypeptides during electrophoresis. Anal Biochem. 1988;173:201–205. doi: 10.1016/0003-2697(88)90179-0. [DOI] [PubMed] [Google Scholar]
- 47.Smith MA, Richey PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Snow AD, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S, Hassell J, Wight TN. The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer's disease. Am J Pathol. 1988;133:456–463. [PMC free article] [PubMed] [Google Scholar]
- 49.Sugioka K, Shimosegawa Y, Nakano M. Estrogens as natural antioxidants of membrane phospholipid peroxidation. FEBS Lett. 1987;210:37–39. doi: 10.1016/0014-5793(87)81293-0. [DOI] [PubMed] [Google Scholar]
- 50.Suo Z, Fang C, Crawford F, Mullan M. Superoxide free radical and intracellular calcium mediate A beta(1–42) induced endothelial toxicity. Brain Res. 1997;762:144–152. doi: 10.1016/s0006-8993(97)00383-1. [DOI] [PubMed] [Google Scholar]
- 51.Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996;348:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- 52.Tappel AL. Will antioxidant nutrients slow aging processes? Geriatrics. 1968;23:97–105. [PubMed] [Google Scholar]
- 53.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. Beta-amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- 54.Thomas T, McLendon C, Sutton ET, Thomas G. Beta-amyloid-induced cerebrovascular endothelial dysfunction. Ann NY Acad Sci. 1997;826:447–451. doi: 10.1111/j.1749-6632.1997.tb48501.x. [DOI] [PubMed] [Google Scholar]
- 55.Toran-Allerand CD. Novel sites and mechanisms of oestrogen action in the brain. Novartis Found Symp. 2000;230:56–69. doi: 10.1002/0470870818.ch6. [DOI] [PubMed] [Google Scholar]
- 56.Valverde MA, Rojas P, Amigo J, Cosmelli D, Orio P, Bahamonde MI, Mann GE, Vergara C, Latorre R. Acute activation of Maxi-K channels (hSlo) by estradiol binding to the beta subunit. Science. 1999;285:1929–1931. doi: 10.1126/science.285.5435.1929. [DOI] [PubMed] [Google Scholar]
- 57.van Duinen SG, Castaño EM, Prelli F, Bots GT, Luyendijk W, Frangione B. Hereditary cerebral hemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer disease. Proc Natl Acad Sci USA. 1987;84:5991–5994. doi: 10.1073/pnas.84.16.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Duinen SG, Maat-Schieman ML, Bruijn JA, Haan J, Roos RA. Cortical tissue of patients with hereditary cerebral haemorrhage with amyloidosis (Dutch) contains various extracellular matrix deposits. Lab Invest. 1995;73:183–189. [PubMed] [Google Scholar]
- 59.van Nostrand WE, Melchor JP, Ruffini L. Pathologic amyloid beta-protein cell surface fibril assembly on cultured human cerebrovascular smooth muscle cells. J Neurochem. 1998;70:216–223. doi: 10.1046/j.1471-4159.1998.70010216.x. [DOI] [PubMed] [Google Scholar]
- 60.Verbeek MM, Otte-Holler I, Veerhuis R, Ruiter DJ, De Waal RM. Distribution of A beta-associated proteins in cerebrovascular amyloid of Alzheimer's disease. Acta Neuropathol (Berl) 1998;96:628–636. doi: 10.1007/s004010050944. [DOI] [PubMed] [Google Scholar]
- 61.Vinters HV, Pardridge WM, Secor DL, Ishii N. Immunohistochemical study of cerebral amyloid angiopathy. Am J Pathol. 1988;133:150–162. [PMC free article] [PubMed] [Google Scholar]
- 62.Watson DJ, Lander AD, Selkoe DJ. Heparin-binding properties of the amyloidogenic peptides Abeta and amylin Dependence on aggregation state and inhibition by Congo red. J Biol Chem. 1997;272:31617–31624. doi: 10.1074/jbc.272.50.31617. [DOI] [PubMed] [Google Scholar]
- 63.Wisniewski HM, Wegiel J. Beta-amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol (Berl) 1994;87:233–241. doi: 10.1007/BF00296738. [DOI] [PubMed] [Google Scholar]
- 64.Wisniewski T, Ghiso J, Frangione B. Peptides homologous to the amyloid protein of Alzheimer's disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem Biophys Res Commun. 1991;179:1247–1254. doi: 10.1016/0006-291x(91)91706-i. [DOI] [PubMed] [Google Scholar]
- 65.Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer's disease. Acta Neuropathol (Berl) 1992;84:117–127. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- 66.Yallampalli S, Micci MA, Taglialatela G. Ascorbic acid prevents beta-amyloid-induced intracellular calcium increase and cell death in PC12 cells. Neurosci Lett. 1998;251:105–108. doi: 10.1016/s0304-3940(98)00515-1. [DOI] [PubMed] [Google Scholar]
- 67.Zhang WW, Lempessi H, Olsson Y. Amyloid angiopathy of the human brain: immunohistochemical studies using markers for components of extracellular matrix smooth muscle actin and endothelial cells. Acta Neuropathol (Berl) 1998;96:558–563. doi: 10.1007/s004010050935. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Z, Rydel RE, Drzewiecki GJ, Fuson K, Wright S, Wogulis M, Audia JE, May PC, Hyslop PA. Amyloid beta-mediated oxidative and metabolic stress in rat cortical neurons: no direct evidence for a role for H2O2 generation. J Neurochem. 1996;67:1595–1606. doi: 10.1046/j.1471-4159.1996.67041595.x. [DOI] [PubMed] [Google Scholar]