

Abstract
The α6 subunit of the nicotinic acetylcholine receptor (nAChR) is expressed at very high levels in dopaminergic (DA) neurons. However, because of the lack of pharmacological tools selective for α6-containing nAChRs, the role of this subunit in the etiology of nicotine addiction remains unknown. To provide new tools to investigate this issue, we generated an α6 nAChR knock-out mouse. Homozygous null mutants (α6−/−) did not exhibit any gross neurological or behavioral deficits. A careful anatomic and molecular examination of α6−/− mouse brains demonstrated the absence of developmental alterations in these animals, especially in the visual and dopaminergic pathways, where the α6 subunit is normally expressed at the highest levels. On the other hand, receptor autoradiography revealed a decrease in [3H]nicotine, [3H]epibatidine, and [3H]cytisine high-affinity binding in the terminal fields of retinal ganglion cells of α6−/− animals, whereas high-affinity [125I]α-conotoxinMII (αCtxMII) binding completely disappeared in the brain. Moreover, inhibition of [3H]epibatidine binding on striatal membranes, using unlabeled αCtxMII or cytisine, revealed the absence of αCtxMII-sensitive and cytisine-resistant [3H]epibatidine binding sites in α6−/− mice, although the total amount of binding was unchanged. Because αCtxMII, a toxin formerly thought to be specific for α3β2-containing nAChRs, is known to partially inhibit nicotine-induced dopamine release, these results support the conclusion that α6 rather than α3 is the partner of β2 in the nicotinic modulation of DA neurons. They further show that α6−/− mice might be useful tools to understand the mechanisms of nicotine addiction, although some developmental compensation might occur in these mice.
Keywords: nicotinic acetylcholine receptor, knock-out, α6 subunit, α-conotoxinMII, dopaminergic system, visual system
As many drugs of addiction, nicotine is thought to exert its addictive properties through stimulation of dopamine release in the nucleus accumbens (Nac) (Pontieri et al., 1996;Di Chiara, 2000). This effect of nicotine has been attributed to a direct activation of nAChRs located on dopaminergic (DA) neurons of the ventral tegmental area (VTA). Indeed, nicotine is known to increase the firing rate of these neurons, both in vitro (Pidoplichko et al., 1997) and in vivo (Grenhoff et al., 1986). Moreover, nicotinic antagonists infused into the VTA prevent nicotine elicited dopamine release in the Nac and disrupt systemic nicotine self-administration in rats (Corrigall et al., 1994; Nisell et al., 1994). Therefore, nAChRs expressed by DA neurons represent valuable targets for the design of new pharmacological agents for the treatment of nicotine abuse, and much research has been devoted to the identification of their subunit composition.
Neuronal nAChRs are composed of five subunits belonging to a family of at least 12 genes exhibiting a discreet expression pattern (Sargent, 1993). DA neurons express α3, α4, α5, α6, α7, β2, and β3 subunits (Le Novère et al., 1996; Charpantier et al., 1998; Klink et al., 2001). Furthermore, it was recently demonstrated that mice lacking the β2 subunit are no longer sensitive to nicotine-mediated dopamine release in the Nac and fail to self-administer nicotine (Picciotto et al., 1998). The same study demonstrated that β2-containing nAChRs, present on the soma of DA neurons, mediate a nicotine-induced increase of their firing rate. Among the other subunits expressed in VTA, only α3, α4, and α6 are likely to associate with the β2 subunit to form a ligand binding site [β3 and α5 are structural subunits, whereas α7 forms homomeric pentamers (Le Novère and Changeux, 1995)]. Although pharmacological evidence suggests α4 and α3 subunits as likely partners of the β2 subunit (Kulak et al., 1997; Kaiser et al., 1998;Sharples et al., 2000), the existence of functional α6-containing nAChRs on DA neurons should not be overlooked. Indeed, the α6 subunit is specifically and highly expressed in catecholaminergic neurons and retina (Le Novère et al., 1996; Vailati et al., 1999). In addition, α6 was recently implicated in the stimulating effect of nicotine on habituated locomotion (Le Novère et al., 1999), an effect known to be blocked by lesions of midbrain DA neurons (Boye et al., 2001). However, the absence of pharmacological information about α6-containing nAChRs has been a deterrent to determining the exact contribution of these receptors to the nicotinic modulation of DA neurons.
To investigate this issue, we generated a strain of mice lacking the α6 nAChR subunit gene. We describe the generation of these animals and their anatomical characterization. We first focus on the detection of possible developmental abnormalities in dopaminergic and visual pathways, because such alterations might secondarily affect behavior and have already been detected in β2 nAChR knock-out mice (Rossi et al., 2001). Then, we perform ligand binding experiments to investigate the pharmacology and distribution of native α6-containing nAChRs and evaluate the representation of these receptors in the dopaminergic system.
MATERIALS AND METHODS
All animals were used in accordance with the Centre National de la Recherche Scientifique guidelines for care and use of laboratory animals.
Construction of the targeting vector
We used a PCR-based strategy (Israel, 1993) to screen a mouse genomic DNA library prepared from the 129Sv/d3 embryonic stem (ES) cells strain (Stratagene, La Jolla, CA), using primers derived from the α6 cDNA rat sequence. An 18 kb clone was isolated, and homologous flanking regions were cloned in KS bluescript (Stratagene), on either side of a Neomycin resistance gene. Ultimately, a diphtheria toxin expression cassette was introduced outside of the recombination arms. The construct was designed to generate a nonfunctioning allele by deleting a 4 kb EcoRI–HindIII fragment including exons 1 and 2 of the α6 gene. The 5′ flanking arm was a 5 kbEcoRI–EcoRI fragment, and the 3′ flanking arm was a 2.4 kb HindIII-SalI fragment (Fig.1).
Fig. 1.

Gene targeting of the neuronal nAChR α6 subunit. a, Construction of the targeting vector. The homologous recombination event generated a 4 kb genomic deletion that removes exons 1 and 2. The targeting vector was designed to obtain a replacement mutation and contains a neomycin resistance gene (Neor) as a positive selection marker and the diphtheria toxin gene (DTA) as a negative selection marker. The expected wild-type and mutant restriction fragments after PstI digest and hybridization with the Southern blot probe are shown as dotted lines. Restriction enzymes are as follows: E,EcoRI; H, HindIII;P, PstI; S,SalI. b, Southern blot analysis of wild-type (Wt), heterozygous (α6+/−), and homozygous null mutant mice (α6−/−). Genomic tail DNA was digested withPstI and hybridized with anSalI–PstI probe, external to the targeting vector. The 4 kb band corresponds to the mutated allele.c, Detection of α6 mRNA in the brain of Wt and α6−/− animals, by in situ hybridization with an oligonucleotide located in the sixth exon of the α6 gene. In Wt animals, α6 mRNA is detected in SN–VTA neurons, LC and RGC. No signal above background levels is detected in α6−/− animals.
Generating the chimeras
The targeting vector was linearized by ScaI and electroporated in ES cells derived from the 129Sv/Pas mouse strain (Kress et al., 1998). After 8 d of geneticin selection (Invitrogen, Gaithersburg, MD), 200 resistant clones were screened by genomic Southern blot for homologous recombination. Three recombinant ES cells clones were obtained, one of which was used to generate mouse chimeras by aggregation (Khillan and Bao, 1997) with morulas obtained from CD1 mice (Charles River, Wilmington, MA). One single germline transmitter was obtained and bred with CD1 mice.
Maintaining the colony
One F1 heterozygous male and one F1 heterozygous female were then mated to establish a set of F2 breeder animals (5 males and 10 females) of each genotype (mutant or wild-type). All the animals used in this study were derived either from the F2 or the F3 generation. Genetic diversity was maintained by randomly interchanging breeders.
Genomic Southern blot
Genomic Southern blot was performed according to standard methods (Sambrook et al., 1989). A PstI digest of mouse tail-derived genomic DNA probed with a SalI–PstI fragment (Fig. 1) was used to check the recombination at the 3′ end (normal allele fragment is 8.1 kb and recombinant is 4.0 kb). Another probe (EcoRI–EcoRV fragment of the α6 promoter), internal to the 5′ recombination arm, was used to check for correct recombination on the 5′ end, using an XbaI digest of genomic DNA. (Normal allele fragment is 9.9 kb and recombinant is 12.2 kb). This also excluded the eventuality of an additional random integration (data not shown).
In situ hybridization
Animals were killed by cervical dislocation, and the brain was quickly dissected out and frozen in dry ice powder. The tissues were stored at −80°C until cut on a cryostat (Leica, Deerfield, IL) (14-μm-thick sections). In situ hybridization was performed according to Zoli et al. (1995), using synthesized oligonucleotides (Eurogentec, Brussels, Belgium) according to rat or mouse sequence, when available (Table 1). The slides were then exposed for 15 d to Hyperfilm-3H (Amersham Biosciences, Arlington Heights, IL) together with microscale radioactive standards (Amersham Biosciences) to correct for nonlinearity of the film response. The semiquantitative analysis was performed on autoradiography films. On each section, an averaged optical density of the background level (obtained by cold competition with unlabeled oligonucleotide) was subtracted from that of the sampled area to obtain specific optical density value.
Table 1.
Oligodeoxynucleotide probes used for in situhybridization
| Target | Species | Probe position | Mismatch | Sequence of the oligonucleotide | Tm (°C) |
|---|---|---|---|---|---|
| α3 | Rat | 138–182 | ? | 5′-GGACACCTCAAACTGGATGATGACTGGATGGGACACATTAGCCAC-3′ | 65 |
| α4 | Mouse | 570–614 | 0 | 5′-GAAGTCCAGTTGGTCCACACGGCTGTGCATGCTCACCAAGTCAAT-3′ | 71 |
| α5 | Rat | 1077–1121 | 5 | 5′-CCGAGATTTAGGTCCAgCccCacTCTCGGCTTCTTCTCTCTGAGT-3′ | 73 |
| α6 | Mouse | 1451–1497 | 0 | 5′-GGGACAATACCAATTAAGAGTTTCCTGTGTTCCCAAGCAGTGGCTGC-3′ | 66 |
| α7 | Rat | 1206–1250 | 2 | 5′-ATCATGTGTTGGGGAGCAGGCCAAACGgCCACAtACGACCCCAGA-3′ | 71 |
| β2 | Mouse | 1269–1313 | 0 | 5′-TCGCATATGGTCCGCAATGAAGCGTACACCGTCCACAGCTTCCCG-3′ | 71 |
| β4 | Rat | 1261–1305 | ? | 5′-AGCTGACACCCTCTAATGCTTCCTGTAGATCTTCCCGGAACCTCC-3′ | 71 |
| TH | Rat | 1429–1468 | 2 | 5′-AGGTGTGCAGCTCATCCTGGACCCCCTCcAAGGAGCGCt-3′ | 79 |
| D1 | Mouse | 592–638 | 0 | 5′-GCTGACGATCATAATGGCTACGGGGATGTAAAAGCTTATGAGGGAGG-3′ | 63 |
| D2 | Mouse | 928–972 | 0 | 5′-GTTTGGCAGGACTGTCAGGGTTGCTATGTAGACCGTGGTGGGATG-3′ | 70 |
| CCK | Rat | 264–308 | 3 | 5′-gAAATCCATCCAGCCCATGTAGTCCCGGTCACTTATcCTgTGGCT-3′ | 74 |
| PPE | Rat | 386–430 | 3 | 5′-GCATCCTTCTTCATGAAagCGCCATACCTCTTGGCAAGGATCTCg-3′ | 67 |
| PPT | Rat | 123–167 | 1 | 5′-GGCGATTCTCTGaAGAAGATGCTCAAAGGGCTCCGGCATTGCCTC-3′ | 68 |
The melting temperature (Tm) is calculated for the most stringent wash condition (0.01 m NaCl, 0% formamide) as described byLe Novère et al. (1996). The indicated position is in reference to the A of the ATG. When the oligonucleotide was designed according to the rat cDNA sequence, and when corresponding mouse cDNA sequence was available, we indicated the number and position (lowercase characters) of the mismatches. (“?” means that the mouse cDNA sequence was not available).
For nAChR subunits (α3, α4, α5, α6, α7, β2, β4), the experiment was performed using 3–6 animals of each genotype. Sections were taken at 17 coronal levels (bregma levels: 3.0, 1.5, 0.9, 0.1, −0.6, −1.0, −1.8, −2.2, −2.7, −2.9, −3.1, −3.5, −3.8, −4.0, −4.8, −5.4, and −7.5 mm according to Franklin and Paxinos (1997).
For dopamine receptors (D1, D2), tyrosine hydroxylase (TH), and neuropeptides [cholecystokinin (CCK), preprotachykinin (PPT), and preproenkephalin (PPE)], the experiment was performed using seven animals of each genotype. Coronal sections were cut from bregma level 1.0–0.7 mm (striatal level) and from bregma level −3.1 to −3.5 mm (mesencephalic level). For each level, three adjacent sections per animal were used for quantitative analysis.
Spontaneous locomotor activity
The open-field apparatus consists of a transparent 31-cm-high × 49-cm-long × 25.4-cm-wide Plexiglas cage. Movements were detected by a set of 9 × 19 infrared photobeam detectors, 1.5 cm above the floor of the apparatus. Tests were performed in a quiet room with ambient fluorescent lighting. For this experiment we used 15 male α6−/− mice and 15 wild-type (Wt) littermates controls derived from F2 heterozygous mating. Mice naive to the apparatus were placed in the cage at time point t = 0, and their movements were recorded for a 2 hr period, with time points taken at intervals of 5 min.
Intraocular injections
Mice were anesthetized with a solution of 1.5% ketamine and 0.05% xylazine in PBS (150 μl/30 gm body weight). Intraocular injections were performed using a glass needle inserted just behind the corneoscleral margin of the eye. One microliter of a 10 μg/μl solution of cholera toxin subunit B, Alexa Fluor 488 conjugate (Molecular Probes, Eugene, OR) was injected into the vitreous chamber of one eye and Alexa Fluor 594 into the other. After 24 hr, mice were anesthetized and perfused with 0.9% saline, followed by 4% paraformaldehyde in PBS. Cryoprotected brains were cut on a freezing microtome into 40 μm coronal sections. Sections were mounted on gelatin-coated slides, air-dried, coverslipped, and then examined under an epifluorescent microscope.
Receptor autoradiography
Nicotinic receptor ligands. For [3H]cytisine (5 nm), [3H]nicotine (5 nm), [3H]epibatidine (400 pm), [125I]α-bungarotoxin (1.5 nm), and [125I]α-conotoxinMII (αCtxMII) (500 pm), binding procedures were performed as described in Zoli et al. (1998) and Whiteaker et al. (2000). For each ligand, 17 coronal levels (see in situ hybridization procedures), and one or two sections per level were examined, from three to five animals of each genotype. Nonspecific binding was determined by using a 1000× excess of unlabeled ligand.
Cytisine displacement of [3H]epibatidine binding was performed by adding 50 nm of cold cytisine to the incubation mix, according to Marks et al. (1998).
Except for [125I]-αCtxMII, which was a generous gift from Dr. J. Michael McIntosh (Departments of Biology and Psychiatry, University of Utah, Salt Lake City, UT), all radiolabeled ligands were purchased from Amersham Biosciences. Unlabeled cytisine, nicotine, epibatidine, and α-bungarotoxin were purchased from Sigma (St. Louis, MO); αCtxMII was obtained from Eurogentec.
Dopamine receptor ligands. Levels of D1 and D2 dopamine receptors were determined by equilibrium binding of [3H]SCH23390 (NEN, Boston, MA) and [3H]raclopride (NEN), respectively. Sections were preincubated for 30 min at room temperature in 50 mm Tris 7.4, 120 mm NaCl, 5 mm KCl, 2 mmCaCl2, 2 mmMgCl2 (SCH23390), or 50 mmTris 7.4, 120 mm NaCl, and 5 mm KCl (raclopride). Slides were then incubated for 1 hr at room temperature in preincubation buffer plus 1.5 nm (SCH23390) or 3 nm(raclopride) of tritiated ligand. Nonspecific binding was determined in the presence of an excess (100×) of cold ligand (Research Biochemicals, Natick, MA). After incubation, the sections were washed three times for 5 min each in ice-cold 50 mm Tris and briefly rinsed in ice-cold water. Slides were exposed for 20 d (D1) or 40 d (D2) to Kodak (Eastman Kodak, Rochester, NY) BioMax MS films (Amersham Biosciences). Eight animals of each genotype were used for quantification. Measures were made on three adjacent sections at mesencephalic and striatal levels (bregma −3.4 and 0.9 mm, respectively).
Histological analysis
Immunocytochemistry. Mice were anesthetized with 4% chloral hydrate (12 μl/gm) and transcardially perfused with 20 ml of 0.9% NaCl (37°C) followed by 100 ml of a 4% paraformaldehyde–PBS solution. Brains were dissected out and post-fixed overnight in the same fixative, rinsed in 12% sucrose–PBS at 4°C over 2 d, and frozen on dry ice powder. The brains were cut from bregma level 1.0–0.7 mm and from bregma level −3.1 to −3.5 mm on a cryostat (20-μm-thick sections). Sections were then processed according to standard immunoperoxidase protocols, using the avidin–biotin complex technique (Pedrazzi et al., 1998). Antibodies against rat tyrosine hydroxylase (TH) (Calbiochem, La Jolla, CA), and dopamine transporter (DAT) (MAB369; Chemicon, Temecula, CA) were used at a 1:2000 dilution for TH and 1:1000 for DAT. Briefly, slides were incubated overnight at 4°C, in the presence of the primary antibody diluted in PBS supplemented with 0.2% Triton X-100 and 1% normal goat serum (NGS). After washing with PBS containing 0.1% NGS, sections were incubated for 1 hr at room temperature, with a biotinylated anti-rabbit (TH) or anti-rat (DAT) secondary antibody (Vector Laboratories, Burlingame, CA) diluted 1:200 in PBS/1% NGS. Sections were washed in PBS and incubated with an avidin biotinylated peroxidase complex (ABC) according to manufacturer's instructions (Vector Laboratories). After peroxidase detection using diaminobenzidine, sections were washed, dehydrated, and coverslipped.
Nissl staining. Rehydrated, fixed (4% PFA) brain sections were incubated with toluidine blue (0.1%), washed, dehydrated, and coverslipped.
Analysis. The semiquantitative evaluation of the intensity of TH and DAT immunostainings as well as automatic cell profile count of Nissl-stained preparations was performed by means of an automatic image analyzer (KS300; Zeiss Kontron, Munich, Germany) according to previously published methods (Zoli et al., 1992). Microdensitometric analysis of immunostaining was performed with a CCD camera connected to a microscope using a 2.5× objective. In mesencephalon, a thresholding procedure was used to determine the integrated optical density value. TH-immunoreactive cell counting in substantia nigra pars compacta (SNc) was performed manually on coded slides under a 20× objective using a calibrated eyepiece grid, at −3.5 mm bregma level. For each staining, the analysis was performed on two sections per animal. The average value per animal was used for statistical purposes.
For Nissl-stained preparations, automatic cell profile count was performed according to Zoli et al. (1993) with a CCD camera connected to a microscope using a 20× objective (rectangular sampled field, 310 × 235 μm). Briefly, stained profiles were separated from the background by means of a dynamic thresholding procedure. Then, a spatial threshold was placed to separate glial (diameter < 7.5 μm) from neuronal (diameter ≥ 7.5 μm) profiles, and the number of neuronal profiles per sampled field was measured.
Membrane binding. Membranes from striatum (dorsal and ventral) and superior colliculus (SC) were prepared according to Marks et al. (1998). Incubations were performed in 6 ml glass tubes, using 50 (Superior colliculus) or 200 μg (striatum) of membrane protein per tube. Membranes were diluted in 500 μl of binding buffer (NaCl 144 mm; KCl 1.5 mm; CaCl2 2 mm; MgSO4 1 mm; HEPES 20 mm, pH 7.5), containing [3H]epibatidine (500 pm) and the appropriate concentration of unlabeled ligand (300 pm to 10 μm for αCtxMII, and 30 pm to 3 μm for cytisine). For αCtxMII displacement, binding buffer was supplemented with BSA (0.1% w/v), 5 mm EDTA, 2 mm EGTA, and 10 μg/ml each of aprotinin, pepstatin A, and leupeptin (Sigma), to protect the ligand from endogenous proteases. After 2 hr at room temperature, incubations were terminated by filtration on polyethilenimine-soaked (0.3%, Sigma) glass fiber filters (type GF/C; Whatman) and washing with 10 ml of ice-cold binding buffer. Nonspecific binding was determined by adding an excess (1000×) of unlabeled epibatidine to the incubation mix. All measures were made in triplicate. The epibatidine concentration and the volume used for the incubation ensured minimal ligand depletion (<5%). Results were first fitted to the Hill equation [B = Bo/(1 + (I/IC50)n) ] to determine the likeliness of a monophasic inhibition (B is the number of binding sites at inhibitor concentration, I,Bo is the binding in the absence of inhibitor, and n is the Hill coefficient). Depending onn value, results were fitted using one of the following equations for one or two binding sites: B =Bo/(1+ (I/IC50)) or B =I B1/(1 + (I/IC50–1)) +B2/(1+ (I/IC50–2)), respectively. IC50 and Bovalues are expressed as the means ± SEM of six independent determinations. The corresponding Kivalue was determined assuming competitive inhibition:Ki = IC50/(1 +L/Kd), where Lis the concentration of [3H]epibatidine (500 pm), and Kd is the equilibrium dissociation constant of [3H]epibatidine in striatum (19.3 μm, according to Marks et al. (1998).
Statistical analysis. Statistical analysis was done with StatView software (SAS Institute). Data are given as means ± SEM; means were compared using a Student's t test.
RESULTS
Generation of α6 knock-out mice
Knock-out mice for the α6 subunit of the nAChRs were generated by replacing the first two exons of the gene with a Neomycin resistance (Neor) cassette (Fig. 1a). These exons encode the ATG initiator codon, the signal peptide, and part of the N-terminal extracellular domain of the mature subunit. Southern blot analysis using a flanking genomic probe confirmed that the expected homologous-recombination had occurred (Fig. 1b). In situ hybridization using an oligonucleotide probe located in the sixth exon (downstream to the deletion) detected α6 mRNA in retinal ganglion cells (RGCs), locus coeruleus (LC), substantia nigra (SN) and VTA neurons of Wt animals, in agreement with previous observations in rat or chicken (Le Novère et al., 1996; Vailati et al., 1999). In heterozygous mice, the intensity of the signal decreased and completely disappeared in α6−/− mice, demonstrating that the deletion of exon 1 and 2, and the insertion of the Neorcassette, is sufficient to prevent transcription (Fig. 1c). We then compared the levels of expression of α3, α4, α5, α7, β2, and β4 nAChR subunit mRNA in Wt and α6−/− animals. Quantitative analysis of film autoradiography revealed no alteration in the relative abundance of the mRNA of these subunits to compensate for the loss of the α6 subunit (Fig. 2). [Note that in agreement with Ross et al. (2000), α3 mRNA was not detected in SN–VTA neurons.]
Fig. 2.

Semiquantitative analysis of α3, α4, α5, α7, β2, and β4 nAChR subunit mRNAs in Wt and Mt mice usingin situ hybridization. For each subunit, optical density value of the signal was measured and corrected for background and film linearity. This value was then normalized to 100 in the region where the signal was maximal. Results are expressed as means ± SEM of at least three animals. Statistical analysis was performed using an unpaired Student's t test (p < 0.05). Regions quantified were medial habenula (MHb), interpeduncular nucleus (IPN), thalamus (Th), cortex (Cx), VTA, and hippocampus (Hp). Note the absence of significant differences in the relative abundance of the mRNA of these subunits, in any brain region.
α6 knock-out mice are viable and do not exhibit major defects
Homozygous null mutant mice are viable, present in the expected Mendelian proportion in litters, grow to normal size, and do not exhibit major physical or neurological defects. Home cage behavior of mutant mice appeared normal, and spontaneous locomotor activity in an open-field observed over a 2 hr period did not reveal significant differences between Wt and α6−/− animals at any time point, either during the nonhabituated (first hour) or habituated period (second hour) of the experiment (data not shown). Furthermore, histological examination of toluidine blue-stained sections of α6−/− mice did not reveal alterations in the size, shape, or location of the major brain nuclei (data not shown).
Absence of developmental adaptations in the dopaminergic and visual pathways of α6−/− animals
One major drawback of knock-out animal studies is the possible existence of developmental abnormalities in mutant mice that might interfere with the interpretation of the results obtained from these studies. For instance, in mice lacking the β2 nAChR subunit, impairment of visual acuity derives from abnormal segregation of retinogeniculate projections during development (Rossi et al., 2001). To examine whether such alterations exist in α6−/− animals, we performed a detailed examination of the anatomical regions where α6 is known to be expressed.
In the visual system, histological staining of the retina showed that the cell layering and the RGC number is normal in α6−/− mice compared with their Wt controls (data not shown). Moreover, we studied the distribution pattern of RGC projections in their thalamic target, the dorsolateral geniculate nucleus (dLGN), in young (postnatal day 15) and adult animals. In Wt mice, retinal projections from the two eyes terminate into separated areas of the dLGN: an outer shell, that receives afferents exclusively from the contralateral eye and an inner core that is innervated by the ipsilateral eye. In α6−/− mice the pattern of retinogeniculate labeling was not different from the one observed in Wt at the same age (data not shown).
The anatomy of the mesostriatal dopaminergic system was studied at both mesencephalic and striatal levels.
At the striatal level, the semiquantitative analysis of TH and DAT immunoreactivity failed to detect any alteration in DA nerve terminals of α6−/− mice in the anatomical regions examined [caudate–putamen (Cpu), Nac, and olfactory tubercle (Tu)]. Intrinsic striatal neurons also appeared normal in terms of number and morphology, as assessed by Nissl staining. Using in situ hybridization and receptor autoradiography, we examined the expression profile of several markers in these neurons: dopamine receptors, D1 and D2, and neuropeptide precursors, PPT and PPE. In Wt mice, D1 and D2 mRNA levels were found to be homogeneously expressed in Cpu, and Tu, and to a lesser extent in the Nac. The study of [3H]raclopride (D2) and [3H]SCH23390 (D1) binding revealed a similar pattern of expression at the protein level. PPT mRNA, coding for the precursor peptide of substance P, was strongly expressed in Tu and Nac shell and to a lesser extent in Cpu and Nac core. Finally, PPE mRNA, coding for the precursor peptide of methionine-enkephalin, was homogeneously expressed in Cpu, Nac, and Tu. In α6−/− mice, the pattern and level of expression of these markers were not altered (Table 2).
Table 2.
Comparison of the expression levels of dopaminergic markers in Cpu and SNc of α6−/− and Wt mice
| Wt (Cpu) | α6−/− (Cpu) | Wt (SNc) | α6−/− (SNc) | |
|---|---|---|---|---|
| Cell count | 82.6 ± 19.4 | 89.3 ± 14.4 | 163.7 ± 5.5 | 158.0 ± 5.0 |
| TH (ICC) | 0.039 ± 0.002 | 0.037 ± 0.002 | 0.26 ± 0.03 | 0.25 ± 0.03 |
| DAT (ICC) | 0.098 ± 0.002 | 0.100 ± 0.002 | 0.31 ± 0.02 | 0.32 ± 0.02 |
| TH (ISH) | ND | ND | 0.83 ± 0.02 | 0.82 ± 0.01 |
| D1 (ISH) | 0.57 ± 0.05 | 0.56 ± 0.02 | ND | ND |
| D2 (ISH) | 0.70 ± 0.01 | 0.68 ± 0.01 | 0.53 ± 0.01 | 0.53 ± 0.02 |
| PPE (ISH) | 0.62 ± 0.01 | 0.58 ± 0.02 | ND | ND |
| PPT (ISH) | 0.70 ± 0.02 | 0.70 ± 0.01 | ND | ND |
| CCK (ISH) | ND | ND | 0.33 ± 0.01 | 0.33 ± 0.01 |
| D1(LBA) | 1.06 ± 0.03 | 1.02 ± 0.04 | ND | ND |
| D2 (LBA) | 0.60 ± 0.02 | 0.58 ± 0.01 | 0.041 ± 0.001 | 0.044 ± 0.001 |
In situ hybridization (ISH), immunocytochemistry (ICC), and ligand binding autoradiography (LBA) were used to compare the levels of expression of various markers in Wt and α6−/− mice at the striatal and mesencephalic levels. Only the results of semiquantitative analysis in Cpu and SNc are presented here. Except for the cell number, each value represents the mean specific optical density ± SEM, obtained from eight animals of each genotype. Cell count was performed on TH-immunostained (SNc) or Nissl-stained (Cpu) sections. [3]SCH23390 and [3H]raclopride were used for the determination of D1 and D2 dopamine receptor levels (respectively). ND, Not detected.
At the mesencephalic level, the morphology and number of DA neurons did not differ in Wt and α6−/− mice when visualized by TH or DAT immunoreactivity. We then examined the expression of TH, dopamine receptors D1, D2, and neuropeptide CCK at the mRNA level. In Wt mice, TH and D2 mRNA was detected in VTA and substantia nigra pars compacta (SNc) only, whereas CCK was also expressed in cortex and hippocampus. D1 mRNA was not detected at the mesencephalic level. Ligand binding studies further revealed low levels of [3H]raclopride (D2) binding sites in VTA and SNc, whereas high levels of [3H]SCH23390 (D1) binding sites could be seen in SN reticulata. Statistical analysis did not reveal any significant difference in the expression of these markers, between Wt and α6−/− animals (Table 2).
Distribution of high-affinity binding sites for nicotinic ligands in the brain of α6−/− mice
We investigated the binding profile of several nicotinic ligands on coronal sections of α6−/− mouse brains. Whereas the distribution of α-bungarotoxin binding sites was unchanged in α6−/− compared with Wt animals, a decrease in the abundance of high-affinity binding sites for [3H]nicotine, [3H]epibatidine, and [3H]cytisine was found in the terminal regions of RGC (SC and dLGN) (Fig.3a–c). Surprisingly, no other differences were detected, especially in the retina, SN–VTA, and striatum, brain regions containing cell bodies or terminals of neurons that express α6. Small decreases, however, might have been masked because of the relative scarcity of α6-containing nAChRs with respect to other major nAChR populations (e.g., α4β2 containing nAChRs) that also bind these ligands. As shown in a previous paper (Marks et al., 1998), a subpopulation of [3H]epibatidine binding sites, enriched in the mesostriatal and visual pathways, has a relatively low affinity for cytisine (“cytisine-resistant” sites). We therefore, compared the distribution of [3H]epibatidine binding sites after displacement by low cytisine concentration (50 nm), in Wt and α6−/− mice. In these conditions, autoradiography quantification revealed a significant decrease of the cytisine-resistant fraction of [3H]epibatidine binding sites in most regions of the visual and dopaminergic systems of mutant animals compared with Wt mice (Figs. 3d,4).
Fig. 3.

Quantitative autoradiography for nicotinic agonists in the mouse brain. Quantitative analysis of [3H]cytisine (a), [3H]nicotine (b), [3H]epibatidine (c), and [3H]epibatidine in the presence of 50 nm unlabeled cytisine (d), in Wt and α6−/− mice. The results are expressed as the mean specific optical density ± SEM, normalized to 100 in the dLGN of Wt animals. AU, Arbitrary unit. Statistical analysis was performed using an unpaired Student's t test (*p < 0.05; **p < 0.01) versus Wt controls. Regions quantified were SC, dLGN, retina (Ret), VTA, Cpu, IPN, MHb, and Cx.
Fig. 4.

Autoradiograms of cytisine-resistant [3H]epibatidine binding in the mouse brain. Sections were incubated with 400 pm[3H]epibatidine in the presence of 50 nm unlabeled cytisine. See Figure 3 for quantification.
Finally, we examined the distribution of [125I]αCtxMII binding sites. In Wt mice, our results were in agreement with those of Whiteaker et al. (2000). Briefly, the highest density of [125I]αCtxMII binding sites was found in the SC (zonal and superficial gray layer), optic tract (opt), geniculate nucleus, and olivary pretectal nucleus (OPN). Lower levels were found in the basal ganglia (including Cpu, Tu, and Nac), SN–VTA, interpeduncular nucleus (IPN), medial habenula (MHb), lateral habenula, fasciculus retroflexus (fr), and retina. It should be noted that these regions correspond to α6-expressing neurons (Le Novère et al., 1996) and to their terminal fields. In α6−/− mice, no signal was detected above background level, indicating a complete disappearance of high-affinity αCtxMII binding sites (Fig.5).
Fig. 5.
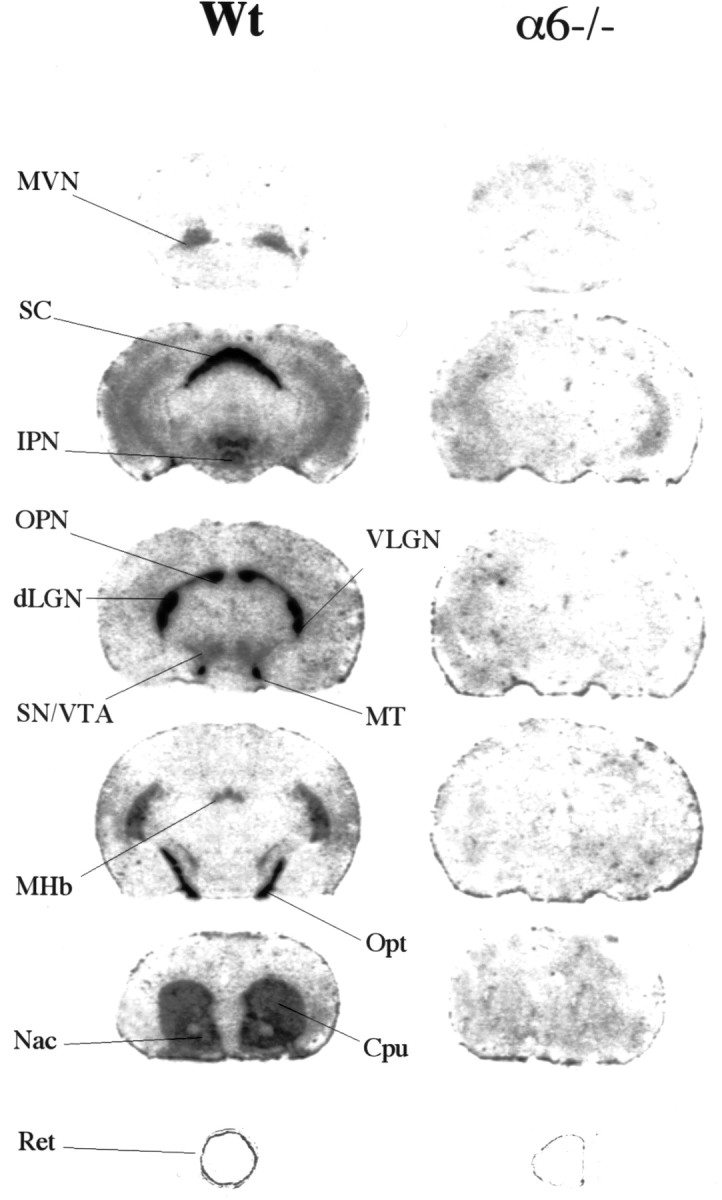
Autoradiograms of [125I]αCtxMII binding in the mouse brain. Sections were incubated with 0.5 nm[125I]αCtxMII, as described in Materials and Methods. Residual signal seen on the eye section of α6−/− animals corresponds to nonspecific binding to pigmented epithelium. MT, Medial terminal nucleus of the accessory tract; MVN, medial vestibular nucleus; VLGN, ventrolateral geniculate nucleus.
Disappearance of the αCtxMII-sensitive and the cytisine-resistant fractions of [3H]epibatidine binding sites in the striatum of α6−/− mice
Receptor autoradiography experiments showed a high selectivity of αCtxMII for α6-containing nAChRs (see above). Still, previous experiments showed that this ligand is a potent inhibitor of α3β2-containing nAChRs (Cartier et al., 1996). We, therefore, examined whether some low-affinity αCtxMII binding sites remained in the striatum of mutant mice. We displaced [3H]epibatidine binding by unlabeled αCtxMII, in striatal membrane preparations. Consistent with our observation in autoradiography experiments, total [3H]epibatidine binding was not significantly different between Wt and α6−/− animals (57.9 ± 2.0 and 53.8 ± 1.7 fmol/mg, respectively). In Wt mice, ∼21% of [3H]epibatidine binding sites could be displaced by αCtxMII, in a monophasic manner (Hill coefficient, 0.96 ± 0.12), and results were fitted to a one site model to determine the Ki value (Table3). On the contrary, in α6−/− animals, αCtxMII had no effect on epibatidine binding at any concentration (up to 10 μm). Strikingly, the total number of αCtxMII-resistant epibatidine binding sites was significantly upregulated in α6−/− mice (p< 0.01) (Fig. 6). We also performed the same experiment on SC membrane preparations and found residual αCtxMII sensitivity in α6−/− mice, representing 16% of the αCtxMII-sensitive [3H]epibatidine binding population in Wt animals (Table 3). However, the low amount of these sites and problems with material availability prevented us from providing an accurate inhibition curve that could have demonstrated the low-affinity nature of these sites.
Table 3.
αCtxMII inhibition of [3H]epibatidine binding on superior colliculus and striatal membranes
| Total [3H]epibatidine binding (fmol/mg protein) | αCtxMII sensitive population (fmol/mg protein) | Ki (nm) | |
|---|---|---|---|
| Wt (SC) | 173.4 ± 9.6 | 59.7 ± 3.8 | ND |
| α6−/−(SC) | 102.0 ± 6.8 | 9.7 ± 1.8 | ND |
| Wt(Str) | 57.9 ± 2.0 | 12.1 ± 1.0 | 1.4 ± 0.2 |
| α6−/−(Str) | 53.8 ± 1.7 | −1.2 ± 0.8 | NA |
Membranes were prepared from SC and striatum (Str) of Wt and α6−/− mice. αCtxMII-sensitive sites were measured using 10 μm unlabeled αCtxMII with 500 pm[3H]epibatidine. Each value represents the mean ± SEM of six separate experiments (expressed in femtomoles per milligram of protein). For Ki determination, data were fitted to a one site model (see Material and Methods). NA, Not applicable. ND, Not determined.
Fig. 6.
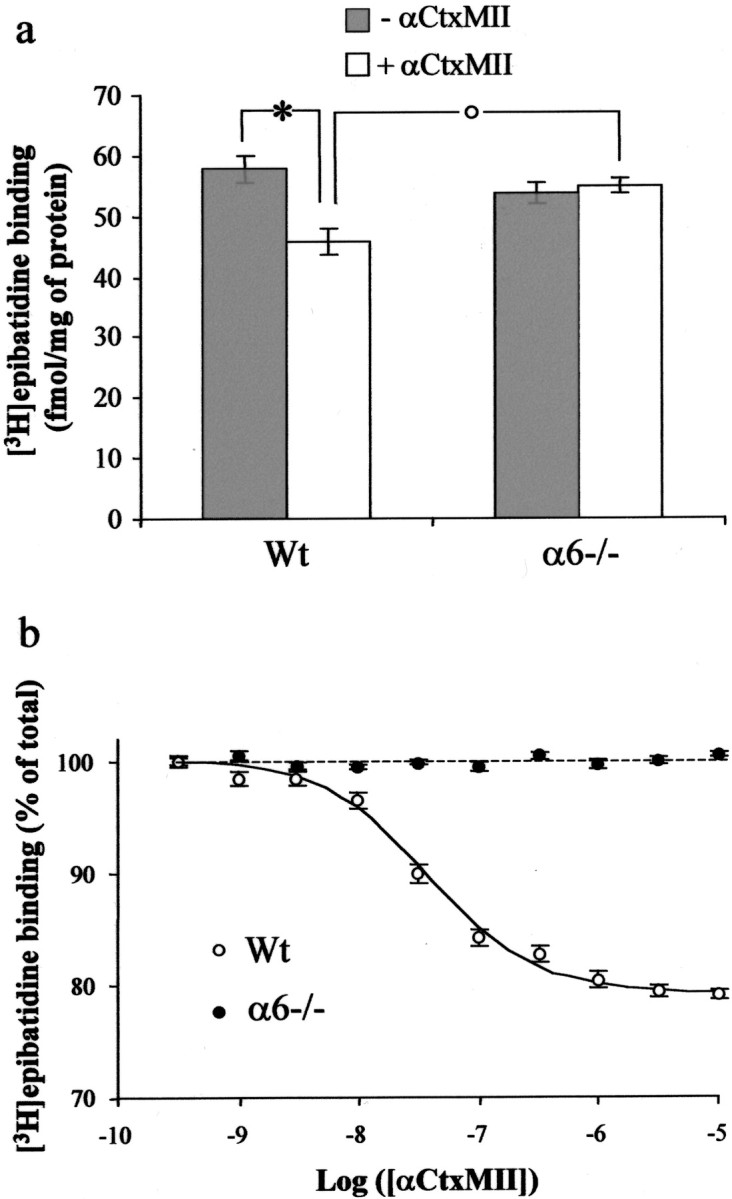
Displacement of [3H]epibatidine binding by αCtxMII in mouse striatal membranes. a, Comparison of total (gray bars) and αCtxMII-resistant (10 μm, white bars) [3H]epibatidine binding sites in Wt and α6−/− animals. Each point represents the mean ± SEM of six separate determinations, expressed in femtomoles per milligram of protein. Statistical analysis was performed using an unpaired Student'st test. A statistically significant fraction (21%; *p < 0.01) of [3H]epibatidine binding sites is sensitive to αCtxMII displacement in Wt but not in α6−/− animals. αCtxMII-resistant [3H]epibatidine binding sites are significantly (°p < 0.01) upregulated in α6−/− animals compared with their Wt controls, whereas total [3H]epibatidine binding is unaffected by the genotype. b, Inhibition curve of [3H]epibatidine (500 pm) binding by αCtxMII (30 pm to 3 μm) in striatal membrane preparations of α6−/− (filled symbols) and Wt (open symbols) animals. Nonspecific binding, determined in the presence of 500 nmunlabeled epibatidine, was subtracted from each measurement. Results were then expressed as a percentage of specific binding in the absence of αCtxMII. Each value is the mean ± SEM of six separate determinations. See also Table 3.
Because αCtxMII-sensitive and cytisine-resistant [3H]epibatidine binding sites have been suggested to represent the same receptor population (Whiteaker et al., 2000), we investigated the displacement of epibatidine binding by unlabeled cytisine. The resulting inhibition curves are shown in Figure7. In Wt animals, the Hill coefficient was significantly <1 (n = 0.76 ± 0.03), suggesting that the inhibition of epibatidine binding by cytisine was not monophasic. Results were thus fitted using a two sites model that revealed the existence of cytisine-sensitive and cytisine-resistant epibatidine binding sites (Table 4), in agreement with the results of Marks et al. (1998). On the contrary, in α6−/− animals, the inhibition curve appeared monophasic (Hill coefficient, 0.94 ± 0.05), and we could fit the data to a one site model to determine Ki value (Table 4). Using a two sites model to fit the data gave similar results because the calculated cytisine-resistant population represented <0.3% of total binding in α6−/− animals. Moreover, the measuredKi value for cytisine inhibition of epibatidine binding in α6−/− animals was comparable with theKi value of the cytisine-sensitive fraction in Wt mice (0.32 ± 0.03 nmcompared with 0.23 ± 0.03 nm, respectively).
Fig. 7.
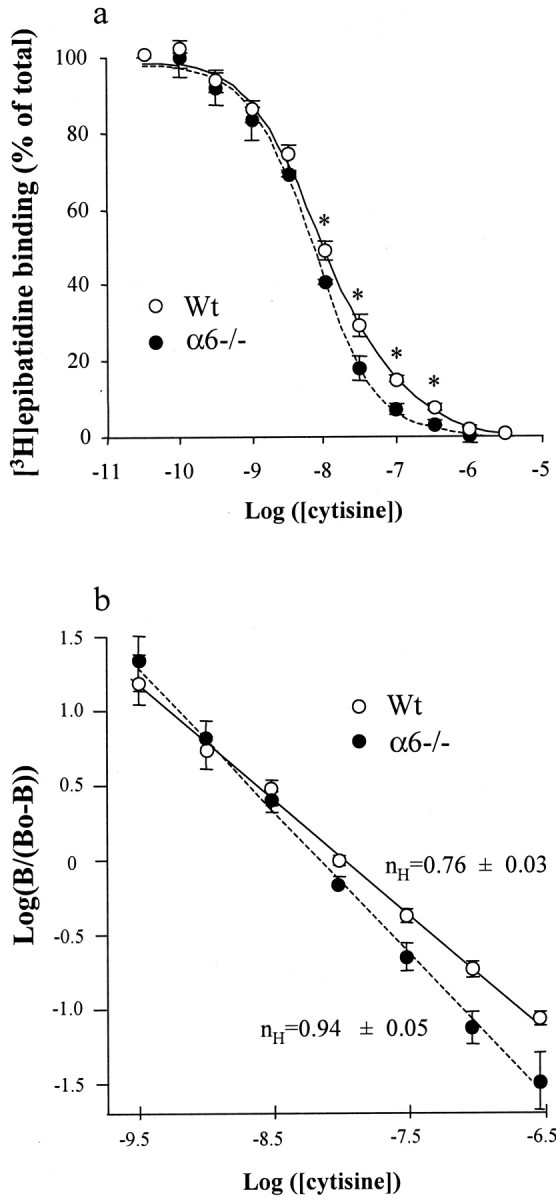
Displacement of [3H]epibatidine binding by cytisine in mouse striatal membranes. a, Inhibition curve of [3H]epibatidine binding by cytisine: membrane samples were incubated with 500 pm[3H]epibatidine in the presence of (30 pm to 3 μm) unlabeled cytisine. Nonspecific binding, determined in the presence of 500 nmunlabeled epibatidine, was subtracted from each measurement. Results were then expressed as a percentage of specific binding in the absence of cytisine. Each point represents the mean ± SEM of five separate determinations. Data obtained from α6−/− (filled symbols) or Wt (open symbols) striatal membranes preparations were fitted to a one- (dotted line) or two-site inhibition model (see Material and Methods), respectively, depending on the value of the Hill number (see below). Statistical analysis was performed using an unpaired Student's t test (*p < 0.05).b, Hill plot representation. Log(B/(Bo −B)) is plotted as a function of log([cytisine]), whereB is the amount of specific [3H]epibatidine binding andBo is the specific binding in the absence of cytisine. In these conditions, the slope of the curve represents the Hill number, nH. In Wt animals,nH is significantly <1 (p < 0.05), suggesting the existence of an heterogeneous population of binding sites. Conversely, in α6−/− animals, nH = 0.94 ± 0.05, which suggests a homogeneous population of binding sites. See also Table4.
Table 4.
Cytisine inhibition of [3H]epibatidine binding on striatal membranes
| B1 (fmol/mg protein) | Ki1(nM) | B2 (fmol/mg protein) | Ki2 (nM) | |
|---|---|---|---|---|
| Wt | 45.9 ± 3.4 | 0.23 ± 0.03 | 12.8 ± 2.8 | 4.0 ± 1.3 |
| α6−/− | 56.5 ± 1.8 | 0.32 ± 0.03 | — | — |
Membranes were prepared from striatum of Wt and α6−/− mice. Membrane samples were incubated in the presence of 500 pm[3H]epibatidine with 30 pm to 3 μm unlabeled cytisine. Results were fitted to a one (α6−/−) or two (Wt) site model (see Material and Methods). Each value represents the mean ± SEM of five separate experiments.
DISCUSSION
We describe the generation and phenotypic characterization of an α6 nAChR subunit knock-out mouse, focusing on the anatomy of neuronal structures where α6 is normally expressed and on the pharmacological properties of native α6-containing nAChRs. Deletion of the first two exons of the α6 gene was sufficient to impair its transcription, as evidenced by the absence of residual α6 mRNA in homozygous null mutant mice. These animals exhibit normal general appearance, survival, growth, anatomy, and locomotor behavior.
The anatomy of the dopaminergic and visual pathways is preserved in α6−/− animals
Because α6 mRNA is abundantly expressed in catecholaminergic neurons and RGCs, we examined whether developmental alterations occurred in the dopaminergic and visual systems of α6−/− animals as a consequence of the loss of the α6 subunit.
In the dopaminergic system, no anatomical alterations could be detected in either midbrain DA neurons at the somatic and terminal levels, or in intrinsic striatal neurons. Moreover, the expression of various molecular markers, including the dopamine biosynthetic enzyme and transporter (TH and DAT, respectively), dopamine receptors (D1, D2) and neuropeptides (CCK, PPT, PPE) were normal in α6−/− mice, although the expression of some of these markers has been reported to be modulated by nicotine or endogenous acetylcholine (Wiener et al., 1989;Smith et al., 1991; Houdi et al., 1998; Drew et al., 2000). In the visual system, retinal cell layering as well as the organization of retinothalamic projections were preserved in α6−/− animals. Relying on our results, we conclude that α6 is not necessary for normal development of visual and mesostriatal dopaminergic pathways. Therefore, these mutant animals are likely to be suitable models for studying the function of α6 in adults.
Moreover, our results contribute to the identification of the subunit composition of nAChRs responsible for the segregation of retinothalamic projections. In the visual system of mammals, the projections of the RGC from each eye are initially intermixed in their thalamic target, the dLGN, and subsequently segregate into eye-specific layers. This phenomenon is dependent on spontaneous retinal activity, termed retinal waves, driven by the activation of nicotinic receptors located on RGC (Feller et al., 1996; Penn et al., 1998). It was recently demonstrated that mice lacking the β2 but not the α4 nAChR subunit, display abnormal segregation of retinogeniculate projections (Rossi et al., 2001). In this context, our finding that adult α6−/− mice exhibit normal segregation, suggests that nAChRs involved in the segregation process do not contain α6, although the substitution of α6 by another α subunit cannot be ruled out (see below). These results further support the hypothesis of Bansal et al. (2000), who proposed α3 as the likely partner of β2 in the genesis of normal retinal waves.
The α6 nAChR subunit is a critical component of native αCtxMII binding sites
Null mutant mice exhibit a complete disappearance of high-affinity [125I]αCtxMII binding sites. Therefore, all these sites appear to correspond to α6-containing nAChRs. Three distinct anatomical systems contain detectable levels of [125I]αCtxMII binding. High levels were shown in the visual system, including retina, RGC fibers (opt), and their terminal fields (geniculate nucleus, SC, OPN). Lower binding density was found in two other systems: the mesostriatal dopaminergic system, which includes SN–VTA neurons and their projection structures (Tu, Nac, Cpu, lateral habenula), and the habenulo-interpeduncular system (MHb, fr, and IPN). On the basis of the distribution of α6 mRNA-expressing neurons, we infer that α6-containing nAChRs in the SN–VTA, MHb, and retina are somatodendritic receptors. In other regions, these receptors are likely to be transported to a presynaptic or preterminal localization, as confirmed by the presence of high levels of [125I]αCtxMII binding in axon systems (optic nerve and tract, fr). Our results expand those ofGoldner et al. (1997), who detected α6 immunoreactivity only in the SN–VTA. However, it should be noted that αCtxMII-insensitive α6-containing nAChRs might also exist, as suggested by the fact that α6 mRNA was detected in areas, such as the LC (Le Novère et al., 1996; Lena et al., 1999), where no [125I]αCtxMII binding was detected.
Although αCtxMII was thought to be a specific inhibitor of α3β2-containing nAChRs (Cartier et al., 1996), the sensitivity of α6-containing nAChRs to this toxin is not surprising. Indeed, α3 and α6 are highly homologous subunits with 67.3% residue identity in their mature N-terminal domains, and critical residues for αCtxMII selectivity are conserved in α6 (Harvey et al., 1997). Moreover, it was recently demonstrated that native α6β4-containing nAChRs, purified from chick retina, are blocked by this toxin (Vailati et al., 1999). Finally, chimeric subunits containing the extracellular domain of the human α6 subunit fused to the rest of α3 or α4 subunits can form functional nAChRs, sensitive to αCtxMII, when expressed with β2 or β4 in Xenopus oocytes (Kuryatov et al., 2000). More intriguing, however, is the fact that αCtxMII binding sites completely disappear in α6−/− mice, although α3 is still expressed. It is possible that α3β2-containing nAChRs are not detected because of the scarcity of these sites. In the SN–VTA, α3 mRNA is barely detectable using in situ hybridization (Le Novère et al., 1996; Ross et al., 2000; present study), whereas in regions where α3 mRNA is abundant (e.g., in MHb), αCtxMII-insensitive α3β4-containing nAChRs are likely to form (Quick et al., 1999). Another explanation would be that α3β2-containing nAChRs form low-affinity αCtxMII binding sites, which could not be detected using receptor autoradiography. In the SC, this hypothesis is supported by our results on αCtxMII displacement of [3H]epibatidine binding on membrane preparations and the findings of Dougherty et al. (2000), which suggest that some non-α6-containing nAChRs with low affinity for αCtxMII are present in this region. In the striatum however, such sites are unlikely to exist because in Wt mice, in agreement with Whiteaker et al., (2000), we report a monophasic of inhibition of [3H]epibatidine binding by αCtxMII, and because, in α6−/− animals, residual [3H]epibatidine binding is insensitive to αCtxMII displacement (up to 10 μm).
Overall, our results prompt a new interpretation of previous works describing the pharmacological actions of αCtxMII on DA neurons. This toxin partially inhibits nicotine-induced dopamine release in striatal synaptosomes (Kulak et al., 1997; Kaiser et al., 1998). Moreover, patch-clamp recordings revealed αCtxMII-sensitive nAChR-mediated currents in midbrain DA neurons (Klink et al., 2001). Relying on the supposed selectivity of αCtxMII, it was proposed that α3β2-containing nAChRs mediate these nicotinic effects. We demonstrate here that this is unlikely to be the case. First, the selectivity of αCtxMII for α3β2-containing nAChRs is questionable because native α6-containing nAChRs exhibit nanomolar affinity for this toxin. Moreover the absence of residual αCtxMII binding sites in the striatum of α6−/− animals, and the low abundance of α3 mRNA in the SN/VTA suggest that α3β2-containing nAChRs are very scarce in the dopaminergic system. Consequently, the role of the α6 subunit in the nicotinic modulation of DA neurons and its importance for nicotine addiction should be reevaluated taking α6β2-containing nAChR rather than α3β2-containing nAChR as the likely candidate for these effects.
Evidence for the existence compensatory mechanisms in α6−/− animals
Pharmacological characterization of native α6-containing nAChRs was completed using various nicotinic agonists. Cytisine displacement of [3H]epibatidine binding on striatal membranes strongly suggested the identity of cytisine-resistant and αCtxMII-sensitive epibatidine binding sites in the striatum, because both types of sites represent the same proportion of total epibatidine binding (∼20%), and disappear in α6−/− animals. Furthermore, autoradiography experiments demonstrated the existence of α6-containing binding sites with high affinity for nicotine, epibatidine, and cytisine, exclusively in SC and dLGN of Wt animals. In the mesostriatal dopaminergic system, however, no significant difference could be detected between Wt and α6−/− animals. This is unlikely to be simply attributable to a lack of sensitivity of our assay because displacement of [3H]epibatidine binding by αCtxMII on striatal membranes revealed an increase of αCtxMII resistant sites in α6−/− animals compared with Wt mice. This observation suggests the existence of a developmental compensation in the striatum of α6−/− mice, where α6-containing nAChRs are replaced by new nicotinic receptors with a different pharmacology. To our knowledge this is the first demonstration of the existence of compensatory mechanisms in nAChRs subunit knock-out mice, although a similar phenomenon has been suggested in α4 knock-out mice (Klink et al., 2001). The exact molecular mechanism of this compensation is unknown, although simple transcriptional upregulation seems unlikely according to our in situ hybridization results. Klink et al. (2001) suggested the existence of α4α6α5(β2)2 nAChRs in DA neurons. In the absence of α6, one may plausibly support the idea that this receptor assembles by recruiting a second α4 subunit. This would lead to the formation of a new cytisine-sensitive, αCtxMII-resistant, epibatidine binding site, without modifying the total amount of binding. Additional experiments however, are required to test this possibility and to evaluate the functional relevance of these new nAChRs in α6−/− animals.
Finally, our results, in combination with those of Klink et al. (2001)and Kulak et al. (1997), demonstrate the importance of the α6 nAChR subunit in the effects of nicotine on DA neurons. Provided that compensating mechanisms are taken into account, α6−/− mice might offer useful tools to further evaluate the contribution of the α6-containing nAChRs to nicotine addiction.
Footnotes
This work was supported by the Collège de France, the Centre National de la Recherche Scientifique, the Institut Pasteur, the Association Française contre les Myopathies, and CommunautéEconomique Européene Contract Numbers 097038 and 097038. N.C. is a recipient of grants from the Direction Générale de l'Armement and the Association pour la Recherche contre le Cancer. M.Z. is a recipient of grants from Italian Telethon and Ministero dell'Università della Ricerca Scientifica e Technologica Cofin 2000. Z.-Y.H. is a recipient of a grant from The Société de Tabacologie (France). J.M.M. is a recipient of National Institutes of Health (NIH) Grants MH 53631, DA 12242, and GM48677. L.M.M. was supported by NIH Grant PO1 DA 12661-2. F.M.R. is supported by European Molecular Biology Organization Fellowship ALTF457. We thank Clément Léna, Thomas Grutter, and Nicolas Polounovsky for critical reading of this paper.
Correspondence should be addressed to Nicolas Champtiaux, Institut Pasteur, Laboratoire de Neurobiologie Moléculaire, 25-28 Rue du Dr Roux, 75724 Paris, France. E-mail: champtia@pasteur.fr.
REFERENCES
- 1.Bansal A, Singer JH, Hwang BJ, Xu W, Beaudet A, Feller MB. Mice lacking specific nicotinic acetylcholine receptor subunits exhibit dramatically altered spontaneous activity patterns and reveal a limited role for retinal waves in forming ON and OFF circuits in the inner retina. J Neurosci. 2000;20:7672–7681. doi: 10.1523/JNEUROSCI.20-20-07672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boye SM, Grant RJ, Clarke PB. Disruption of dopaminergic neurotransmission in nucleus accumbens core inhibits the locomotor stimulant effects of nicotine and d-amphetamine in rats. Neuropharmacology. 2001;40:792–805. doi: 10.1016/s0028-3908(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 3.Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. A new alpha-conotoxin which targets alpha3beta2 nicotinic acetylcholine receptors. J Biol Chem. 1996;271:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- 4.Charpantier E, Barneoud P, Moser P, Besnard F, Sgard F. Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. NeuroReport. 1998;9:3097–3101. doi: 10.1097/00001756-199809140-00033. [DOI] [PubMed] [Google Scholar]
- 5.Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653:278–284. doi: 10.1016/0006-8993(94)90401-4. [DOI] [PubMed] [Google Scholar]
- 6.Di Chiara G. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur J Pharmacol. 2000;393:295–314. doi: 10.1016/s0014-2999(00)00122-9. [DOI] [PubMed] [Google Scholar]
- 7.Dougherty JJ, Nayak SV, McIntosh JM, Nichols RA (2000) α-Conotoxin MII (α-CTXMII) interacts with two distinct binding sites in mouse brain. Neuronal nicotinic receptors, The 10th Neuropharmacology Conference, Abstract Book, 122.
- 8.Drew AE, Derbez AE, Werling LL. Nicotinic receptor-mediated regulation of dopamine transporter activity in rat prefrontal cortex. Synapse. 2000;38:6–10. doi: 10.1002/1098-2396(200010)38:1<10::AID-SYN2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 9.Feller MB, Wellis DP, Stellwagen D, Werblin FS, Shatz CJ. Requirement for cholinergic synaptic transmission in the propagation of spontaneous retinal waves. Science. 1996;272:1182–1187. doi: 10.1126/science.272.5265.1182. [DOI] [PubMed] [Google Scholar]
- 10.Franklin K, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 11.Goldner FM, Dineley KT, Patrick JW. Immunohistochemical localization of the nicotinic acetylcholine receptor subunit alpha6 to dopaminergic neurons in the substantia nigra and ventral tegmental area. NeuroReport. 1997;8:2739–2742. doi: 10.1097/00001756-199708180-00019. [DOI] [PubMed] [Google Scholar]
- 12.Grenhoff J, Aston-Jones G, Svensson TH. Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand. 1986;128:351–358. doi: 10.1111/j.1748-1716.1986.tb07988.x. [DOI] [PubMed] [Google Scholar]
- 13.Harvey SC, McIntosh JM, Cartier GE, Maddox FN, Luetje CW. Determinants of specificity for alpha-conotoxin MII on alpha3beta2 neuronal nicotinic receptors. Mol Pharmacol. 1997;51:336–342. doi: 10.1124/mol.51.2.336. [DOI] [PubMed] [Google Scholar]
- 14.Houdi AA, Dasgupta R, Kindy MS. Effect of nicotine use and withdrawal on brain preproenkephalin A mRNA. Brain Res. 1998;799:257–263. doi: 10.1016/s0006-8993(98)00454-5. [DOI] [PubMed] [Google Scholar]
- 15.Israel DI. A PCR-based method for high stringency screening of DNA libraries. Nucleic Acids Res. 1993;21:2627–2631. doi: 10.1093/nar/21.11.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaiser SA, Soliakov L, Harvey SC, Luetje CW, Wonnacott S. Differential inhibition by alpha-conotoxin-MII of the nicotinic stimulation of [3H]dopamine release from rat striatal synaptosomes and slices. J Neurochem. 1998;70:1069–1076. doi: 10.1046/j.1471-4159.1998.70031069.x. [DOI] [PubMed] [Google Scholar]
- 17.Khillan JS, Bao Y. Preparation of animals with a high degree of chimerism by one-step coculture of embryonic stem cells and preimplantation embryos. Biotechniques. 1997;22:544–549. doi: 10.2144/97223rr06. [DOI] [PubMed] [Google Scholar]
- 18.Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–1463. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kress C, Vandormael-Pournin S, Baldacci P, Cohen-Tannoudji M, Babinet C. Nonpermissiveness for mouse embryonic stem (ES) cell derivation circumvented by a single backcross to 129/Sv strain: establishment of ES cell lines bearing the Omd conditional lethal mutation. Mamm Genome. 1998;9:998–1001. doi: 10.1007/s003359900914. [DOI] [PubMed] [Google Scholar]
- 20.Kulak JM, Nguyen TA, Olivera BM, McIntosh JM. Alpha-conotoxin MII blocks nicotine-stimulated dopamine release in rat striatal synaptosomes. J Neurosci. 1997;17:5263–270. doi: 10.1523/JNEUROSCI.17-14-05263.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuryatov A, Olale F, Cooper J, Choi C, Lindstrom J. Human alpha6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology. 2000;39:2570–2590. doi: 10.1016/s0028-3908(00)00144-1. [DOI] [PubMed] [Google Scholar]
- 22.Le Novère N, Changeux JP. Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells. J Mol Evol. 1995;40:155–172. doi: 10.1007/BF00167110. [DOI] [PubMed] [Google Scholar]
- 23.Le Novère N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci. 1996;8:2428–2439. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 24.Le Novère N, Zoli M, Lena C, Ferrari R, Picciotto MR, Merlo-Pich E, Changeux JP. Involvement of alpha6 nicotinic receptor subunit in nicotine-elicited locomotion, demonstrated by in vivo antisense oligonucleotide infusion. NeuroReport. 1999;10:2497–2501. doi: 10.1097/00001756-199908200-00012. [DOI] [PubMed] [Google Scholar]
- 25.Lena C, de Kerchove D'Exaerde A, Cordero-Erausquin M, Le Novère N, del Mar Arroyo-Jimenez M, Changeux JP. Diversity and distribution of nicotinic acetylcholine receptors in the locus ceruleus neurons. Proc Natl Acad Sci USA. 1999;96:12126–12131. doi: 10.1073/pnas.96.21.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marks MJ, Smith KW, Collins AC. Differential agonist inhibition identifies multiple epibatidine binding sites in mouse brain. J Pharmacol Exp Ther. 1998;285:377–386. [PubMed] [Google Scholar]
- 27.Nisell M, Nomikos GG, Svensson TH. Systemic nicotine-induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse. 1994;16:36–44. doi: 10.1002/syn.890160105. [DOI] [PubMed] [Google Scholar]
- 28.Pedrazzi P, Cattaneo L, Valeriani L, Boschi S, Cocchi D, Zoli M. Hypothalamic neuropeptide Y and galanin in overweight rats fed a cafeteria diet. Peptides. 1998;19:157–165. doi: 10.1016/s0196-9781(97)00258-1. [DOI] [PubMed] [Google Scholar]
- 29.Penn AA, Riquelme PA, Feller MB, Shatz CJ. Competition in retinogeniculate patterning driven by spontaneous activity. Science. 1998;279:2108–2112. doi: 10.1126/science.279.5359.2108. [DOI] [PubMed] [Google Scholar]
- 30.Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 31.Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- 32.Pontieri FE, Tanda G, Orzi F, Di Chiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature. 1996;382:255–257. doi: 10.1038/382255a0. [DOI] [PubMed] [Google Scholar]
- 33.Quick MW, Ceballos RM, Kasten M, McIntosh JM, Lester RA. Alpha3beta4 subunit-containing nicotinic receptors dominate function in rat medial habenula neurons. Neuropharmacology. 1999;38:769–783. doi: 10.1016/s0028-3908(99)00024-6. [DOI] [PubMed] [Google Scholar]
- 34.Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, Drago J. Phenotypic characterization of an alpha 4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci. 2000;20:6431–6441. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossi FM, Pizzorusso T, Porciatti V, Marubio LM, Maffei L, Changeux JP. Requirement of the nicotinic acetylcholine receptor beta 2 subunit for the anatomical and functional development of the visual system. Proc Natl Acad Sci USA. 2001;98:6453–6458. doi: 10.1073/pnas.101120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrook J, Fritsch E, Maniatis T. A laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 37.Sargent PB. The diversity of neuronal nicotinic acetylcholine receptors. Annu Rev Neurosci. 1993;16:403–443. doi: 10.1146/annurev.ne.16.030193.002155. [DOI] [PubMed] [Google Scholar]
- 38.Sharples CGV, Kaiser S, Soliakov L, Marks MJ, Collins AC, Washburn M, Wright E, Spencer JA, Gallagher T, Whiteaker P, Wonnacott S. UB-165: a novel nicotinic agonist with subtype selectivity implicates the α4β2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J Neurosci. 2000;20:2783–2791. doi: 10.1523/JNEUROSCI.20-08-02783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith KM, Mitchell SN, Joseph MH. Effects of chronic and subchronic nicotine on tyrosine hydroxylase activity in noradrenergic and dopaminergic neurones in the rat brain. J Neurochem. 1991;57:1750–1756. doi: 10.1111/j.1471-4159.1991.tb06377.x. [DOI] [PubMed] [Google Scholar]
- 40.Vailati S, Hanke W, Bejan A, Barabino B, Longhi R, Balestra B, Moretti M, Clementi F, Gotti C. Functional alpha6-containing nicotinic receptors are present in chick retina. Mol Pharmacol. 1999;56:9–11. doi: 10.1124/mol.56.1.11. [DOI] [PubMed] [Google Scholar]
- 41.Whiteaker P, McIntosh JM, Luo S, Collins AC, Marks MJ. 125I-alpha-conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol. 2000;57:913–925. [PubMed] [Google Scholar]
- 42.Wiener HL, Lajtha A, Sershen H. Dopamine D1 receptor and dopamine D2 receptor binding activity changes during chronic administration of nicotine in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. Neuropharmacology. 1989;28:535–537. doi: 10.1016/0028-3908(89)90091-9. [DOI] [PubMed] [Google Scholar]
- 43.Zoli M, Guidolin D, Agnati LF. Morphometric evaluation of populations of neuronal profiles (cell bodies, dendrites, and nerve terminals) in the central nervous system. Microsc Res Tech. 1992;21:315–337. doi: 10.1002/jemt.1070210408. [DOI] [PubMed] [Google Scholar]
- 44.Zoli M, Ferraguti F, Toffano G, Fuxe K, Agnati LF. Neurochemical alterations but not nerve cell loss in aged rat neostriatum. J Chem Neuroanat. 1993;6:131–145. doi: 10.1016/0891-0618(93)90023-w. [DOI] [PubMed] [Google Scholar]
- 45.Zoli M, Le Novère N, Hill JA, Jr, Changeux JP. Developmental regulation of nicotinic ACh receptor subunit mRNAs in the rat central and peripheral nervous systems. J Neurosci. 1995;15:1912–1939. doi: 10.1523/JNEUROSCI.15-03-01912.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zoli M, Lena C, Picciotto MR, Changeux JP. Identification of four classes of brain nicotinic receptors using beta2 mutant mice. J Neurosci. 1998;18:4461–4472. doi: 10.1523/JNEUROSCI.18-12-04461.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]