

Abstract
Synaptic release of Zn2+ and its translocation into postsynaptic neurons probably contribute to neuronal injury after ischemia or epilepsy. Studies in cultured neurons have revealed that of the three major routes of divalent cation entry, NMDA channels, voltage-sensitive Ca2+ channels (VSCCs), and Ca2+-permeable AMPA/kainate (Ca-A/K) channels, Ca-A/K channels exhibit the highest permeability to exogenously applied Zn2+. However, routes through which synaptically released Zn2+ gains entry to postsynaptic neurons have not been characterized in vivo. To model ischemia-induced Zn2+ movement in a system approximating the in vivo situation, we subjected mouse hippocampal slice preparations to controlled periods of oxygen and glucose deprivation (OGD). Timm's staining revealed little reactive Zn2+ in CA1 and CA3 pyramidal neurons of slices exposed in the presence of O2 and glucose. However, 15 min of OGD resulted in marked labeling in both regions. Whereas strong Zn2+ labeling persisted if both the NMDA antagonist MK-801 and the VSCC blocker Gd3+ were present during OGD, the presence of either the Ca-A/K channel blocker 1-naphthyl acetyl spermine (NAS) or the extracellular Zn2+chelator Ca2+ EDTA substantially decreased Zn2+ accumulation in pyramidal neurons of both subregions. In parallel experiments, slices were subjected to 5 min OGD exposures as described above, followed 4 hr later by staining with the cell-death marker propidium iodide. As in the Timm's staining experiments, substantial CA1 or CA3 pyramidal neuronal damage occurred despite the presence of MK-801 and Gd3+, whereas injury was decreased by NAS or by Ca2+ EDTA (in CA1).
Keywords: zinc, ischemia, glutamate, AMPA, naphthyl acetyl spermine, Timm's stain, pyramidal neuron, neurotoxicity, hippocampal slice
Transient global ischemia causes degeneration of certain hippocampal pyramidal neurons, particularly in the CA1 subregion (Pulsinelli et al., 1982). Recent studies implicate Zn2+ ions as likely contributors to this injury. Zn2+ is sequestered at high concentrations in presynaptic boutons of many excitatory synapses, with particularly high levels in the hippocampus; when released with neuronal activity, it is estimated to achieve peak synaptic concentrations of several hundred micromoles per liter (Frederickson et al., 2000). In vivo, both transient global ischemia and seizure activity have been associated with a depletion of presynaptic Zn2+ and concomitant Zn2+ accumulation in degenerating postsynaptic neurons (“Zn2+translocation”) (Sloviter, 1985; Frederickson et al., 1989; Tonder et al., 1990; Koh et al., 1996). Additional support for a direct injurious role for Zn2+ in these conditions is provided by observations that extracellular Zn2+ chelators decrease both the appearance of Zn2+ in postsynaptic neurons and the resultant selective neuronal loss (Koh et al., 1996; Suh et al., 1996).
Because presynaptic Zn2+ is coreleased with glutamate from excitatory terminals and appears to gain direct entry into certain postsynaptic neurons, it is reasonable to consider that Zn2+ might permeate postsynaptic glutamate-activated channels. Indeed, in vitro studies have indicated that Zn2+ is potently neurotoxic (Choi et al., 1988) and is able to gain entry to neurons through voltage-sensitive Ca2+ channels (VSCCs), NMDA channels, or Ca2+-permeable AMPA/kainate (Ca-A/K) channels (Weiss et al., 1993; Yin and Weiss, 1995; Sensi et al., 1997). However, neurotoxicity and imaging studies have suggested that of these routes, Ca-A/K channels have the greatest permeability to Zn2+ (Yin and Weiss, 1995;Sensi et al., 1999), with intermediate VSCC and minimal NMDA channel permeability (and Zn2+ actually being an effective NMDA channel blocker) (Peters et al., 1987; Westbrook and Mayer, 1987).
Although culture studies would favor the possibility that synaptically released Zn2+ might preferentially pass through Ca-A/K channels (Yin and Weiss, 1995; Sensi et al., 1999), their presence on pyramidal neurons has not been substantiated by most electrophysiological studies. However, certain histochemical and electrophysiological evidence suggests that Ca-A/K channels might often be present in hippocampal pyramidal neurons, but with preferential localization in the distal dendrites, where they are hard to detect by recording on or near the soma (Pruss et al., 1991; Williams et al., 1992; Toomim and Millington, 1998; Yin et al., 1999; Lerma et al., 1994).
Most models of ischemic neurodegeneration have focused on the putative role of NMDA receptor activation. However, use of NMDA antagonists in animal models of ischemia as well as in human clinical trials has not generally shown the anticipated robust efficacy (Lee et al., 1999). One possible factor is that certain environmental perturbations associated with acute ischemia, specifically synaptic Zn2+ elevations and tissue acidosis, each can decrease NMDA channel activity (Peters et al., 1987; Westbrook and Mayer, 1987; Tang et al., 1990; Traynelis and Cull-Candy, 1990). The present study is motivated by the hypothesis that Ca-A/K channels, which share high Ca2+ permeability with NMDA channels but are unique in their high permeability to Zn2+, contribute to ischemic neurodegeneration by serving as routes through which synaptically released Zn2+ gains entry to hippocampal pyramidal neurons. To address this hypothesis, we used acute hippocampal slice preparations from adult mice subjected to brief periods of oxygen and glucose deprivation (OGD) (Kass and Lipton, 1982;Monette et al., 1998) as a model of trans-synaptic Zn2+ movement occurring under conditions of ischemia.
MATERIALS AND METHODS
Chemicals and reagents. Propidium iodide (PI) and Newport Green were purchased from Molecular Probes (Eugene, OR). 1-Naphthyl acetyl spermine (NAS) was kindly provided by Daicel Chemical (Tokyo, Japan). MK-801 was purchased from Research Biochemicals (Natick, MA). Tissue culture media and serum were supplied by Invitrogen (Grand Island, NY). Most other chemicals and reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Animal usage and tissue preparations. All animal procedures were conducted in accordance with the National Institutes of HealthGuide for the Care and Use of Laboratory Animals and were approved by the University of California Irvine Institutional Animal Care and Use Committee. Adult Swiss-Webster mice (8–10 weeks of age; weight 25–30 gm) from Simonsen Laboratories (Gilroy, CA) were deeply anesthetized with halothane and decapitated; their brains were rapidly removed, and coronal slices (400 μm) were cut with a vibratome. (Thus, all slice manipulations were effectively performed in duplicate, with effects on each hemisphere averaged before compilations across experiments.)
Murine forebrain cultures, derived from embryonic day 15 embryos, were plated on previously established astrocytic monolayers and used between 13 and 16 d in vitro (Yin and Weiss, 1995).
Oxygen–glucose deprivation of slices. All slice manipulations (including equilibration) were performed in covered chambers containing 6 ml of buffer, with slices completely submerged and protected from the vigorous bubbling in the chamber by a semipermeable nylon mesh (Millicell CM inserts; Millipore, Bedford, MA) through which small needle holes were made to facilitate solution exchange. All chamber solutions were prebubbled with either O2/5% CO2 or N2/5% CO2 gas for 30 min before slice immersion to ensure O2 saturation or O2 removal as desired. Drugs were all dissolved in water at high concentrations (NAS, MK-801 and Gd3+ at 15 mm; Ca2+EDTA at 100 mm) and added to buffers immediately before experiments.
Immediately after vibratome sectioning, coronal slices (the most anterior slice was discarded) were transferred to a chamber with 6 ml of cold (4°C) Ca2+-free equilibration buffer (in mm: 126 NaCl, 24 NaHCO3, 1 NaH2PO4, 2.5 KCl, 10 MgSO4, 10 glucose, pH 7.4) bubbled with 95% O2 and 5% CO2 and containing the NMDA blocker MK-801 (15 μm), the VSCC blocker Gd3+ (20 μm), and the Ca-A/K channel blocker NAS (300 μm) for 25 min. Consistent with a recent report (Suh et al., 2000), longer periods of equilibration were associated with depletion of much of the endogenous Zn2+ stores.
At the end of the 25 min of equilibration, slices were transferred to separate chambers, each containing oxygenated equilibration buffer (4°C), but only with antagonists to be used with that slice during the OGD exposure. Specifically, for the primary set of experiments presented, two slices were transferred to buffer alone (for the OGD and the +O2 conditions) and one each to chambers containing MK-801 (15 μm) with Gd3+ (20 μm), NAS (300 μm), or Ca2+ EDTA (3 mm). After 2 min, slices were subjected to OGD (for 5 or 15 min, see below) by transfer to chambers containing warmed (37°C) glucose-free artificial CSF (ACSF) (in mm: 126 NaCl, 24 NaHCO3, 1 NaH2PO4, 2.5 KCl, 2 MgCl2, 1 CaCl2, 7.0 sucrose, pH 7.4) bubbled with 95% N2 and 5% CO2 either alone or with the same antagonists as present in the previous step. In each experiment, one other matched slice was exposed to oxygenated ACSF with glucose (OG-ACSF; 10 mm instead of sucrose; +O2 condition) in place of the OGD exposure.
To assess Zn2+ translocation, OGD exposures occurred for 15 min, followed immediately by Timm's staining as described. To assess injury, 5 min OGD exposures were used, followed by additional incubation of slices at 22°C in OG-ACSF containing the same antagonists as present during the OGD exposure. [This temperature was selected as one that permits evolution of injury without causing the rapid release of Zn2+ from slices that has been reported to occur at warmer temperatures (Suh et al., 2000).] After 3.5 hr, the cell-death marker PI (5 μg/ml) was added to the bath for 30 min before fixation in 4% paraformaldehyde (in PBS; 3 hr at room temperature, then 12 hr at 4°C) and visualization of PI staining under confocal microscopy. For all experiments,p < 0.05 was preselected as a cutoff point for significance.
For control experiments examining the effects of NAS on transmitter release after equilibration, slices were loaded with [3H]-d-aspartate (2.5 μCi/ml) in OG-ACSF buffer (37°C, 30 min) with or without NAS (300 μm). (MK-801 was included in each condition to help maintain slice viability during the prolonged loading and wash steps.) After washout of extracellular isotope (20 min, 22°C, in the presence of the same antagonists), slices either were subjected to OGD as above (37°C, 15 min) in the presence of the same antagonists or were identically incubated in OG-ACSF with MK-801. Immediately after OGD, the slice was removed from the buffer and solubilized in 20% HCl; isotope accumulation was then counted in both the slice and the bathing OGD buffer. The percentage of [3H]-d-aspartate released in each condition was calculated as the counts in the buffer divided by the total counts for that slice (buffer + slice).
Timm's staining. Immediately after the OGD exposures, slices were incubated in 0.1% (NH4)2S in oxygenated equilibration buffer (containing the same antagonists as present during OGD) for 10 min to precipitate intracellular Zn2+, followed by fixation in 4% paraformaldehyde (3 hr at room temperature). Slices were then incubated in the dark in a solution consisting of 1 part solution A (1m AgNO3), 20 parts solution B (2% hydroquinone and 5% citric acid in water), and 100 parts solution C (20% gum arabic in water). Development took ∼1 hr, was monitored by periodic evaluations under low light, and was terminated by washing in water. Slices were then incubated overnight in 30% sucrose (4°C) before frozen sections (25 μm) were made for dehydration and permanent mount.
To assess the degree of labeling, the top and bottom three sections from each slice were discarded because of frequent nonspecific trauma-induced stain, and the remaining 7–10 sections were kept for examination. In each section, pyramidal neuronal labeling in each of the two hemispheres was visually assessed on a four-point scale (near absence, light, strong, and very strong staining) in two regions of CA3 and three regions of CA1 by careful matching with previously established standards. Thus, in each experiment, values for each condition were derived in 42–60 regions for CA1 and 28–40 regions for CA3. Values for each condition were then averaged within each experiment before averaging across experiments.
Kainate-stimulated Co2+ uptake labeling. Kainate-induced Co2+ uptake labeling of cultured neurons (used for the experiments summarized in Fig. 1) was performed generally as described previously (Yin et al., 1994, 1999). After Co2+ loading (by exposure to 100 μm kainate with 5 mmCo2+ for 10 min), intracellular Co2+ was precipitated with 0.05% (NH4)2S, the cultures were fixed, and the stain silver was enhanced by a modified Timm's stain procedure, largely as described above.
Fig. 1.

NAS blocks Zn2+ entry through Ca-A/K channels. Cultures were loaded with the Zn2+-sensitive fluorescent probe Newport Green and exposed to kainate (KA; 100 μm) in the presence of Zn2+ (300 μm), MK-801 (MK; 10 μm), and NAS (300 μm) for 5 min. After 20 min, the cultures were identically re-exposed without NAS. After imaging, the subpopulation of neurons possessing large numbers of Ca-A/K(+) neurons was identified by kainate-stimulated Co2+ uptake labeling [n = 3 experiments; 325 total neurons; 28 Ca-A/K(+) neurons; calibrated [Zn2+]ivalues are ± SEM]. Note the high Zn2+increases occurring in Ca-A/K(+) neurons after removal of the NAS block. In contrast, the NAS had little effect on Zn2+ increases in other neurons, which result primarily from slower influx through VSCCs.
Imaging studies. Forebrain cultures (plated on coverslips) were loaded with the low-affinity Zn2+selective probe Newport Green diacetate by adding 5 μl of a 1 mm stock (in DMSO) with 0.2% pluronic acid per milliliter of buffer (30 min, room temperature), followed by washing and incubation in the dark for an additional 30 min. Images were obtained (excitation, 490 nm; emission, 530 nm) using a 12 bit digital CCD camera (Roper Scientific, Tucson, AZ) attached to a Nikon Diaphot inverted microscope (Nikon USA, New York, NY) equipped with a 40× (NA, 1.3) epifluorescence oil immersion objective. Experiments were analyzed using Metafluor 4.0 software (Universal Imaging, West Chester, PA), and [Zn2+]i was determined, after background subtraction, as:
using Kd of 1 μm. Fmax was obtained at the end of each experiment by adding the Zn2+-selective ionophore Na+ pyrithione (10 μm) in the presence of 500 μm Zn2+ (the fluorescence rapidly approached a maximum) andFmin was obtained (after Zn2+ washout) by adding the cell-permeable Zn2+ chelatorN,N,N′,N′-tetrakis (2-pyridylmethyl)ethylenediamine (50 μm) (Sensi et al., 1999).
For imaging of slices, we used a Bio-Rad MRC 600 confocal system (Bio-Rad Laboratories, Hercules, CA) attached to an inverted Nikon Diaphot microscope (Nikon USA) equipped with a krypton laser (excitation, 568 nm; emission, >648 nm) and a 20× epifluorescence objective. In each experiment, the OGD condition was initially scanned to determine a depth halfway between the slice surface and loss of fluorescence near midslice, and the identical depth was used for other conditions (between 75 and 100 μm from the slice surface). PI staining intensity in each condition was quantified as the background-subtracted average pixel intensity in the CA1 or CA3 pyramidal cell layers of each image, averaged between the two hemispheres. Because of a moderate degree of experiment to experiment variability intrinsic to this paradigm, values in each condition were first normalized to the OGD condition of that experiment (=100%) before averaging across experiments, and statistical assessment was by a paired t test against the OGD condition.
RESULTS
NAS blocks Zn2+ entry through Ca-A/K channels on cultured neurons
To block Ca-A/K channels, we made use of the polyamine Ca-A/K channel pore blocker NAS, a synthetic analog of joro spider toxin (Koike et al., 1997) that has been used previously, much as in this study, to block injury resulting from Ca-A/K channel activation in a hippocampal slice model (Oguro et al., 1999). NAS has been found by electrophysiological studies to block Ca2+entry through Ca-A/K channels in a voltage- and use-dependent manner, with potent (low micromolar) block at resting potentials and decreasing efficacy on depolarized neurons (Koike et al., 1997). Thus, before NAS was used in slice OGD studies, control experiments were performed to characterize its utility as a blocker of Zn2+ entry into neurons through Ca-A/K channels.
Because NAS block is highly voltage dependent, we first examined concentrations needed to block Ca-A/K channels on neurons depolarized by ongoing glutamate receptor activation (as would be expected to be the case during OGD). To screen its efficacy, we made use of a histochemical procedure based on kainate-stimulated uptake of Co2+ ions that identifies the subpopulation of neurons possessing large numbers of Ca-A/K channels [Ca-A/K(+) neurons] (Pruss et al., 1991; Yin et al., 1994). The specificity of the stain depends on selective permeability of Co2+ ions through Ca-A/K channels and has been demonstrated by the inability of NMDA or high-K+ depolarization (to activate VSCCs) to trigger comparable Co2+ uptake. We found that 300 μm but not 100 μm NAS effectively blocked the Co2+ uptake (data not shown); thus, we used this concentration in subsequent studies. Additional control experiments were undertaken to rule out the possibility that NAS chelates extracellular Zn2+ and prevents Zn2+ entry through that mechanism rather than by blocking channels. Cortical neuronal cultures were loaded with the Zn2+-sensitive (and Ca2+-insensitive) fluorescent probe Newport Green and exposed for 5 min to 300 μmZn2+ in 60 mmK+ buffer to induce neuronal depolarization and consequent entry of Zn2+ through VSCCs. With repeat exposures (after 15 min of recovery) in the additional presence of excess NAS (1 mm), there was no decrement in the [Zn2+]i response [peak fluorescence increase of 36 ± 2.1% (SEM) without NAS and 40 ± 3.1% with NAS; n = 44 neurons], indicating that NAS neither appreciably chelates Zn2+nor interferes with Newport Green fluorescence.
Subsequent experiments examined the ability of NAS to specifically block Zn2+ entry through Ca-A/K channels. Newport Green-loaded cultures were exposed to kainate (100 μm) in the presence of Zn2+(300 μm), the NMDA channel blocker MK-801 (10 μm), and NAS (300 μm) for 5 min. After 20 min, the cultures were subjected to a second 5 min exposure to an identical solution lacking NAS. After imaging, Ca-A/K(+) neurons were identified by kainate-stimulated Co2+uptake labeling, as described above. In the presence of NAS, all neurons showed moderate increases in [Zn2+]i, indicative of depolarization and Zn2+entry through VSCCs. After removal of the NAS, however, unblocking of Ca-A/K channels resulted in greater [Zn2+]i increases in Ca-A/K(+) neurons (Fig. 1). Thus, NAS appears to be able to effectively block Zn2+ entry through Ca-A/K channels of depolarized neurons, while having little effect on influx through VSCCs.
As a final control, we examined possible effects of NAS on presynaptic release, an effect that could also contribute to observed sequelae of OGD in slices. Coronal brain slices were loaded with [3H]-d-aspartate before being subjected to OGD in the presence or absence of NAS (300 μm) or to incubation in the presence of O2 and glucose (+O2). We found that both OGD and OGD plus NAS induced significantly more release than +O2([3H]-d-aspartate release of 21.2 ± 6% and 17.1 ± 3.5% in OGD and OGD+NAS, respectively, vs 3.1 ± 0.3% in +O2), but that NAS had no significant effect on the OGD-induced release (n = 4 experiments; OGD and OGD plus NAS different from +O2 at p< 0.05 by ANOVA with a Student–Newman–Keuls post hoctest), indicating the paucity of presynaptic action of NAS.
Ca-A/K channels are a major route for OGD-induced Zn2+ translocation into hippocampal pyramidal neurons
To model ischemia-induced Zn2+translocation under simplified conditions, acute hippocampal slice preparations have distinct advantages: they can be subjected to well-controlled environmental and pharmacological manipulations while maintaining the synaptic connectivity and presynaptic Zn2+ stores present in the animals from which they are derived. Conversely, a potential disadvantage is that Zn2+ is rapidly depleted after slice cutting and incubation (Suh et al., 2000). Thus, certain aspects of the experimental protocol were designed to minimize this nonspecific Zn2+ loss: the slice-stabilization step (in cold buffer) is of limited duration, followed by rapid transitioning to the OGD treatment (at 37°C), and slices are subsectioned after OGD exposures to visualize Zn2+ accumulation deep in the slices, where direct trauma-induced release is minimized.
Coronal slices (400 μm) were obtained from adult (8–10 week old) Swiss-Webster mice and immediately placed in “equilibration chambers” containing cold, Ca2+-free equilibration buffer, with the additional presence of NAS (300 μm), MK-801 (15 μm), and the broad-spectrum VSCC antagonist Gd3+ (20 μm) (Canzoniero et al., 1993; Sensi et al., 1997) to allow slices to stabilize and equilibrate in the presence of channel blockers. After 25 min, slices were transferred for 2 min to “pre-OGD chambers,” allowing each slice to partially equilibrate with the drugs with which it is to be subjected to OGD. Slices were then transferred to “OGD chambers” containing glucose-free, O2-depleted OGD buffer (37°C) containing no drugs, MK-801 and Gd3+, NAS, or Ca2+ EDTA (3 mm). One additional slice in each set was handled exactly as the OGD condition but was exposed in the presence of O2 and glucose (+O2). Slices were then subjected to a modified Timm's staining procedure to visualize histochemically reactive intracellular Zn2+ (Yin et al., 1999) and also cut to 25 μm sections for assessment of labeling intensity, as described previously. [For comparison, in some experiments in which slices were stained immediately after removal from the cold equilibration buffer, labeling was comparable with that seen in +O2 (data not shown).]
Slices that were incubated in the presence of O2and glucose showed characteristic strong Zn2+ labeling in the mossy fiber projections from dentate granule cells to CA3 pyramidal neurons (indicative of the high Zn2+ content of this pathway) but little or no staining in pyramidal neurons. In contrast, after OGD, distinct Zn2+staining was consistently evident in pyramidal neurons of both CA1 and CA3 subregions. Although strong staining was also seen in the MK-801 and Gd3+ condition, slices that were treated with either Ca2+ EDTA or NAS showed significantly less Zn2+accumulation in pyramidal neurons of both subregions (Figs.2, 3). Because it is cell impermeant, the block by Ca2+ EDTA strongly supports an extracellular origin for much of the Zn2+, consistent with its derivation from presynaptic release. The additional observation of block by NAS suggests that much of this Zn2+ enters through Ca-A/K channels.
Fig. 2.
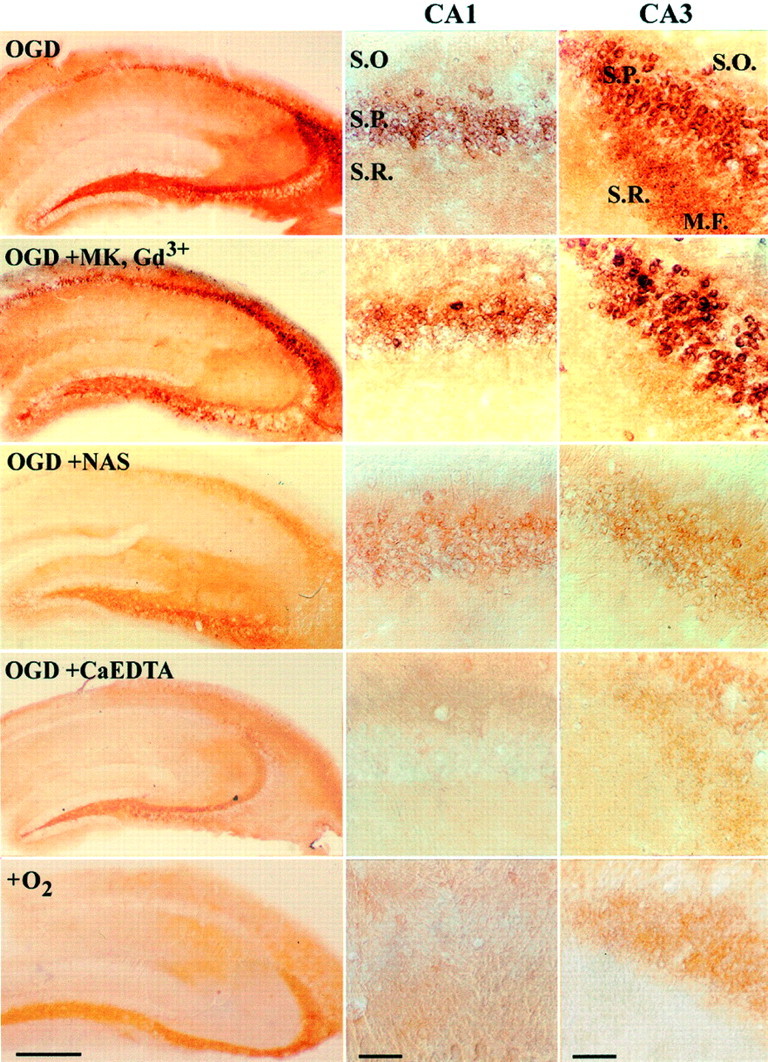
OGD causes translocation of endogenous Zn2+ through Ca-A/K channels on hippocampal pyramidal neurons in slice. After equilibration, coronal slices of adult mouse hippocampus were exposed for 15 min (37°C) to OGD alone (OGD), with the NMDA antagonist MK-801 (15 μm) and the VSCC antagonist Gd3+ (20 μm) (OGD+MK, Gd3+), with the Ca-A/K channel blocker NAS (300 μm) (OGD+NAS), or with the extracellular Zn2+ chelator Ca2+ EDTA (OGD+CaEDTA). One other slice was exposed in the absence of drugs but in the presence of oxygen and glucose-containing media (+O2). After exposures, intracellular Zn2+ was visualized by Timm's staining; high-magnification photomicrographs show detail of the CA1 (middle column) and CA3 (right column) pyramidal layers. M.F., Mossy fibers;S.O., Stratum oriens; S.P., stratum pyramidale; S.R., stratum radiatum. Scale bar, 500 μm (low-power views) or 50 μm (CA1 and CA3 details). Note the paucity of Zn2+ labeling in pyramidal neurons in the absence of OGD exposure, in contrast to the strong accumulation in both CA1 and CA3 pyramidal neurons after OGD. Note also that whereas strong staining occurred despite the presence of NMDA and VSCC blockers, the presence of either NAS or Ca2+ EDTA substantially decreased Zn2+ labeling in both CA1 and CA3 subfields.
Fig. 3.

OGD causes translocation of endogenous Zn2+ through Ca-A/K channels on hippocampal pyramidal neurons in slice: quantitative assessment. Hippocampal slices were exposed for 15 min (37°C) to each of the conditions described above. The graph shows mean Timm's staining intensity (mean ± SEM) of neurons in CA1 and CA3 with each exposure (n = 9; * and # indicate difference from staining intensity in same hippocampal region after OGD; p< 0.05 by ANOVA with Student–Newman–Keuls test).A.U., Arbitrary units.
NAS and Ca2+ EDTA decrease pyramidal neuronal damage resulting after OGD
Subsequent experiments examined the extent of pyramidal neuronal injury resulting after OGD exposure. These studies used the fluorescent cell-death marker PI to assess injury. PI enters injured neurons in which the plasma membrane is disrupted and preferentially accumulates in the nucleus of dying neurons. After OGD, the slices were incubated for 4 hr (22°C) in oxygenated media containing the same antagonists that were present during the exposures to permit evolution of the injury. PI was added for the last 30 min of the incubation, followed by fixation and subsequent visualization of PI fluorescence under confocal microscopy. Images were obtained deep (75–100 μm) within the slice, where injury resulting from the trauma of slicing is minimized.
Because 15 min OGD exposures resulted in severe neurodegeneration in all conditions, these studies used a shorter (5 min) period of OGD. Paralleling the degree of Zn2+accumulation observed in the translocation experiments, relatively little PI fluorescence was seen in the CA1 and CA3 pyramidal cell layers of the +O2 condition, but strong signal was present in the OGD condition. Although strong labeling was also seen with OGD in the presence of MK-801 and Gd3+, PI labeling was decreased substantially by NAS and to a smaller degree by Ca2+ EDTA (significant only in CA1) (Figs. 4, 5). After imaging, in some experiments slices were resectioned to 25 μm and stained with toluidine blue to evaluate structural changes (Fig. 4). Note that the OGD caused extensive distortion, loss, and swelling of CA1 pyramidal neurons and that addition of NAS or Ca2+EDTA resulted in substantial preservation of architecture. Thus, these observations suggest that the Zn2+ that enters postsynaptic pyramidal neurons during OGD contributes to the resultant injury.
Fig. 4.
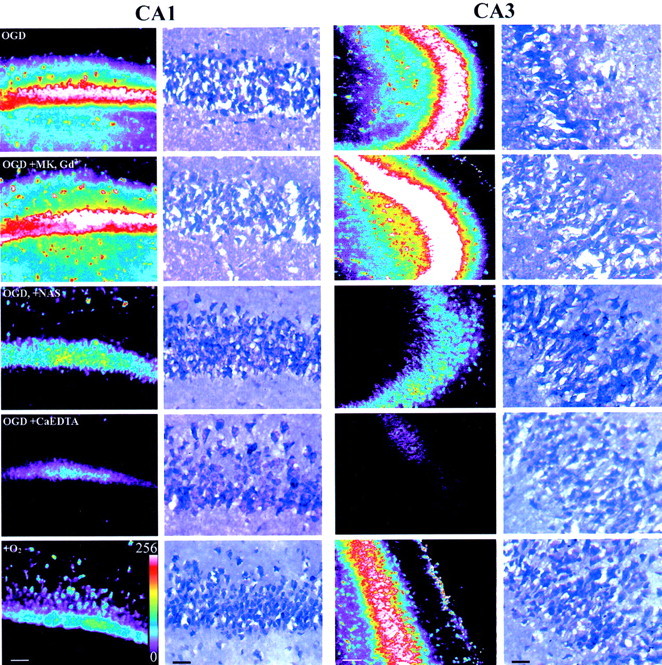
Ca-A/K channel blockade attenuates OGD-induced pyramidal neuronal damage. After equilibration, coronal slices of adult mouse hippocampus were exposed for 5 min (37°C) to OGD alone, with the NMDA antagonist MK-801 (15 μm) and the VSCC antagonist Gd3+ (20 μm) (OGD+MK, Gd3+), with the Ca-A/K channel blocker NAS (300 μm) (OGD+NAS), or with the extracellular Zn2+ chelator Ca2+ EDTA (OGD+CaEDTA). One other slice was exposed in the absence of drugs but in the presence of oxygen and glucose-containing media (+O2). After exposures, slices were incubated for 4 hr at 22°C, and injury was evaluated by confocal imaging of PI labeling in the CA1 and CA3 pyramidal cell layers. In each hippocampal region, the left column shows a set of confocal images from a single experiment displayed on an eight bit pseudocolor scale. After imaging, matched slices from a single experiment were sectioned to 25 μm and stained with toluidine blue (right columns). Note the disruption of neuronal morphology and loss of neurons (indicated by voids) in the CA1 pyramidal cell layer after OGD alone or with MK-801 and Gd3+ and the relatively preserved morphology in other conditions. Scale bars, 50 μm.
Fig. 5.

Quantification of neuronal injury. After equilibration, coronal slices of adult mouse hippocampus were exposed for 5 min (37°C) to OGD alone, with the NMDA antagonist MK-801 (15 μm) and the VSCC antagonist Gd3+ (20 μm) (+MK, Gd3+), with the Ca-A/K channel blocker NAS (300 μm) (+NAS), or with the extracellular Zn2+ chelator Ca2+ EDTA (+Ca-EDTA). One other slice was exposed in the absence of drugs but in the presence of oxygen and glucose-containing media (+O2). After exposures, slices were incubated for 4 hr at 22°C, and injury was evaluated by confocal imaging of PI labeling in the CA1 and CA3 pyramidal cell layers. The graph shows mean PI fluorescence (±SEM) in the CA1 (left bars) and CA3 (right bars) pyramidal cell layers with each exposure, scaled to the fluorescence of the same hippocampal subregion in the OGD condition (n = 8–9; * and # indicate difference from OGD in the same subregion;p < 0.05 by paired t test).
DISCUSSION
Despite the considerable evidence supporting a role for Zn2+ translocation in the hippocampal pyramidal neuronal injury resulting after transient global ischemia or prolonged seizures, routes through which synaptic Zn2+ might enter postsynaptic neuronsin vivo have not been examined. Using the system of acute hippocampal slice as an in vitro model of ischemic injury, we found that brief periods of OGD induce substantial accumulation of histochemically detectable Zn2+ in hippocampal pyramidal neurons and the subsequent degeneration of these neurons. Strong Zn2+ accumulation and pyramidal neuronal injury were still observed if OGD exposures were performed in the presence of combined NMDA channel and VSCC blockade. However, both of these measures were significantly decreased by the presence of either the extracellular Zn2+chelator Ca2+ EDTA or the Ca-A/K channel blocker NAS.
The positive findings of the present studies need to be considered in the context of certain ongoing areas of uncertainty regarding ways in which Zn2+ contributes to hippocampal neurodegeneration after ischemia or epilepsy. The first concerns the origin of the reactive Zn2+ accumulation in pyramidal neurons occurring in these conditions. It is clear that Zn2+ is present in vesicles and is released in response to presynaptic activity (Assaf and Chung, 1984;Howell et al., 1984; Thompson et al., 2000). But recent generation of transgenic mice that appear to completely lack vesicular Zn2+ (in which the vesicular Zn2+ transporter ZnT3 is knocked out) has raised questions about the source of the postsynaptic Zn2+ accumulation. In these mice, prolonged seizures still caused the appearance of reactive Zn2+ accumulation in and degeneration of hippocampal pyramidal neurons (Cole et al., 2000; Lee et al., 2000). Although the source of the Zn2+ is uncertain, it is likely that much of it comes from release from intracellular stores after strong excitotoxic activation. Zn2+ is a component of many metalloenzymes (Vallee and Falchuk, 1993), and Zn2+binding proteins such as metallothioneins are hypothesized to be important endogenous Zn2+ buffers (Aschner et al., 1997). Indeed, a recent study found that oxidant exposure caused elevation of cytosolic Zn2+ levels in cultured forebrain neurons. The additional observation of that study that Zn2+ chelators decreased the injury suggested that intracellular Zn2+ release might even contribute to neuronal injury (Aizenman et al., 2000).
Thus, one highly relevant question is the degree to which the Zn2+ that appears in pyramidal neurons after OGD represents translocation from presynaptic terminals or cytosolic release of Zn2+ already present in the neurons. The observation that Ca2+EDTA decreased Zn2+ accumulation as assessed immediately after a 15 min period of OGD provides strong evidence that a substantial portion of the Zn2+ is of extracellular origin, as would be predicted by the Zn2+ translocation model. The additional observation that Ca2+ EDTA appeared to mildly decrease injury in the CA1 subfield 4 hr after an OGD exposure also suggests that the Zn2+ entering the pyramidal neurons contributes to their injury. The fact that the protection by Ca2+ EDTA appears to be relatively mild might be compatible with an increased Ca2+-dependent component to the injury. Indeed, because Zn2+ is a potent antagonist of NMDA channels (Peters et al., 1987; Westbrook and Mayer, 1987), removal of synaptic Zn2+ by Ca2+ EDTA (or its absence in ZnT3 knock-out mice) could result in increased Ca2+ entry through NMDA channels. Alternatively, previous studies (Vogt et al., 2000; Li et al., 2001) have suggested that Ca2+ EDTA may be slow to chelate rapid synaptic Zn2+ increases and thus may fail to fully prevent Zn2+interaction with postsynaptic receptors.
A second critical question concerns the route through which Zn2+ gains entry to the pyramidal neurons. As discussed in the introductory remarks, we thought it unlikely that NMDA channels, which are poorly Zn2+permeable and are potently blocked by Zn2+, could permit much Zn2+ entry; thus, we set out to examine the potential role of highly Zn2+-permeable Ca-A/K channels. Indeed, observations that NAS attenuates both Zn2+accumulation and the subsequent neuronal cell death to a far greater degree than potent combined blockade of VSCC and NMDA channels implicate Ca-A/K channel activation in both of these events. However, the presence of Ca-A/K channels on pyramidal neurons is controversial; although most electrophysiological studies in slice have failed to detect them, one in which glutamate was locally applied to acutely dissociated pyramidal neurons found evidence for their presence in distal dendrites (Lerma et al., 1994). Other evidence favoring their presence has been based on kainate-stimulated Co2+ uptake labeling (Pruss et al., 1991;Williams et al., 1992; Toomim and Millington, 1998; Yin et al., 1999) and on immunostaining for AMPA subunits. Although AMPA channels are made up of combinations of subunits (GluR1–4), the Ca2+ permeability of these channels is regulated by the GluR2 subunit, the presence of which in a heteromeric channel blocks Ca2+ permeability. Consistent with a preferential dendritic localization of Ca-A/K channels, several studies have reported an apparent gradient in the intensity of the GluR2 label, with strong somatic staining and less in the distal dendrites (Vickers et al., 1993; Ikonomovic et al., 1995;Yin et al., 1999). Thus, our present observations extend these previous studies in providing new evidence for the presence of functional Ca-A/K channels in postsynaptic membranes of hippocampal pyramidal neurons adjacent to sites of Zn2+ release. Other recent studies have raised the intriguing possibility that the numbers of Ca-A/K channels on pyramidal neurons might not be constant. Indeed, observations that GluR2 mRNA and protein may be selectively decreased in hippocampal pyramidal neurons after transient global ischemia or prolonged seizures led Zukin and colleagues (Bennett et al., 1996;Pellegrini-Giampietro et al., 1997) to propose the “GluR2 hypothesis,” which suggests that these decreases in GluR2 result in increased numbers of Ca-A/K channels, thereby permitting more Ca2+ or Zn2+entry and contributing to the delayed neurodegeneration often seen in these conditions.
Although an ischemia-induced increase in the numbers of Ca-A/K channels might be expected to play a late role in injury, the present results suggest that with strong presynaptic activation, basal numbers of Ca-A/K channels permit sufficient Zn2+entry to mediate rapid neuronal damage. Although it is of interest to consider the potential physiological significance of Ca-A/K channel regulation and trans-synaptic Zn2+signaling, the simple demonstration that endogenous Zn2+ permeates through Ca-A/K channels of adult brain in an in vitro model of ischemia could have important therapeutic implications. Indeed, neuroprotective trials with NMDA antagonists have been generally disappointing, whereas AMPA/kainate receptor antagonists have demonstrated surprisingly good efficacy in certain ischemia models (Diemer et al., 1992). The poor efficacy of NMDA antagonists could be explained in part if NMDA channels were already substantially blocked by synaptic Zn2+, whereas the efficacy of AMPA/kainate antagonists might reflect the presence of functionally significant numbers of Ca-A/K channels on hippocampal pyramidal neurons and their selective high permeability to Zn2+. The present observations may thus provide new rationale for neuroprotective strategies targeting Ca-A/K channels and Zn2+ passage through them in conditions of ischemia or epilepsy, which are associated with rapid synaptic Zn2+ release.
Footnotes
This work was supported by National Institutes of Health Grants NS30884 and AG00836 (J.H.W.), AG00919 (S.L.S.), and 5T32NS07444 (F.O.), and by a grant from the Alzheimer's Association (J.H.W.). We thank Simin Amindari and Dien Ton-That for expert assistance with the cell cultures.
Correspondence should be addressed to John H. Weiss, Departments of Neurology, Anatomy and Neurobiology, and Neurobiology and Behavior, University of California, Irvine, Irvine, CA 92697-4292. E-mail:jweiss@uci.edu.
REFERENCES
- 1.Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- 2.Aschner M, Cherian MG, Klaassen CD, Palmiter RD, Erickson JC, Bush AI. Metallothioneins in brain: the role in physiology and pathology. Toxicol Appl Pharmacol. 1997;142:229–242. doi: 10.1006/taap.1996.8054. [DOI] [PubMed] [Google Scholar]
- 3.Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–736. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- 4.Bennett MV, Pellegrini-Giampietro DE, Gorter JA, Aronica E, Connor JA, Zukin RS. The GluR2 hypothesis: Ca++-permeable AMPA receptors in delayed neurodegeneration. Cold Spring Harb Symp Quant Biol. 1996;61:373–384. [PubMed] [Google Scholar]
- 5.Canzoniero LM, Taglialatela M, Di Renzo G, Annunziato L. Gadolinium and neomycin block voltage-sensitive Ca2+ channels without interfering with the Na+-Ca2+ antiporter in brain nerve endings. Eur J Pharmacol. 1993;245:97–103. doi: 10.1016/0922-4106(93)90116-q. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW, Yokoyama M, Koh J. Zinc neurotoxicity in cortical cell culture. Neuroscience. 1988;24:67–79. doi: 10.1016/0306-4522(88)90312-0. [DOI] [PubMed] [Google Scholar]
- 7.Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 2000;39:153–169. doi: 10.1016/s0920-1211(99)00121-7. [DOI] [PubMed] [Google Scholar]
- 8.Diemer NH, Jorgensen MB, Johansen FF, Sheardown M, Honore T. Protection against ischemic hippocampal CA1 damage in the rat with a new non-NMDA antagonist, NBQX. Acta Neurol Scand. 1992;86:45–49. doi: 10.1111/j.1600-0404.1992.tb08052.x. [DOI] [PubMed] [Google Scholar]
- 9.Frederickson CJ, Hernandez MD, McGinty JF. Translocation of zinc may contribute to seizure-induced death of neurons. Brain Res. 1989;480:317–321. doi: 10.1016/0006-8993(89)90199-6. [DOI] [PubMed] [Google Scholar]
- 10.Frederickson CJ, Suh SW, Silva D, Frederickson CJ, Thompson RB. Importance of zinc in the central nervous system: the zinc-containing neuron. J Nutr. 2000;130:1471S–1483S. doi: 10.1093/jn/130.5.1471S. [DOI] [PubMed] [Google Scholar]
- 11.Howell GA, Welch MG, Frederickson CJ. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- 12.Ikonomovic MD, Sheffield R, Armstrong DM. AMPA-selective glutamate receptor subtype immunoreactivity in the aged human hippocampal formation. J Comp Neurol. 1995;359:239–252. doi: 10.1002/cne.903590205. [DOI] [PubMed] [Google Scholar]
- 13.Kass IS, Lipton P. Mechanisms involved in irreversible anoxic damage to the in vitro rat hippocampal slice. J Physiol (Lond) 1982;332:459–472. doi: 10.1113/jphysiol.1982.sp014424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 15.Koike M, Iino M, Ozawa S. Blocking effect of 1-naphthyl acetyl spermine on Ca2+-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci Res. 1997;29:27–36. doi: 10.1016/s0168-0102(97)00067-9. [DOI] [PubMed] [Google Scholar]
- 16.Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 17. Lee JY, Cole TB, Palmiter RD, Koh JY. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci 20 2000. RC79:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerma J, Morales M, Ibarz JM, Somohano F. Rectification properties and Ca2+ permeability of glutamate receptor channels in hippocampal cells. Eur J Neurosci. 1994;6:1080–1088. doi: 10.1111/j.1460-9568.1994.tb00605.x. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Hough CJ, Frederickson CJ, Sarvey JM. Induction of mossy fiber → Ca3 long-term potentiation requires translocation of synaptically released Zn2+. J Neurosci. 2001;21:8015–8025. doi: 10.1523/JNEUROSCI.21-20-08015.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monette R, Small DL, Mealing G, Morley P. A fluorescence confocal assay to assess neuronal viability in brain slices. Brain Res Brain Res Protoc. 1998;2:99–108. doi: 10.1016/s1385-299x(97)00020-2. [DOI] [PubMed] [Google Scholar]
- 21.Oguro K, Oguro N, Kojima T, Grooms SY, Calderone A, Zheng X, Bennett MV, Zukin RS. Knockdown of AMPA receptor GluR2 expression causes delayed neurodegeneration and increases damage by sublethal ischemia in hippocampal CA1 and CA3 neurons. J Neurosci. 1999;19:9218–9227. doi: 10.1523/JNEUROSCI.19-21-09218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–470. doi: 10.1016/s0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
- 23.Peters S, Koh J, Choi DW. Zinc selectively blocks the action of N-methyl-d-aspartate on cortical neurons. Science. 1987;236:589–593. doi: 10.1126/science.2883728. [DOI] [PubMed] [Google Scholar]
- 24.Pruss RM, Akeson RL, Racke MM, Wilburn JL. Agonist-activated cobalt uptake identifies divalent cation-permeable kainate receptors on neurons and glial cells. Neuron. 1991;7:509–518. doi: 10.1016/0896-6273(91)90302-g. [DOI] [PubMed] [Google Scholar]
- 25.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 26.Sensi SL, Canzoniero LM, Yu SP, Ying HS, Koh JY, Kerchner GA, Choi DW. Measurement of intracellular free zinc in living cortical neurons: routes of entry. J Neurosci. 1997;17:9554–9564. doi: 10.1523/JNEUROSCI.17-24-09554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sloviter RS. A selective loss of hippocampal mossy fiber Timm stain accompanies granule cell seizure activity induced by perforant path stimulation. Brain Res. 1985;330:150–153. doi: 10.1016/0006-8993(85)90017-4. [DOI] [PubMed] [Google Scholar]
- 29.Suh SW, Koh JH, Choi DW. Extracellular zinc mediates selective neuronal death in hippocampus and amygdala following kainate-induced seizure. Soc Neurosci Abstr. 1996;22:823.6. [Google Scholar]
- 30.Suh SW, Danscher G, Jensen MS, Thompson R, Motamedi M, Frederickson CJ. Release of synaptic zinc is substantially depressed by conventional brain slice preparations. Brain Res. 2000;879:7–12. doi: 10.1016/s0006-8993(00)02675-5. [DOI] [PubMed] [Google Scholar]
- 31.Tang CM, Dichter M, Morad M. Modulation of the N-methyl-d-aspartate channel by extracellular H+. Proc Natl Acad Sci USA. 1990;87:6445–6449. doi: 10.1073/pnas.87.16.6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thompson RB, Whetsell WO, Jr, Maliwal BP, Fierke CA, Frederickson CJ. Fluorescence microscopy of stimulated Zn(II) release from organotypic cultures of mammalian hippocampus using a carbonic anhydrase-based biosensor system. J Neurosci Methods. 2000;96:35–45. doi: 10.1016/s0165-0270(99)00183-1. [DOI] [PubMed] [Google Scholar]
- 33.Tonder N, Johansen FF, Frederickson CJ, Zimmer J, Diemer NH. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci Lett. 1990;109:247–252. doi: 10.1016/0304-3940(90)90002-q. [DOI] [PubMed] [Google Scholar]
- 34.Toomim CS, Millington WR. Regional and laminar specificity of kainate-stimulated cobalt uptake in the rat hippocampal formation. J Comp Neurol. 1998;402:141–154. [PubMed] [Google Scholar]
- 35.Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-d-aspartate receptors in cerebellar neurons. Nature. 1990;345:347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- 36.Vallee BL, Falchuk KH. The biochemical basis of zinc physiology. Physiol Rev. 1993;73:79–118. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- 37.Vickers JC, Huntley GW, Edwards AM, Moran T, Rogers SW, Heinemann SF, Morrison JH. Quantitative localization of AMPA/kainate and kainate glutamate receptor subunit immunoreactivity in neurochemically identified subpopulations of neurons in the prefrontal cortex of the macaque monkey. J Neurosci. 1993;13:2982–2992. doi: 10.1523/JNEUROSCI.13-07-02982.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- 39.Weiss JH, Hartley DM, Koh JY, Choi DW. AMPA receptor activation potentiates zinc neurotoxicity. Neuron. 1993;10:43–49. doi: 10.1016/0896-6273(93)90240-r. [DOI] [PubMed] [Google Scholar]
- 40.Westbrook GL, Mayer ML. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature. 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 41.Williams LR, Pregenzer JF, Oostveen JA. Induction of cobalt accumulation by excitatory amino acids within neurons of the hippocampal slice. Brain Res. 1992;581:181–189. doi: 10.1016/0006-8993(92)90707-g. [DOI] [PubMed] [Google Scholar]
- 42.Yin H, Turetsky D, Choi DW, Weiss JH. Cortical neurones with Ca2+ permeable AMPA/kainate channels display distinct receptor immunoreactivity and are GABAergic. Neurobiol Dis. 1994;1:43–49. doi: 10.1006/nbdi.1994.0006. [DOI] [PubMed] [Google Scholar]
- 43.Yin HZ, Weiss J. Zn2+ permeates Ca2+ permeable AMPA/kainate channels and triggers selective neural injury. NeuroReport. 1995;6:2553–2556. doi: 10.1097/00001756-199512150-00025. [DOI] [PubMed] [Google Scholar]
- 44.Yin HZ, Sensi SL, Carriedo SG, Weiss JH. Dendritic localization of Ca2+-permeable AMPA/kainate channels in hippocampal pyramidal neurons. J Comp Neurol. 1999;409:250–260. [PubMed] [Google Scholar]