

Abstract
The basolateral amygdala (BLA) is believed to be involved in schizophrenia, depression, and other disorders that display affective components. The neuronal activity of the BLA, and BLA-mediated affective behaviors, are driven by sensory stimuli transmitted in part from sensory association cortical regions. These same behaviors may be regulated by prefrontal cortical (PFC) inputs to the BLA. However, it is unclear how two sets of glutamatergic inputs to the BLA can impose opposing actions on BLA-mediated behaviors; specifically, it is unclear how PFC inputs exert inhibitory actions over BLA projection neurons. Dopamine (DA) receptor activation enhances BLA-mediated behaviors. Although we have demonstrated that DA suppresses medial PFC inputs to the BLA and enhances sensory cortical inputs, the precise cellular mechanisms for its actions are unknown. In this study we usein vivo intracellular recordings to determine the means by which glutamatergic inputs from the PFC inhibit BLA projection neurons, contrast that with glutamatergic inputs from the association sensory cortex (Te3) that drive BLA projection neurons, and examine the effects of DA receptor activation on neuronal excitability, spontaneous postsynaptic potentials (PSPs), and PFC-evoked PSPs. We found that PFC stimulation inhibits BLA projection neurons by three mechanisms: chloride-mediated hyperpolarization, a persistent decrease in neuronal input resistance, and shunting of PSPs; all effects are possibly attributable to recruitment of inhibitory interneurons. DA receptor activation enhanced neuronal input resistance by a postsynaptic mechanism (via DA D2 receptors), suppressed spontaneously occurring and PFC-evoked PSPs (via DA D1 receptors), and enhanced Te3-evoked PSPs.
Keywords: dopamine, basolateral amygdala, prefrontal cortex, sensory association cortex, Te3, in vivointracellular
Cortical inputs to the basolateral amygdala (BLA) are believed to regulate amygdala-mediated behaviors. Sensory cortical areas drive amygdala-mediated behaviors in response to sensory stimuli laden with affective value (LeDoux et al., 1990;Campeau and Davis, 1995; Uwano et al., 1995; Armony et al., 1997;Poremba et al., 1998), whereas prefrontal cortical (PFC) areas suppress some of these behaviors (Al Maskati and Zbrozyna, 1989; Zbrozyna and Westwood, 1991; Jaskiw and Weinberger, 1992; Morgan and LeDoux, 1995;Dias et al., 1996; Jinks and McGregor, 1997). Using extracellular recording techniques, we have demonstrated previously (Rosenkranz and Grace, 2001) that PFC inputs excite inhibitory interneurons of the BLA (composed of the lateral nucleus, basal nucleus, and accessory basal nucleus) (Alheid et al., 1995), and this may be a mechanism by which the PFC regulates BLA output and thus suppresses BLA-mediated behaviors. However, inputs from sensory association cortical areas (Te3, entorhinal and perirhinal cortices) also activate interneurons and produce inhibition (Lang and Pare, 1997, 1998; Szinyei et al., 2000; Rosenkranz and Grace, 2001), but instead of suppression of BLA-mediated behaviors, these inputs appear to drive them. Inhibition may be used to completely suppress output of projection neurons, or it may be able to increase the fidelity of projection neuron firing in response to an input (Hata et al., 1988; Nelson et al., 1994; Vidyasagar et al., 1996; Shevelev et al., 1998; Anderson et al., 2001). On a cellular level, this may appear as cortically evoked feedforward inhibition that excludes projection neuron firing, or as excitation followed by inhibition, respectively (Freund and Buzsaki, 1996; Chung and Ferster, 1998; Anderson et al., 2000). Functionally, the former completely suppresses BLA output in a time-locked manner, whereas the latter sharpens, or tunes, the transmitted signal. To understand the mechanism by which glutamatergic PFC and sensory association cortical inputs implement distinct behavioral actions, the effects of the inputs on BLA neuronal membrane potential and excitability should be examined. In this paper, we have examined the means by which PFC stimulation inhibits BLA neuronal firing while sensory cortical stimulation drives it.
Dopamine (DA) receptor activation alters neuronal activity in a complex manner via effects on afferents as well as seemingly opposing effects on somatodendritic conductances (Cepeda et al., 1993; Geijo-Barrientos and Pastore, 1995; Yang and Seamans, 1996; Flores-Hernandez et al., 1997; Behr et al., 2000). However, from a functional standpoint, this modulation of afferents and somatic excitability may in fact be complementary. We have demonstrated previously that DA receptor activation attenuates PFC inputs to the BLA while enhancing sensory cortical inputs (Rosenkranz and Grace, 2001). Thus, the overall effect of DA is the removal of PFC-induced suppression of neuronal output and enhanced activity driven by sensory cortical inputs. Furthermore, DA receptor activation increases the firing rate of local inhibitory interneurons, inhibiting BLA projection neurons (Ben-Ari and Kelly, 1976; Spehlmann and Norcross, 1984; Rosenkranz and Grace, 1999) and perhaps decreasing spurious responses of BLA projection neurons. Thus, via seemingly opposing effects on inputs, BLA interneurons, and projection neurons, DA may in fact aid in the coordination of an affective response to a sensory stimulus. Although DA appears to exert such actions on a behavioral level (Willick and Kokkinidis, 1995;Borowski and Kokkinidis, 1996; Harmer et al., 1997; Lamont and Kokkinidis, 1998; Guarraci et al., 1999; Nader and LeDoux, 1999; See et al., 2001), the cellular mechanisms and receptor pharmacology that underlie this modulation are unclear. Here we have examined the effects of DA receptor manipulations on neuronal excitability and spontaneously occurring as well as cortically evoked postsynaptic potentials (PSPs), using in vivo intracellular recordings to further elucidate the sites at which DA exerts its actions. A portion of these results have been published previously in abstract form (Rosenkranz and Grace, 2000).
MATERIALS AND METHODS
Materials. Eticlopride, apomorphine, quinpirole, SKF-81297, and SKF-38393 were purchased from Sigma (St. Louis, MO) and dissolved in 0.9% saline to a concentration of 0.5, 1.0, or 3.0 mg/ml. Haloperidol was a generous gift from McNeil Laboratories (Spring House, PA); it was dissolved in dilute lactic acid and then diluted further with 0.9% saline to a concentration of 0.5 mg/ml.
Animal preparation. All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Male Sprague Dawley rats (250–410 gm weight) were housed in pairs in a temperature-controlled environment with a 12 hr on/off light/dark schedule. Food and water were available ad libitum. Rats were anesthetized with an initial injection of 8% chloral hydrate (400 mg/kg, i.p.). Supplemental anesthesia (8% chloral hydrate) was delivered via a lateral tail-vein catheter as necessary to maintain suppression of hindlimb withdrawal reflex. Rats were placed on a temperature-controlled heating pad (Heater VL-20F; Fintronics, Orange, CT) and their temperature was monitored (Precision Thermometer 4600; Yellow Springs Instruments, Yellow Springs, CO) and maintained at ∼37°C. The rats were placed in a stereotaxic device (Kopf Instruments, Tujunga, CA), and incisions were made in the scalp to expose the skull. Burr holes were drilled and the dura was removed overlying the BLA and medial PFC (mPFC) or Te3. Coordinates were determined using a stereotaxic atlas (Paxinos and Watson, 1997), as follows: BLA, −5.0 lateral, −3.3 caudal from bregma; mPFC (prelimbic/infralimbic cortex), −0.7 lateral, +2.2 to +3.0 rostral from bregma; Te3, −6.0 lateral, +2.2 rostral from the interaural line. Concentric bipolar stimulating electrodes (Rhodes Medical Instruments, Inc., obtained from Kopf Instruments) were lowered into the mPFC or Te3 (at the coordinates listed above) to a depth of 5.0 mm. Experiments started no earlier than 45 min after stimulating electrode placement. Neurons were recorded from the lateral and basal nuclei of the amygdala.
Intracellular recordings. Electrodes were constructed using borosilicate glass tubing (1.5 mm outer diameter, 0.84 mm inner diameter; World Precision Instruments, Sarasota, FL) pulled with a Flaming–Brown micropipette puller (model P-80/PC; Sutter Instruments, Novato, CA). Electrodes were filled with 2% biocytin in 3m potassium acetate (Sigma). Impedances were measured in situ and ranged from 45 to 100 MΩ. Electrodes were slowly lowered to the BLA via a hydraulic micromanipulator (model 640, Kopf Instruments). A stimulator (Grass S88; Grass Instruments, Quincy, MA) in series with an intracellular recording amplifier (IR-283; Neuro Data Instruments, New York, NY) and a stimulus isolation unit (PSIU6; Grass Instruments) was used to deliver pulses of constant DC (150 msec, 0.07 nA). Electric potentials were monitored visually with an oscilloscope (Hitachi V134; Hitachi, Tokyo, Japan) and a multimeter (model 179A; Keithley, Cleveland, OH) and audially with a Grass AM5 audio monitor (Grass Instruments). Data were fed to a PC, monitored using custom-designed software (Neuroscope), and stored on a hard disk for analysis offline. Data were also digitized (Neuro-corder DR-484; Neuro Data Instruments) and saved on video tapes using a videocassette recorder (Panasonic AG-1280; Panasonic, Secaucus, NJ). At the completion of experiments, neurons were hyperpolarized with negative current to below spike threshold, and positive DC pulses (0.1–1.0 nA, 250 msec, 2 Hz) were used to eject biocytin from the electrode. Rats were perfused transcardially with 0.85% cold saline followed by 4% paraformaldehyde in 0.1m phosphate buffer. Brains were removed, stored in the perfusion fixative, and then cryoprotected in a 25% sucrose 0.1m phosphate buffer solution. Brains were sliced on a freezing microtome into 60- to 80-μm-thick sections and labeled for biocytin using the Vectastain Elite avidin–biotin complex peroxidase kit (Vector Laboratories Inc., Burlingame, CA). Sections were counterstained with a custom mixture of cresyl violet and neutral red stains, and stimulating electrode and recording electrode placements were identified as described previously (Rosenkranz and Grace, 2001).
Data collection and analysis. After collection of several minutes of baseline data, DC pulses were injected into the neurons (±0.02–1.2 nA; 250–350 msec; 0.2–0.4 Hz). Hyperpolarizing pulses were used to determine the input resistance of the neurons. In some experiments the mPFC or Te3 was stimulated (0.7–1.2 mA; 0.3 msec; Grass S88 stimulator; Grass PSIU6 stimulus isolation unit) at the last 100 msec of the DC pulse to examine the effects of membrane potential on evoked PSPs. For data analysis, a stimulation intensity of 0.7 mA was used for comparisons because it was consistently below action-potential threshold at resting membrane potentials and it evoked responses that were ∼50–75% of maximum PSP amplitude. Drugs were administered intravenously, and the above procedure was repeated.
Mean resting membrane potential was determined from 30 sec sampling periods, and action potentials were eliminated from this analysis. Neuronal input resistance was determined from the voltage deflections resulting from injection of DC pulses. Only hyperpolarizing DC pulses were used for this analysis, and only the linear portion of the plot was included to determine input resistance. In addition, the input resistance was determined only from traces that displayed no co-occurring membrane fluctuations/PSPs. The peak amplitude of stimulus-evoked PSPs was examined as a function of membrane potential. The peak amplitude was defined as the largest difference from prestimulus baseline in the absence of coincident spontaneous PSPs. The peak latency was measured from the time of stimulation. Action-potential threshold was defined as the onset of the rapid transition in membrane potential at the initiation of the spike. The amplitude of the action potential was the voltage difference between the threshold and the action-potential peak. The duration of action potentials was measured as the time between action-potential initiation at spike threshold and the return to baseline preceding the afterhyperpolarization, if present. The membrane potential calculated during intracellular recording was adjusted based on the difference obtained after withdrawal of the electrode from the neuron after the recording. In an attempt to quantify spontaneously occurring PSPs, the SD of the membrane potential was quantified [similar to Pare et al. (1998)]. The SD of the mean membrane potential was calculated from 30 sec epochs. Action potentials were removed from this analysis if they were observed in the 30 sec epoch by setting a cutoff at action-potential threshold. To examine the effect of membrane potential on SD, 15 sec epochs were used while the neuron was directed to different membrane potentials with constant intracellular current injection. The normalized SD (SD at given membrane potential/largest SD measured for that neuron) was plotted against membrane potential and fit with a linear function. The reversal potential was extrapolated to the x-intercept of this plot. Using similar methods, the reversal potentials of components of evoked and spontaneous PSPs were examined. Neurons were excluded from analysis if their action potentials did not overshoot a 0 mV membrane potential or if they were less polarized than −55 mV. Paired t tests were used to examine the effects of drugs on resting membrane potential, SD, action-potential parameters, firing rate, and PSP amplitude. For all other analyses, t tests on grouped data or Pearson correlations were used. If data did not fit a normal distribution, a Mann–Whitney U test was used. For post hoccomparisons, Bonferroni corrections of p values were performed.
RESULTS
Basic electrophysiological properties of BLA neurons
All recordings were obtained from neurons of the lateral or basal nuclei of the BLA. No differences in the physiological properties were observed, except as noted. Projection neurons of the BLA could be distinguished from interneurons based on morphology, longer action-potential duration, and the ability to be antidromically activated (Washburn and Moises, 1992; Rainnie et al., 1993; Sugita et al., 1993; Gaudreau and Pare, 1996). Using a mixture of these criteria, a total of 139 neurons were categorized as projection neurons and were located in the BLA. When recovered, the projection neurons had morphologies consistent with previously described BLA projection neurons (McDonald, 1985; Washburn and Moises, 1992; Rainnie et al., 1993; Sugita et al., 1993), as determined by biocytin labeling (n = 9). They were typically pyramidal-like or oblong neurons, most of which displayed a primary dendrite (Fig.1). When secondary dendrites were filled, dendritic spines were observed. Some of the recorded neurons could be antidromically activated from mPFC stimulation (in that the action potentials exhibited constant latency and arose directly from the resting membrane potential with no evidence of an underlying EPSP, and followed 300 Hz stimulation when tested) (Fig. 1). In addition, these neurons displayed electrophysiological characteristics similar to projection neurons described previously (Washburn and Moises, 1992;Rainnie et al., 1993; Sugita et al., 1993). This consisted of action potentials with a duration of 2.1 ± 0.06 msec (mean ± SEM), 69.0 ± 0.8 mV amplitude, a slow firing rate of 0.09 ± 0.04 Hz (range, 0–0.62 Hz), and a hyperpolarized mean membrane potential (−78.4 ± 0.9 mV). These projection neurons also had a mean action-potential threshold of −55.8 ± mV and input resistances of 38.9 ± 1.4 MΩ (ranging from 15.9 to 79.6 MΩ) (Fig. 1). Most projection neurons displayed clear spike accommodation in response to suprathreshold depolarizing current injection (97 of 114 neurons tested), and delayed rectification of the membrane potential in response to hyperpolarizing current pulses was observed in a subset of neurons.
Fig. 1.

BLA projection neurons. Projection neurons of the BLA can be identified by antidromic activation (A), demonstrated by constant response latency (top, arrow 1) in the absence of a PSP (top, arrow 2), and by the ability to follow high-frequency stimulation (A,bottom). The excitability of these neurons can be estimated by membrane deflections in response to intracellular current injection (B, projection neuron; C, interneuron), yielding a measurement of input resistance (D, black circles, projection neurons, 32 MΩ; white circles, interneurons, 46 MΩ). Also note that projection neurons display spike accommodation, whereas interneurons of the BLA do not (B, C), and interneurons exhibit shorter duration action potentials (E, bottom trace) when compared with projection neurons (E, top trace). Neurons identified post hoc had morphologies consistent with projection neurons (F); Scale bar, 20 μm.
The level of spontaneous membrane fluctuations varied dramatically between BLA neurons; some displayed little fluctuation, whereas others displayed activity reminiscent of the bistable membrane states observed in cortical and striatal neurons (Steriade et al., 1993; Wilson, 1993;O'Donnell and Grace, 1995; Stern et al., 1997), with transitions of close to 10 mV. However, unlike the bistable neurons of the striatum, BLA neurons were not truly bistable. Thus, although membrane depolarizations were observed, the membrane potential of these plateaus varied in an almost continuous nature, stable plateaus were rare, and the duration was highly variable (Fig.2). The frequency and magnitude of membrane potential fluctuations in the BLA neurons can be roughly estimated with the SD of the membrane potential (Pare et al., 1998). The mean SD was 2.69 ± 0.13 mV, and ranged from 0.58 to 7.10. These fluctuations may be attributable to barrages of PSPs, intrinsic subthreshold oscillations, or a combination of the two. There appeared to be little or no effect of membrane voltage (−120 to −70 mV) on the frequency of these oscillations, whereas the amplitude was sensitive to the membrane potential (Fig. 2), indicating that the occurrence of these fluctuations is not attributable to intrinsic oscillations of the membrane potential (Pare et al., 1995) but is probably synaptic in nature. Under the assumption that the SD approaches zero near the reversal potential of the membrane fluctuations represented by the SD, additional evidence that these membrane fluctuations are attributable to synaptic potentials is derived from extrapolation of the reversal potential of the normalized SD as a function of membrane potential (Fig. 2) (n = 14 neurons). The reversal potential was extrapolated to −23 mV (using the linear portion of the plot of SD as a function of membrane potential and non-normalized data), consistent with PSPs. To further validate this possibility, the amplitudes of large spontaneous PSPs were measured over varying membrane potentials and analyzed as a function of membrane potential. The reversal potential was extrapolated to −25 mV. In addition, the difference between the mean membrane potential and the resting membrane potential was plotted as a function of the resting membrane potential (the reversal potential was extrapolated to −36 mV) (Fig.2F, inset). Thus, the magnitude of the spontaneous fluctuations decreased with depolarizing membrane potentials, consistent with a reduction of driving force for spontaneous PSPs. However, it should be noted that in two neurons depolarization above approximately −60 mV resulted in fast (∼10 Hz) oscillations similar to oscillations reported previously (Pape et al., 1998). Only a weak, insignificant correlation between the SD of the membrane potential and the input resistance was seen here (Pearsonr2 = 0.03; p > 0.05) (Fig. 2), indicating that our measurement of input resistance was not dependent on the level of synaptic activity.
Fig. 2.

Spontaneous activity of BLA projection neurons. BLA neurons display spontaneous membrane deflections (A) that can be estimated by the SD of the membrane potential (SD is 2.8 mV in this case); this SD can be demonstrated by a histogram of the time spent at a given membrane potential (B). The fluctuations are not systematically associated with input resistance (C), but the amplitude is dependent on the membrane potential (D) (also note the reversal of many PSPs near −65 mV, as demonstrated by hyperpolarizing PSPs), as indicated by a plot of normalized SD as a function of membrane potential (E) (group data, n= 14). This indicates that the fluctuations are likely to reflect spontaneous inputs. The inset in E plots the difference between the mean membrane potential and the resting membrane potential in several neurons (n = 14) as a function of membrane potential. Voltages listed beside intracellular traces are the resting membrane potentials.
Fewer neurons that fit the classification of interneurons were successfully recorded (n = 6). These neurons displayed shorter duration action potentials (1.1 ± 0.1 msec), less polarized resting membrane potentials (−66.0 ± 3.6), a spike threshold closer to the resting membrane potential (−61 ± 2 mV), nonaccommodating spike trains in response to depolarizing current injection, and a tendency toward larger input resistance values (46 ± 12 MΩ). The spontaneous membrane fluctuations at resting membrane potentials could not be accurately measured because of the significantly higher frequency of action-potential discharge (2.2 ± 0.9 Hz, p < 0.05 t testt = 8.1) that was often followed by long-duration hyperpolarizations that may be afterhyperpolarizing potentials and/or feedback-collateral inhibition.
Properties of Te3- and PFC-evoked PSPs
As may be expected based on behavioral evidence (see the introductory remarks) and as extrapolated from our previous extracellular recording studies (Rosenkranz and Grace, 2001), there were dramatic differences between the PFC- and Te3-evoked actions.
Stimulation of the mPFC resulted in PSPs with an amplitude that was dependent on stimulus intensity (Fig. 3). At resting membrane potentials, the response was a depolarizing potential (6.5 ± 0.8 mV; n = 39; peak latency, 29.4 ± 1.5 msec) that was sometimes followed by a longer duration hyperpolarization. However, it became evident that the initial depolarizing potential was not excitatory in nature when the membrane potential was directed to less polarized potentials using intracellular current injection (Fig. 3). Thus, at depolarized membrane potentials (typically less than −60 mV), PFC stimulation resulted in a hyperpolarization of the membrane potential. The reversal potential of the first component was −66.6 mV (n = 39), consistent with a GABA-activated chloride conductance (Fig. 3). The second component was typically of a small magnitude and could not be measured accurately; however, it appeared to reverse between −90 and −100 mV, consistent with a potassium conductance. In some instances, a depolarizing potential (8.3 ± 1.6 mV at resting membrane potentials; peak latency, 19.9 ± 1.5 msec; 0.7 mA stimulation) could be seen preceding (n = 3 of 39 neurons; all 3 neurons in the lateral nucleus) or within (n = 6 of 39 neurons) the initial, presumably GABAergic, hyperpolarizing potential. This small depolarizing potential did not reverse over the membrane potentials tested (−140 to −50 mV; reversal potential extrapolated to −34.2 mV) and may reflect a glutamatergic component that is entirely overwhelmed by concurrent GABAergic effects. PFC stimulation evoked a monosynaptic spike in 3 of 39 neurons only, and this spike occurred at the peak of this latter depolarizing component. Attempts to examine the EPSP component at the reversal potential of the IPSP were not successful because of the small amplitude of the EPSP at this membrane potential. Thus, mPFC stimulation typically resulted in an IPSP that precluded spike firing and usually occluded any EPSP if present. This mPFC-evoked inhibition was present in the vast majority of neurons encountered, regardless of recording location within the BLA.
Fig. 3.

Response to mPFC stimulation.A, The PSP amplitudes evoked by mPFC stimulation are stimulus intensity-dependent. (Voltage traces of responses to 0.1, 0.4, and 0.7 mA of stimulation are displayed.) Intracellular voltage traces indicate that mPFC stimulation evokes PSPs that reverse close to the chloride reversal potential (B, arrow 2) and sometimes PSPs that are extrapolated to reverse near −30 mV (B, arrow 1).C, The reversal potential of the primary PSP evoked in the neuron in B, demonstrated by the intersection of baseline membrane potential (filled circles) and PSP amplitude (open circles) in a plot of PSP amplitude as a function of membrane potential, is −67 mV. The grouped data (n = 39) plot of the PSP amplitude by membrane potential (D) for the EPSPs (white circles) and the IPSPs (black circles) demonstrates that the IPSPs reverse near the chloride reversal potential (dashed line), whereas the EPSPs are extrapolated to reverse near −20 mV (solid line). In contrast, mPFC-evoked responses in striatal neurons do not reverse near the chloride reversal potential (E). Upward arrows indicate mPFC stimulation.
The mPFC-evoked IPSP is probably disynaptic, and not caused by stimulation of an as yet unknown GABAergic projection, because PSPs evoked from the same stimulation sites, recorded from striatal neurons, do not reverse between −50 and −130 mV (Fig. 3). Therefore, this IPSP is probably the result of mPFC excitation of BLA inhibitory interneurons. This is supported by mPFC-evoked depolarizations and bursts of action potentials at resting membrane potentials in BLA neurons tentatively identified as interneurons (n = 3 of 3 neurons) (Fig. 4) based on the criteria described above. Additional evidence that the mPFC-evoked IPSP is not monosynaptic is derived from the decrease in onset latency with increased stimulation intensity and from the additional components of the response that are evoked in a nonlinear manner with increasing stimulation intensity (Fig. 3).
Fig. 4.
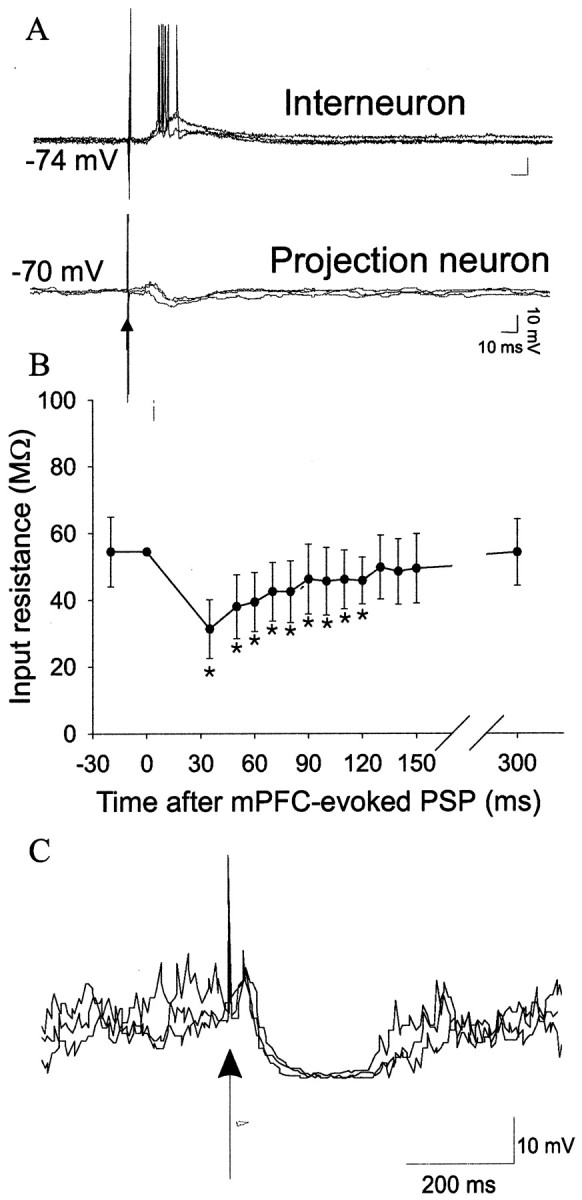
Inhibition evoked by mPFC stimulation. mPFC stimulation evokes a depolarizing potential and spikes in interneurons (A, top) as well as hyperpolarization in projection neurons (A, bottom). In addition to the hyperpolarization, mPFC stimulation produces a prolonged decrease in neuronal input resistance (B) and shunts spontaneous PSPs (C), as reflected by the lack of spontaneous PSPs after mPFC stimulation. Upward arrows indicate mPFC stimulation. *Significant difference from baseline; p < 0.05; paired t test.
Injections of brief hyperpolarizing current pulses (0.3–0.7 nA, 70–200 msec; n = 5) after mPFC stimulation demonstrated a decrease in input resistance, lasting ∼100 msec from the onset of the mPFC-evoked PSP (Fig. 4). The input resistance remained significantly decreased for 120 msec compared with prestimulus input resistance (paired t tests, p < 0.05 with Bonferroni correction; df = 4) and returned to near baseline input resistance values by 300 msec, indicating that mPFC inputs inhibit BLA projection neurons by the chloride-mediated hyperpolarization detailed above as well as by a persistent attenuation of neuronal somatic excitability. It was also noted that mPFC stimulation often resulted in a period of reduced spontaneous PSPs (Fig. 4), perhaps indicative of GABAergic shunting of dendritic events.
Stimulation of Te3 also evoked PSPs with an amplitude that was dependent on stimulation intensity (Fig.5). Most Te3-evoked responses (17 of 18 responses) were recorded from neurons of the lateral nucleus. Neurons outside of the lateral nucleus did not display robust monosynaptic responses to Te3 stimulation. Similar to mPFC stimulation, a depolarizing potential was evoked at resting membrane potentials. However, as became evident with manipulations of the membrane potential, there was an initial component that did not reverse at the chloride reversal potential (15.2 ± 1.8 mV; n = 18; reversal potential extrapolated to −16.6 mV; peak latency, 16.5 ± 0.8 msec) and often resulted in action potentials at more depolarized membrane potentials, usually (11 of 18 neurons) followed by a component that reversed at −64.6 mV (n = 18; 8.2 ± 1.9 mV at resting membrane potentials; peak latency, 29.4 ± 1.6 msec). Thus, Te3 stimulation typically resulted in an EPSP that was truncated by an IPSP. Although truncated by the IPSP, the EPSP was always of sufficient magnitude to evoke action-potential discharge.
Fig. 5.

Excitatory response to Te3 stimulation.A, Te3 stimulation evokes an intensity-dependent PSP. (Voltage traces of responses to 0.1, 0.4, and 0.7 mA of stimulation are displayed.) This PSP is composed of a component that depolarizes at all membrane potentials tested (B,arrow 1) and a component that appears to reverse near the chloride reversal potential (B, arrow 2). In this neuron, the second component reverses at −65 mV, as determined by the intersection of the membrane potential (C, dashed line, time point 2 inB) and the IPSP amplitude in response to hyperpolarizing pulses of current (C, solid line, time point 1 in B). D, In grouped data (n = 18 neurons), it can be demonstrated that the IPSPs evoked by Te3 stimulation (black circles) reverse near −65 mV (dashed line), whereas the evoked EPSPs (white circles) reverse near −20 mV (solid line) in a plot of PSP amplitude as a function of membrane potential. Upward arrows indicate Te3 stimulation in all traces.
DAergic modulation of neuronal properties
Administration of the nonselective DA agonist apomorphine (1.0 ± 0.15 mg/kg, i.v.; n = 8) consistently increased input resistance (mean of 29%; 7 of 7 neurons; before apomorphine, 40.7 ± 2.8 MΩ; after apomorphine, 52.0 ± 5.4 MΩ; p < 0.05; paired t testt = −2.88; df = 6) (Fig.6) and decreased the amplitude of spontaneous membrane fluctuations (mean of −31%; 6 of 7 neurons; SD before apomorphine, 2.87 ± 0.17 mV; SD after apomorphine, 1.99 ± 0.14 mV; p < 0.05; pairedt test; t = 4.80; df = 6) (Fig.7). No significant or consistent changes were seen in action-potential threshold, action-potential duration or amplitude, resting membrane potential, or firing rate (Table1). There was only a weak, nonsignificant, correlation between the change in input resistance and the change in SD after apomorphine administration (Pearsonr2 = 0.27).
Fig. 6.

DA receptor activation enhances neuronal excitability. Apomorphine (1.2 mg/kg, i.v.) increases measured voltage deflections in response to pulses of current injection (A, left, before apomorphine) resulting in enhanced input resistance and depolarization-evoked spike discharge (A, right, after apomorphine). This was reflected as an increase in input resistance (B) (before apomorphine, solid circles,Rin = 38 MΩ; after apomorphine,open circles, Rin = 47 MΩ).C, In all neurons tested, apomorphine administration increased neuronal input resistance; *p < 0.05.
Fig. 7.

DAergic effects on spontaneous PSPs. DA receptor activation (apomorphine, 1.2 mg/kg, i.v.) suppresses spontaneous PSPs (A, before apomorphine, 3.1 mV, after apomorphine, 1.7 mV, measured as SD of membrane potential), whereas DA receptor blockade (haloperidol, 0.8 mg/kg, i.v.) enhances spontaneous PSPs (B, before haloperidol, 1.6 mV, after haloperidol, 2.3 mV). C, Apomorphine consistently reduced spontaneous PSPs (six of seven neurons), whereas haloperidol consistently enhanced spontaneous PSPs (six of seven neurons); *p < 0.05.
Table 1.
DA receptor activation alterations of neuronal membrane properties
| DRUG | Input resistance (MΩ) | Membrane fluctuation (SD in mV) | AP threshold (mV) | AP duration (msec) | AP amplitude (mV) | Membrane potential (mV) | Firing rate (Hz) |
|---|---|---|---|---|---|---|---|
| Control | 40.7 ± 2.8 | 2.87 ± 0.17 | −54 ± 4 | 2.0 ± 0.2 | 61 ± 2 | −73.7 ± 1.5 | 0.23 ± 0.26 |
| Apomorphine | 52.0 ± 5.4* | 1.99 ± 0.14* | −57 ± 5 | 2.1 ± 0.1 | 60 ± 1 | −68.7 ± 2.8 | 0.05 ± 0.04 |
| Control | 50.0 ± 6.6 | 1.64 ± 0.34 | −55 ± 3 | 2.1 ± 0.1 | 60 ± 1 | −80.0 ± 1.7 | 0.10 ± 0.07 |
| Haloperidol | 54.0 ± 4.1 | 2.41 ± 0.39* | −58 ± 3 | 2.2 ± 0.2 | 61 ± 1 | −80.4 ± 1.6 | 0.19 ± 0.08* |
| Control | 35.3 ± 5.2 | 2.81 ± 0.36 | −64 ± 3 | 2.0 ± 0.2 | 71 ± 3 | −78.9 ± 3.5 | 0.44 ± 0.24 |
| Quinpirole | 49.1 ± 5.6* | 3.16 ± 0.64 | −65 ± 1 | 2.1 ± 0.3 | 69 ± 3 | −79.8 ± 3.4 | 1.23 ± 0.691-a |
| Control | 35.6 ± 4.1 | 3.14 ± 0.80 | −54 ± 2 | 2.1 ± 0.2 | 68 ± 2 | −76.0 ± 2.2 | 0.10 ± 0.07 |
| SKF-38393/81297 | 38.1 ± 5.4 | 2.42 ± 0.81* | −55 ± 4 | 2.1 ± 0.2 | 66 ± 3 | −75.6 ± 3.4 | 0.09 ± 0.07 |
p < 0.05, paired t test. Firing rate data were analyzed using Mann–Whitney U tests.
NS, Not significant, difference largely attributable to single neuron.
To determine whether tonic DA levels exert actions and may occlude the actions of apomorphine, the DA antagonist haloperidol was administered. Haloperidol administration (0.63 ± 0.06 mg/kg, i.v.) consistently increased the amplitude of membrane fluctuations (47%; 6 of 7 neurons; before haloperidol, 1.64 ± 0.34 mV; after haloperidol, 2.41 ± 0.39 mV; p < 0.05; paired t test;t = −2.57; df = 6) (Fig. 7) and increased the neuronal firing rate (5 of 7 neurons; before haloperidol, 0.10 ± 0.07 Hz; after haloperidol, 0.19 ± 0.08 Hz; p < 0.05; paired t test; t = −2.55; df = 6). By chance, the neurons recorded while administration of haloperidol occurred displayed a lower level of membrane fluctuations compared with other groups, as measured by their SD. However, it is unlikely that the opposite effects of apomorphine and haloperidol administration on SD were entirely attributable to this difference in baseline SD, because even neurons with a small SD (<0.9 mV) displayed a decreased SD in response to apomorphine administration, whereas neurons with a higher baseline SD (>3.3 mV) displayed an increase after haloperidol administration. Changes in action-potential parameters, input resistance, and membrane potential were not consistent or significant (Table 1). These data indicate that tonic levels of DA may suppress spontaneous membrane fluctuations, but do not necessarily exert tonic actions on membrane excitability or action-potential parameters.
In an attempt to determine which DA receptors are responsible for alterations in neuronal excitability and spontaneous membrane fluctuations, specific DA receptor agonists were used. Administration of the DA D2 agonist quinpirole (0.79 ± 0.09 mg/kg, i.v.) increased neuronal input resistance (39%; 6 of 7 neurons; before quinpirole, 35.3 ± 5.2 MΩ; after quinpirole, 49.1 ± 5.6 MΩ; p < 0.05; paired t test;t = 2.52; df = 6) (Fig.8) but had no significant actions on any other measurement (Table 1). In contrast, administration of the DA D1 agonists SKF-38393 (0.49 ± 0.15 mg/kg, i.v.; n = 3) or SKF-81296 (6.1 ± 0.28 mg/kg, i.v.; n = 4) had no effect on input resistance (Fig.9) but consistently decreased the amplitude of spontaneous membrane fluctuations (−23%; 7 of 7 neurons; before SKF, 3.14 ± 0.80 mV; after SKF, 2.43 ± 0.81 mV;p < 0.05; paired t test; t= 3.41; df = 6) (Fig. 10); no other consistent or significant measured actions were observed (Table1). Thus, different DA receptor subtypes exert actions on somatic input resistance and spontaneous PSPs.
Fig. 8.

DA D2 receptor activation enhances neuronal excitability. Administration of the DA D2 agonist quinpirole (0.6 mg/kg, i.v.) enhanced membrane deflections in response to intracellular current injection (A, before quinpirole, 58 MΩ; B, after quinpirole, 71 MΩ). This is demonstrated by overlaying voltage traces in response to 1 nA of current injection before and after quinpirole (C) showing an enhanced response after quinpirole, as well as by a plot of input resistance (D; dashed lineafter quinpirole). E, Quinpirole consistently increased neuronal input resistance (six of seven neurons); *p < 0.05.
Fig. 9.

DA D1 receptor activation does not enhance neuronal excitability. Administration of the DA D1 agonist SKF-81297 (6 mg/kg, i.v.) did not alter neuronal responses to current injection (A, before SKF-81297, 28 MΩ;B, after SKF-81297, 26 MΩ). Responses to 1.0 nA of current injection (C) as well as input resistance plots (D) overlay closely. E, Administration of the DA D1 agonists SKF-81297 or SKF-38393 did not have a significant effect on neuronal input resistance.
Fig. 10.

DA D1 receptor activation but not D2 stimulation suppresses spontaneous PSPs. Administration of the DA D1 agonist SKF-81297 (6 mg/kg, i.v.) suppresses spontaneous PSPs (A), as demonstrated by decreased fluctuation of the membrane potential in a histogram of percentage of time spent at a given membrane potential (B, SD of membrane potential before SKF-81297, 1.8 mV; SD of membrane potential after SKF-81297, 0.6 mV). Administration of the DA D2 agonist quinpirole (1.2 mg/kg, i.v.) did not suppress spontaneous PSPs (C), nor did it alter the distribution of membrane potential over time (D, SD before quinpirole, 3.4 mV; SD after quinpirole, 3.3 mV). E, Whereas administration of the DA D1 agonists attenuated spontaneous PSPs (seven of seven neurons), the DA D2 agonist quinpirole had no significant effect; *p < 0.05.
DAergic modulation of PFC inputs
We have demonstrated previously that apomorphine suppresses mPFC-evoked inputs to the BLA (Rosenkranz and Grace, 2001). To determine whether this suppression is mediated by DA D1 or DA D2 receptors, specific agonists were used. To minimize the effects of somatic membrane potential on the evoked PSP, all comparisons were performed between PSPs measured at the same membrane potential.
The suppressive action of apomorphine on mPFC inputs appears to be mediated by DA D1 receptors, because administration of the DA D1 agonists SKF-38393 (0.5 ± 0.01 mg/kg, i.v.; n = 3) or SKF-81297 (6.3 ± 0.7 mg/kg, i.v.; n = 3) produced a powerful attenuation of the mPFC-evoked PSP (6 of 6 neurons; mean of −70%; before SKF, 6.3 ± 1.5 mV; after SKF, 1.9 ± 0.1 mV; p < 0.05; paired ttest; t = 2.8; df = 5) (Fig.11). Administration of quinpirole (0.8 ± 0.2 mg/kg, i.v.; n = 4) enhanced the amplitude of mPFC-evoked PSPs (Fig. 11), perhaps because of increased somatic excitability, as demonstrated above (4 of 4 neurons; before quinpirole, 13.3 ± 3.1 mV; after quinpirole, 15.4 ± 3.3 mV;p < 0.05; paired t test; t= −4.3; df = 3). To determine whether a portion of the response to quinpirole may have been obscured because of pre-existing tonic activation of DA D2 receptors, the specific DA D2 antagonist eticlopride was administered (0.7 ± 0.01 mg/kg, i.v.;n = 5). At resting membrane potentials, eticlopride did not have a significant effect on the mPFC-evoked PSP, although there was a trend toward increased peak amplitude (before eticlopride, 5.4 ± 2.1 mV; after eticlopride, 8.6 ± 2.4 mV;p > 0.05; paired t test; t= 1.5).
Fig. 11.

DA D1 receptor activation but not D2 stimulation suppresses mPFC-evoked PSPs. Administration of the DA D1 agonist SKF-81297 (6 mg/kg, i.v.) suppressed mPFC-evoked PSPs (A, before SKF-81297, 11 mV; after SKF-81297, 0 mV). Administration of the DA D2 agonist quinpirole (0.6 mg/kg, i.v.) slightly enhanced mPFC-evoked responses (B, before quinpirole, 12 mV; after quinpirole, 14 mV). C, The DA D1 agonists consistently suppressed mPFC-evoked responses (six of six neurons), whereas the DA D2 agonist consistently enhanced mPFC-evoked responses (four of four neurons); *p < 0.05.
To demonstrate that other, sensory-related inputs are not suppressed, but in fact enhanced, by DA receptor activation, the effects of apomorphine on Te3 inputs were examined. Apomorphine administration (1.2 ± 0.1 mg/kg, i.v.; n = 4) enhanced Te3-evoked EPSPs by 25% (4 of 4 neurons; before apomorphine, 10.7 ± 3.5 mV; after apomorphine, 13.3 ± 4.2 mV; paired ttest; p < 0.05; df = 3).
DISCUSSION
This study illustrates potential mechanisms by which glutamatergic inputs from the mPFC are able to suppress BLA output, and the means by which DA receptor activation suppresses the influence of mPFC inputs while enhancing other inputs. On the whole, the data support the hypothesis that mPFC inputs recruit BLA inhibitory interneurons to suppress output, and that DA receptor activation removes mPFC-evoked inhibition and enhances association sensory cortical inputs via postsynaptic increases in neuronal excitability. Furthermore, these data provide a mechanism by which mPFC inputs and DA receptor activation exert such actions.
Inhibition of BLA projection neurons by mPFC inputs
It has been demonstrated previously that the PFC exerts a role in behavior that goes beyond sensory processing, such as temporal organization and selection of behavior (Watanabe, 1986; Goldman-Rakic, 1995; Sawaguchi and Yamane, 1999; Miller and Cohen, 2001). The data presented here demonstrate a means by which the PFC glutamatergic inputs may exert a functionally distinct action in the BLA, when compared with other glutamatergic inputs related to sensory processing. Thus, the mPFC inputs recruit inhibitory interneurons that result in a threefold inhibition: chloride-mediated hyperpolarization or clamping of the membrane potential to the chloride reversal potential, persistent decreases in input resistance, and related shunting of spontaneously occurring events. The contention that the mPFC-evoked inhibition is mediated by BLA interneurons is consistent with what is known about this system. Thus, there are few extrinsic sources of GABAergic inputs to the BLA. Although one such source may be from the ventral tegmental area (VTA) (Swanson, 1982), which receives excitatory inputs from the mPFC (Tong et al., 1996, 1998; Carr and Sesack, 2000), this disynaptic projection to the midbrain and back to the BLA would probably take too long to account for the short-latency inhibition observed here. In addition, the BLA has a large number of highly collateralizing inhibitory interneurons (McDonald, 1982, 1985; McDonald and Betette, 2001) that respond potently to mPFC stimulation (Rosenkranz and Grace, 2001), and these intrinsic GABAergic inputs would presumably overwhelm a much more sparse GABAergic input from the VTA. Furthermore, we observed a close temporal relationship between the latencies of the mPFC-evoked excitation of BLA interneurons and inhibition of BLA projection neurons.
In contrast to mPFC inputs, Te3 inputs, as well as other sensory-related inputs such as perirhinal and entorhinal cortical inputs (Lang and Pare, 1997), drive BLA projection neurons. Although these inputs also excite inhibitory interneurons (Lang and Pare, 1997), the excitation on interneurons is smaller in magnitude than on projection neurons (Rosenkranz and Grace, 2001), and furthermore, the temporal EPSP/IPSP sequence is such that inhibition occurs after BLA projection neurons are given the opportunity to fire (Fig. 5). It is clear that differences observed between mPFC- and Te3-evoked PSPs do not reflect the effects of cortically evoked PSPs on different sets of BLA projection neurons, because our previous study (Rosenkranz and Grace, 2001) demonstrated that mPFC stimulation will also suppress BLA neurons that receive Te3 inputs. Thus, the effects uncovered in this study provide a mechanism by which mPFC inputs inhibit BLA spontaneous and Te3-driven activity. This effect of mPFC inputs is not observed in all regions, because mPFC-evoked responses recorded from striatal neurons are excitatory.
It is important to note that the normal function of the mPFC is likely to be more complex than a simple inhibition of behavior. By stimulating the mPFC, we are likely to activate a large population of fibers that probably does not reflect the typical output of this system. In contrast, it is possible that clusters of mPFC neurons fire together during the course of PFC-related tasks, causing a selective inhibition to a subpopulation of BLA output neurons. The mPFC would thereby play a role in the selection of the appropriate affective behavior. However, in exceptional circumstances a large degree of mPFC–BLA axons may be activated, resulting in suppression of all affective behavior. Similarly, the role of the mPFC is not limited to the BLA, and afferents to association sensory cortical areas, for example, may select what sensory information reaches the BLA. Therefore, the interactions between the mPFC and the BLA are likely to be highly complex when considered in the context of ongoing behavior. In addition, it is unclear what relationship other regions of the mPFC (i.e., orbitofrontal and cingulate cortices) have with the BLA.
Modulation of neuronal excitability and spontaneous events by DA
Activation of DA receptors enhanced BLA neuronal excitability and suppressed spontaneous PSPs. Initially, this would appear paradoxical, because increases in input resistance would lead to amplification of PSP size. However, only somatic excitability is measured with the methods used here, and the suppressive actions of DA may be limited to dendrites (Herrling and Hull, 1980; Yang and Seamans, 1996; Hoffman and Johnston, 1999) or may be presynaptic in nature (Hsu et al., 1995; Behr et al., 2000). In contrast, the excitatory actions of DA are likely to be postsynaptic effects for at least two reasons. First, some neurons displayed delayed rectification in response to current injection. This delayed rectification was removed by DA receptor activation, indicating that DA suppresses somatic potassium conductances. Second, evoked PSPs often displayed a sublinearity when examined as a function of membrane potential. DA receptor activation linearized this relationship, again indicative of a suppressive action of DA on somatic voltage-dependent potassium conductances that reduce PSP amplitude. Furthermore, it has been reported that DA reduces a slow inhibitory potential mediated by a postsynaptic calcium-activated potassium conductance (Danober and Pape, 1998). The increases in neuronal excitability are not the indirect result of decreased spontaneous PSPs, as demonstrated by the data indicating that our measurement of input resistance was not correlated with SD, and the magnitude of the increases in input resistance after DA receptor activation was not correlated with the magnitude of the changes in SD. Furthermore, DA D2 receptor activation enhanced input resistance without significant alterations of spontaneous PSP amplitude.
The spontaneous PSPs modulated by DA receptor manipulations are probably of synaptic origin. Thus, their amplitude is dependent on the membrane potential, but no evidence was seen for a voltage dependence of their frequency. The reversal potential of these events as a whole (approximately −25 mV) is between the reversal potential for chloride and sodium, indicating that they may be a combination of glutamatergic and GABAergic events. This is supported by the demonstration that a portion, but not all, of these events reverse close to −65 mV. Furthermore, it is expected that even if these events were entirely glutamatergic in nature, the reversal potential would still be shifted, because the majority of excitatory inputs are to more distally located spines. Thus, altering somatic membrane potential by current injection is not likely to cause an equivalent change at distal dendritic regions, in contrast to the proximally located GABAergic inputs.
Differential modulation by DA receptor subtypes
The actions of DA on spontaneous PSPs, which may be presynaptic or dendritic, appear to be mediated by the DA D1 receptor, whereas alterations of somatic excitability are mediated by the DA D2 receptor. As a consequence, DA would be expected to suppress spontaneous or weaker signals by DA D1 activation, while enhancing large, coordinated inputs via D2 receptor stimulation. Furthermore, DA D1 receptor activation attenuates mPFC-evoked inhibition of BLA projection neurons. Removal of this inhibition will further enhance the response to nonattenuated inputs via a postsynaptic effect of DA.
It is unclear how DA receptor activation suppresses the majority of spontaneously occurring PSPs yet enhances Te3-evoked responses. One possibility is that the majority of spontaneously occurring PSPs are the result of mPFC inputs, suggesting that, at least in the anesthetized rat, inputs from Te3 do not appear to provide a tonic input to this system. Alternatively, the DA D1 agonist may indirectly affect activity within Te3.
The results described above demonstrate that, and provide a mechanism by which, mPFC inputs exert a qualitatively different action on BLA projection neurons when compared with inputs from Te3. Furthermore, DA D1 and D2 receptors exert actions that would appear to counterbalance each other with respect to effects on general excitability. However, from a functional standpoint, DA D1 receptors suppress mPFC-evoked inhibition to such a degree that DA D2-mediated enhancement of neuronal excitability is negated with regard to this input. This would allow DA D2 receptor stimulation to selectively enhance other inputs, such as those arising from Te3.
Functional consequences
These data support our hypothesis that mPFC inputs inhibit BLA projection neurons and thus suppress BLA-mediated behaviors by the recruitment of inhibitory BLA interneurons. In this manner, limbic inputs appear to exert a suppressive effect over sensory-evoked emotional responses. In contrast, DA receptor stimulation exerts effects in an opposite direction; this type of stimulation enhances BLA output by (1) a presynaptic removal of mPFC-evoked inhibition and (2) a postsynaptic enhancement of neuronal excitability that amplifies sensory-related inputs, thereby amplifying affective responses to sensory stimuli. DA appears to allow an animal to make a transition to a state in which affective behavior is driven by salient sensory stimuli. Lending support to this hypothesis is the observation that DA is released in the BLA in the presence of affective or stressful stimuli (Coco et al., 1992; Hori et al., 1993; Inglis and Moghaddam, 1999) and is necessary for the production of a BLA-dependent response to these stimuli (Lamont and Kokkinidis, 1998; Guarraci et al., 1999; Greba and Kokkinidis, 2001; Greba et al., 2001; Guarraci et al., 2000; See et al., 2001). Furthermore, artificial elevations of DA, such as those that occur after amphetamine or cocaine administration, also result in inappropriate BLA-mediated affective behaviors (Willick and Kokkinidis, 1995; Harmer et al., 1997; Harmer and Phillips, 1999a,b). Indeed, an emotional reaction to nonsalient sensory stimuli in the absence of PFC-induced suppression could be interpreted as a type of “paranoia,” an effect often associated with amphetamine administration (Angrist and Gershon, 1970; Janowsky and Risch, 1979;Hall et al., 1988).
Footnotes
This work was supported by National Institutes of Health Grants MH57440, MH45156 (A.A.G.), and MH12533 (J.A.R.). We thank Nicole MacMurdo, Christy Wyant, and Brian Lowry for excellent technical assistance. We also thank Dr. Anthony R. West for enlightening discussions.
Correspondence should be addressed to J. Amiel Rosenkranz, Department of Neuroscience, 446 Crawford Hall, University of Pittsburgh, Pittsburgh, PA 15260. E-mail: rosenk@bns.pitt.edu.
REFERENCES
- 1.Alheid G, de Olmos JS, Beltramino CA. Amygdala and extended amygdala. In: Paxinos G, editor. The rat nervous system, Ed 2. Academic; Sydney: 1995. pp. 495–572. [Google Scholar]
- 2.Al Maskati HA, Zbrozyna AW. Stimulation in prefrontal cortex area inhibits cardiovascular and motor components of the defense reaction in rats. J Auton Nerv Syst. 1989;28:117–126. doi: 10.1016/0165-1838(89)90084-2. [DOI] [PubMed] [Google Scholar]
- 3.Anderson JS, Carandini M, Ferster D. Orientation tuning of input conductance, excitation, and inhibition in cat primary visual cortex. J Neurophysiol. 2000;84:909–926. doi: 10.1152/jn.2000.84.2.909. [DOI] [PubMed] [Google Scholar]
- 4.Anderson JS, Lampl I, Gillespie DC, Ferster D. Membrane potential and conductance changes underlying length tuning of cells in cat primary visual cortex. J Neurosci. 2001;21:2104–2112. doi: 10.1523/JNEUROSCI.21-06-02104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angrist BM, Gershon S. The phenomenology of experimentally induced amphetamine psychosis: preliminary observations. Biol Psychiatry. 1970;2:95–107. [PubMed] [Google Scholar]
- 6.Armony JL, Servan-Schreiber D, Romanski LM, Cohen JD, LeDoux JE. Stimulus generalization of fear responses: effects of auditory cortex lesions in a computational model and in rats. Cereb Cortex. 1997;7:157–165. doi: 10.1093/cercor/7.2.157. [DOI] [PubMed] [Google Scholar]
- 7.Behr J, Gloveli T, Schmitz D, Heinemann U. Dopamine depresses excitatory synaptic transmission onto rat subicular neurons via presynaptic D1-like dopamine receptors. J Neurophysiol. 2000;84:112–119. doi: 10.1152/jn.2000.84.1.112. [DOI] [PubMed] [Google Scholar]
- 8.Ben-Ari Y, Kelly JS. Dopamine evoked inhibition of single cells of the feline putamen and basolateral amygdala. J Physiol (Lond) 1976;256:1–21. doi: 10.1113/jphysiol.1976.sp011308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borowski TB, Kokkinidis L. Contribution of ventral tegmental area dopamine neurons to expression of conditional fear: effects of electrical stimulation, excitotoxin lesions, and quinpirole infusion on potentiated startle in rats. Behav Neurosci. 1996;110:1349–1364. doi: 10.1037//0735-7044.110.6.1349. [DOI] [PubMed] [Google Scholar]
- 10.Campeau S, Davis M. Involvement of subcortical and cortical afferents to the lateral nucleus of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2312–2327. doi: 10.1523/JNEUROSCI.15-03-02312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr DB, Sesack SR. Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci. 2000;20:3864–3873. doi: 10.1523/JNEUROSCI.20-10-03864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cepeda C, Buckwald NA, Levine MS. Neuromodulatory action of DA in the neostriatum is dependent on the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci USA. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung S, Ferster D. Strength and orientation tuning of the thalamic input to simple cells revealed by electrically evoked cortical suppression. Neuron. 1998;20:1177–1189. doi: 10.1016/s0896-6273(00)80498-5. [DOI] [PubMed] [Google Scholar]
- 14.Coco ML, Kuhn CM, Ely TD, Kilts CD. Selective activation of mesoamygdaloid dopamine neurons by conditioned stress: attenuation by diazepam. Brain Res. 1992;590:39–47. doi: 10.1016/0006-8993(92)91079-t. [DOI] [PubMed] [Google Scholar]
- 15.Danober L, Pape H-C. Mechanisms and functional significance of a slow inhibitory potential in neurons of the lateral amygdala. Eur J Neurosci. 1998;10:853–867. doi: 10.1046/j.1460-9568.1998.00092.x. [DOI] [PubMed] [Google Scholar]
- 16.Dias R, Robbins TW, Roberts AC. Dissociation in prefrontal cortex of affective and attentional shifts. Nature. 1996;380:69–72. doi: 10.1038/380069a0. [DOI] [PubMed] [Google Scholar]
- 17.Flores-Hernandez J, Galarraga E, Bargas J. Dopamine selects glutamatergic inputs to neostriatal neurons. Synapse. 1997;25:185–195. doi: 10.1002/(SICI)1098-2396(199702)25:2<185::AID-SYN9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 18.Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 19.Gaudreau H, Pare D. Projection neurons of the lateral amygdaloid nucleus are virtually silent throughout the sleep–waking cycle. J Neurophysiol. 1996;75:1301–1305. doi: 10.1152/jn.1996.75.3.1301. [DOI] [PubMed] [Google Scholar]
- 20.Geijo-Barrientos E, Pastore C. The effects of dopamine on the subthreshold electrophysiological responses of rat prefrontal cortex neurons in vitro. Eur J Neurosci. 1995;7:358–366. doi: 10.1111/j.1460-9568.1995.tb00331.x. [DOI] [PubMed] [Google Scholar]
- 21.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 22.Grace AA. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev. 2000;31:330–341. doi: 10.1016/s0165-0173(99)00049-1. [DOI] [PubMed] [Google Scholar]
- 23.Greba Q, Kokkinidis L. Peripheral and intraamygdalar administration of the dopamine D1 receptor antagonist SCH23390 blocks fear-potentiated startle but not shock reactivity or the shock sensitization of acoustic startle. Behav Neurosci. 2001;114:262–272. doi: 10.1037//0735-7044.114.2.262. [DOI] [PubMed] [Google Scholar]
- 24.Greba Q, Gifkins A, Kokkinidis L. Inhibition of amygdaloid dopamine D2 receptors impairs emotional learning measured with fear-potentiated startle. Brain Res. 2001;899:218–226. doi: 10.1016/s0006-8993(01)02243-0. [DOI] [PubMed] [Google Scholar]
- 25.Guarraci FA, Frohardt RJ, Kapp BS. Amygdaloid D1 dopamine receptor involvement in Pavlovian fear conditioning. Brain Res. 1999;827:28–40. doi: 10.1016/s0006-8993(99)01291-3. [DOI] [PubMed] [Google Scholar]
- 26.Guarraci FA, Frohardt RJ, Falls WA, Kapp BS. The effects of intra-amygdaloid infusions of a D2 dopamine receptor antagonist on Pavlovian fear conditioning. Behav Neurosci. 2000;114:647–651. doi: 10.1037//0735-7044.114.3.647. [DOI] [PubMed] [Google Scholar]
- 27.Hall RC, Popkin MK, Beresford TP, Hall AK. Amphetamine psychosis: clinical presentations and differential diagnosis. Psychiatr Med. 1988;6:73–79. [PubMed] [Google Scholar]
- 28.Harmer CJ, Phillips GD. Enhanced conditioned inhibition following repeated pretreatment with d-amphetamine. Psychopharmacology. 1999a;142:120–131. doi: 10.1007/s002130050870. [DOI] [PubMed] [Google Scholar]
- 29.Harmer CJ, Phillips GD. Enhanced dopamine efflux in the amygdala by a predictive, but not a non-predictive, stimulus: facilitation by prior repeated d-amphetamine. Neuroscience. 1999b;90:119–130. doi: 10.1016/s0306-4522(98)00464-3. [DOI] [PubMed] [Google Scholar]
- 30.Harmer CJ, Hitchcott PK, Morutto SL, Phillips GD. Repeated d-amphetamine enhances stimulated mesoamygdaloid dopamine transmission. Psychopharmacology. 1997;132:247–254. doi: 10.1007/s002130050342. [DOI] [PubMed] [Google Scholar]
- 31.Hata Y, Tsumoto T, Sato H, Hagihara K, Tamura H. Inhibition contributes to orientation selectivity in visual cortex of cat. Nature. 1988;335:815–817. doi: 10.1038/335815a0. [DOI] [PubMed] [Google Scholar]
- 32.Herrling PL, Hull CD. Iontophoretically applied dopamine depolarizes and hyperpolarizes the membrane of cat caudate neurons. Brain Res. 1980;192:441–462. doi: 10.1016/0006-8993(80)90896-3. [DOI] [PubMed] [Google Scholar]
- 33.Hoffman DA, Johnston D. Neuromodulation of dendritic action potentials. J Neurophysiol. 1999;81:408–411. doi: 10.1152/jn.1999.81.1.408. [DOI] [PubMed] [Google Scholar]
- 34.Hori K, Tanaka J, Nomura M. Effects of discrimination learning on the rat amygdala dopamine release: a microdialysis study. Brain Res. 1993;621:296–300. doi: 10.1016/0006-8993(93)90119-8. [DOI] [PubMed] [Google Scholar]
- 35.Hsu KS, Huang CC, Yang CH, Gean PW. Presynaptic D2 dopaminergic receptors mediate inhibition of excitatory synaptic transmission in rat neostriatum. Brain Res. 1995;690:264–268. doi: 10.1016/0006-8993(95)00734-8. [DOI] [PubMed] [Google Scholar]
- 36.Inglis FM, Moghaddam B. Dopaminergic innervation of the amygdala is highly responsive to stress. J Neurochem. 1999;72:1088–1094. doi: 10.1046/j.1471-4159.1999.0721088.x. [DOI] [PubMed] [Google Scholar]
- 37.Janowsky DS, Risch C. Amphetamine psychosis and psychotic symptoms. Psychopharmacology. 1979;65:73–77. doi: 10.1007/BF00491982. [DOI] [PubMed] [Google Scholar]
- 38.Jaskiw GE, Weinberger DR. Ibotenic acid lesions of the medial prefrontal cortex augment swim-stress-induced locomotion. Pharmacol Biochem Behav. 1992;41:607–609. doi: 10.1016/0091-3057(92)90380-x. [DOI] [PubMed] [Google Scholar]
- 39.Jinks AL, McGregor IS. Modulation of anxiety-related behaviors following lesions of the prelimbic or infralimbic cortex in the rat. Brain Res. 1997;772:181–190. doi: 10.1016/s0006-8993(97)00810-x. [DOI] [PubMed] [Google Scholar]
- 40.Lamont EW, Kokkinidis L. Infusion of the dopamine D1 receptor antagonist SCH 23390 into the amygdala blocks fear expression in a potentiated startle paradigm. Brain Res. 1998;795:128–136. doi: 10.1016/s0006-8993(98)00281-9. [DOI] [PubMed] [Google Scholar]
- 41.Lang EJ, Pare D. Similar inhibitory processes dominate the responses of cat lateral amygdaloid projection neurons to their various afferents. J Neurophysiol. 1997;77:341–352. doi: 10.1152/jn.1997.77.1.341. [DOI] [PubMed] [Google Scholar]
- 42.Lang EJ, Pare D. Synaptic responsiveness of interneurons of the cat lateral amygdaloid nucleus. Neuroscience. 1998;83:877–889. doi: 10.1016/s0306-4522(97)00420-x. [DOI] [PubMed] [Google Scholar]
- 43.LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci. 1990;10:1062–1069. doi: 10.1523/JNEUROSCI.10-04-01062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald AJ. Neurons of the lateral and basolateral amygdaloid nuclei: a Golgi study in the rat. J Comp Neurol. 1982;212:293–312. doi: 10.1002/cne.902120307. [DOI] [PubMed] [Google Scholar]
- 45.McDonald AJ. Immunohistochemical identification of γ-aminobutyric acid-containing neurons in the rat basolateral amygdala. Neurosci Lett. 1985;53:203–207. doi: 10.1016/0304-3940(85)90186-7. [DOI] [PubMed] [Google Scholar]
- 46.McDonald AJ, Betette RL. Parvalbumin-containing neurons in the rat basolateral amygdala: morphology and co-localization of Calbindin-D(28k). Neuroscience. 2001;102:413–425. doi: 10.1016/s0306-4522(00)00481-4. [DOI] [PubMed] [Google Scholar]
- 47.Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- 48.Morgan MA, LeDoux JE. Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci. 1995;109:681–688. doi: 10.1037//0735-7044.109.4.681. [DOI] [PubMed] [Google Scholar]
- 49.Nader K, LeDoux JE. Inhibition of the mesoamygdala dopaminergic pathway impairs the retrieval of conditioned fear associations. Behav Neurosci. 1999;113:891–901. doi: 10.1037//0735-7044.113.5.891. [DOI] [PubMed] [Google Scholar]
- 50.Nelson S, Toth L, Sheth B, Sur M. Orientation selectivity of cortical neurons during intracellular blockade of inhibition. Science. 1994;265:774–777. doi: 10.1126/science.8047882. [DOI] [PubMed] [Google Scholar]
- 51.O'Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pape H-C, Pare D, Driesang RB. Two types of oscillations in neurons of the lateral and basolateral nuclei of the amygdala. J Neurophysiol. 1998;79:205–216. doi: 10.1152/jn.1998.79.1.205. [DOI] [PubMed] [Google Scholar]
- 53.Pare D, Pape H-C, Dong J. Bursting and oscillating neurons of the cat basolateral amygdaloid complex in vivo: electrophysiological properties and morphological features. J Neurophysiol. 1995;74:1179–1191. doi: 10.1152/jn.1995.74.3.1179. [DOI] [PubMed] [Google Scholar]
- 54.Pare D, Shink E, Gaudreau H, Destexhe A, Lang EJ. Impact of spontaneous synaptic activity on the resting membrane properties of cat neocortical pyramidal neurons in vivo. J Neurophysiol. 1998;79:1450–1460. doi: 10.1152/jn.1998.79.3.1450. [DOI] [PubMed] [Google Scholar]
- 55.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 3. Academic; San Diego: 1997. [DOI] [PubMed] [Google Scholar]
- 56.Poremba A, Jones D, Gonzalez-Lima F. Classical conditioning modifies cytochrome oxidase activity in the auditory system. Eur J Neurosci. 1998;10:3035–3043. doi: 10.1046/j.1460-9568.1998.00304.x. [DOI] [PubMed] [Google Scholar]
- 57.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- 58.Rosenkranz JA, Grace AA. Modulation of basolateral amygdala neuronal firing and afferent drive by dopamine receptor activation in vivo. J Neurosci. 1999;19:11027–11039. doi: 10.1523/JNEUROSCI.19-24-11027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosenkranz JA, Grace AA. Dopamine attenuates the ability of the prefrontal cortex to modulate sensory cortical inputs to basolateral amygdala neurons: in vivo intracellular and extracellular studies. Soc Neurosci Abstr. 2000;26:1726. [Google Scholar]
- 60.Rosenkranz JA, Grace AA. Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J Neurosci. 2001;21:4090–4103. doi: 10.1523/JNEUROSCI.21-11-04090.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sawaguchi T, Yamane I. Properties of delay-period neuronal activity in the monkey dorsolateral prefrontal cortex during a spatial delayed matching-to-sample task. J Neurophysiol. 1999;82:2070–2080. doi: 10.1152/jn.1999.82.5.2070. [DOI] [PubMed] [Google Scholar]
- 62.See RE, Kruzich PJ, Grimm JW. Dopamine, but not glutamate, receptor blockade in the basolateral amygdala attenuates conditioned reward in a rat model of relapse to cocaine-seeking behavior. Psychopharmacology. 2001;154:301–310. doi: 10.1007/s002130000636. [DOI] [PubMed] [Google Scholar]
- 63.Shevelev IA, Eysel UT, Lazareva NA, Sharaev GA. The contribution of intracortical inhibition to dynamics of orientation tuning in cat striate cortex neurons. Neuroscience. 1998;84:11–23. doi: 10.1016/s0306-4522(97)00363-1. [DOI] [PubMed] [Google Scholar]
- 64.Spehlmann R, Norcross K. Decreased sensitivity of neurons in the basolateral amygdala to dopamine and noradrenaline iontophoresis after a kindling stimulus. Exp Neurol. 1984;83:204–210. doi: 10.1016/0014-4886(84)90059-1. [DOI] [PubMed] [Google Scholar]
- 65.Steriade M, Nunez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stern EA, Kincaid AE, Wilson CJ. Spontaneous subthreshold membrane potential fluctuations and action potential variability of rat corticostriatal and striatal neurons in vivo. J Neurophysiol. 1997;77:1697–1715. doi: 10.1152/jn.1997.77.4.1697. [DOI] [PubMed] [Google Scholar]
- 67.Sugita S, Tanaka E, North RA. Membrane properties and synaptic potentials of three types of neurones in the rat lateral amygdala. J Physiol (Lond) 1993;460:705–718. doi: 10.1113/jphysiol.1993.sp019495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull. 1982;9:321–353. doi: 10.1016/0361-9230(82)90145-9. [DOI] [PubMed] [Google Scholar]
- 69.Szinyei C, Heinbockel T, Montagne J, Pape H-C. Putative cortical and thalamic inputs elicit convergent excitation in a population of GABAergic interneurons of the lateral amygdala. J Neurosci. 2000;20:8909–8915. doi: 10.1523/JNEUROSCI.20-23-08909.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tong ZY, Overton PG, Clark D. Stimulation of the prefrontal cortex in the rat induces patterns of activity in midbrain dopaminergic neurons which resemble natural burst events. Synapse. 1996;22:195–208. doi: 10.1002/(SICI)1098-2396(199603)22:3<195::AID-SYN1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 71.Tong ZY, Overton PG, Martinez-Cue C, Clark D. Do non-dopaminergic neurons in the ventral tegmental area play a role in the responses elicited in A10 dopaminergic neurons by electrical stimulation of the prefrontal cortex? Exp Brain Res. 1998;118:466–476. doi: 10.1007/s002210050303. [DOI] [PubMed] [Google Scholar]
- 72.Uwano T, Nishijo H, Ono T, Tamura R. Neuronal responsiveness to various sensory stimuli and associative learning in the rat amygdala. Neuroscience. 1995;68:339–361. doi: 10.1016/0306-4522(95)00125-3. [DOI] [PubMed] [Google Scholar]
- 73.Vidyasagar TR, Pei X, Volgushev M. Multiple mechanisms underlying the orientation selectivity of visual cortical neurones. Trends Neurosci. 1996;19:272–277. doi: 10.1016/S0166-2236(96)20027-X. [DOI] [PubMed] [Google Scholar]
- 74.Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci. 1992;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watanabe M. Prefrontal unit activity during delayed conditional Go/No-Go discrimination in the monkey. II. Relation to Go and No-Go responses. Brain Res. 1986;382:15–27. doi: 10.1016/0006-8993(86)90105-8. [DOI] [PubMed] [Google Scholar]
- 76.Willick ML, Kokkinidis L. Cocaine enhances the expression of fear-potentiated startle: evaluation of state-dependent extinction and the shock-sensitization of acoustic startle. Behav Neurosci. 1995;109:929–938. doi: 10.1037//0735-7044.109.5.929. [DOI] [PubMed] [Google Scholar]
- 77.Wilson CJ. The generation of natural firing patterns in neostriatal neurons. Prog Brain Res. 1993;99:277–297. doi: 10.1016/s0079-6123(08)61352-7. [DOI] [PubMed] [Google Scholar]
- 78.Yang CR, Seamans JK. Dopamine D1 receptor actions in Layers V-VI rat prefrontal cortex neurons in vitro: modulation of dendritic–somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zbrozyna AW, Westwood DM. Stimulation in prefrontal cortex inhibits conditioned increase in blood pressure and avoidance bar pressing in rats. Physiol Behav. 1991;49:705–708. doi: 10.1016/0031-9384(91)90306-9. [DOI] [PubMed] [Google Scholar]