

Abstract
The physical interaction between the presynaptic vesicle release complex and the large cytoplasmic region linking domains II and III of N-type (Cav2.2) calcium channel α1B subunits is considered to be of fundamental importance for efficient neurotransmission. By PCR analysis of human brain cDNA libraries and IMR32 cell mRNA, we have isolated novel N-type channel variants, termed Cav2.2-Δ1 and Δ2, which lack large parts of the domain II–III linker region, including the synaptic protein interaction site. They appear to be widely expressed across the human CNS as indicated by RNase protection assays. When expressed in tsA-201 cells, both novel variants formed barium-permeable channels with voltage dependences and kinetics for activation that were similar to those observed with the full-length channel. All three channel types exhibited the hallmarks of prepulse facilitation, which interestingly occurred independently of G-protein βγ subunits. By contrast, the voltage dependence of steady-state inactivation seen with both Δ1 and Δ2 channels was shifted toward more depolarized potentials, and recovery from inactivation of Δ1 and Δ2 channels occurred more rapidly than that of the full-length channel. Moreover, the Δ1 channel was dramatically less sensitive to both ω-conotoxin MVIIA and GVIA than either the Δ2 variant or the full-length construct. Finally, the domain II–III linker region of neither variant was able to effectively bind syntaxin in vitro. These results suggest that the structure of the II–III linker region is an important determinant of N-type channel function and pharmacology. The lack of syntaxin binding hints at a unique physiological function of these channels.
Keywords: human brain, class B calcium channel, alternative splicing, synprint site, ω-conotoxins, syntaxin, Gβγ
N-type (Cav2.2) calcium channels are highly concentrated in presynaptic nerve terminals and dendrites (Westenbroek et al., 1992,1998), and they mediate multiple cellular functions in neurons, including neurotransmitter release (Wheeler et al., 1994). The activity of N-type channels is extensively regulated by protein kinases (Swartz, 1993; Stea et al., 1995), Gβγ subunits (Zamponi and Snutch, 1998a; Kaneko et al., 1999), and SNARE proteins (for review, see Jarvis and Zamponi, 2001a). Whereas G-protein βγ subunits inhibit N-type channels by binding to the α1B domain I–II linker and the C-terminus regions (DeWaard et al., 1997; Page et al., 1997; Qin et al., 1997; Zamponi et al., 1997), SNARE proteins such as syntaxin 1A, SNAP-25, and cysteine string protein tightly interact with the large cytoplasmic loop connecting domains II and III of both N-type and P/Q-type calcium channels (Sheng et al., 1994; Leveque et al., 1998; Magga et al., 2000), and it is believed that their interaction with this region of the channel is a key step in neurotransmission (Mochida et al., 1996; Rettig et al., 1997). Moreover, the association of these proteins with the channels modulates their inhibition by G-proteins (Jarvis et al., 2000; Magga et al., 2000; Lü et al., 2001) and can decrease channel availability (Bezprozvanny et al., 1995, 2000; Jarvis and Zamponi, 2001b). Finally, both the domain I–II and II–III linker regions are targets for protein kinase C-dependent phosphorylation (Yokoyama et al., 1997;Hamid et al., 1999). Hence, cytosolic structures of the N-type calcium channel α1 subunit are important regulatory elements of channel activity, and any sequence variation in those regions might be expected to alter channel function and regulation.
Several different isoforms of Cav2.2 have now been cloned and functionally characterized. A single base change in the rat sequence results in a glutamine to glycine switch in the domain IS3 region that induces a permanent reluctant gating mode (Zhong et al., 2001). In the Gβγbinding motif contained within the domain I–II linker, an alanine residue can be inserted or deleted (Lin et al., 1997). An insertion of 21 amino acids in the domain II–III linker has been shown to be preferentially expressed in monoamine neurons in the rat brain and to alter channel function in a calcium channel β subunit-dependent manner (Coppola et al., 1994; Ghasemzadeh et al., 1999; Pan and Lipscombe, 2000). An insertion of four amino acids into the extracellular linker between the domain III S3 and S4 is predominantly expressed in the rat CNS (Lin et al., 1997) and changes the activation range of the channel (Stea et al., 1999). An insertion of a Glu-Thr motif into the extracellular linker between IVS3 and IVS4 produces a dramatic slowing of channel activation (Lin et al., 1999). Finally, an alternative selection of a splice acceptor site produces different C termini in human Cav2.2 channels (Williams et al., 1992), and Cav2.2 proteins with different C termini have also been found in rat brain (Westenbroek et al., 1992;Hell et al., 1994) and chick dorsal root ganglion cells (Lü and Dunlap, 1999).
Here, we have identified two uniquely novel α1Bcalcium channel variants (termed Cav2.2-Δ1 and -Δ2) that contain large sequence deletions in the domain II–III linker, including the synaptic protein interaction (synprint) site. Functional expression studies revealed that these variants display unique biophysical and pharmacological characteristics and do not effectively bind syntaxin in vitro. Overall, this suggests that these variants may mediate a unique physiological role in the human CNS.
MATERIALS AND METHODS
Reverse transcription-based PCR analyses of human neuronal mRNA
Three types of human neuronal mRNA/cDNA were used as the templates for PCR analyses of the human Cav2.2 α1B channel. Human neuroblastoma IMR32 cells were cultured and differentiated with dibutyryl-cAMP. Total RNA was isolated from the differentiated cells, and poly(A)+ mRNA was purified by an oligo(dT) cellulose column. A random-hexamer and Moloney murine leukemia virus reverse transcriptase (Amersham Biosciences) was used for the synthesis of cDNA. The single-strand cDNA was stored at −4°C and used for PCR analysis within 1 week. Human brain poly(A)+ mRNA was purified from frozen tissue of adult human occipital cortex, and an oligo(dT)-primed, double-stranded cDNA library was synthesized. A >3 kb fraction was selected by sucrose density sedimentation and inserted into a λZAPII vector (Stratagene). The λ-cDNA library was amplified once, and the lysate was diluted 1:1000 and boiled for 5 min before adding to the PCR mixture. A commercial human brain cDNA plasmid library (catalog #9603) was obtained from Takara Biomedicals (Ohtsu, Japan).
PCR analyses of the human α1B channel II–III linker region were conducted using the primer pairs listed in Table 1. A 50 μl reaction mix containing <100 ng cDNA, 2.5 U enzyme mix, 400 μm dNTP, 1.5 mm MgCl2, 0.4 μm of each primer and 1× PCR buffer was used with either Takara LA Taq DNA polymerase with GC buffer I, ELONGase enzyme mix (Life Technologies BRL), or Expand long-template PCR system 1 (Boehringer Mannheim, Laval, Quebec) to amplify the II–III linker fragment. The PCR-derived cDNAs were analyzed and purified by agarose gel electrophoresis, and the identity of the product was confirmed by a combination of restriction digestion analysis and complete DNA sequencing after cloning into a pGEM-T easy vector (Promega).
Table 1.
PCR analyses of the human Cav2.2 domain II-III linker region
| Template | Primer 1 | Primer 2 | Product size | Sequence |
|---|---|---|---|---|
| IMR 32 cDNA | ||||
| Bp21 (2329 → 2358) | Bp31 (−3854 → −3830) | 1.5 kb (62°C) | FL | |
| Bp21 | Bp31 | 0.7 kb (65°C) | Δ2 | |
| Bp22 (2314 → 2338) | Bp34 (−4357 → −4333) | 0.9 kb | Δ1 | |
| Bp23 (2308 → 2332) | Bp33 (−4335 → −4311) | 0.9 kb | Δ1 | |
| Bp24 (2293 → 2317) | Bp33 | 1.3 kb | Δ2 | |
| Human brain cDNA λ library | ||||
| Bp22 | Bp34 | 0.87 kb | Δ1 | |
| Bp23 | Bp33 | 0.85 kb | Δ1 | |
| Human brain cDNA plasmid library | ||||
| Bp25 (2285 → 2309) | Bp34 | 1.3 kb | Δ2 | |
| Bp26 (2263 → 2288) | Bp32 (−3860 → −3836) | 1.5 kb, 0.4 kb | FL, Δ1 | |
A random-hexamer-primed reverse transcription product using differentiated human neuroblastoma IMR32 cell mRNA, a size-selected, human cerebral cortical cDNA λZAPII library, and a commercial human brain cDNA plasmid library (Takara) were used as templates. PCR primers were synthesized as indicated by nucleotide numbers in parentheses (negative numbers reflect the antisense strand), according to the sequence of the original human α1Bsubunit [Williams et al. (1992); GenBank accession number M94172]. Listed are combinations in which the indicated size of one or two PCR products was obtained. For the Bp21 × Bp31 combination, the IMR32 cDNA yielded different products when the annealing temperature was varied (see temperature value parentheses).
Ribonuclease protection assays
Sequence-specific oligonucleotides were synthesized for use as primers in PCR to generate a 180 bp DNA fragment from bases 2301–2481 of the human α1B calcium channel. The fragment was gel isolated and subcloned into the pGEM-T easy vector (Promega).32P-labeled antisense riboprobe was synthesized by reverse transcription on a SpeI-linearized subclone using T7 RNA polymerase (Amersham Biosciences) in the presence of 50 μCi (800 Ci/mmol) α32P-UTP (NEN Life Science, Boston, MA). The resultant transcript was correctly sized (235 bp including vector sequence) and isolated by PAGE. The riboprobe was eluted in 0.5 mNH4OAc, 1 mm EDTA, 0.2% SDS, and then precipitated in isopropanol and washed twice with 70% ethanol. Riboprobe resuspended in RNase-free water was immediately used in assays. A sense transcript was synthesized from aSacI-linearized subclone using SP6 RNA polymerase to act as positive control in ribonuclease protection assays (RPAs).
For the RPAs, 1.0 μg of each poly(A)+ mRNA sample (Clontech) was resuspended in 30 μl of hybridization buffer containing 77% (v/v) formamide, 300 mm PIPES, 5m NaCl, 50 mm EDTA, and 2.5 × 106 cpm riboprobe, then denatured at 85°C and incubated at 60°C. After 15–18 hr of hybridization, the samples were cooled to 30°C and incubated for 1 hr in 350 μl of digestion buffer containing 300 mm NaCl, 10 mmTris, 5 mm EDTA, 4 μg/μl RNase A, and 10 U/μl RNase T1 (Boehringer Mannheim). In the negative control hybridization containing glycogen, we observed complete digestion of the riboprobe within 45–60 min, and in the positive control hybridization containing the sense transcript, no undigested probe could be detected after 45–60 min. Digestion was stopped by incubation at 37°C for 15 min after addition of 25 μg proteinase K, 0.5% SDS. Samples were isolated by phenol/chloroform-isoamyl alcohol extraction followed by isopropanol precipitation and two 70% ethanol washes. Air-dried samples were resuspended in 8 μl of formamide loading buffer. Samples were then denatured at 95°C and loaded onto a 7 m urea, 6% polyacrylamide gel for electrophoretic separation. The gel was analyzed by autoradiography immediately after electrophoresis. The positive control sample was expected to exhibit a protected band of 190 bp, including the full 180 bp probe, plus 5 bp of vector sequence at each end. By estimates from PCR-generated fragments, the probe was chosen to detect variant channels in RNA samples by exhibiting protected bands of 180 bp [for full-length (FL)], 110 bp (Δ1, deletion at nucleotides 2413–3558), and 55 bp (Δ2, deletion at nucleotides 2357–3145).
Genomic analysis
The IIS6–IIIS1 region of the human Cav2.2 gene was analyzed by genomic PCR. Primer pairs were directed to the coding regions that were presumed to reside in the 5′ and 3′ exons flanking the deletion boundaries of Δ1 and Δ2 variants. PCR was conducted in a 50 μl reaction mix containing 10 ng human whole-blood genomic DNA (Clontech catalog #6550-1), 200 μm dNTP, 1.5 mm MgCl2, 0.4 μm of each primer, and 1× GC buffer I. After preincubation for 5 min at 99°C, 2.5 U Takara LA Taq enzyme was added to start the amplification by 35 cycles with 30 sec at 94°C, 1 min at 62°C, 2 min at 72°C. The resultant PCR products were gel purified, cloned into pGEM-T easy, and sequenced. When products were too large for the T-vector, they were cut into fragments by a rare-cutter restriction enzyme, cloned into pBluescript II (Stratagene), and sequenced.
Construction of cDNAs
Plasmids encoding entire open reading frames of human Cav2.2 variants were constructed as follows. A 2.2 kb fragment encoding the entire sequence between the N terminal and the IIS6 region and a 3.4 kb fragment encoding the entire sequence downstream of the IIIS3 region (to a SphI site at nucleotide 7191) were cloned from human neuroblastoma IMR32 cell mRNA by RT-PCR. An EcoRI site was inserted upstream of the start codon in the 2.2 kb fragment as part of the forward primer sequence. Compared with the original sequence of M94172 (Williams et al., 1992), a GCA (Ala415) insert between nucleotides 1387 and 1388 was present in the 2.2 kb clone. The 2.2 kb EcoRI–HindIII fragment, a 0.1 kb HindIII–SacI fragment from Δ2, and the 3.4 kb SacI–SphI fragment were ligated to aSphI–EcoRI fragment of pcDNA1.1/Amp (Invitrogen) to produce pHCa1B-D2 encoding the entire open reading frame of the Δ2 variant. After complete digestion of pHCaB-D2 with HindIII and NarI, the resulting 2.4 kbHindIII–HindIII and 8.0 kbNarI–HindIII fragments were ligated to a 0.4 kbHindIII–NarI fragment from Δ1 or a 1.5 kbHindIII–NarI partial digestion fragment from FL to produce pHCa1B-D1 (as Δ1) and pHCa1B (as FL), respectively.
The human calcium channel α2-δ1 subunit was cloned as follows. Based on the published sequence [GenBank M76559;Williams et al. (1992)], a 1.6 kb fragment encoding the N-terminal half (nucleotides 13–1590) with a KpnI site upstream of the start codon and a 1.7 kb fragment encoding the C-terminal half (nucleotides 1563–3315) with a NotI site downstream of the stop codon were cloned from a human brain cDNA plasmid library via PCR. The products were gel purified, cloned into a pGEM-T easy vector, and sequenced. The 1.6 kb KpnI–ClaI fragment and the 1.7 kb ClaI–NotI fragment were ligated to produce pHCa2d1 encoding the entire open reading frame of the α2-δ1 subunit. Compared with the original sequence, a base pair change at nucleotide 329 from A to C (resulting in a conversion of Ser99 to Arg) was found in three of three independent clones. The KpnI–NotI fragment from pHCa2d1 was subcloned into pcDNA3 (Invitrogen) for subsequent transfection studies.
A plasmid encoding the human β1b subunit was constructed by subcloning of a 3.3 kbEcoRI–EcoRI fragment of pHCaB into pcDNA3. The construct was originally cloned from a library of human brain cDNA and exhibited the following differences relative to published sequence (GenBank M92303): Arg434 and Arg435 to Ala-Ala; Gly538 and Ala539 to Gly-Gly-Thr-Pro; Trp571 and Pro572 to Cys-Ala. The new sequence has been deposited into GenBank as AB054985 (Fukuda et al., 1996).
Biochemistry
Preparation of 6xHis fusion proteins. We used PCR to generate domain II–III linker cDNA fragments from each of the three splice variants. For the two short variants, the entire II–III linker regions were synthesized, whereas for the full-length variant, a fragment corresponding only to the first domain of the synprint site was generated (residues 711–862), which has been shown to bind syntaxin 1A in the rat isoform (Yokoyama et al., 1997). The fragments were cloned in frame into the pTrcHis fusion vector by restriction sites engineered into the oligonucleotides and sequenced. Proteins were grown and purified using conditions adapted from the manufacturer. For growth, constructs in pTrcHis were transformed into Escherichia coli TOP10 cells (Invitrogen). A fresh colony was picked and grown overnight in 2xYT broth. Two hundred milliliters of SOB broth were inoculated with 4 ml of the 2xYT culture and grown toA600 = 0.5. Protein expression was induced with 100 mmisopropyl-1-thio-β-d-galactoside (IPTG) and grown for 4 hr. Cells were harvested and lysed by three cycles of sonication/freeze-thaw in 30 ml of native binding buffer (NBB7.8; 20 mmNa2HPO4, pH 7.8, 500 mm NaCl) supplemented with 100 μg/μl egg white lysozyme (Sigma, St. Louis, MO), and 20 μl protease inhibitor mixture containing 4-(2-aminoethyl)benzensulfonyl fluoride, bestatin, pepstatin, E-64, and phosphoramidon (P8849, Sigma). The lysate was incubated with RNase (5 μg/ml), centrifuged to remove insoluble debris, and passed through a 0.8 μm filter. Lysates were used immediately or stored at −80°C. For purification, 1 vol of 50% Ni-NTA agarose (Qiagen) was incubated with 3 vol of lysate, 12 mm imidazole, 10 mmβ-mercaptoethanol, and 0.1% Triton X-100 for 30 min at 4°C, and washed with NBB7.8. This was repeated a second time. The beads were then washed at room temperature with 50 bed vol of wash buffer consisting of 20 mmNaH2PO4, pH 6.0, 500 mm NaCl, 21 mm imidazole, 10 mm β-ME, 0.1% Triton X-100. Proteins were eluted by incubation at 4°C for 30 min with 20 mmNaH2PO4, pH 6.0, 500 mm NaCl, 500 mm imidazole. Eluted proteins were dialyzed overnight at 4°C against PBS (137 mm NaCl, 2.7 mm KCl, 4.3 mmNa2HPO4·7H2O, 1.4 mmKH2PO4). 6xHis proteins were use immediately or stored at −80°C.
Preparation of syntaxin 1A-GST. Syntaxin 1A (residues 1–268) in pGEX-4T-3 (Amersham Biosciences) was transformed intoE. coli BL21 (Amersham Biosciences) as described previously (Jarvis and Zamponi, 2001b). A fresh colony was grown in 7 ml of 2xYT. Three hundred milliliters of this starter culture were used to inoculate 100 ml LB broth that was grown overnight and then added to 900 ml of LB broth. The 1 l culture was grown toA600 = 0.5, at which point protein expression was induced by the addition of 0.1 mmIPTG. Cells were grown for 4 hr and harvested by centrifugation, resuspended in 35 ml resuspension buffer [PBS supplemented with 0.1% Tween 20, 2 mm EDTA, 350 mmNaCl, 0.1% β-ME, and 20 μl protease inhibitor mixture P8849 (Sigma)], and passed twice through a French press. Cellular debris was removed by centrifugation, and the lysate was either used immediately or stored at −80°C. For purification, 1 vol of 50% glutathione Sepharose beads (Sigma) was incubated with 2 vol of lysate at 4°C for 1 hr. This was repeated a second time. The beads were subsequently washed at room temperature with 10 bed vol of MKM buffer [10 mm MOPS, pH 7.5, 150 mmKCl, 4.5 mmMg(CH3COO)2, 0.2% Triton X-100] and 45 bed vol of PBS with 0.1% Tween 20 (PBST). The washed bead/protein slurry was then used for in vitro binding assays.
In vitro binding assays and Western blot analysis. In vitro binding between immobilized syntaxin 1A-GST and 6xHis-tagged II–III linkers was conducted under the following conditions, in a total volume of 600 μl. Syntaxin 1A-GST immobilized on glutathione Sepharose (varying amounts; see Fig. 8 legend), 1 nmol 6xHis-II–III linker, 137 mm NaCl, 2.7 mm KCl, 4.3 mmNa2HPO4·7H2O, 1.4 mmKH2PO4, 0.1% Tween 20. The assay proceeded for 2.5 hr at 4°C with rotation. After the incubation, the beads were washed twice with 20 vol of PBST, resuspended in 40 μl of 2× Laemmli sample buffer, and rotated for 1 hr at 4°C.
Fig. 8.
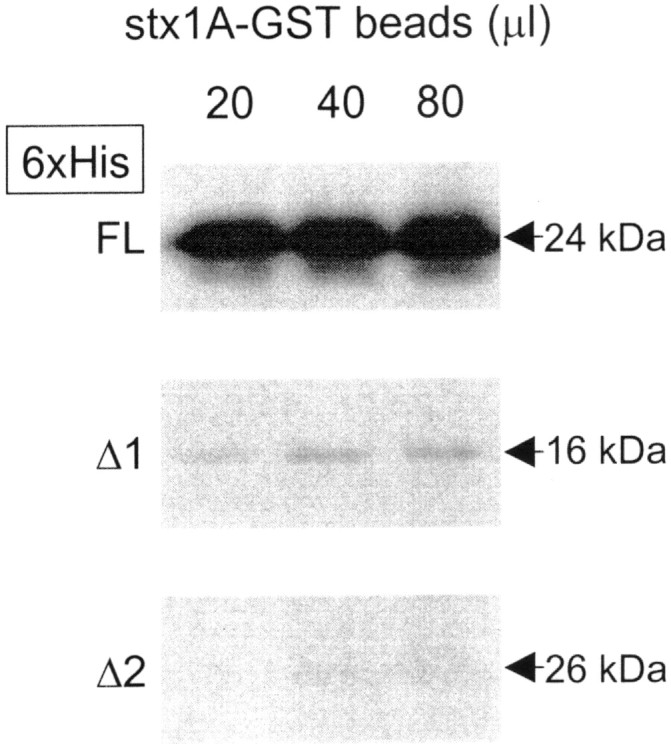
Western blots illustrating the in vitro affinities of 6xHis fusion proteins of the three N-type channel domain II–III linker regions for rat syntaxin 1A-GST (stx1A-GST). 6xHis-tagged constructs containing a 5′ Xpress epitope were incubated with 20, 40, or 80 μl of a 50% slurry of syntaxin 1A immobilized on glutathione agarose. An anti-Xpress antibody was used to detect bound N-type II–III linker FL, Δ1, and Δ2. Note that fusion proteins comprising part of the synprint region of the FL variant interact strongly with syntaxin 1A, whereas the Δ1 variant interacts only very weakly, and the Δ2 variant does not interact at all. 6xHis linker peptides bound at relatively low background levels to naked 50% glutathione agarose (data not shown). The lower molecular weight band (∼22 kDa) in the top FL row is most likely a prematurely truncated FL linker, attributable to a rare codon not handled 100% of the time byE. coli during peptide production. Interestingly, this suggests that syntaxin 1A will bind to an even shorter stretch of the human II–III FL linker. All blots were subjected to the same treatment and exposure times. This figure is representative of three experiments.
Western blots were performed as described previously (Jarvis et al., 2000). Briefly, samples were run on SDS-PAGE and transferred to Hybond ECL nitrocellulose at 100 V for 1 hr, and the membranes were blocked for 2 hr in 5% skim milk powder in PBST at room temperature. Primary incubation was as follows: anti-Xpress (Invitrogen) 1:3000, 2 hr, room temperature. Secondary incubation was as follows: anti-mouse (Amersham Biosciences) 1:2000, 1 hr, room temperature. Blots were subjected to ECL plus (Amersham Biosciences) and detected on Kodak Biomax ML film.
Functional assessment of the Ca2+ channel α1B cDNA constructs
Human embryonic kidney (HEK) tsA-201 cells were grown to 80% confluence in DMEM medium supplemented with 10% fetal bovine serum and 0.06% kanamycin. Cells were split and plated on glass coverslips at 10% confluence 12 hr before transfection. Immediately before transfection the medium was renewed, and a calcium phosphate transfection procedure was used to transfect cDNAs encoding human α1B FL, Δ1, or Δ2 together with human α2-δ1 and β1b subunits, and the reporter gene EGFP (Clontech) at a molar ratio of 1:1:1:0.5. Cells were washed after 12 hr and maintained at 37°C for an additional 12 hr before being moved into a CO2 incubator set at 28°C. The cells were maintained under those conditions for 24–72 hr before recording. During this time period, current densities appeared to be stable for each of the three constructs.
Glass coverslips carrying transfected cells were transferred to a recording chamber perfused with recording solution consisting of (in mm): 20 BaCl2, 1 MgCl2, 10 HEPES, 40 tetraethylammonium (TEA)-Cl, 10 glucose, and 65 CsCl, pH 7.2, with TEA-OH. Borosilicate glass-patch pipettes were pulled with a microelectrode puller (Narishige PP-83 or Sutter P87) and fire polished. The internal pipette solution contained (in mm): 105 Cs methanesulfonate, 25 TEA-Cl, 1 MgCl2, 11 EGTA, 10 HEPES, pH 7.2, and 4 mm Mg-ATP. The electrode showed typical resistances of 3–4 MΩ. Whole-cell patch-clamp recordings were performed with a List EPC-7 amplifier (List, Darmstadt, Germany) linked to a Power Macintosh computer (Apple) equipped with an ITC-16 A/d converter (Instrutech Corp., New York, NY) and AxoGraph 4.6 (Axon Instruments, Union City, CA). Alternatively, recordings were performed with an Axopatch 200 B amplifier linked to a personal computer equipped with pCLAMP v 6.0. Data were filtered at 1 kHz and recorded directly onto the hard drive of the computer. Unless stated otherwise, currents were evoked by stepping from a holding potential of −90 mV to a test potential of 0 mV. Current densities were measured via Axograph 4.6, which automatically provides picoAmpere/picoFarad values for each trace. Fitting of the raw data, least-square fittings of activation, inactivation, and dose–response curves, statistical analyses, and all figures for electrophysiological data were performed using Prism 3.01 software (Graphpad, San Diego, CA) or Sigmaplot v. 4.0 (Jandel Scientific). The activation time constants were determined by fitting the raw current data with the equation: I(t) = Imax (1 − exp(−t/τa)) exp(−t/τh)), whereI(t) indicates the amplitude of current at timet, Imax is the maximum amplitude, and τa and τh are the time constants for activation and inactivation, respectively. This was done using the nonlinear fitting function in Graphpad Prism 3.01. Each trace was fitted separately, and the averaged values were plotted. We note that this equation assumes that both activation and inactivation occur with monoexponential time courses that nicely described the data. The error bars that are given reflect SEs. Statistical significance was evaluated using ANOVA andpost hoc Tukey's tests.
RESULTS
Identification of novel human Cav2.2 domain II–III linker variants
During the process of cloning a human Cav2.2 cDNA construct via RT-PCR from a human fetal brain cDNA library, we unexpectedly encountered two large in-frame deletions in the domain II–III linker region of the channel: a 1146 base pair deletion between nucleotides 2412 and 3559 that corresponded to a 382 amino acid deletion between Arg756 and Leu1139 (designated Δ1), and a second deletion (designated Δ2) of 789 base pairs between nucleotides 2356 and 3146 that corresponds to a 263 amino acid deletion between Lys737 and Ala1001 (Fig. 1A). To determine whether these deletions were cloning artifacts or truly novel variants of Cav2.2, we conducted a PCR-based analysis of this region using various sets of primers, PCR enzymes, and cDNA sources. As shown in Table1, six forward primers (named Bp21-Bp26) and four reverse primers (Bp31-Bp34) flanking the II–III linker synprint site were designed to detect specific variants, and PCR reactions were performed using three different types of PCR enzyme systems and three distinct kinds of neuronal cDNA sources. Using reverse-transcribed IMR32 cell mRNA as a template, the primer pair Bp21–Bp31 amplified an expected 1.5 kb product that corresponded perfectly to the original, full-length (FL) Cav2.2 α1B subunit identified by Williams et al. (1992). However, in two of five independent clones, an AGG triplet (nucleotides 2413–2416) was absent, indicating the lack of Arg756. In addition, shorter products were obtained reproducibly with different combinations of primer pairs (i.e., Bp22 + Bp34, Bp23 + Bp33, and Bp24 + Bp33) and different PCR polymerases, which corresponded to the Δ1 and Δ2 variants isolated from the fetal brain library. The deletion variant Δ1 was also amplified from a human cerebral cortical cDNA λ library using the same sets of primers (Bp22 + Bp34 and Bp23 + Bp33) (Table 1) and from a commercial human brain cDNA plasmid library; the two deletion variants as well as the FL clone were amplified by additional pairs of primers (Bp25 + Bp34). Overall, these results are consistent with the existence of two novel Cav2.2 variants in human brain that lack large parts of the domain II–III linker region including the synprint site (Fig. 1B).
Fig. 1.
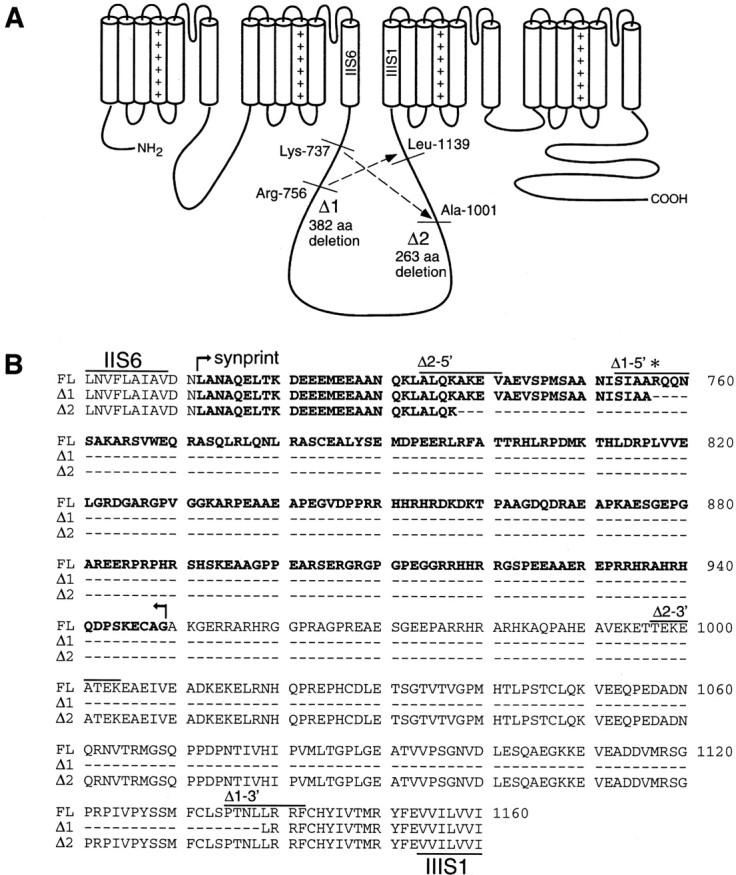
Deletion sites in the human N-type calcium channel α1B subunit. Nucleotide and amino acid numbers correspond to those of the original human α1B subunit [Williams et al. (1992); GenBank accession number M94172]. A, Putative membrane topology of the α1B subunit and two patterns of deletion that occurred in the cytoplasmic domain II–III linker region. In the Δ1 variant, 382 amino acids between Arg756 and Leu1139 are deleted. Variant Δ2 lacks 263 amino acids between Lys737 and Ala1001. B, Alignment of the amino acid sequence of the full-length (FL) and Δ1 and Δ2 variants of the human α1B subunit, indicating the deleted sequence relative to the location of the synaptic protein interaction site (synprint, boldface).
Genomic analysis of the deletion variants
To determine whether the variants arose from mRNA splicing, we analyzed the human genome sequence encoding the α1B II–III linker region by genomic PCR (Fig.2A). The PCR primer pairs were directed to the coding regions that were presumed to reside in the 5′ and 3′ exons flanking the deletion boundaries of Δ1 and Δ2 variants. At the presumed 5′ end of the Δ1 deletion, primer pairs that spanned a 180 base pair segment of coding sequence produced a roughly 9 kb genomic DNA product that included a 9 kb intron with consensus GT-AG terminals (Mount, 1982). The 5′ end of the intron corresponded to the N-terminal boundary of the Δ1 deletion (i.e., after nucleotide 2412). The 3′ end of the intron sequence can account for the observation that Arg756 was absent in two of five PCR products of the FL variant via use of two possible 3′ acceptor sites (nucleotide 2413 or 2416) (Fig. 2A). Within the intron, we found another putative exon cassette encoding a 21 amino acid insert (FVKQARGTVSRSSSVSSVNSP), which would predict the possible existence of a human splice variant analogous to one found previously in mouse (Coppola et al., 1994) and rat (Pan and Lipscombe, 2000). When using human cDNA libraries as the template, PCR analysis using a primer sequence specific to the 21 amino acid insert, however, produced no fragments under a number of different PCR conditions. At the predicted 3′ end of the Δ1 deletion, a 3 kb intron was detected immediately before the C-terminal boundary of the deletion (nucleotide 3559). Thus, the genomic DNA layout is consistent with a typical RNA splicing mechanism as a basis for generating the Δ1 variant.
Fig. 2.

A, Genomic DNA sequence at boundary sites of the deletion variants. Genomic sequence analysis via PCR reveals an intron sequence with GT-AG sites (Mount, 1982), suggesting that nucleotides between positions 2412 and 3559 may be spliced out to produce the Δ1 variant. The intron sequence at the 5′ end of Δ1 also explains the observation that Arg756 (indicated by ∗ in Fig. 7B) was absent in two of five PCR products of the full-length α1B subunit (via the use of alternative 3′ splice acceptors). In contrast, there were no intron sequences around the deletion loci in variant Δ2. B, Combinations of exon cassettes encoding the II–III linker region of the human Cav2.2 variants. The complete II–III linker from Asp710 to Glu1153 is encoded by six exon cassettes (A toF) that are separated by five introns. A very long (∼40 kb) intron was found between nucleotides 3431 and 3432. All of these exons are involved in generating the FL α1B subunit (boundary amino acids are indicated at the top). In variant Δ1, the exons C, D, and E and flanking four introns (total 54.1 kb) are spliced out to link exons A-B-F. In variant Δ2, a 9.8 kb region between nucleotide 2356 in Exon b and nucleotide 3146 in Exon c including a 9.0 kb intron is spliced out to link exons A-b-c-D-E-F, in which b and c would represent truncated exons. Note that another putative exon cassette encoding a 21 amino acid insert was found within the 9.0 kb intron between Exons b and c (see Results).
The entire genomic structure of the human α1Bsubunit II–III linker region is illustrated in Figure2B. Because a very long (∼40 kb) intron was found between nucleotides 3431 and 3432, a 1.5 kb mRNA encoding the domain II–III linker region consists of six exons that are spliced from a gene in total >55 kb length. All of these exons are used in the FL variant, and another longer variant having a 21 amino acid insert is predicted from the sequence of the 9 kb intron between exons B and C. In the variant Δ1, exons C, D, and E and the flanking four introns (total 54.1 kb) may be spliced to yield a shorter string of exons (A-B-F). We note that the exon–intron structure has been confirmed recently by a working draft sequence (as of June 2001) published by the human genome project (GenBank accession number 13639614).
In contrast, there appear to be no intron–exon boundaries corresponding to the deletion sites in the Δ2 variant. As shown in Figure 2B, in the Δ2 variant, a 9.8 kb region from nucleotide 2356 in exon B to nucleotide 3146 in exon C (including a 9.0 kb intron) appears to be “spliced out” to yield a string of exons (A-b-c-D-E-F) in which exons b and c, however, would have to be somehow truncated. No duplication of exonic sequence was found in the genomic PCR products. Because the variants were isolated from mRNA via RT-PCR, they could not have arisen from intein splicing. Instead, this suggests the possibility that perhaps an unknown mechanism of RNA splicing or editing may be responsible for the Δ2 variant. Although at this point we are not able to pinpoint a plausible mechanism, the fact that the deletion was found under a range of different conditions, and using various template sources (see also below), supports the validity of the Δ2 variant.
Expression of the deletion variants in human brain
Several previously identified N-type calcium channel splice variants have been shown to be expressed in a region-specific manner (Lin et al., 1997). Furthermore, there is evidence that the expression of certain types of calcium channel subunits changes during development (McEnery et al., 1998). To determine whether this might occur for the two novel II–III linker variants, we performed ribonuclease protection assays using commercially available RNA from whole fetal and adult human brain, as well as from several individual brain subregions (Fig.3). As shown in Figure 3, in all sources of mRNA examined, three bands corresponding to the FL channel as well as the two variants were detected, with the FL variant yielding the most intense bands. However, because the probe for the FL variant is longer, it is expected to contain more32P, and hence the higher intensity seen with the corresponding band may not necessarily reflect a greater abundance of the FL transcript. Nonetheless, these data further support the physiological relevance of the two deletion mutants and indicate that these variants may be a global feature in the human CNS.
Fig. 3.

Tissue-specific transcription of the α1B channel splice variants in the human brain. Autoradiograph of a ribonuclease protection assay using a32P-labeled RNA probe transcribed from a 180 bp PCR fragment of the full-length α1B variant. Poly(A)+ mRNA (1.0 μg) from each of total fetal brain, total adult brain, and various regions of adult brain as indicated was hybridized with the radiolabeled riboprobe. Three protected products of the expected sizes (180 base pairs for FL, 115 base pairs for Δ1, 60 base pairs for Δ2) were detected in both fetal and adult human brain and across all tissues tested. We note that faint bands can be observed in the water control that were likely caused by spillover from the adjacent lane, which was not seen in other repetitions of the assay.
Biophysical properties of the novel human Cav2.2 variants
To characterize the biophysical properties of the deletion variants, we generated expression constructs encoding the entire open reading frame of the human FLα1B subunit as well as of the variants, all of which contained a previously reported alanine insertion in position 415 in the domain I–II linker region of the channel (Lin et al., 1997). The cDNAs were transfected into tsA-201 cells together with the ancillary human β1b and α2-δ1 subunits, and barium currents were recorded via whole-cell patch clamp (Fig.4). Under these conditions all three variants yielded functional channels but produced different levels of current activity (Fig. 4A). Compared with the current density seen with the FL channel (23.1 ± 2.6 pA/pF;n = 35), the average current density of the Δ2 channel was significantly larger (36.6 ± 4.8 pA/pF;n = 34) and that of the Δ1 variant was significantly reduced (8.4 ± 4.8 pA/pF; n = 46). A similar trend was also observed when channels were expressed inXenopus oocytes (data not shown), indicating that the observed effects were not an artifact of our expression system. The half-activation voltages (Va) estimated from the current–voltage relations were shifted to slightly more hyperpolarized potentials in Δ2 and to more depolarized potentials in Δ1 (FL: −8.7 ± 1.3 mV; Δ1: −5.3 ± 1.4 mV; Δ2: −11.6 ± 1.0 mV) (Fig. 4B). There was no significant difference in the time constant for activation among the three variants (Fig. 4C). In contrast, the time constant for inactivation was
Fig. 4.

Macroscopic currents recorded from HEK tsA-201 cells expressing human α1B FL, Δ1, and Δ2 channels (coexpressed with β1b and α2-δ1 subunits). A, Representative current responses obtained with the three variants. Tight-seal, whole-cell voltage clamp was used in measuring Ca2+ channel current using external 20 mmbarium as the charge carrier. Cells were held at −90 mV, and a 100 msec step depolarization to 0 mV was applied. B, Averaged current–voltage relations for the three variants for activation in the form of current densities. Cells expressing FL (○,n = 16), Δ1 (●, n = 14), and Δ2 (▴, n = 12) were held at −90 mV, and 100 msec step depolarizations from −50 to +50 mV were applied.C, D, Comparisons of the activation time constants (C) and inactivation time constants (D) determined as outlined in Materials and Methods. Note that there was no significant difference in the time constants of activation, whereas the Δ2 variant inactivated significantly more quickly than the other two channel isoforms. Theasterisk indicates statistical significance relative to the FL variant (p < 0.05; Tukey's multiple comparison test).
significantly smaller in Δ2 than in either FL or Δ1 (Fig.4D), indicating that the Δ2 channel inactivates more rapidly than the other variants.
Figure 5 illustrates the differences in the voltage dependence of steady-state inactivation and the recovery from inactivation. Either deletion resulted in a dramatic positive shift in the half-inactivation potential. Whereas the FL variant displayed a half-inactivation potential of −47.8 ± 1.3 mV, variants Δ1 and Δ2 were half-inactivated at −23.2 ± 0.8 mV and −30.1 ± 1.1 mV, respectively, indicating that deletions in the domain II–III linker can mediate a shift of as much as 25 mV in the voltage dependence of inactivation. Furthermore, compared with the FL channel, the slope factors of the inactivation curve were significantly (p < 0.05) reduced (FL: 13.4 ± 1.3; Δ1: 7.8 ± 0.6; Δ2: 8.4 ± 0.9). A comparison of the time course of recovery from inactivation also indicated profound differences among the three variants. At a recovery potential of −90 mV, the two deletion variants recovered significantly quicker than FL channels (Fig. 5B), such that the time constant for recovery from inactivation (when fitted monoexponentially) was 320.2 ± 36.4 msec for the FL channel but only 21.9 ± 2.5 msec and 37.9 ± 2.6 msec for the Δ1 and Δ2 variants, respectively. This trend was observed over a range of recovery potentials with the difference relative to the FL channel increasing at more depolarized potentials (Fig. 5C). Overall, these data indicate that the deletion in the domain II–III linker regions mediate profound effects on the inactivation characteristics of the channel.
Fig. 5.
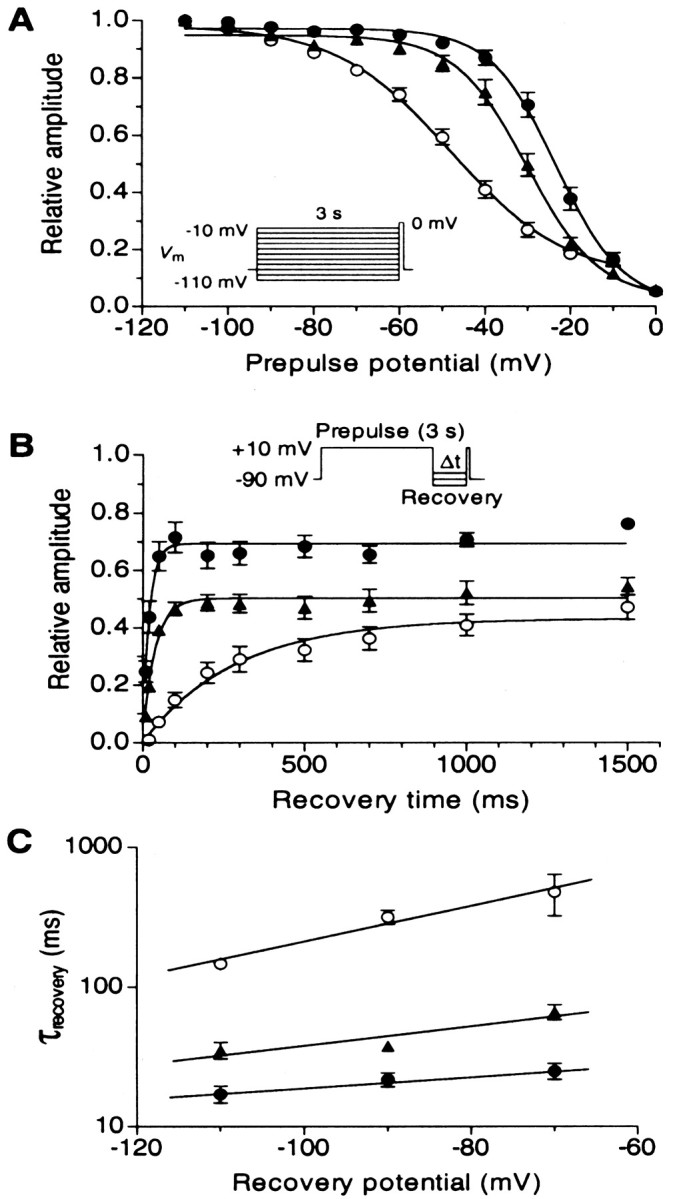
Differences in the inactivation properties among human α1B FL, Δ1, and Δ2 channels coexpressed with human β1b and α2-δ1 subunits. A, Steady-state inactivation curves for FL (○, n = 17), Δ1 (●, n = 13), and Δ2 (▴,n = 12) channels. Cells were held at −90 mV, and 3 sec conditioning depolarizations from −110 to −10 mV were applied, followed by a test depolarization to 0 mV for 100 msec. Current amplitudes were normalized to those obtained at a conditioning potential of −110 mV. Data points were fit with the standard Boltzmann equation, I/Imax = (1 + exp(Vm −Vh)/k)−1, where Vm is prepulse potential,Vh is the half-inactivation potential, andk is a slope factor. B, Time-dependent recovery from inactivation for FL (n = 6), Δ1 (n = 6), and Δ2 (n = 6) channels. After a 3 sec conditioning depolarization to +10 mV, a recovery hyperpolarization to −90 mV was applied for the indicated period (Δt, 20–1500 msec), followed by a 200 msec test depolarization to +10 mV. Time constant of recovery (τrecovery) was determined from a monoexponential fitIt = Imax × (1 − exp(−τrecovery × IΔt)), where It andImax indicate the current amplitude at recovery time Δt and the maximum current amplitude, respectively. The use of symbols was the same as that inA. C, Semi-logarithmic plots of the average recovery time constants, obtained at three different recovery potentials (−110, −90, and −70 mV) for five to six experiments each.
G-protein inhibition of human N-type calcium channels
It is well established that N-type calcium channel activity is inhibited by G-protein βγ subunits (Herlitze et al., 1996; Ikeda, 1996). This inhibition can be relieved by application of strong depolarizing prepulses (Bean, 1989; Zamponi and Snutch, 1998b) such that immediately after such a strong membrane depolarization, current amplitudes appear “facilitated.” To determine whether the sequence deletions in the domain II–III loop affected the abilities of G-proteins to inhibit the channels, we added 200 μmGTPγS to the internal recording solution and used a 50 msec prepulse to 150 mV to assess the degree of tonic G-protein inhibition. As seen from Figure 6, in the presence of GTPγS, all three channels displayed a similar degree of prepulse relief, which would suggest that G-protein inhibition was not affected by the II–III linker deletions. However, when we applied prepulses in the absence of G-protein activators, a robust prepulse relief was seen with all three channel variants (Fig. 6). Because we do not typically observe such a behavior for the rat Cav2.2 channel isoform (Arnot et al., 2000), it is unlikely that this effect is mediated by an excess of free endogenous G-protein βγ subunits in tsA-201 cells. To confirm this, we coexpressed the three variants with the C-terminal fragment of the β adrenergic receptor kinase (βARK-ct), a known Gβγ sink (Koch et al., 1994), which we have shown previously to remove tonic Gβγ-mediated inhibition of rat N-type calcium channels (Jarvis and Zamponi, 2001b). As evident from Figure 6, the prepulse effect persisted in the presence of βARK-ct. A significant variability among the treatment groups occurred only for Δ1 (p = 0.02), whereas no difference was observed for the Δ2 (p = 0.11) and FL (p = 0.11) channels. Overall, we conclude that the prepulse relief occurred independently of G-proteins and is therefore likely an intrinsic feature of the human Cav2.2 α1B subunit similar to what was described recently for a specific point mutant variant of the rat Cav2.2 channel (Zhong et al., 2001). Nonetheless, the observation that all three variants responded similarly to prepulses indicates that the deletions in the domain II–III linker do not affect this process.
Fig. 6.

Effect of depolarizing prepulses on the activities of the three α1B channel variants.A, Prepulse paradigm and a representative set of current records before and after the prepulse is shown. A 50 msec prepulse to +150 mV results in a large degree of facilitation of the FL channel in the absence of G-protein activators. B, Bar graph showing the normalized increase in current amplitude in response to the prepulse under control conditions, in the presence of 200 μm GTPγS, and in the presence of coexpressed βARK-ct. For the FL channel, no significant difference (p > 0.05) in facilitation ratios was seen under the three experimental conditions. For the deletion variants, the only significant differences occurred between GTPγS and βARK-ct for Δ1 and control versus GTPγS for Δ2. There was no statistically significant difference in the prepulse facilitation ratios among the three channels (p > 0.05) across the whole data set (ANOVA).
The domain II–III linker region affects block by external peptide toxins
To determine whether the deletion variants exhibited typical N-type channel pharmacology, we investigated the sensitivity of the three channel isoforms to marine snail peptide toxins known to potently and specifically block the N-type channels, ω-conotoxins MVIIA and GVIA. As seen in Figure 7, FL and Δ2 channel currents were both potently and equally effectively inhibited by ω-conotoxin MVIIA in a dose-dependent manner with IC50 values of ∼1 nm under our recording conditions. Surprisingly, the Δ1 channel was significantly more resistant to the blockade by ω-conotoxin MVIIA with an apparent IC50 value of 15 nm (Fig.7A). Similarly, block by ω-conotoxin GVIA, the binding site of which overlaps with that of MVIIA (Feng et al., 2001), was substantially less pronounced for the Δ1 variant compared with FL and Δ2 channels (Fig. 7B). Hence, structural alterations in one of the cytoplasmic regions of the N-type calcium channel α1 subunit can exert a pronounced affect on the ability of externally acting peptide toxins to interact with the channel.
Fig. 7.
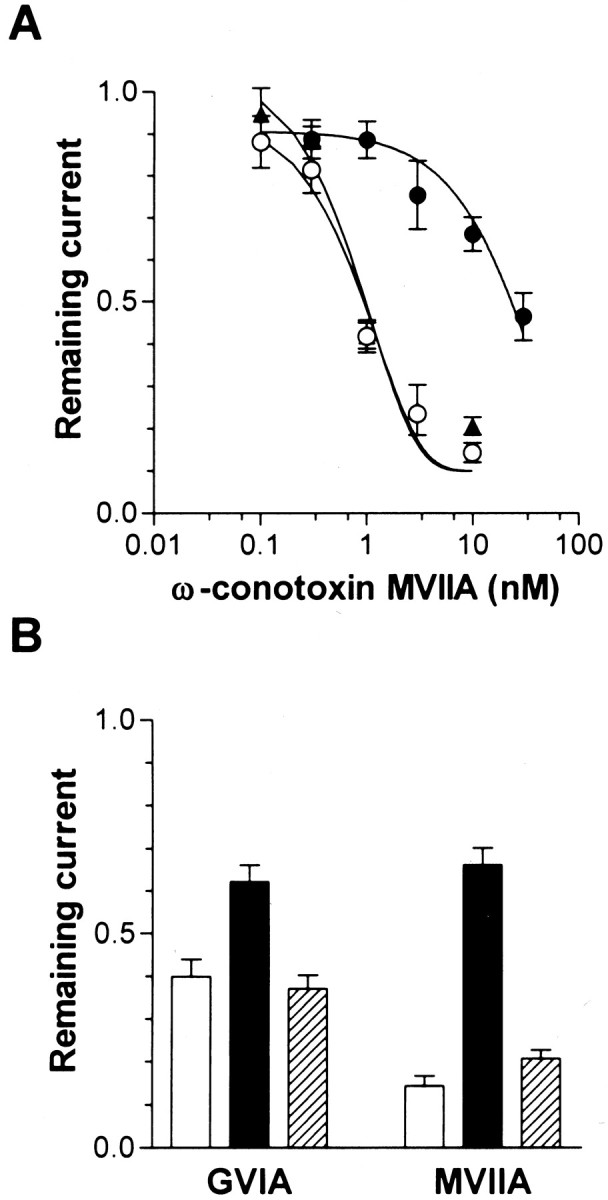
Effect of ω-conotoxins on the three human α1B variants. Cells expressing FL (○), Δ1 (●), and Δ2 (▴) together with human β1b and α2-δ1 subunits were held at −90 mV in 20 mm external Ba2+, and 100 msec depolarizations to 0 mV were applied every 15 sec and ω-conotoxins were perfused onto the cell by a microperfusion system.A, Dose dependence of ω-conotoxin MVIIA block of the three variants. The dose–response curves were fitted with the Hill equation; the numbers of experiments were between 5 and 7. Note that the dose–response curve obtained with the Δ1 variant is shifted toward higher concentrations. B, Comparison of the effects of ω-conotoxin MVIIA and GVIA on N-type channel activity at a fixed toxin concentration of 10 nm. For both types of conotoxins, the Δ1 variant is significantly less effectively inhibited, but the degree of this effect varies with toxin species (p < 0.05).
Interactions between syntaxin and human N-type channel variants
The lack of most of the synprint region in both Δ1 and Δ2 variants would imply that syntaxin 1A should not be able to functionally interact with these channels. For rat N-type calcium channels, the coexpression of syntaxin 1A with N-type calcium channels expressed in Xenopus oocytes or tsA-201 cells has been shown to mediate a 10–15 mV negative shift in half-inactivation potential (Bezprozvanny et al., 1995; Jarvis et al., 2000; Jarvis and Zamponi, 2001b), and one might thus predict that a deletion of the synprint region should no longer render the channels sensitive to these effects of syntaxin. To investigate this possibility, we first coexpressed syntaxin 1A with the human FL variants; however, to our surprise, we found that the midpoint of the steady-state inactivation curve observed with the human FL variant did not differ significantly in the presence and absence of syntaxin 1A (data not shown). Evidently, one or more of the amino acid differences present in the human N-type calcium channel domain II–III linker region abolish the effect of syntaxin 1A on steady-state inactivation seen with the rat isoform. Although this is consistent with our previous observation that the syntaxin 1A-mediated shifts in half-inactivation are easily disrupted (Jarvis and Zamponi, 2001b), this finding precludes the use of the inactivation effect as a discriminating factor among the three variants.
Instead, we performed in vitro binding assays between GST-syntaxin 1A and 6xHis fusion proteins of the entire domain II–III linker regions of the Δ1 and Δ2 variants and of the first domain of the synprint region of the FL variant (residues 711–862) that has been shown to bind syntaxin 1A in rat (Yokoyama et al., 1997). As shown in Figure 8, whereas the synprint motif of the FL variant effectively bound syntaxin 1A in vitro, only little if any binding could be detected to the domain II–III linker region of the Δ1 variant, and no binding was observed with Δ2. Identical results were obtained for a syntaxin 1B-GST construct (data not shown). These data indicate that the novel N-type channel variants cannot effectively associate with key components of the synaptic release machinery and are thus unlikely to mediate a role in fast synaptic transmission.
DISCUSSION
Novel N-type calcium channel variants
Our findings demonstrate that the human brain expresses two previously unidentified N-type calcium channel variants that lack large portions of the cytoplasmic domain II–III linker region. Of all the N-type channel splice isoforms identified to date, the variants reported here display the largest sequence deletions and, perhaps with the exception of the slowly activating splice isoform identified in rat (Lin et al., 1999), appear to mediate the most profound effects on N-type channel function and particularly on channel availability. Furthermore, both of the variants identified here essentially lack the synaptic protein interaction site that appears to be critically involved in docking the channels to the exocytotic machinery, and hence in efficient neurotransmitter release (Sheng et al., 1994;Mochida et al., 1996; Rettig et al., 1997). From the lack of the synprint site, one might expect that these variants may mediate a unique role in neuronal function.
Our genomic analysis strongly supports the idea that the Δ1 isoform is a naturally occurring splice variant. The deletion is initiated and terminates at intron–exon boundaries containing the appropriate splice donor and acceptor sites (Mount, 1982), as indicated by our own PCR data, as well as information obtained from the human genome project. In addition, a classical splice mechanism can nicely account for the variable presence of residue Arg756. Together with our results obtained from RNase protection assays, this strongly supports mRNA splicing as the basis of variant Δ1.
In contrast, the deletion pattern of Δ2 does not fit with classical mRNA splicing at exon–intron boundaries. Interestingly, the deletion pattern of the Δ2 variant resembles that of rabbit α1A (Cav2.1) variants CBP103 and CBP107, which were previously found during cloning using hybridization screening of a rabbit brain cDNA λ-phage library (Mori et al., 1991) and which showed 349 and 280 amino acid deletions, respectively (see Swiss-Prot accession number P27884). Although the rabbit genome sequence is unknown, the deletion occurs at positions different from the presumed exon–intron boundaries in the human and mouse Cav2.1 genome. Although at this time we cannot provide a possible mechanism to account for the generation of these variants and our Δ2 isoform, this does not rule out the possibility that this variant could be generated under physiological conditions. First, we identified this variant via PCR using different human cDNA libraries as a template. Second, the isoform was abundantly detected in RT-PCR reactions performed on IMR32 cell mRNA, as well as using human brain mRNA as a template. Third, the variant could be reliably detected using various different PCR enzyme systems. Finally, an appropriate band was consistently detected in RNase protection assays using commercially available mRNA from a number of human brain tissues. Hence, it appears likely that Δ2 is indeed a naturally occurring N-type channel variant that is generated by an as yet to be determined mechanism.
There is some evidence in the literature that N-type calcium channels expressed at different parts of neurons can differ in their functional properties. For example, in rat brain, antibodies raised against the II–III linker region immunoprecipitated less than half of brain ω-conotoxin receptor sites, suggesting the possible existence of ω-conotoxin-sensitive α1B subunits that may lack parts of the domain II–III linker (Westenbroek et al., 1992). Moreover, in rat sympathetic neurons, dendritic N-type calcium channels were hypersensitive to neurotransmitters and G-proteins but also showed a lower sensitivity to block by ω-conotoxins (Kavalali et al., 1997;Delmas et al., 2000). Although this may well be attributable to differential second messenger regulation or to association with different calcium channel β subunits, such distinct functional properties would also be consistent with alternate splicing. The observation that our two variants do not interact in vitrowith synaptic proteins such as syntaxin 1A and 1B would suggest that they are unlikely to participate in fast neurotransmitter release. This therefore raises the intriguing prospect that the domain II–III linker could be involved in targeting the channels to their appropriate subcellular locations with channels containing the synprint site being preferentially directed toward presynaptic nerve termini, whereas variants lacking this region could perhaps be targeted to dendrites. However, further experimentation will be required to substantiate such a possibility.
Implications for calcium channel structure and function
The sequence deletions in the domain II–III linker region produced three major effects on N-type channel function. First, a significant change in current density was observed. Second, the voltage dependence of inactivation was shifted toward more positive potentials by ∼20 mV. Third, the recovery from inactivation was accelerated dramatically in the deletion variants. Finally, the sensitivity of the Δ1 isoform to pore-blocking ω-conotoxins was reduced by more than one order of magnitude. It is interesting to note that the current densities observed with the two deletion mutants were in opposing directions. This might suggest that specific portions of the domain II–III linker that are differentially absent in the two deletion variants may serve to either enhance or depress current densities. The altered current densities could be caused by a change in single-channel conductance, the number of functional channels in the membrane, or the maximum open probability. Consequently, to distinguish among the alternatives, and to gain detailed mechanistic insights into the basis of these differential effects on current density, single-channel experiments will be required.
The altered inactivation profile is consistent with our recent work showing that the domain IIS6 region that immediately precedes the domain II–III linker is a key inactivation determinant in R-type and L-type calcium channels (Stotz et al., 2000). The deletions may affect the coupling of the domain II–III linker to the IIS6 region, thereby altering the voltage dependence of, and recovery from, inactivation. A key role of the N-type channel domain II–III linker in the voltage dependence of inactivation is also supported by the observation that the association of syntaxin 1A and SNAP-25 with this region mediates a hyperpolarizing shift in the voltage dependence of inactivation in rat Cav2.2 channels (Bezprozvanny et al., 1995;Jarvis and Zamponi, 2001b).
The reduced toxin sensitivity of the Δ1 variant is perplexing. It is commonly thought that ω-conotoxins MVIIA and GVIA physically occlude the pore from the extracellular side of the channel (Ellinor et al., 1994; Olivera et al., 1994; McDonough et al., 1996; Feng et al., 2001). The fact that sequence deletions in a large cytoplasmic region of the channel can affect the action of external pore blockers would be consistent with an allosteric coupling between the domain II–III and key residues forming the conotoxin receptor site. However, compared with the Δ2 variant that exhibits a normal conotoxin sensitivity, the additional sequence missing in the Δ1 isoform results in a net loss of 12 negative charges (Fig. 1). Given the net positive charge of the toxins (+4 for GVIA and +5 for MVIIA) (Olivera et al., 1994) and that electrostatic trans-channel interactions between external conotoxins and cytoplasmic channel blockers have been reported in sodium channels (French et al., 1996), it is conceivable that the reduction in toxin affinity could be mediated by an electrostatic mechanism (i.e., a reduced attraction of the toxin to its binding site caused by a loss of negative charges). In such a scenario, the larger net positive charge of MVIIA could perhaps account for the greater effect of the sequence deletion on this toxin isoform.
The observation that all three variants exhibited an intrinsic voltage-dependent facilitation indicates that the domain II–III linker is not an important determinant of this process and is consistent with data showing that G-protein inhibition of rat N-type channels is not affected by deletions of large parts of the II–III linker region (Meza and Adams, 1998). An intrinsic ability of N-type calcium channels to undergo voltage-dependent facilitation is not without precedent. Zhong et al. (2001) identified a single residue in the domain IS3 region of the rat Cav2.2 isoform (Glu177) that could induce a tonic reluctant gating state in rat Cav2.2 N-type calcium channels. It could be relieved with strong membrane depolarizations, and it rendered the channels insensitive to further G-protein inhibition, similar to what we observed here. When residue 177 was substituted with glycine, this effect was abolished (Zhong et al., 2001). However, our channel variants naturally contain a glycine residue in this position, and hence, the mechanism identified by Zhong et al. (2001) in the rat isoform cannot account for our observations with the human channel. This suggests that a reluctant gating state can be induced via more than one intrinsic mechanism, and at this point one cannot discount the possibility that the human calcium channel β1b subunit used in our experiments could contribute to these effects. Further experimentation will be required to identify the molecular basis of the effects in the human N-type channel isoform.
In summary, we have identified two novel human N-type calcium channel variants that provide interesting insights into the structural aspects of N-type calcium channel function. Their unique biophysical and pharmacological characteristics in conjunction with the absence of the synaptic protein interaction motif suggest that these channels are likely to mediate a unique role in neuronal physiology.
Footnotes
This work was supported by a Grant-in-Aid (11672168) from the Ministry of Education, Culture, Sports, Science and Technology (S.K.) and by an operating grant (G.W.Z.) from the Canadian Institutes of Health Research (CIHR). Portions of this work were supported by funding provided by Ono Pharmaceuticals, Osaka, Japan (S.K.). G.W.Z. holds faculty Scholarships from the Alberta Heritage Foundation for Medical Research (AHFMR), the CIHR, and the EJLB Foundation. C.B.C. holds a studentship award from the Heart and Stroke Foundation of Canada, and S.E.J. holds an AHFMR studentship award.
Correspondence should be addressed to Dr. Shuji Kaneko, Department of Neuropharmacology, Graduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail:skaneko@pharm.kyoto-u.ac.jp.
REFERENCES
- 1.Arnot MI, Stotz SC, Jarvis SE, Zamponi GW. Differential modulation of N-type α1B and P/Q-type α1A calcium channels by different G protein β subunit isoforms. J Physiol. 2000;527:203–212. doi: 10.1111/j.1469-7793.2000.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage-dependence. Nature. 1989;340:153–155. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 3.Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 4.Bezprozvanny I, Zhong P, Scheller RH, Tsien RW. Molecular determinants of the functional interaction between syntaxin and N-type Ca2+ channel gating. Proc Natl Acad Sci USA. 2000;97:13943–13948. doi: 10.1073/pnas.220389697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coppola T, Waldmann R, Borsotto M, Heurteaux C, Romey G, Mattéi M-G, Lazdunski M. Molecular cloning of a murine N-type calcium channel α1 subunit. Evidence for isoforms, brain distribution, and chromosomal localization. FEBS Lett. 1994;338:1–5. doi: 10.1016/0014-5793(94)80105-3. [DOI] [PubMed] [Google Scholar]
- 6.Delmas P, Abogadie FC, Buckley NJ, Brown DA. Calcium channel gating and modulation by transmitters depend on cellular compartmentalization. Nat Neurosci. 2000;3:670–678. doi: 10.1038/76621. [DOI] [PubMed] [Google Scholar]
- 7.DeWaard M, Liu H, Walker D, Scott VES, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 8.Ellinor PT, Zhang JF, Horne WA, Tsien RW. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature. 1994;372:272–275. doi: 10.1038/372272a0. [DOI] [PubMed] [Google Scholar]
- 9.Feng ZP, Hamid J, Doering CJ, Bosey GM, Snutch TP, Zamponi GW. Residue G1326 of the N-type calcium channel α1B subunit controls reversibility of ω-conotoxin GVIA and MVIIA block. J Biol Chem. 2001;276:15728–15735. doi: 10.1074/jbc.M100406200. [DOI] [PubMed] [Google Scholar]
- 10.French RJ, Prusak-Sochaczewski E, Zamponi GW, Becker S, Kularatna AS, Horn R. Interactions between a pore-blocking peptide and the voltage-sensor of a sodium channel: an electrostatic approach to channel geometry. Neuron. 1996;16:407–413. doi: 10.1016/s0896-6273(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda K, Kaneko S, Yada N, Kikuwaka M, Akaike A, Satoh M. Cyclic AMP-dependent modulation of N- and Q-type Ca2+ channels expressed in Xenopus oocytes. Neurosci Lett. 1996;217:13–16. doi: 10.1016/0304-3940(96)13055-x. [DOI] [PubMed] [Google Scholar]
- 12.Ghasemzadeh MB, Pierce RC, Kalivas PW. The monoamine neurons of the rat brain preferentially express a splice variant of α1B subunit of the N-type calcium channel. J Neurochem. 1999;73:1718–1723. doi: 10.1046/j.1471-4159.1999.731718.x. [DOI] [PubMed] [Google Scholar]
- 13.Hamid J, Nelson D, Spaetgens R, Dubel SJ, Snutch TP, Zamponi GW. Identification of an integration center for crosstalk between protein kinase C and G protein modulation of N-type calcium channels. J Biol Chem. 1999;278:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- 14.Hell JW, Appleyard SM, Yokoyama CT, Warner C, Catterall WA. Differential phosphorylation of two size forms of the N-type calcium channel α1 subunit which have different COOH termini. J Biol Chem. 1994;269:7390–7396. [PubMed] [Google Scholar]
- 15.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 17.Jarvis SE, Zamponi GW. Interactions between presynaptic calcium channels, cytoplasmic messengers, and proteins of the synaptic vesicle release complex. Trends Pharmacol Sci. 2001a;22:519–525. doi: 10.1016/s0165-6147(00)01800-9. [DOI] [PubMed] [Google Scholar]
- 18.Jarvis SE, Zamponi GW. Distinct molecular determinants govern syntaxin 1A-mediated inactivation and G-protein inhibition of N-type calcium channels. J Neurosci. 2001b;21:2939–2948. doi: 10.1523/JNEUROSCI.21-09-02939.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jarvis SE, Magga JM, Beedle AM, Braun JEA, Zamponi GW. G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gβγ. J Biol Chem. 2000;275:6388–6394. doi: 10.1074/jbc.275.9.6388. [DOI] [PubMed] [Google Scholar]
- 20.Kaneko S, Akaike A, Satoh M. Receptor-mediated modulation of voltage-dependent Ca2+ channels via heterotrimeric G-proteins in neurons. Jpn J Pharmacol. 1999;81:324–331. doi: 10.1254/jjp.81.324. [DOI] [PubMed] [Google Scholar]
- 21.Kavalali ET, Zhuo M, Bito H, Tsien RW. Dendritic Ca2+ channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. [DOI] [PubMed] [Google Scholar]
- 22.Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein coupled receptor kinase attenuates G beta gamma-mediated signaling. J Biol Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- 23.Leveque C, Pupier S, Marqueze B, Geslin L, Kataoka M, Takahashi M, De Waard M, Seagar M. Interaction of cysteine string proteins with the α1A subunit of the P/Q-type calcium channel. J Biol Chem. 1998;273:13488–13492. doi: 10.1074/jbc.273.22.13488. [DOI] [PubMed] [Google Scholar]
- 24.Lin Z, Haus S, Edgerton J, Lipscombe D. Identification of functionally distinct isoforms of the N-type Ca2+ channel in rat sympathetic ganglia and brain. Neuron. 1997;18:153–166. doi: 10.1016/s0896-6273(01)80054-4. [DOI] [PubMed] [Google Scholar]
- 25.Lin Z, Lin Y, Schorge S, Pan JQ, Beierlein M, Lipscombe D. Alternative splicing of a short cassette exon in α1B generates functionally distinct N-type calcium channels in central and peripheral neurons. J Neurosci. 1999;19:5322–5331. doi: 10.1523/JNEUROSCI.19-13-05322.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lü Q, Dunlap K. Cloning and functional expression of novel N-type Ca2+ channel variants. J Biol Chem. 1999;274:34566–34575. doi: 10.1074/jbc.274.49.34566. [DOI] [PubMed] [Google Scholar]
- 27.Lü Q, Atkisson MS, Jarvis SE, Feng Z-P, Zamponi GW, Dunlap KD. Syntaxin 1A supports a voltage-dependent inhibition of α1B calcium channels by Gβγ in chick sensory neurons. J Neurosci. 2001;21:2949–2957. doi: 10.1523/JNEUROSCI.21-09-02949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magga JM, Jarvis SE, Arnot MI, Zamponi GW, Braun JEA. Cysteine string protein regulates G protein modulation of N-type calcium channels. Neuron. 2000;28:195–204. doi: 10.1016/s0896-6273(00)00096-9. [DOI] [PubMed] [Google Scholar]
- 29.McDonough SI, Swartz KJ, Mintz IM, Boland LM, Bean BP. Inhibition of calcium channels in rat central and peripheral neurons by ω-conotoxin MVIIC. J Neurosci. 1996;16:2612–2623. doi: 10.1523/JNEUROSCI.16-08-02612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McEnery MW, Vance CL, Begg CM, Lee WL, Choi Y, Dubel SJ. Differential expression and association of calcium channels subunits in development and disease. J Bioenerg Biomembr. 1998;30:409–418. doi: 10.1023/a:1021997924473. [DOI] [PubMed] [Google Scholar]
- 31.Meza U, Adams B. G-protein-dependent facilitation of neuronal α1A, α1B, and α1E Ca channels. J Neurosci. 1998;18:5240–5252. doi: 10.1523/JNEUROSCI.18-14-05240.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mochida S, Sheng Z-H, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron. 1996;17:781–788. doi: 10.1016/s0896-6273(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 33.Mori Y, Friedrich T, Kim M-S, Mikami A, Nakai J, Ruth P, Bosse E, Hofman F, Flockerzi V, Furuichi T, Mikoshiba K, Imoto K, Tanabe T, Numa S. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 34.Mount SM. A catalogue of splice junction sequences. Nucleic Acids Res. 1982;10:459–472. doi: 10.1093/nar/10.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olivera BM, Miljanich GP, Ramachandran J, Adams ME. Calcium channel diversity and neurotransmitter release: the omega-conotoxins and omega-agatoxins. Annu Rev Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- 36.Page KM, Stephens GJ, Berrow NS, Dolphin AC. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J Neurosci. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan JQ, Lipscombe D. Alternative splicing in the cytoplasmic II–III loop of the N-type Ca channel α1B subunit: functional differences are β subunit-specific. J Neurosci. 2000;20:4769–4775. doi: 10.1523/JNEUROSCI.20-13-04769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a C-terminal Gβγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc Natl Acad Sci USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rettig J, Heinemann C, Ashery U, Sheng ZH, Yokoyamam CT, Catterall WA, Neher E. Alteration of Ca2+ dependence of neurotransmitter release by disruption of Ca2+ channel/syntaxin interaction. J Neurosci. 1997;17:6647–6656. doi: 10.1523/JNEUROSCI.17-17-06647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheng ZH, Rettig T, Takahashi M, Catterall WA. Identification of a syntaxin-binding site on N-type calcium channels. Neuron. 1994;13:1303–1313. doi: 10.1016/0896-6273(94)90417-0. [DOI] [PubMed] [Google Scholar]
- 41.Stea A, Soong TW, Snutch TP. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;15:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- 42.Stea A, Dubel SJ, Snutch TP. Alpha 1B N-type calcium channel isoforms with distinct biophysical properties. Ann NY Acad Sci. 1999;868:118–130. doi: 10.1111/j.1749-6632.1999.tb11282.x. [DOI] [PubMed] [Google Scholar]
- 43.Stotz S, Hamid J, Spaetgens RL, Jarvis SE, Zamponi GW. Fast inactivation of voltage-dependent calcium channels: a hinged lid mechanism? J Biol Chem. 2000;275:24575–24582. doi: 10.1074/jbc.M000399200. [DOI] [PubMed] [Google Scholar]
- 44.Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- 45.Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel α1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- 46.Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci. 1998;18:6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- 48.Williams ME, Brust PF, Feldman DH, Patthi S, Simerson S, Maroufi A, McCue AF, Veliçelebi G, Ellis SB, Harpold MM. Structure and functional expression of an ω-conotoxin-sensitive human N-type calcium channel. Science. 1992;257:389–395. doi: 10.1126/science.1321501. [DOI] [PubMed] [Google Scholar]
- 49.Yokoyama CT, Sheng Z, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17:6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. 1998a;8:351–356. doi: 10.1016/s0959-4388(98)80060-3. [DOI] [PubMed] [Google Scholar]
- 51.Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gβγ subunits. Proc Natl Acad Sci USA. 1998b;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- 53.Zhong H, Li B, Scheuer T, Catterall WA. Control of gating mode by a single amino acid residue in transmembrane segment IS3 of the N-type Ca2+ channel. Proc Natl Acad Sci USA. 2001;98:4705–4709. doi: 10.1073/pnas.051629098. [DOI] [PMC free article] [PubMed] [Google Scholar]