

Abstract
12-Hydroxyeicosatetraenoic acid (12-HETE) is a neuromodulator that is synthesized during ischemia. Its neuronal effects include attenuation of calcium influx and glutamate release as well as inhibition of AMPA receptor (AMPA-R) activation. Because 12-HETE reduces ischemic injury in the heart, we examined whether it can also reduce neuronal excitotoxicity. When treated with 12-(S)HETE, cortical neuron cultures subjected to AMPA-R-mediated glutamate toxicity suffered up to 40% less damage than untreated cultures. The protective effect of 12-(S)HETE was concentration-dependent (EC50 = 88 nm) and stereostructurally selective. Maximal protection was conferred by 300 nm12-(S)HETE; 300 nm15-(S)HETE was similarly protective, but 300 nm 5-(S)HETE was less effective. The chiral isomer 12-(R)HETE offered no protection; neither did arachidonic acid or 12-(S)hydroperoxyeicosatetraenoic acid. Excitotoxicity was calcium-dependent, and 12-(S)HETE was demonstrated to protect by inactivating N and L (but not P) calcium channels via a pertussis toxin-sensitive mechanism. Calcium imaging demonstrated that 12-(S)HETE also attenuates glutamate-induced calcium influx into neurons via a pertussis toxin-sensitive mechanism, suggesting that it acts via a G-protein-coupled receptor. In addition, 12-(S)HETE stimulates GTPγS binding (indicating G-protein activation) and inhibits adenylate cyclase in forskolin-stimulated cultures over the same concentration range as it exerts its anti-excitotoxic and calcium-influx attenuating effects. These studies demonstrate that 12-(S)HETE can protect neurons from excitotoxicity by activating a Gi/o-protein-coupled receptor, which limits calcium influx through voltage-gated channels.
Keywords: HETE, hydroxyeicosatetraenoic acid, lipoxygenase, ischemia, AMPA, eicosanoid, G-protein, VSCCs, glutamate, excitotoxicity
12-(S)Hydroxyeicosatetraenoate (12-(S)HETE) is a bioactive lipid formed by the action of a 12-lipoxygenase (12-LO) enzyme on arachidonic acid. Lipoxygenases are highly expressed by blood and immune cells (Yamamoto et al., 1997), but 12-LOs are also found in a number of other tissues, including the heart and the brain (Nishiyama et al., 1992, 1993). Notably, 12-HETE synthesis occurs in the heart during ischemic preconditioning, a phenomenon wherein a brief ischemic period “primes” the heart so that a later prolonged ischemia (Yellon and Dana, 2000) is less damaging and functional recovery is more rapid (Edwards et al., 2000). In rat and rabbit in vivo models, 12-HETE synthesis has been shown to be important in the development of ischemic preconditioning (Murphy et al., 1995; Chen et al., 1999). In isolated rat hearts, preconditioning did not develop when 12-HETE synthesis was prevented. Furthermore, preconditioning was potentiated when endogenous 12-HETE levels were elevated by inhibiting breakdown (Murphy et al., 1995). Ischemic preconditioning also occurs in the brain (Barone et al., 1998; Kawai et al., 1998), although the factors that control cerebral ischemic preconditioning are less well understood than those that underlie cerebral ischemia itself.
One of the critical components of cerebral ischemic pathology is the excitotoxic activation of glutamatergic receptors. Neurons, when starved of oxygen and glucose, indiscriminately release neurotransmitter stores (Jorgensen and Diemer, 1982), resulting in calcium influx through NMDA receptors (Garthwaite et al., 1986), AMPA receptors (AMPA-Rs) (Arias et al., 1999; Weiss and Sensi, 2000), and voltage-sensitive calcium channels (VSCCs) (Osuga and Hakim, 1996;Hampson et al., 1998).This excitotoxic influx of calcium overwhelms and damages mitochondrial calcium buffers (Fiskum et al., 1999) and indiscriminately activates cytosolic enzymes (Luiten et al., 1997). One of the enzymes activated during ischemia, phospholipase A2, induces massive arachidonic acid release from membrane stores (Gardiner et al., 1981; Dumuis et al., 1988), which is then rapidly converted to biologically active metabolites. Two of the principal arachidonate metabolites formed during ischemia are 12-(S)HETE and its precursor, 12-(S)hydroperoxyeicosatetraenoic acid (12-(S)HPETE) (Miyamoto et al., 1987; Dumuis et al., 1988; Wolfe et al., 1990), agents that are known to act as inhibitory neuromodulators by reducing VSCC activity (Ruehr et al., 1997), attenuating glutamate release (Freeman et al., 1991; Manzoni and Williams, 1999), and decreasing the affinity of AMPA-Rs for glutamate (Chabot et al., 1998). Because of such inhibitory effects as well as the protective role of 12-HETE against cardiac ischemia and its synthesis under excitotoxic conditions (Dumuis et al., 1988; Wolfe et al., 1990)], this study hypothesized that 12-HETE production during ischemia might protect neurons from excitotoxic injury. This proposal was examined using an in vitro (rat) cortical neuron model of glutamate excitotoxicity.
MATERIALS AND METHODS
Materials. All reagents other than those specifically listed below were purchased from Sigma (St. Louis, MO). Cyclothiazide and MK-801 were obtained from Tocris Cookson (Bristol, UK). 12-HETE and other eicosanoids were products of Cayman Chemicals (Ann Arbor, MI). Agatoxin IVa and conotoxin GVIa were purchased from Peptide International (Louisville, KY), and fura-2 AM was obtained from Molecular Probes (Eugene, OR). Radiolabeled GTPγS was purchased from New England Nuclear (Boston, MA). All culture media were from Life Technologies (Gaithersburg, MD). Assay kits for cAMP (RPN225) were purchased from Amersham Pharmacia Biotech (Piscataway, NJ).
Solution preparation. Solutions of HETEs and other lipophilic compounds were prepared by evaporating a 10 mm ethanolic solution (under a stream of nitrogen) in a siliconized microcentrifuge tube. Dimethyl sulfoxide (<0.05% of final volume) was added to ethanol to prevent the lipophil from completely drying onto the tube wall. After evaporation, 1 ml of culture medium was added and the drug was dispersed using a high-power sonic probe. Special attention ensured that the solutions did not generate foam or become hot to the touch. After dispersal, all solutions were brought to their final volume in siliconized glass tubes by mixing with an appropriate quantity of culture medium.
Neuronal cultures. Primary cortical neuron cultures were prepared according to a previously published method (Grimaldi and Cavallaro, 1999). Briefly, fetuses were extracted from Wistar rats that had been pregnant for 17 d. The cortices were then dissected out, cut into small pieces, and incubated with papain (1 mg/ml) for 9 min at 37°C. After this time the tissue was dissociated by passage through a fire-polished Pasteur pipette, and the resultant cell suspension was separated by centrifugation over a gradient consisting of 10 mg/ml bovine serum albumin (BSA) and 10 mg/ml ovomucoid (a trypsin inhibitor) in Earle's balanced salt solution. The pellet was then resuspended in high-glucose, phenol red-free DMEM containing 10% fetal bovine serum, 2 mm glutamine, 100 IU of penicillin, and 100 μg/ml streptomycin. Cells were counted and tested for viability using the trypan blue exclusion test; next, 3.2 × 105 cells/well were seeded onto the inner 24 wells of a poly-d-lysine-coated 48 well plate. The outer wells of the plate were filled with water to reduce evaporation from the cultures. Ninety-six hours after seeding, a cocktail containing 10 μm fluorodeoxyuridine and 10 μm uridine was added to block the growth of glial cells. After 7 d, 100 μl of fresh medium containing the glial-cell-inhibiting cocktail was added again to compensate for nutrient depletion.
NMDA-R-mediated toxicity procedure. Cortical neurons were cultured for 13–15 d in vitro. Before the experiment, half of the medium in which the cells were maintained was removed and kept for later use. Glutamate toxicity was examined by exposing the cultures to 200 μm glutamate for 15 min in a magnesium-free saline solution composed of (in mm): 125 NaCl, 25 glucose, 10 HEPES, pH 7.4, 5 KCl, and 1.8 calcium chloride, as well as 2.5% fatty-acid-free BSA. After exposure, cells were washed twice with saline and incubated in a medium composed of 70% original culture medium (in which the neurons had been cultured for the past 13–15 d) and 30% fresh medium. The cells were then incubated for 18 hr, after which time lactate dehydrogenase (LDH) levels in the media were examined and used as an index of cell toxicity. Preliminary studies demonstrated that glutamate toxicity was prevented by 500 nm MK-801, confirming an NMDA-R-mediated mechanism (data not shown).
AMPA- and kainate receptor-mediated toxicity procedures. To examine AMPA- and kainate receptor-mediated toxicity, neurons were cultured for 6–9 d and then exposed to 100 μmglutamate and 25 μm cyclothiazide (used to prevent AMPA-R desensitization) and 500 nm MK-801 for 18–20 hr before analysis.
The neuron preparation technique described above results in a primarily neuronal culture, although a limited number of astrocytes remain. This cell type is resistant to glutamate toxicity (Amin and Pearce, 1997), although such cell death has been reported in the presence of cyclothiazide (David et al., 1996). Initial studies were performed to confirm that astrocytes did not significantly contribute to AMPA–kainate toxicity in our cultures. When astrocytes were exposed to glutamate under the same conditions used on neuron preparations and left for 20 hr, LDH levels increased to only 105% of background, compared with 150–200% in neuron-enriched cultures (data not shown). Therefore, it was concluded that astrocyte contamination did not substantially contribute to the effects of glutamate in our neuronal cultures.
Toxicity assay. Cell toxicity was assessed 18–20 hr after insult by measuring LDH release into the phenol red-free culture media according to the method of Decker and Lohmann-Matthes (1988). Experiments were conducted with quadruplicate values at each point, and all plates contained glutamate (positive) and nonglutamate (negative) controls. The required assay development time varied between culture batches and number of days in vitro, and values were recorded when the positive control wells demonstrated absorbance values at 495–650 nm of 0.3–0.4 au. The assay was validated by comparison with a mitochondrial function viability assay [2,3-bis-(2-methoxy-4-nitro-5-sulphenyl)-(2H)-tetrazolium-5-carboxanilide (XTT) assay; Roehm et al., 1991]. The results obtained with the two systems were similar, although LDH release was used in this study because it provided a greater signal-to-noise ratio than the XTT assay. The ratio of mean LDH levels in negative and positive controls was used as an index of culture quality. Data from trials in which the mean LDH level in the negative controls exceeded 60% of the maximum (positive control) values were not included.
Measurement of single neuron, cytosolic calcium concentrations ([Ca2+]i).Neurons were seeded onto glass coverslips at a density of 1 × 106/cm2. After 8 d in culture, the cells were washed twice in a HEPES-buffered Krebs–Ringer saline (KRS) containing (in mm): 125 NaCl, 5 KCl, 1 Na2HPO4, 1 MgSO4, 1 CaCl2, 5.5 glucose, and 20 HEPES, pH 7.2. Coverslips were then placed in a 4 μm solution of fura-2 AM in KRS for 22 min at room temperature with continuous gentle agitation. Next, cells were washed and incubated for an additional 22 min in fresh KRS (Grimaldi and Cavallaro, 1999). The coverslips were then mounted in a chamber and perfused with KRS containing 0.002% methylcellulose (vehicle) at a flow rate of ∼800 μl/min. Images were acquired by an inverted microscope equipped with a 40× neofluar lens and an intensified CCD camera attached to a desktop computer. Metafluor software (Universal Imaging Corporation, West Chester, PA) was used to analyze experimental data. Ratio values, obtained by alternately exciting the preparations at wavelengths of 340 and 380 nm and recording the emitted light at 510 nm, were converted into calcium concentrations using the Grynkiewicz equation (subtracted equation form) (Grynkiewicz et al., 1985). Calibration values for this equation (Rmax − R,R − Rmin) were acquired by exposing neurons either to 10 μmionomycin in KRS containing 10 mm calcium or to 20 mm EGTA in calcium-free KRS, respectively.
Intracellular cAMP measurements. Intracellular cAMP levels were measured using a commercially available enzyme immunoassay kit. Before assay, cortical neuron cultures were exposed to 100 μm isobutylmethylxanthine and varying concentrations of 12-HETE for 10 min before and for 15 min during exposure to 5 μm forskolin. After this time cells were lysed with a detergent solution provided by the assay manufacturer and intracellular cAMP concentrations were assessed. Assays were conducted according to the manufacturers' instructions. Concentrations of cAMP in each sample were calculated using a standard curve and the data analysis software Prism (Graphpad Software, San Diego, CA).
Agonist-stimulated [35S] GTPγS binding studies. Rat forebrains (minus the cerebellum) were disrupted on ice in a buffer containing 0.32m sucrose, 50 mm Tris–HCl, pH 7.4, 3 mm MgCl2, and 1 mm EGTA, pH 7.4. The homogenates were centrifuged for 10 min at 1000 × g, the pellets were discarded, and the supernatants were then recentrifuged for 30 min at 3000 ×g. The resulting pellet was triturated in ice-cold Tris buffer containing (in mm): 50 Tris–HCl, pH 7.4, 3 MgCl2, and 1 EGTA, pH 7.4; it was then recentrifuged for 30 min at 3000 × g. The pellet was then suspended in 10 vol of Tris buffer containing 0.03% digitonin, incubated at room temperature for 15 min, and centrifuged for 30 min at 3000 × g. Membranes were then dispersed in Tris buffer; the protein concentration was assayed using Bradford's reagent (Bio-Rad, Hercules, CA) and the membranes were then frozen in aliquots at −80°C. On the day of the assay, an aliquot was thawed and diluted in assay buffer containing (in mm): 20 HEPES, 100 NaCl, 3 MgCl2, and 0.5 EGTA, as well as 0.1% (w/v) BSA, pH 7.4. The aliquot was then centrifuged for 30 min at 3000 × g. Membranes were subaliquoted into 1 ml of assay buffer containing 15 μg of protein, 30 μm GDP, 160 pm[35S]GTPγS, and varying concentrations of 12-(S)HETE. Nonspecific binding was determined in the presence of 30 μm “cold” GTPγS. Samples were incubated for 1 hr at 23°C, after which time the reaction was terminated by rapid filtration under vacuum through Whatman GF/B glass fiber filters (Whatman, Maidstone, UK), followed by four washes with cold assay buffer. Radioactivity bound to the filters was determined by liquid scintillation counting. The data were plotted and EC50 values were calculated using the Prism software.
Data analysis. All data are reported as mean percentages of the positive controls, ±SE. In the toxicity assays, statistical significance was examined using the two-tailed Student's ttest (unless otherwise indicated). Single-cell calcium values were analyzed by ANOVA together with a post hoc ttest. In all cases, significance was attributed when p< 0.05. In all cases, data was taken from experiments performed on at least three different neuron preparations.
RESULTS
Glutamate release during ischemia injures neurons by overstimulating NMDA (Choi et al., 1988) and AMPA–kainate-type (Calabresi et al., 2000; Weiss and Sensi, 2000) receptors. Although these receptor types are all ionotropic, they possess significantly different properties and require separate in vitro protocols to model their behavior. In the AMPA toxicity model, 12-(S)HETE reduced LDH release by up to 40%. This protection was concentration-dependent, with an EC50 of ∼80 nm, and was maximal in the presence of 300 nm HETE (Fig.1). Similar protection was also achieved with 1.5 μm 12-(S)HETE in an NMDA toxicity protocol (LDH reduction of 37 ± 6%;n = 20), although concentration dependence was not as evident as with the AMPA-R-dependent system. Consequently, AMPA-R-mediated toxicity was used in subsequent studies to characterize and examine the molecular mechanism by which 12-(S)HETE protects neurons.
Fig. 1.

Concentration-dependent effect of 12-HETE on glutamate toxicity. The effect of 12-HETE on AMPA-type toxicity is shown. Cortical neurons were exposed to 100 μm glutamate in the presence of 25 μm cyclothiazide and 500 nm MK-801 for 18–20 hr (n = 7 × 3). Values are means ± SEM compared with negative controls. *p < 0.05 compared with positive controls.
Structural specificity
To examine the structural requirements for HETE protection, 300 nm 12-(S)HETE was compared with equal concentrations of other structurally related arachidonate compounds. The isomer with opposing chirality, 12-(R)HETE, had no protective effect (Fig.2A), but 15-(S)HETE, an isomer with the same chirality and a structure similar to that of 12-(S)HETE, protected neurons to a degree similar to that seen for 12-(S)HETE (Fig. 2A). Another HETE isomer with a less similar structure, 5-(S)HETE, was also examined. This compound was somewhat protective (Fig.2B), although it was notably less effective than either 12-(S)HETE or 15-(S)HETE (p < 0.05 by one-tailed t test). In addition, arachidonic acid, which is the substrate for HETE synthesis, and 12-(S)HPETE, which is the hydroperoxy precursor of 12-(S)HETE, were also examined. Neither of these compounds protected cultures from glutamate toxicity (Fig.2B). The apparent relationship between structure and effect (as well as the low concentration required for maximal effect) suggests that 12-(S)HETE probably protects through a stereochemically sensitive mechanism such as a receptor rather than by a nonspecific mechanism such as membrane perturbation.
Fig. 2.
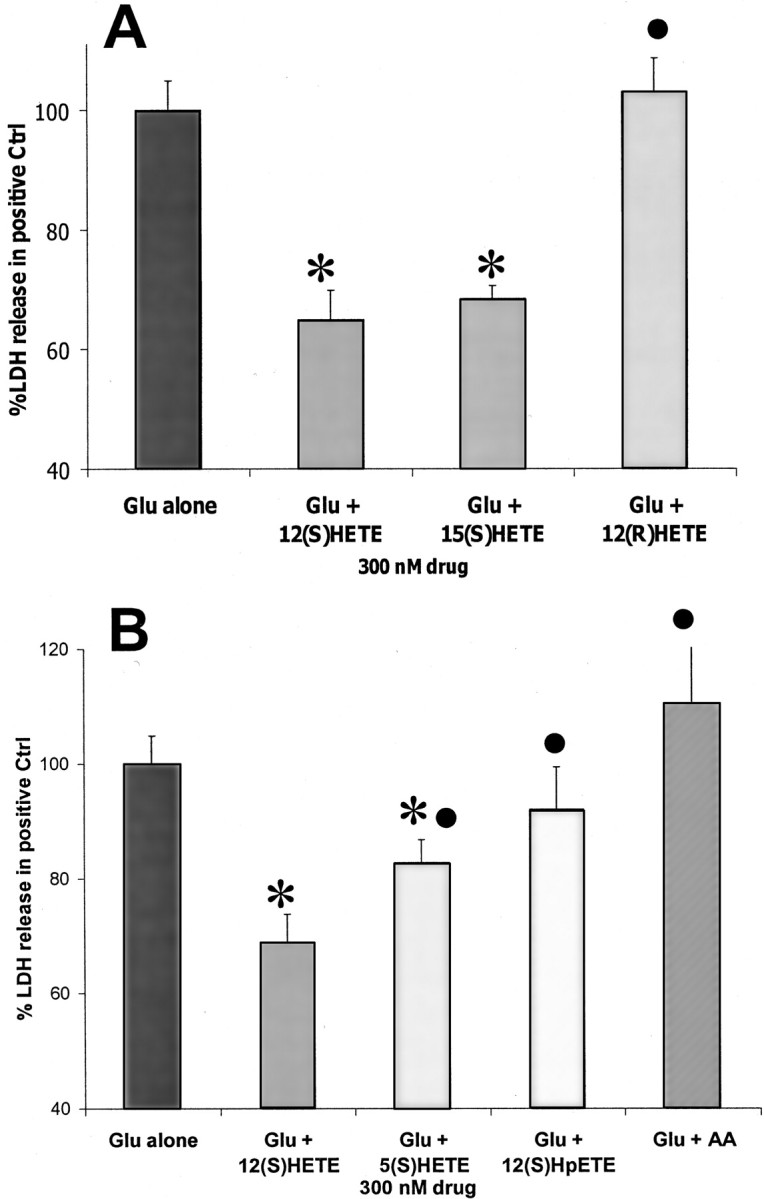
Comparison of the effect of 12-(S)HETE and similar molecules on AMPA-type toxicity. A, Comparison of the effect of 300 nm 12-(S)HETE with that of 300 nm 15-(S)HETE and 12-(R)HETE (n = 9 × 3).B, Comparison of the effect of 300 nm12-(S)HETE with that of 300 nm5-(S)HETE, 12-HPETE, or arachidonic acid (AA) (n = 6 × 3). The values for 12-(S)HETE in A andB are taken from the same data sets and included in the two sections for comparative purposes. Values are means ± SEM compared with negative controls. *p < 0.05 compared with positive controls. Protective agents with an effect that is significantly (p = 0.05 by one-tailedt test) different from 12-(S)HETE are marked with a ●.
Potassium channel studies
Previous reports have indicated that 12-LO products activate potassium channels sensitive to 4-aminopyridine (4-AP) (Volterra et al., 1991; Yu, 1995) and tetraethylammonium (TEA) (Lopes et al., 1998). Potassium influx through such channels hyperpolarizes neurons; therefore, this influx could reduce the depolarizing effects of glutamate. To investigate whether 12-HETE reduces excitotoxicity by activating these channels, 12-(S)HETE protection was examined in the presence of 2 mm TEA or 500 μm 4-AP. Figure 3demonstrates that 12-HETE protection was unaffected by the presence of either agent, suggesting that 12-HETE protection is not mediated by TEA- or 4-AP-sensitive potassium channels.
Fig. 3.

The effect of potassium-channel inhibitors on 12-HETE protection. Dark bars indicate the control condition; light bars represent inhibitor-treated cultures. A, Effect of 2 mm TEA on protection offered by 300 nm12-(S)HETE in an AMPA-type neurotoxicity model (n = 5 × 3). B, Effect of 500 μm 4-AP on protection offered by 300 nm12-(S)HETE in an AMPA-type neurotoxicity model (n = 5 × 3). Values are means ± SEM compared with negative controls. *p < 0.05 compared with positive controls; #p < 0.05 compared with glutamate plus inhibitor.
12-(S)HETE and calcium channels
To confirm that in vitro AMPA-R-mediated toxicity is calcium-dependent, the calcium chelator EDTA was demonstrated to reduce LDH production in a concentration-dependent manner (Fig.4). To examine whether 12-(S)HETE might also affect calcium influx during AMPA-R activation ([Ca2+]i), fura-2 AM fluorometric imaging was used. When cortical neurons were exposed to glutamate (in the presence of MK-801), [Ca2+]iimmediately rose to a peak value and then fell to a lower, long-lasting plateau phase. When 300 nm12-(S)HETE was included in the perfusion medium before glutamate exposure, glutamate-induced [Ca2+]i elevation at the peak was significantly reduced (Fig.5A,C), demonstrating that 12-(S)HETE reduces glutamate-induced [Ca2+]i in neurons.
Fig. 4.

Concentration-dependent effect of EDTA on AMPA-type toxicity. Cortical neurons were exposed to 100 μm glutamate in the presence of 25 μmcyclothiazide and 500 nm MK-801 for 18–20 hr (n = 5 × 3). Values are means ± SEM compared with negative controls. *p < 0.05 compared with positive controls.
Fig. 5.
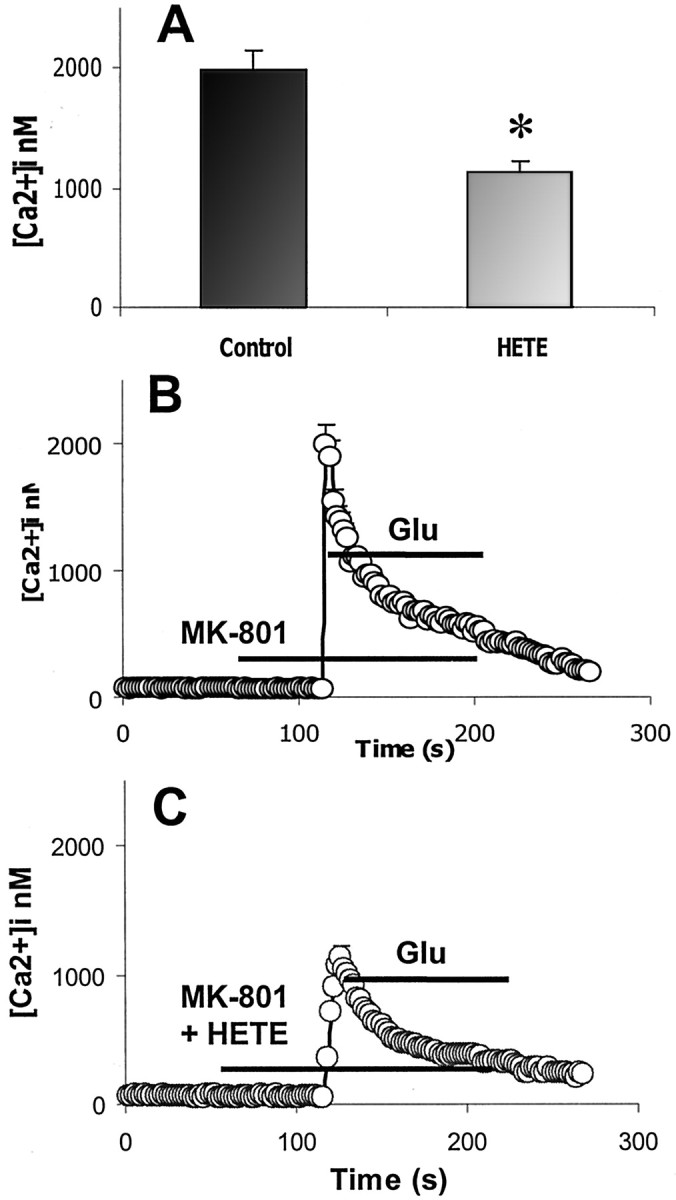
Effect of 300 nm12-(I)HETE on glutamate-induced [Ca2+]i elevation into cortical neurons. A, Mean (± SEM) values of [Ca2+]i at the peak in cells exposed to 100 μm glutamate in the absence or presence of 300 nm 12-HETE. *p < 0.05.B, Effect of 100 μm glutamate on calcium influx (n = 578, 7 replicates) C, Effect of 100 μm glutamate on calcium influx in the presence of 300 nm 12-(S)HETE (n = 488, 8 replicates).
In the AMPA-R-mediated toxicity model, calcium can enter neurons through both AMPA-Rs and VSCCs (Colwell and Levine, 1999; Leski et al., 1999). To examine whether 12-HETE protection is mediated by such depolarization-sensitive channels, toxicity studies were conducted in the presence of 250 nm calcicludine, a toxin that inactivates L-, N-, and P-type calcium channels (Schweitz et al., 1994). Calcicludine treatment reduced the overall level of LDH released after glutamate stimulation (Fig.6A) but also completely prevented 12-(S)HETE protection, suggesting that 12-HETE and calcicludine may share a site of action. To confirm this result and further characterize the population(s) of calcium channel(s) that are sensitive to 12-HETE inhibition, the study was repeated with channel-specific inhibitors. In the presence of the N-type blocker conotoxin GVIa (500 nm, Fig.6B) or the synthetic L-type channel blocker nifedipine (2 μm, Fig. 6C), the results achieved were similar to those observed with calcicludine (i.e., the VSCC blockers reduced toxicity and 12-HETE did not provide additional protection). In contrast, although the P/Q-type calcium-channel toxin agatoxin IVa (250 nm) reduced glutamate toxicity, its protective effect was additive with that of 12-HETE (Fig. 6D), suggesting that agatoxin and 12-HETE affect different targets. These data strongly suggest that 12-HETE prevents glutamate-induced excitotoxicity by inhibiting N- and L-type VSCCs but does not affect P-type channel function.
Fig. 6.

Effect of calcium-channel inhibitors on 12-HETE protection. Dark gray bars represent the control condition; light gray bars represent inhibitor-treated cultures. A, Effect of 250 nmcalcicludine on protection offered by 300 nm12-(S)HETE in an AMPA-type neurotoxicity model (n = 7 × 3). B, Effect of 500 nm conotoxin GVIa on protection offered by 300 nm 12-(S)HETE in an AMPA-type neurotoxicity model (n = 6 × 3).C, Effect of 2.5 μm nifedipine on protection offered by 300 nm12-(S)HETE in an AMPA-type neurotoxicity model (n = 14 × 3). D, Effect of 250 nm agatoxin IVa on protection offered by 300 nm 12-(S)HETE in an AMPA-type neurotoxicity model (n = 4 × 3). Values are means ± SEM compared with negative controls. *p < 0.05 compared with positive controls; #p < 0.05 compared with glutamate plus inhibitor.
Pertussis toxin studies
Pertussis toxin (PTx, an agent that inactivates Gi/o proteins) was used to investigate whether 12-HETE exerts its effect by direct action on N- and L-type VSCCs or by activating a Gi/o-protein-coupled receptor (Zamponi and Snutch, 1998; Kaneko et al., 1999). After treatment with 100 ng/ml PTx overnight, the anti-excitotoxic effect of 12-(S)HETE was eliminated (Fig.7), which suggests that 12-(S)HETE inhibits N- and L-type VSCCs via Gi/o-protein linkage rather than through a direct action. The effect of PTx on 12-(S)HETE-inhibited calcium influx was also examined by calcium imaging. As with the glutamate toxicity studies, PTx treatment eliminated the inhibitory effect of 12-HETE on glutamate-induced [Ca2+]i (Fig.8A,C), which suggests that 12-(S)HETE inhibits glutamate-stimulated calcium influx via a Gi/o-protein-coupled mechanism.
Fig. 7.

Effect of 24 hr of pretreatment with 100 ng/ml PTx on protection offered by 300 nm12-(S)HETE against AMPA-type toxicity.Dark gray bars represent the control condition;light gray bars represent PTx-treated cultures (n = 4 × 3). Values are means ± SEM compared with negative controls. *p < 0.05 compared with positive controls.
Fig. 8.
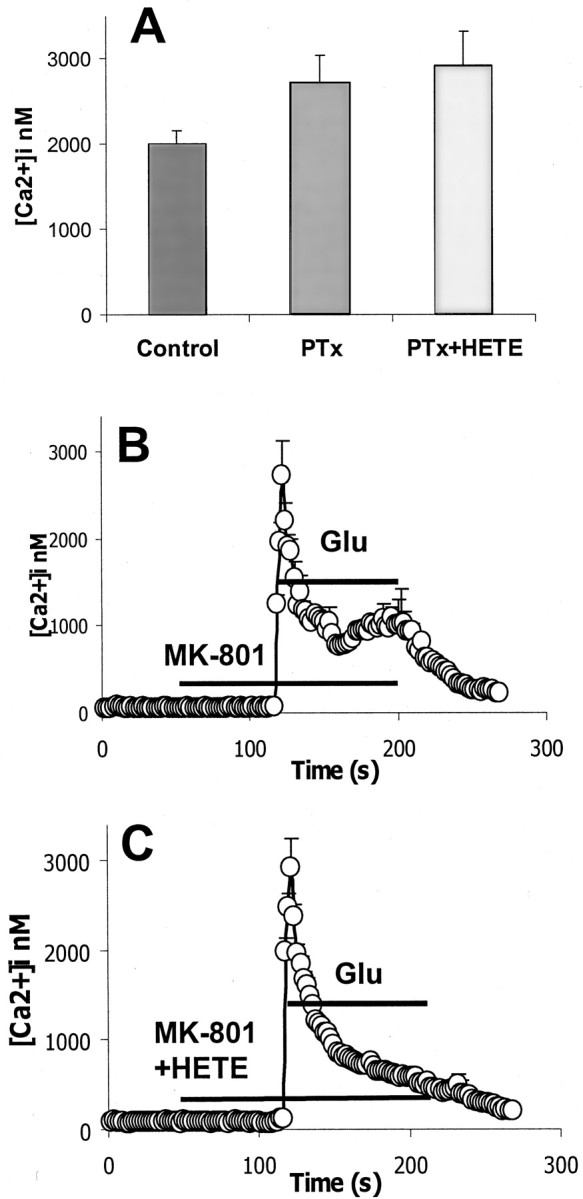
Effect of 300 nm12-(S)HETE on glutamate-induced [Ca2+]i elevation in cortical neurons pretreated with 100 ng/ml pertussis toxin for 24 hr. A, Mean values of [Ca2+]i at peak in cells exposed to 100 μm glutamate in the absence or presence of 300 nm 12-HETE. B, Effect of 100 μm glutamate on calcium influx (n = 353, 3 replicates) C, Effect of 100 μmglutamate on calcium influx in the presence of 300 nm12-(S)HETE (n = 303, 3 replicates). Samples with or without HETE were not significantly different (p < 0.05). Error bars represent SEM.
12-(S)HETE stimulation of GTPγS binding
The Gi/o-protein-coupled receptor-mediated effect of 12-(S)HETE suggested by pertussis toxin studies was confirmed by directly examining G-protein activity. Activation of a G-protein-coupled receptor increases the rate of GTP association with G-proteins. In the presence of labeled GTPγS, a GTP analog that is only slowly hydrolyzable, this association of GTP and G-protein can be examined. The effect of 12-(S)HETE on G-protein activation was measured in washed rat forebrain membrane preparations by incubation with [35S]-labeled GTPγS. Figure9A demonstrates that application of 12-(S)HETE increases specific GTPγS binding in preparations of rat forebrain by ∼20%, with an EC50 of 28 nm. The concentration range over which 12-(S)HETE affects GTPγS binding is comparable with that over which it exerts an anti-excitotoxic effect in cortical cultures.
Fig. 9.
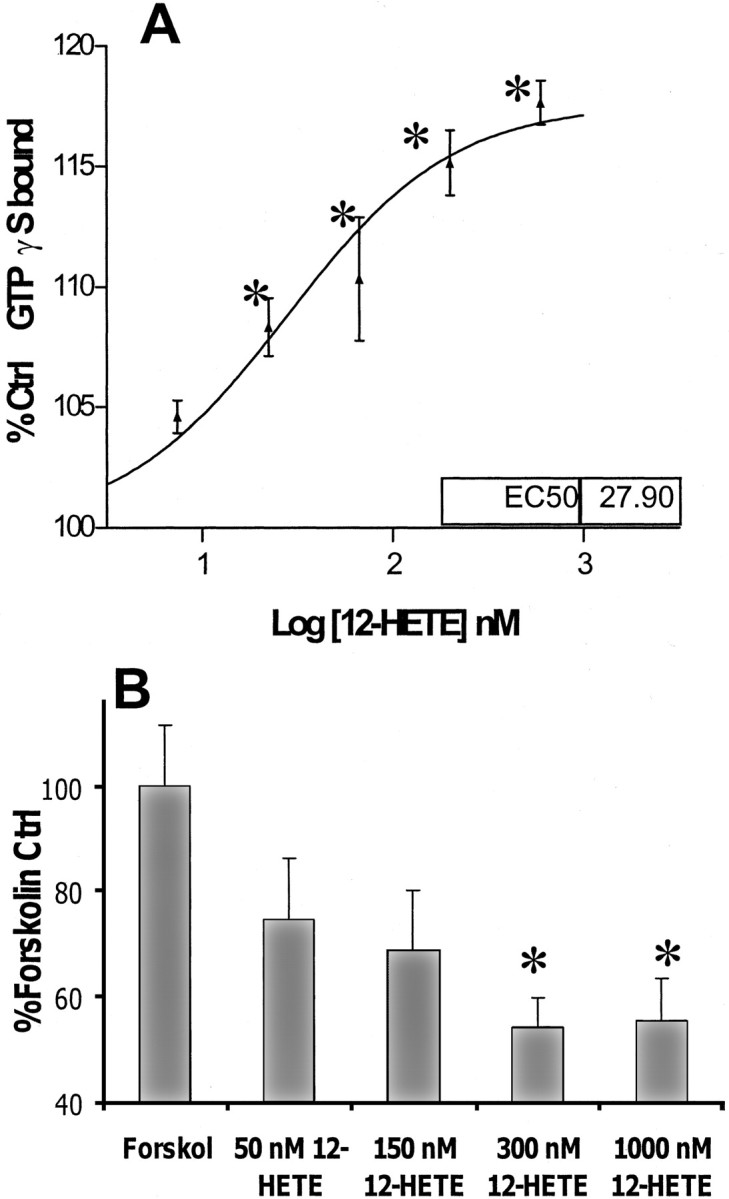
A, The effect of 12-HETE on agonist-stimulated GTPγS binding. 12-(S)HETE exposure increased specific binding of [35S]GTPγS in preparations of rat forebrain membrane. Values are percentages of specific binding in control samples. Representative figure n = 3, experiment repeated three times with similar results. B, The effect of 12-HETE on forskolin-stimulated adenylate cyclase activity. Cortical neurons were exposed to 12-HETE for 10 min before and for 15 min during exposure to 5 μm forskolin. Values are mean percentages of forskolin alone ± SEM compared with negative control values (n = 3 × 3). *p < 0.05 compared with positive controls.
Adenylate cyclase inhibition by 12-(S)HETE
Gi/o-protein-coupled receptor systems characteristically inhibit adenylate cyclase activity in addition to exerting an inhibitory effect on VSCCs. This inhibition can be studied by examining the ability of an agonist to inhibit forskolin-stimulated cAMP formation. Figure 9B shows that the concentration range over which 12-(S)HETE inhibits cAMP formation in forskolin-stimulated cortical cultures is similar to that over which it exerts its anti-excitotoxic effects.
DISCUSSION
The hypothesis that 12-HETE may be an endogenous neuroprotector is based on two previous findings: 12-HETE is known to be an inhibitory neuromodulator (Freeman et al., 1991; Ruehr et al., 1997; Chabot et al., 1998), which is formed in vivo in response to cerebral ischemia (Asano et al., 1985; Ellis et al., 1989) and in vitro in response to excitotoxic conditions (Dumuis et al., 1988;Wolfe et al., 1990). The results of the present study are consistent with this hypothesis in that they demonstrate that 12-(S)HETE reduces the in vitroneurotoxicity caused by glutamate exposure. The concentration range over which 12-(S)HETE attenuates glutamate toxicity (EC50 = 88 nm) (Fig. 1) is also consistent with a neuroprotective role, because 12-(S)HETE is reportedly present in brain tissue during ischemia at similar levels (80 nm) (Ellis et al., 1989).
To examine the mechanism by which 12-(S)HETE prevents excitotoxic damage, preliminary studies were performed with a calcium chelator (Fig. 4), which confirmed that AMPA-R-mediated toxicity was a calcium-dependent process. A calcium imaging study then revealed that calcium influx into neurons during AMPA-R stimulation was substantially reduced by 12-(S)HETE (300 nm) (Fig. 5). These data provided a possible mechanism by which 12-(S)HETE could prevent neuronal toxicity: reduction of calcium overload during glutamate exposure. However, because the majority of AMPA-Rs flux very little calcium (because of the presence of AMPA-R subunit GluR2, which confers calcium impermeability), it was suggested that the calcium influx during AMPA-R stimulation probably entered the neurons via VSCCs activated by AMPA-R-mediated depolarization. This concept was confirmed by glutamate toxicity studies, which indicated that L-, N-, and P/Q-type VSCC blockers reduce glutamate toxicity in neuronal cultures. However, whereas protection offered by the P/Q-type VSCC toxin, ω-agatoxin-IVa (Fig.6D) was additive with that offered by 12-HETE, additivity was not observed between 12-(S)HETE and N- or L-type blockers (Fig. 6A–C). These results suggest that agatoxin and 12-HETE operate via distinct pathways, whereas 12-HETE shares a common mode of action with conotoxin, calcicludine, and nifedipine. Therefore, it was concluded that 12-(S)HETE reduces glutamate toxicity by attenuating excitotoxic calcium influx through L- and N- but not P/Q-type voltage-gated calcium channels.
Structure–activity studies revealed that 12-(S)HETE protection can be mimicked by 15-(S)HETE, a structurally similar isomer, and to a lesser extent by 5-(S)HETE, an analog with a structure that is more different from 12-HETE than is 15-(S)HETE (Fig. 2). However, the protective effect of 12-(S)HETE was not shared by 12-(R)HETE or by the 12-(S)HETE precursor molecules 12-(S)HPETE or arachidonic acid, indicating that the protection offered by 12-(S)HETE is stereostructurally defined. Together, the specificity and high potency of the actions of 12-HETE suggest that 12-(S)HETE probably acts on cortical neurons via a unique site, such as a receptor or an enzyme.
A previous study demonstrated that lymphocytes contain a specific 12-HETE binding site that modulates phospholipase D activity (Zakaroff-Girard et al., 1999) and can be blocked by PTx (an agent that inactivates Gi/o proteins by ADP ribosylation). In the current study, we similarly demonstrated that PTx pretreatment prevents 12-(S)HETE from attenuating glutamate toxicity (Fig. 7) and eliminates the reductive effect of 12-(S)HETE on glutamate-induced [Ca2+]i(Fig. 8). These data suggest that the inhibitory effect of 12-(S)HETE on VSCCs is not attributable to direct binding to a site on the calcium channel, but rather that 12-(S)HETE inhibits L- and N-type channel opening by activating a PTx-sensitive Gi/o protein that is coupled to the VSCCs. The hypothesis that 12-HETE acts specifically on a Gi/o-protein-coupled receptor was supported by examining the ability of 12-(S)HETE to stimulate G-protein binding of GTP (a step in the signal transduction process that occurs after activation of all G-protein-coupled receptors) in washed and permeabilized preparations of rat forebrain membrane. Figure9A demonstrates that 12-(S)HETE stimulates the association of G-protein with labeled GTPγS, indicating activation of a G-protein-coupled receptor, with an EC50 of 28 nm, a concentration range comparable with its anti-excitotoxic effect. An alternative means by which to examine the activation of an inhibitory G-protein-coupled receptor is to assess the effect of its agonist on forskolin-stimulated adenylate cyclase activity. Figure 9Bdemonstrates that 12-(S)HETE inhibits adenylate cyclase activity over a concentration range similar to that over which it reduces AMPA-R toxicity.
The pertussis toxin sensitivity of the effect of 12-HETE on glutamate-induced [Ca2+]iand AMPA-R-mediated toxicity, when taken together with the ability of 12-HETE to stimulate GTPγS binding and inhibit adenylate cyclase activity (over the same concentration range), provides compelling evidence that rat neurons possess a 12,15-(S)HETE receptor that couples via a Gi/o-protein linkage to L- and N-type calcium channels and adenylate cyclase.
These data indicating the existence of a 12-(S)HETE receptor in neurons adequately explain a number of the reported neuromodulatory effects of 12-HETE. In hippocampal synaptosomes 12-HETE inhibits calcium uptake in a nifedipine-sensitive manner (Ruehr et al., 1997), a finding that is in keeping with the present study, which demonstrates a 12-HETE receptor that negatively modulates L-type calcium channels. In another study, 12-LO metabolites were reported to provide “a presynaptic inhibitory signal that limits neurotransmitter release from hippocampal mossy fiber terminals” (Freeman et al., 1991). Because N-type calcium channels are known to be key regulators of neurotransmitter release (Elliott et al., 1995;Catterall, 1999), this result is also consistent with our data, which show 12-(S)HETE inhibition of N-type calcium channel activity. Furthermore, the ability of 12-LO metabolites to regulate neurotransmitter release in hippocampal presynapses (Normandin et al., 1996; Chabot et al., 1998) has been shown to control long-term depression, suggesting that the putative receptor described in this study may play a significant role in modulating memory formation by regulating N-type calcium-channel activity.
During the drafting of this manuscript a report was published indicating that 12-(S)HETE has some affinity for the low-affinity leukotriene B4 receptor (BLT2) (Yokomizo et al., 2001). However, the features of BLT2 are considerably different from those described here; in particular, this receptor is not pertussis-toxin sensitive, 12-(S)HETE binds to BLT2 with an EC50 in the micromolar rather than nanomolar range, and most importantly, mRNA to BLT2 is found in almost every tissue except the brain (Yokomizo et al., 2000).
In summary, the data presented in this study demonstrate that 12-(S)HETE significantly attenuates neuronal AMPA toxicity by reducing calcium influx through N- and L-type calcium channels during glutamate exposure. The protective effect of 12-HETE is stereostructurally defined and is mediated via a PTx-sensitive Gi/o protein linkage. As with other Gi/o-protein-coupled receptor systems, the putative 12-(S)HETE receptor inhibits adenylate cyclase activity in addition to inhibiting VSCCs. The concentration range over which 12-(S)HETE inhibits cAMP formation and stimulates GTPγS binding is similar to that over which it reduces glutamate toxicity. Together, these data strongly support the existence of a G-protein-coupled 12-(S)HETE receptor in rat cortical neuronal membranes, which may act as a neuroprotective system during excitotoxic events such as ischemia.
Footnotes
Correspondence should be addressed to Dr. Aidan Hampson, Cortex Pharmaceuticals, 15231 Barranca Parkway, Irvine, CA 92618. E-mail:ahampson@cortexpharm.com.
Dr. Hampson's present address: Cortex Pharmaceuticals, Irvine, CA 92618.
Dr. Grimaldi's present address: Department of Neurology, Uniformed Services University of the Health Sciences, Bethesda, MD 20814.
REFERENCES
- 1.Amin N, Pearce B. Glutamate toxicity in neuron-enriched and neuron-astrocyte co-cultures: effect of the glutamate uptake inhibitor L-trans-pyrrolidine-2,4-dicarboxylate. Neurochem Int. 1997;30:271–276. doi: 10.1016/s0197-0186(96)00092-7. [DOI] [PubMed] [Google Scholar]
- 2.Arias RL, Tasse JR, Bowlby MR. Neuroprotective interaction effects of NMDA and AMPA receptor antagonists in an in vitro model of cerebral ischemia. Brain Res. 1999;816:299–308. doi: 10.1016/s0006-8993(98)01051-8. [DOI] [PubMed] [Google Scholar]
- 3.Asano T, Gotoh O, Koide T, Takakura K. Ischemic brain edema following occlusion of the middle cerebral artery in the rat. II. Alteration of the eicosanoid synthesis profile of brain microvessels. Stroke. 1985;16:110–113. doi: 10.1161/01.str.16.1.110. [DOI] [PubMed] [Google Scholar]
- 4.Barone FC, White RF, Spera PA, Ellison J, Currie RW, Wang X, Feuerstein GZ. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke. 1998;29:1937–1951. doi: 10.1161/01.str.29.9.1937. [DOI] [PubMed] [Google Scholar]
- 5.Calabresi P, Picconi B, Saulle E, Centonze D, Hainsworth AH, Bernardi G. Is pharmacological neuroprotection dependent on reduced glutamate release? Stroke. 2000;31:766–773. doi: 10.1161/01.str.31.3.766. [DOI] [PubMed] [Google Scholar]
- 6.Catterall WA. Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann NY Acad Sci. 1999;868:144–159. doi: 10.1111/j.1749-6632.1999.tb11284.x. [DOI] [PubMed] [Google Scholar]
- 7.Chabot C, Gagne J, Giguere C, Bernard J, Baudry M, Massicotte G. Bidirectional modulation of AMPA receptor properties by exogenous phospholipase A2 in the hippocampus. Hippocampus. 1998;8:299–309. doi: 10.1002/(SICI)1098-1063(1998)8:3<299::AID-HIPO11>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 8.Chen W, Glasgow W, Murphy E, Steenbergen C. Lipoxygenase metabolism of arachidonic acid in ischemic preconditioning and PKC-induced protection in heart. Am J Physiol. 1999;276:H2094–H2101. doi: 10.1152/ajpheart.1999.276.6.H2094. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colwell CS, Levine MS. Metabotropic glutamate receptor modulation of excitotoxicity in the neostriatum: role of calcium channels. Brain Res. 1999;833:234–241. doi: 10.1016/s0006-8993(99)01545-0. [DOI] [PubMed] [Google Scholar]
- 11.David JC, Yamada KA, Bagwe MR, Goldberg MP. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods. 1988;115:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- 13.Dumuis A, Sebben M, Haynes L, Pin JP, Bockaert J. NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature. 1988;336:68–70. doi: 10.1038/336068a0. [DOI] [PubMed] [Google Scholar]
- 14.Edwards RJ, Saurin AT, Rakhit RD, Marber MS. Therapeutic potential of ischaemic preconditioning. Br J Clin Pharmacol. 2000;50:87–97. doi: 10.1046/j.1365-2125.2000.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott EM, Malouf AT, Catterall WA. Role of calcium channel subtypes in calcium transients in hippocampal CA3 neurons. J Neurosci. 1995;15:6433–6444. doi: 10.1523/JNEUROSCI.15-10-06433.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellis EF, Police RJ, Rice LY, Grabeel M, Holt S. Increased plasma PGE2, 6-keto-PGF1 alpha, and 12-HETE levels following experimental concussive brain injury. J Neurotrauma. 1989;6:31–37. doi: 10.1089/neu.1989.6.31. [DOI] [PubMed] [Google Scholar]
- 17.Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab. 1999;19:351–369. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Freeman EJ, Damron DS, Terrian DM, Dorman RV. 12-Lipoxygenase products attenuate the glutamate release and Ca2+ accumulation evoked by depolarization of hippocampal mossy fiber nerve endings. J Neurochem. 1991;56:1079–1082. doi: 10.1111/j.1471-4159.1991.tb02032.x. [DOI] [PubMed] [Google Scholar]
- 19.Gardiner M, Nilsson B, Rehncrona S, Siesjo BK. Free fatty acids in the rat brain in moderate and severe hypoxia. J Neurochem. 1981;36:1500–1505. doi: 10.1111/j.1471-4159.1981.tb00592.x. [DOI] [PubMed] [Google Scholar]
- 20.Garthwaite G, Hajos F, Garthwaite J. Ionic requirements for neurotoxic effects of excitatory amino acid analogues in rat cerebellar slices. Neuroscience. 1986;18:437–447. doi: 10.1016/0306-4522(86)90164-8. [DOI] [PubMed] [Google Scholar]
- 21.Grimaldi M, Cavallaro S. Functional and molecular diversity of PACAP/VIP receptors in cortical neurons and type I astrocytes. Eur J Neurosci. 1999;11:2767–2772. doi: 10.1046/j.1460-9568.1999.00693.x. [DOI] [PubMed] [Google Scholar]
- 22.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 23.Hampson AJ, Bornheim LM, Scanziani M, Yost CS, Gray AT, Hansen BM, Leonoudakis DJ, Bickler PE. Dual effects of anandamide on NMDA receptor-mediated responses and neurotransmission. J Neurochem. 1998;70:671–676. doi: 10.1046/j.1471-4159.1998.70020671.x. [DOI] [PubMed] [Google Scholar]
- 24.Jorgensen MB, Diemer NH. Selective neuron loss after cerebral ischemia in the rat: possible role of transmitter glutamate. Acta Neurol Scand. 1982;66:536–546. doi: 10.1111/j.1600-0404.1982.tb03140.x. [DOI] [PubMed] [Google Scholar]
- 25.Kaneko S, Akaike A, Satoh M. Receptor-mediated modulation of voltage-dependent Ca2+ channels via heterotrimeric G-proteins in neurons. Jpn J Pharmacol. 1999;81:324–331. doi: 10.1254/jjp.81.324. [DOI] [PubMed] [Google Scholar]
- 26.Kawai K, Nakagomi T, Kirino T, Tamura A, Kawai N. Preconditioning in vivo ischemia inhibits anoxic long-term potentiation and functionally protects CA1 neurons in the gerbil. J Cereb Blood Flow Metab. 1998;18:288–296. doi: 10.1097/00004647-199803000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Leski ML, Valentine SL, Coyle JT. L-type voltage-gated calcium channels modulate kainic acid neurotoxicity in cerebellar granule cells. Brain Res. 1999;828:27–40. doi: 10.1016/s0006-8993(99)01270-6. [DOI] [PubMed] [Google Scholar]
- 28.Lopes CM, Franks NP, Lieb WR. Actions of general anaesthetics and arachidonic pathway inhibitors on K+ currents activated by volatile anaesthetics and FMRF amide in molluscan neurones. Br J Pharmacol. 1998;125:309–318. doi: 10.1038/sj.bjp.0702069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luiten PG, Stuiver B, DeJong GI, DeKeyser JH. Calcium homeostasis, nimodipine and stroke. In: Ter Horst GJ, Korf J, editors. Clinical pharmacology of cerebral ischemia. Humana; Totowa, NJ: 1997. pp. 67–101. [Google Scholar]
- 30.Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyamoto T, Lindgren JA, Samuelsson B. Isolation and identification of lipoxygenase products from the rat central nervous system. Biochim Biophys Acta. 1987;922:372–378. doi: 10.1016/0005-2760(87)90061-0. [DOI] [PubMed] [Google Scholar]
- 32.Murphy E, Glasgow W, Fralix T, Steenbergen C. Role of lipoxygenase metabolites in ischemic preconditioning. Circ Res. 1995;76:457–467. doi: 10.1161/01.res.76.3.457. [DOI] [PubMed] [Google Scholar]
- 33.Nishiyama M, Okamoto H, Watanabe T, Hori T, Hada T, Ueda N, Yamamoto S, Tsukamoto H, Watanabe K, Kirino T. Localization of arachidonate 12-lipoxygenase in canine brain tissues. J Neurochem. 1992;58:1395–1400. doi: 10.1111/j.1471-4159.1992.tb11355.x. [DOI] [PubMed] [Google Scholar]
- 34.Nishiyama M, Watanabe T, Ueda N, Tsukamoto H, Watanabe K. Arachidonate 12-lipoxygenase is localized in neurons, glial cells, and endothelial cells of the canine brain. J Histochem Cytochem. 1993;41:111–117. doi: 10.1177/41.1.8417106. [DOI] [PubMed] [Google Scholar]
- 35.Normandin M, Gagne J, Bernard J, Elie R, Miceli D, Baudry M, Massicotte G. Involvement of the 12-lipoxygenase pathway of arachidonic acid metabolism in homosynaptic long-term depression of the rat hippocampus. Brain Res. 1996;730:40–46. doi: 10.1016/0006-8993(96)00428-3. [DOI] [PubMed] [Google Scholar]
- 36.Osuga H, Hakim AM. Relationship between extracellular glutamate concentration and voltage-sensitive calcium channel function in focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1996;16:629–636. doi: 10.1097/00004647-199607000-00013. [DOI] [PubMed] [Google Scholar]
- 37.Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J Immunol Methods. 1991;142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- 38.Ruehr ML, Zhang L, Dorman RV. Lipid-dependent modulation of Ca2+ availability in isolated mossy fiber nerve endings. Neurochem Res. 1997;22:1215–1222. doi: 10.1023/a:1021976828513. [DOI] [PubMed] [Google Scholar]
- 39.Schweitz H, Heurteaux C, Bois P, Moinier D, Romey G, Lazdunski M. Calcicludine, a venom peptide of the Kunitz-type protease inhibitor family, is a potent blocker of high-threshold Ca2+ channels with a high affinity for L-type channels in cerebellar granule neurons. Proc Natl Acad Sci USA. 1994;91:878–882. doi: 10.1073/pnas.91.3.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volterra A, Buttner N, Siegelbaum SA. Direct opening of S-type K+ channels of Aplysia sensory neurons by 12-lipoxygenase metabolites. Adv Prostaglandin Thromboxane Leukot Res. 1991;21B:727–730. [PubMed] [Google Scholar]
- 41.Weiss JH, Sensi SL. Ca2+-Zn2+ permeable AMPA or kainate receptors: possible key factors in selective neurodegeneration. Trends Neurosci. 2000;23:365–371. doi: 10.1016/s0166-2236(00)01610-6. [DOI] [PubMed] [Google Scholar]
- 42.Wolfe LS, Pellerin L, Drapeau C, Rostworowski K. Formation of 12-lipoxygenase metabolites in rat cerebral cortical slices: stimulation by calcium ionophore, glutamate and N-methyl-d-aspartate. J Neural Transm Suppl. 1990;29:29–37. doi: 10.1007/978-3-7091-9050-0_4. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto S, Takahashi Y, Hada T, Hagiya H, Suzuki H, Reddy GR, Ueda N, Arakawa T, Nakamura M, Matsuda S, Taketani Y, Yoshimoto T, Azekawa T, Morita Y, Ishimura K, Arase S, Glasgow WC, Brash AR, Anton M, Kuhn H. Mammalian arachidonate 12-lipoxygenases. Adv Exp Med Biol. 1997;400A:127–131. doi: 10.1007/978-1-4615-5325-0_18. [DOI] [PubMed] [Google Scholar]
- 44.Yellon DM, Dana A. The preconditioning phenomenon: a tool for the scientist or a clinical reality? Circ Res. 2000;87:543–550. doi: 10.1161/01.res.87.7.543. [DOI] [PubMed] [Google Scholar]
- 45.Yokomizo T, Kato K, Terawaki K, Izumi T, Shimizu T. A second leukotriene B(4) receptor, BLT2. A new therapeutic target in inflammation and immunological disorders. J Exp Med. 2000;192:421–432. doi: 10.1084/jem.192.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokomizo T, Kato K, Hagiya H, Izumi T, Shimizu T. Hydroxyeicosanoids bind to and activate the low affinity leukotriene B4 receptor, BLT2. J Biol Chem. 2001;276:12454–12459. doi: 10.1074/jbc.M011361200. [DOI] [PubMed] [Google Scholar]
- 47.Yu SP. Roles of arachidonic acid, lipoxygenases and phosphatases in calcium-dependent modulation of M-current in bullfrog sympathetic neurons. J Physiol (Lond) 1995;487:797–811. doi: 10.1113/jphysiol.1995.sp020919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zakaroff-Girard A, Dubois M, Gilbert M, Meskini N, Nemoz G, Lagarde M, Prigent AF. The priming effect of 12(S)-hydroxyeicosatetraenoic acid on lymphocyte phospholipase D involves specific binding sites. Life Sci. 1999;64:2135–2148. doi: 10.1016/s0024-3205(99)00164-2. [DOI] [PubMed] [Google Scholar]
- 49.Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. 1998;8:351–356. doi: 10.1016/s0959-4388(98)80060-3. [DOI] [PubMed] [Google Scholar]