

Abstract
Mitochondria are known to be involved in the early stage of apoptosis by releasing cytochrome c, caspase-9, and the second mitochondria-derived activator of caspases (Smac). We have reported that overexpression of copper/zinc superoxide dismutase (SOD1) reduced superoxide production and ameliorated neuronal injury in the hippocampal CA1 subregion after global ischemia. However, the role of oxygen free radicals produced after ischemia/reperfusion in the mitochondrial signaling pathway has not been clarified. Five minutes of global ischemia was induced in male SOD1-transgenic (Tg) and wild-type (Wt) littermate rats. Cytosolic expression of cytochromec and Smac and activation of caspases were evaluated by immunohistochemistry, Western blot, and caspase activity assay. Apoptotic cell death was characterized by DNA nick end and single-stranded DNA labeling. In the Wt animals, early superoxide production, mitochondrial release of cytochrome c, Smac, and cleaved caspase-9 were observed after ischemia. Active caspase-3 was subsequently increased, and 85% of the hippocampal CA1 neurons showed apoptotic DNA damage 3 d after ischemia. Tg animals showed less superoxide production and cytochrome c and Smac release. Subsequent active caspase-3 expression was not evident, and only 45% of the neurons showed apoptotic DNA damage. A caspase-3 inhibitor (N-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone) reduced cell death only in Wt animals. These results suggest that overexpression of SOD1 reduced oxidative stress, thereby attenuating the mitochondrial release of cytochrome cand Smac, resulting in less caspase activation and apoptotic cell death. Oxygen free radicals may play a pivotal role in the mitochondrial signaling pathway of apoptotic cell death in hippocampal CA1 neurons after global ischemia.
Keywords: superoxide dismutase, oxidative stress, global cerebral ischemia, neuron, apoptosis, cytochrome c, second mitochondrial activator of caspases, caspase
Reactive oxygen radicals have been implicated in the pathophysiology of reperfusion injury after cerebral ischemia (Chan, 1994, 1996). It has been demonstrated that ∼2–5% of the electron flow in isolated brain mitochondria produces superoxide anion radicals and hydrogen peroxide (H2O2) (Boveris and Chance, 1973). These constantly produced reactive oxygen radicals are scavenged by superoxide dismutase (SOD), glutathione peroxidase, and catalase. During reperfusion, these endogenous antioxidative defenses are likely to be perturbed as a result of overproduction of oxygen radicals by cytosolic pro-oxidant enzymes and mitochondria, inactivation of detoxification systems, consumption of antioxidants, and failure to adequately replenish antioxidants in ischemic brain tissue. It has been demonstrated in numerous studies that oxygen radicals are directly involved in oxidative damage of cellular macromolecules such as lipids, proteins, and nucleic acids in ischemic tissues, which lead to cell death. Recent studies have provided evidence that indirect signaling pathways by oxygen radicals can also cause cellular damage and death in cerebral ischemia and reperfusion.
Mitochondria are assumed to be involved in apoptosis by releasing cytochrome c from their intermembrane space to the cytoplasm. If ATP or deoxy-ATP is present, cytochromec binds to the Caenorhabditis elegans gene ced-3 (CED) homolog, Apaf-1, and subsequently, Apaf-1 binds to procaspase-9, resulting in activation of caspase-9, which has been shown to be an initiator of the cytochrome c-dependent caspase cascade (Li et al., 1997; Kuida et al., 1998; Yoshida et al., 1998). Activated caspase-9 directly cleaves procaspase-3, and active caspase-3 triggers activation of additional caspases and leads to apoptosis (Liu et al., 1996; Slee et al., 1999). On the other hand, the inhibitor of apoptosis (IAP) family proteins negatively regulate caspase activation. IAPs suppress apoptosis by inhibiting the enzymatic activity of active caspases (Deveraux and Reed, 1999; Miller, 1999). In the early stage of apoptosis, a newly identified apoptosis regulator, second mitochondria-derived activator of caspases (Smac), is released from mitochondria into the cytosol, concurrently with cytochromec. Smac eliminates the inhibitory effects of many IAPs and promotes caspase activation (Du et al., 2000; Verhagen et al., 2000).
We have shown evidence that copper/zinc-SOD (SOD1), a cytosolic antioxidant, plays a protective role against focal (Kinouchi et al., 1991; Chan et al., 1996) and global (Chan et al., 1998) cerebral ischemia. Our studies showed that the early release of cytochromec from mitochondria to the cytosol and subsequent DNA-fragmented cell death were attenuated in SOD1-overexpressing transgenic (Tg) mice after transient focal cerebral ischemia (Fujimura et al., 2000), and that cytochrome c release corresponded to the selective vulnerability of rat hippocampal CA1 neurons after global ischemia (Sugawara et al., 1999). However, whether SOD1 could affect the mitochondrial signaling pathway after transient global ischemia has not been studied. Using SOD1 Tg rats, we provide evidence that the delayed death of vulnerable hippocampal CA1 neurons is mediated partly by the superoxide radicals–mitochondrial signaling pathway.
MATERIALS AND METHODS
SOD1 Tg rats. Heterozygous SOD1 Tg rats of the SOD1 with Sprague Dawley background, carrying human SOD1 genes with a four- to sixfold increase in copper/zinc-SOD, were derived from the founder stock described previously (Chan et al., 1998). They were further bred with wild-type (Wt) Sprague Dawley rats to generate heterozygous rats. The SOD1 Tg rats were identified by isoelectric focusing gel electrophoresis as described (Chan et al., 1998). There were no observable phenotypic differences, including cerebral vasculature, between the Tg rats and Wt littermates, as reported previously (Chan et al., 1998).
Enzyme activity. The total activity of SOD in non-ischemic tissue was determined in the hippocampus, cortex, striatum, spinal cord, and heart, as described previously (n = 4 each) (Copin et al., 2001). One unit of SOD was defined as the quantity of enzyme necessary to inhibit by 50% the rate of reduction of ferricytochrome c. The measurement was also performed for the ischemic hippocampus at 1, 2, and 3 d after global ischemia (n = 4 each).
Surgery. Five minutes of transient global ischemia was induced by bilateral common carotid artery occlusion and bleeding to lower the mean arterial blood pressure to 30–35 mmHg, using the method described originally by Smith et al. (1984) with some modifications (Sugawara et al., 1999, 2000). Male SOD1 Tg rats (300–350 gm) and their Wt littermates were anesthetized with 5% isoflurane and maintained during surgery at a level of 2.0% isoflurane in 70% N2O and 30% O2 with spontaneous breathing. The rectal temperature was controlled at 37.0 ± 0.5°C during surgery with a feedback-regulated heating pad. The femoral artery was exposed and catheterized with a PE-50 catheter to allow continuous recording of arterial blood pressure and withdrawal of blood samples for blood gas analysis. After recovery of the arterial blood pressure, the arterial blood was collected for blood gas analysis. The animals were maintained in an air-conditioned room at 20°C with ad libitum access to food and water before and after surgery. All animals were treated in accordance with Stanford University guidelines and the animal protocol approved by Stanford University's Administrative Panel on Laboratory Animal Care.
Histological assessment and immunohistochemistry. Anesthetized animals were perfused with 10 U/ml heparin and subsequently with 4% paraformaldehyde in 0.1 mphosphate buffer, pH 7.4. Brains were removed and post-fixed for 24 hr in the same fixative. Post-fixed brains were coronally sectioned on a vibratome at a 25 μm thickness or paraffin embedded and sliced on a microtome at a 6 μm thickness. The sections between 3 and 4 mm posterior of the bregma were used for this study. For histological assessment of damage to the hippocampus, the paraffin-embedded brain sections were stained with hematoxylin and eosin (H&E). The antibodies used for immunohistochemistry and Western blotting are listed in Table1. For cytochrome c and Smac staining, free-floating vibratome sections were processed immunohistochemically. Briefly, the sections were incubated with 3% H2O2 in PBS and appropriate 5% blocking serum and exposed to the primary antibodies for 24 hr at 4°C. The sections were then incubated in the fluorescein-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), and the signal of fluorescein was observed at excitation of 495 nm and emission of >515 nm. For active caspase-3 immunohistochemistry, anesthetized animals were decapitated and removed brains were frozen rapidly in −20°C 2-methylbutane, subsequently placed in −50°C powdered dry ice, and stored at −80°C. The brains were sectioned with a cryostat to a thickness of 20 μm. The sections were fixed for 30 min in 3.7% formaldehyde in PBS and incubated in the primary antibody for 24 hr at 4°C. They were then exposed to biotin-conjugated IgG (Vector Laboratories, Burlingame, CA) against the host of the primary antibody for 1 hr and then incubated with avidin–biotin horseradish peroxidase solution (ABC kit, Vector Laboratories) for 30 min. Then the staining was visualized using 0.025% 3,3′-diaminobenzidine (DAB) and 0.075% H2O2 in PBS.
Table 1.
Antibodies used for immunohistochemistry and Western blot
| Protein | Format | Concentration Immuno/WB | Manufacturer |
|---|---|---|---|
| Cytochromec | Mouse monoclonal | 1:500 /1:1000 | PharMingen1-a |
| Smac | Rabbit polyclonal | 1:200 /1:1000 | Dr. Xiaodong Wang1-b |
| Caspase-9 | Rabbit polyclonal | — /1:1000 | Santa Cruz Biotechnology1-c |
| Caspase-3 | Goat polyclonal | — /1:5000 | Santa Cruz Biotechnology |
| Active caspase-3 | Rabbit polyclonal | 1:200 /— | PharMingen |
| Cleaved PARP | Rabbit polyclonal | — /1:1000 | Cell Signaling Technology1-d |
| NeuN | Mouse monoclonal | 1:200 /— | Chemicon1-e |
| GFAP | Goat polyclonal | 1:200 /— | Santa Cruz Biotechnology |
| β-Actin | Mouse monoclonal | — /1:10000 | Sigma1-f |
| Cytochrome oxidase IV | Mouse monoclonal | — /1:5000 | Molecular Probes |
Immuno, Immunohistochemistry; WB, Western blot.
Hercules, CA.
Howard Hughes Medical Institute and Department of Biochemistry, University of Texas Southwestern, Medical Center at Dallas, TX.
Santa Cruz, CA.
Beverly, MA.
Temecula, CA.
St. Louis, MO.
In situ detection of superoxide anion production. The production of superoxide during cerebral ischemia was investigated by the in situ detection of oxidized hydroethidine (HEt) as described previously (Murakami et al., 1998), with minor modifications. HEt (Molecular Probes, Eugene, OR) is taken up by living cells and oxidized to a red fluorescent dye, ethidium, specifically by superoxide, but not by other reactive oxygen species in the cells (Bindokas et al., 1996). The rats were anesthetized with 2% isoflurane in 30% O2 and 70% N2O. HEt solution (1 ml; 1 mg/ml in 1% dimethylsulfoxide with saline) was administered intravenously 1 hr before animals were killed. The rats were killed 6 and 12 hr and 1 and 3 d after ischemia by transcardiac perfusion fixation, described above. After 24 hr of post-fixation, the brains were cut on a vibratome into slices 50 μm thick at the level of the hippocampus. The sections were then mounted on glass slides and examined under a fluorescent microscope at excitation of 510 nm and emission of >580 nm for detection of ethidium.
To clarify the cell population of the ethidium-expressing cells, double staining of ethidium and a neuronal marker, neuron-specific nuclear protein (NeuN), or an astroglial marker, glial acidic fibrillary protein (GFAP), was performed. Vibratome sections were incubated in a primary antibody (Table 1) for 2 hr and then in fluorescein-conjugated IgG as a secondary antibody (Jackson ImmunoResearch) for 1 hr at room temperature. The signal of fluorescein was observed at excitation of 495 nm and emission of >515 nm. To analyze the spatial relationship between ethidium signals and the mitochondria, vibratome sections were also double stained with a mitochondrial marker, MitoTracker green (Molecular Probes). The sections were incubated in MitoTracker green in normal saline (200 nm) for 1 hr at 37°C, and the signal was observed under the same conditions as with fluorescein detection. The sections for fluorescent staining were mounted with a DNA dye, 4′, 6 diamidino-2-phenylindole (DAPI), containing mounting medium (Vectashield, Vector Laboratories) for nuclear counterstaining.
For quantitative study, the signal intensity of ethidium approximately at the center of the hippocampal CA1 subregion (250 × 250 μm;n = 4 at each time point) was measured using NIH image software (version 1.62; National Institutes of Health, Bethesda, MD).
Western blot analysis. To obtain the whole-cell fraction, ∼50 mg of samples from the CA1 subregion of the hippocampus was processed as described (Kim et al., 2001); ∼30 μg of total protein was loaded per lane. Proteins were separated by SDS-PAGE on a 10–20% Tris-glycine gel (Invitrogen, Carlsbad, CA) and transferred to a polyvinylidene difluoride membrane (Invitrogen). The membrane was incubated in the primary antibody against caspase-3 and poly(ADP-ribose) polymerase (PARP) (Table 1) overnight at 4°C and then incubated in the appropriate peroxidase-conjugated secondary antibody. The signals were detected with a chemiluminescence kit (Amersham Biosciences, Buckinghamshire, UK) and exposed on x-ray film. Protein extraction of both the mitochondrial and cytosolic fractions was performed 6 and 12 hr and 1 d after ischemia using a multiple centrifugation method as described (Fujimura et al., 1998). The primary antibody was against cytochrome c, Smac, or caspase-9 (Table 1), and signals were detected as described above. To confirm a consistent protein loading for each lane, membranes were stained for β-actin (whole-cell and cytosolic fractions) or cytochrome oxidase subunit IV (mitochondrial fraction). After the film was scanned with a GS-700 imaging densitometer (Bio-Rad, Hercules, CA), a quantitative analysis was performed using Multi-Analyst software (Bio-Rad).
In situ labeling of DNA fragmentation. The experimental animals were killed at 1, 2, 3, and 7 d after 5 min of global ischemia/reperfusion. Frozen brain sections (20 μm thick) at the level of the hippocampus were placed on slides and stained using anin situ technique [terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) reaction] to detect the DNA-free 3′-OH ends. Briefly, frozen brain sections were fixed for 30 min in 3.7% formaldehyde in PBS, pH 7.4. The slides were placed in 1× terminal deoxynucleotidyl transferase (TdT) buffer (Invitrogen, Gaithersburg, MD) for 15 min, followed by reaction with a TdT enzyme (Invitrogen) and biotinylated 16-dUTP (Boehringer Mannheim, Indianapolis, IN) at 37°C for 60 min. The slides were then washed in 2× saline-sodium citrate (150 mol/l sodium chloride, 15 mol/l sodium citrate, pH 7.4) for 15 min, followed by washing in PBS two times for 15 min each. ABC solution (Vector Laboratories) was applied to the sections for 30 min, and then the slides were washed for 15 min with 0.175 mol/l sodium acetate. Staining was visualized using 0.025% DAB and 0.075% H2O2 in PBS with 0.4 mg/ml nickel sulfate. The sections were then counterstained with methyl green.
Single-stranded DNA staining. Paraffin-embedded sections were incubated in saponin (0.1 mg/ml in PBS) for 20 min and proteinase K (20 μg/ml in PBS) for 20 min at room temperature. The sections were processed in 50% formamide preheated in a water bath to 60°C for 20 min. After processing in 3% hydrogen peroxide for 5 min to quench endogenous peroxidase, the sections were blocked with 3% nonfat dry milk, exposed to a primary antibody against single-stranded DNA (ssDNA) (1:10, mouse monoclonal IgM; Chemicon, Temecula, CA) for 15 min, and subsequently exposed to peroxidase-conjugated anti-mouse immunoglobulin M (1:20; Jackson ImmunoResearch). The sections were finally exposed to 0.025% DAB and 0.075% H2O2 in PBS and then counterstained with methyl green.
Caspase-3 activity assay. Caspase-3 activity was measured using an activity assay kit (PharMingen, San Diego, CA). Fifty milligrams of the hippocampal CA1 tissue were obtained from non-ischemic and ischemic brains 3 d after ischemia from both the Wt and Tg animals. The tissue was sonicated in lysis buffer (10 mm Tris-HCl, 10 mmNaH2PO4/NaHPO4, 130 mm NaCl, 1% Triton X-100, and 10 mm sodium pyrophosphate, pH 7.5) and centrifuged at 20,800 × g for 15 min at 4°C. The protein concentration of the supernatant was measured by the Bradford method, and exactly 2 μg of the protein was loaded in each well of a 96-well plate. After the caspase-3 fluorogenic substrateN-acetyl-Asp-Glu-Val-Asp-AMC (7-amino-4-methylcoumarin) (PharMingen) was added, the samples were incubated for 1 hr at 37°C. Fluorescent signals of the cleaved substrate were measured (n = 4 in each group) using the Fmax fluorescence plate reader with Softmax Pro software (Molecular Devices, Sunnyvale, CA) with an excitation wavelength of 355 nm and an emission wavelength of 460 nm. Relative fluorescence (n = 4 each) was expressed as the mean ± SD.
Caspase-3 inhibitor study. To examine the effects of a caspase-3 inhibitor,N-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (Z-DEVD-FMK) was administered intraventricularly. Each animal received six injections of Z-DEVD-FMK solution [5 μl of volume, 1 μg Z-DEVD-FMK, and 0.25% dimethylsulfoxide (DMSO) in PBS] or vehicle (5 μl of volume, 0.25% DMSO in PBS). Injections were performed using a 10 μl Hamilton syringe (Hamilton, Reno, NV) through a 21 gauge needle in the right ventricle (1 mm lateral, 1 mm anterior from bregma, and 4 mm deep) 2 hr before and 12, 24, 36, 48, and 60 hr after ischemia under the same anesthesia as the ischemia surgery. The animals were allowed to regain consciousness right after each injection. The paraffin-embedded brain sections were stained with H&E and examined 7 d after ischemia.
Cell-counting procedure and statistical analyses. The number of morphologically damaged, TUNEL-positive, and ssDNA-positive hippocampal CA1 pyramidal neurons was counted in four brain slices (∼3.0, 3.3, 3.6, and 3.9 mm posterior of the bregma) in each animal and expressed as percentage of damaged/positive cells of that in the entire CA1 pyramidal neuron population. The averaged result in each group is presented as the mean ± SD. For Western blot analyses, optical density (OD) of each band was measured on the same membrane at certain time points (n = 4 each), and the results are presented as the mean ± SD. Statistical significance between the two groups was established with an F test followed by an unpaired Student's t test. Significance was accepted withp < 0.05.
RESULTS
Total SOD activity was not altered after ischemia in Wt and Tg animals
As shown in Figure1A, the average total SOD activity in Tg rats ranged from 60.1 to 205.0 U/mg in various tissues and from 14.6 to 51.1 U/mg in their littermates. In Wt animals, the activity was significantly greater in the striatum (p < 0.05), spinal cord (p < 0.001) and heart (p< 0.001) compared with that in the hippocampus. In Tg rats, the SOD activity increased approximately four- to sixfold compared with that in the Wt littermates. The activity in the hippocampal CA1 subregion was not altered after ischemia for at least 3 d in both the Tg animals and the Wt littermates (Fig. 1B).
Fig. 1.

SOD activity in non-ischemic tissue (A) and in the hippocampus (B) before and after ischemia. In Wt tissue, SOD activity in the striatum, spinal cord, and heart was significantly higher than in the hippocampus. SOD activity in the hippocampus was not altered after ischemia until 3 d in both the Wt and Tg animals.Wt, Wild-type littermates; Tg, SOD1 transgenic animals. n = 4 in each group.
Superoxide production was greater in Wt animals than in Tg animals
In the hippocampal CA1 pyramidal neurons of non-Tg littermates, superoxide production was shown by ethidium signals as small particles in the cytosol under normal conditions (Fig.2A). One hour after ischemia, the hippocampal CA1 pyramidal neurons showed a marked increase in punctate cytosolic signals and diffuse cytosolic signals (Fig. 2B). In the Tg animals, small particles of ethidium were also observed in the non-ischemic CA1 subregion (Fig.2C), but the increase in signals 1 hr after ischemia was not as noticeable as in the non-Tg animals (Fig. 2D). Quantitative analysis of these signals confirmed the difference between Tg and non-Tg animals at 1 hr and revealed that the signal intensity gradually decreased until 3 d (Fig. 2E).
Fig. 2.
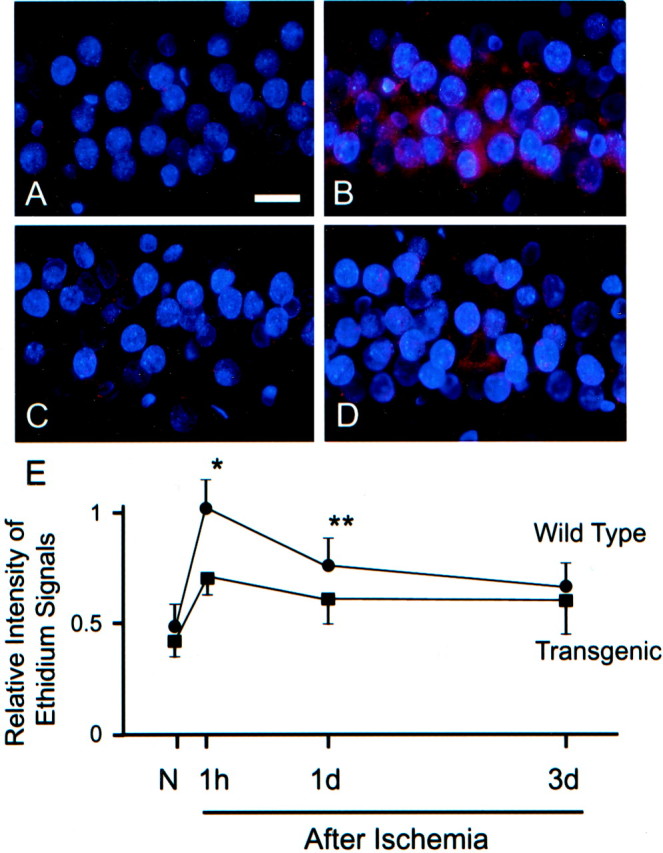
Ethidium signals in the hippocampal CA1 pyramidal cell layer (A–D) and quantitative evaluation of the signals (E). In non-ischemic brains, ethidium signals (red) were observed as small particles in Wt (A) as well as in Tg (C) animals. One hour after ischemia, a marked increase in these punctate signals and also diffuse cytosolic signals were observed in the Wt animals (C), but the increase in signals in the Tg animals (D) was not as obvious as in the Wt rats. A quantitative analysis (n = 4) confirmed that the intensity of the ethidium signals was greater in the Wt animals 1 hr and 1 d after ischemia (*p < 0.001; **p < 0.01). Nuclei were counterstained with DAPI (blue).N, Non-ischemic. Scale bar, 20 μm.
Superoxide production was observed primarily in neuronal mitochondria
Double staining of ethidium and NeuN/GFAP revealed that oxidized HEt signals were almost exclusively in neurons but not in astrocytes (Fig.3A–F). Double staining of ethidium and the mitochondrial marker MitoTracker showed that many of the punctate superoxide signals were colocalized with mitochondria, confirming mitochondria as a major source of superoxide production (Fig. 3G–I).
Fig. 3.

Fluorescent double staining of ethidium signals (red) and NeuN (A–C), GFAP (D–F), or the mitochondrial marker MitoTracker (G–I). Results were confirmed by at least three independent studies. Ethidium signals 1 d after ischemia in Wt animals (A) were almost exclusively localized in the NeuN-positive pyramidal neurons (B). An overlapped photo from the same field (C) confirmed the colocalization. Most of the ethidium signals 1 d after ischemia in the Wt animals (D) were not in the GFAP-positive astrocytes (E). In the overlapped image (F), arrows indicate GFAP-positive astrocytes without the ethidium signals, and thearrowhead indicates one with the ethidium signals. MitoTracker visualized numerous small particles considered to be mitochondria (G), and many of them were colocalized with ethidium signals (H). An overlapped image (I) showed double-stained particles in yellow (arrows). Nuclei were also counterstained with DAPI (blue). Scale bars, 20 μm.
Overexpression of SOD1 protected the hippocampal CA1 neurons from delayed DNA-damaged cell death
Delayed death of the hippocampal CA1 neurons was observed 2–3 d after ischemia. Most of the CA1 neurons in the Wt animals showed shrunken, triangular-shaped, condensed nuclei in H&E-stained sections; however, in a significant number of neurons, the normal features were preserved in the Tg animals (Fig.4A). Most of the morphologically damaged neurons were also positive for TUNEL and ssDNA staining. The cell counting studies (n = 6 each) showed that the damaged neurons accounted for ∼85% of the hippocampal CA1 pyramidal neurons in the Wt rats and 45% in the Tg animals, and that the neuronal damage matured by 3 d after ischemia (Fig.4B).
Fig. 4.

Representative photomicrographs of H&E, TUNEL, and ssDNA staining (A) and cell counting study of morphologically damaged, TUNEL-positive, and ssDNA-positive cells (B). Three days after ischemia, most of the CA1 neurons in the Wt animals showed shrunken, triangular-shaped, condensed nuclei on H&E-stained sections; however, many neurons preserved their normal features of the nuclei in the Tg animals. A majority of these damaged neurons became TUNEL- and ssDNA-positive at the same time (A). Cell-counting analyses (n = 6 each) confirmed that ∼85% of the hippocampal CA1 pyramidal neurons in the Wt rats and 45% in the Tg animals underwent delayed death, and most of these cells were positive for TUNEL and ssDNA. The neuronal damage matured by 3 d after ischemia. There was a significant difference between the Wt and Tg groups. *p < 0.01; **p < 0.001. Scale bar, 20 μm.
Immunohistochemistry demonstrated that the cytosolic expression of cytochrome c and Smac was greater in Wt than in Tg animals
In both the Wt and Tg animals, a significant number of cytochromec-positive cells appeared in the hippocampal CA1 subregion 6 hr after ischemia, and a characteristic cytosolic staining pattern was observed as described previously (Sugawara et al., 1999). Faint punctate immunoreactivity of cytochrome c/Smac in the non-ischemic brains was also seen (Fig.5A). These faintly stained regions were likely to be partially stained mitochondria. As reported previously (Fujimura et al., 1999), thorough fixation of the mitochondria with paraformaldehyde may have prevented the antibody from reaching the mitochondrial intermembrane space but not cytosol. The number of positive cells at 12 hr to 1 d was obviously increased from that at 6 hr, but the characteristic staining pattern was barely observed 2 d or later because of the extracellular immunoreactivity, as described (Sugawara et al., 1999). The number of cytosolic cytochrome c-positive cells was always greater in the Wt than in the Tg animals (Fig. 5A). Smac immunostaining showed a similar time course in the CA1 pyramidal cell layer. It appeared in the cytosol at 6 hr and immunoreactivity increased 12 hr to 1 d after ischemia, and staining intensity was stronger in the Wt than in the Tg animals (Fig. 5A). Cytosolic cytochromec/Smac expression was not observed in the other areas of the hippocampus.
Fig. 5.

Representative photomicrographs of cytochromec and Smac immunohistochemistry in the hippocampal CA1 subregion (A) and Western blot analyses of cytochrome c, Smac, and cleaved caspase-9 in cytosolic (B) and mitochondrial (C) fractions. Cytochrome c staining appeared as an indistinct faint punctate staining in the non-ischemic hippocampus in both the Wt and Tg animals, and it became more intense and some cells showed a diffuse cytosolic staining pattern (arrows). Some of the cytosolic cytochrome c-positive cells showed a long, process-like structure resembling axons or dendrites. The number of the cells with a cytosolic cytochrome cpattern was greater in the Wt animals than in the Tg animals. Smac staining appeared as more diffuse cytosolic staining with a few small dots in non-ischemic brains, and it became more intense 1 d after ischemia. The staining intensity was stronger in Wt animals than in Tg animals. Ethidium signals (red) were overlapped in cytochrome c immunostaining, and nuclei were counterstained with DAPI (blue) in all photomicrographs. Western blot analysis of cytosolic fraction showed that cytochromec and Smac started to increase 6 hr after ischemia and remained elevated until 1 d after ischemia, and cleaved caspase-9 started to increase at 12 hr and also remained increased at 1 d (B). The increase of cytochrome cand Smac was greater in the Wt animals than in the Tg animals. Correspondingly, cytochrome c and Smac both decreased at the same time in the mitochondrial fraction (C). The decrease in cytochrome c in the Wt animals was greater than in the Tg animals. Consistent bands of β-actin and cytochrome oxidase are also shown. Results shown are representative of three independent studies. N, Non-ischemic;CyC, cytochrome c; COX, cytochrome oxidase. Scale bar, 20 μm.
Western blot analysis confirmed the mitochondrial release of cytochrome c and Smac, and subsequent caspase-9 cleavage
Cytochrome c and Smac immunoreactivity was evident as a single band of molecular mass of ∼15 and 21 kDa in the cytosolic fraction of the hippocampal CA1 subregion before and after ischemia, respectively (Fig. 5B). The cytosolic increase of these proteins was observed 6 hr after ischemia in both the Wt and Tg groups; however, the increase in the Wt animals was greater than in the Tg animals (Fig. 5B). Cleaved caspase-9 was also increased in the ischemic brains, but not until 12 hr of ischemia. A corresponding decrease in cytochrome c and Smac was observed in the mitochondrial fraction at the same time points (Fig. 5C). Statistical analyses (n = 4 each) confirmed that cytosolic cytochrome c/Smac in the non-ischemic brain was not significantly different between the Wt and Tg groups (OD of cytochrome c/Smac in Wt and Tg animals was 0.190 ± 0.116/1.245 ± 0.143 and 0.200 ± 0.099/1.188 ± 0.251, respectively); however, 1 d after ischemia, these proteins were more abundant in the Wt animals than in the Tg animals (OD of cytochrome c/Smac in Wt and Tg animals was 1.435 ± 0.383/5.927 ± 0.781, 0.865 ± 0.234/3.308 ± 1.005, respectively; p = 0.0441/0.0062). The mitochondrial cytochrome c/Smac in the non-ischemic brain was not different between the Wt and Tg groups (OD of cytochromec/Smac in Wt and Tg animals was 4.607 ± 0.590/0.792 ± 0.097, 4.525 ± 0.473/0.725 ± 0.176, respectively), but mitochondrial cytochrome c at 1 d was less abundant in the Wt animals than in the Tg animals (OD of cytochrome c in Wt and Tg animals was 0.652 ± 0.183, 2.360 ± 0.597, respectively; p = 0.0016). However, there was no difference in mitochondrial Smac at 1 d between the two groups (OD of Smac in Wt and Tg animals was 0.065 ± 0.021, 0.085 ± 0.025, respectively). There was a consistent amount of β-actin in the cytosolic fraction and cytochrome oxidase IV in the mitochondrial fraction, suggesting that the amount of the loaded protein was consistent (Fig. 5B,C). The purity of the cytosolic fraction was confirmed by the absence of cytochrome oxidase IV bands.
Caspase-3 and PARP cleavage were evident in the CA1 neurons in Wt animals but not in Tg animals
Cleaved caspase-3-positive cells were barely seen in the Wt hippocampal CA1 pyramidal cell layer until 1 d after ischemia; however, a majority of these neurons became strongly positive at 3 d. In contrast, the Tg hippocampal pyramidal cells became weakly caspase-3 positive at 3 d (Fig.6A). Positive cells appeared only in the CA1 region in the hippocampus. Western blot analysis of caspase-3 showed an increase in cleaved caspase-3 (p20) and caspase-3 proform (p32) at 3 d only in the Wt animals, and furthermore, cleaved PARP was clearly observed in the same samples (Fig. 6B). A consistent β-actin immunoreactivity suggested that the loaded protein was consistent (Fig.6B). Caspase-3 activity (n = 4 each) in the Wt hippocampal CA1 subregion was significantly increased (p < 0.001) only in Wt animals 3 d after ischemia, whereas no change was observed in the Tg animals (Fig.6C).
Fig. 6.
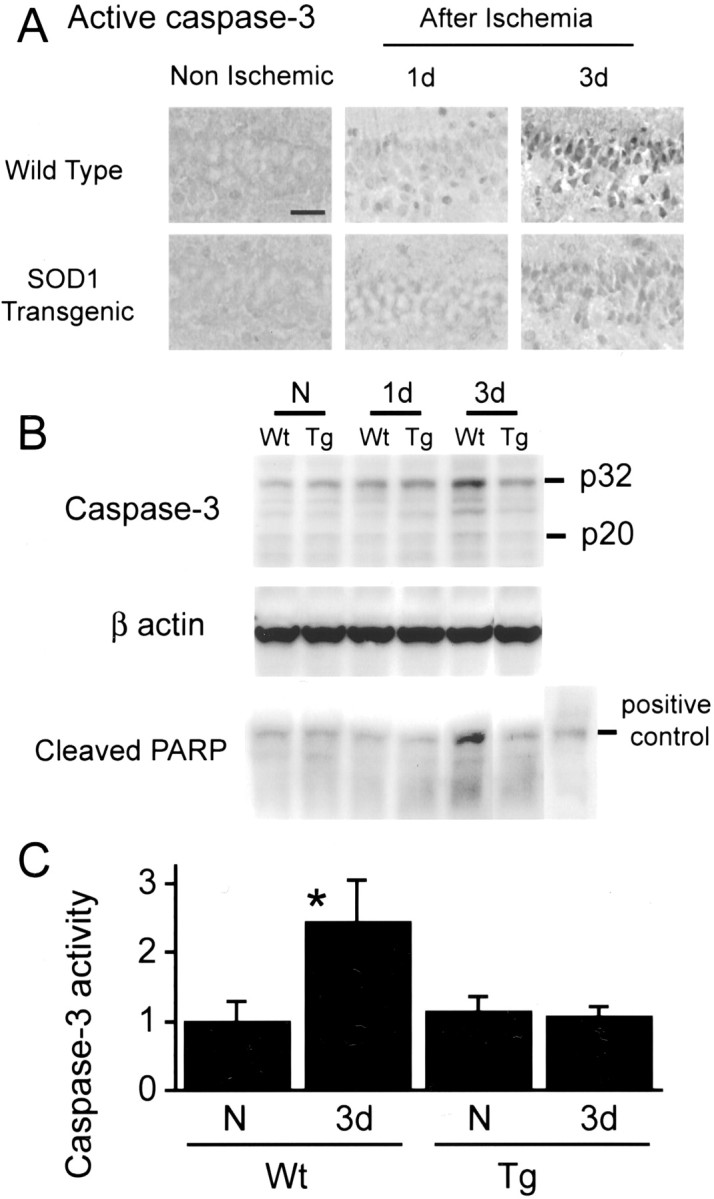
Active caspase-3 immunohistochemistry (A), Western blot studies of caspase-3 and PARP (B), and caspase-3 activity assay (C). A majority of the CA1 pyramidal neurons became strongly active caspase-3 positive at 3 d only in the Wt animals (A). Western blot analysis also showed that procaspase-3, active caspase-3, and PARP were more abundant at 3 d in the Wt animals than in other samples (B). These data are representative of two independent studies. Caspase-3 activity was increased at 3 d compared with non-ischemic brains in Wt animals, but there was no increase in Tg animals (n = 4 each). *p < 0.001. N, Non-ischemic;Wt, wild-type; Tg, SOD1 transgenic. Scale bar, 20 μm.
Caspase-3 inhibitor reduced the hippocampal CA1 neuronal injury only in Wt animals but not in Tg animals
In the caspase-3 inhibitor Z-DEVD-FMK-treated Wt animals, the hippocampal CA1 neuronal injury was significantly reduced (p < 0.05) compared with the vehicle-treated animals (n = 6 each). In contrast, there was no difference in neuronal damage between the Z-DEVD-FMK-treated and vehicle-treated Tg animals (Fig.7A). Expression of caspase-3 proform and active form 3 d after ischemia was decreased only in Z-DEVD-FMK-treated Wt animals (Fig. 7B). Cytosolic cytochrome c expression was not altered by Z-DEVD-FMK or the vehicle compared with noninjection groups as shown in Figure 5.
Fig. 7.

Cell-counting study of damaged neurons (A) and Western blot analyses of cytosolic cytochrome c and caspase-3 (B) in the caspase-3 inhibitor study. The number of damaged CA1 neurons was significantly smaller in the Z-DEVD-FMK-treated Wt group than in the vehicle-treated Wt group (n = 6 each), whereas no difference was observed in the Tg group (A). Z-DEVD-FMK did not affect cytosolic cytochrome c at 1 d but reduced procaspase-3 and active caspase-3 only in the Wt animals at 3 d (B). The data shown are representative of two independent studies. *p < 0.05. N, Non-ischemic; Veh, vehicle;DEVD, Z-DEVD-FMK.
DISCUSSION
Overexpression of SOD1 reduced superoxide production in mitochondria after global cerebral ischemia
It is well established that the rat transient global ischemia model mimics the condition after transient cardiac arrest and causes selective neuronal death in vulnerable regions, such as hippocampal CA1 pyramidal cells, Purkinje cells of the cerebellum, and neurons in the third to fifth layers of the cerebral cortex (Kirino, 1982; Pulsinelli et al., 1982). The vulnerability has been attributed to many factors, such as glutamate neurotoxicity, calcium, expression of cell suicide genes, activation of apoptotic proteins, mitochondrial dysfunction, and oxygen free radicals (Abe et al., 1995; Chan, 1996; Sugawara et al., 1999). The oxygen free radical hypothesis seems to be especially important because superoxide production is thought to be closely associated with reperfusion injury (McCord, 1985; Chan, 1996). We previously developed an in situ imaging method for superoxide radical measurement (Murakami et al., 1998). This method is based on the selective oxidation of HEt by superoxide radicals (Bindokas et al., 1996). In Figure 2, ethidium was observed as numerous punctate signals, and the quantitative study revealed that the intensity of these signals peaked 1 hr after ischemia and gradually returned toward the normal level thereafter. The signal intensity was significantly lower in the Tg animals 1 hr and 1 d after ischemia. The highest signal intensity of ethidium immediately after reperfusion is consistent with the hypothesis that superoxide is a major contributor to reperfusion injury. A significantly lower production of superoxide in Tg than in Wt animals suggests that overexpression of SOD in the cytosol can eliminate a significant amount of superoxide. Furthermore, unequal activity of the total SOD in various regions of the CNS (Fig. 1A) may help explain the vulnerability of the cells in the hippocampus and cortex. Consistent SOD activity in both the Wt and Tg animals after ischemia (Fig. 1B) suggests the stability of this enzyme after ischemia/reperfusion and that SOD can play a major role in detoxifying superoxide for at least 3 d after ischemia.
Mitochondrial superoxide production and the mitochondrial release of cytochrome c and Smac
In the present study, our findings (Fig. 3) suggest that neuronal mitochondria were a major source of superoxide production. Because mitochondria are known as the site of superoxide production under normal or pathological conditions such as focal cerebral ischemia (Piantadosi and Zhang, 1996), excessive superoxide production may cause mitochondrial injury that leads to the release of proteins such as cytochrome c and Smac from their intermembrane space. Our study also demonstrated that cytochrome c and Smac are released from mitochondria to the cytosol in CA1 cells after transient global ischemia. Numerous faint punctate staining of cytochromec and Smac in the non-ischemic CA1 subregion conceivably shows these proteins in the intact mitochondria (Fig. 4). The subcellular distribution of cytochrome c and Smac was confirmed by Western blot analyses (Fig. 5). A significant amount of mitochondrial cytochrome c and Smac was detected in the normal hippocampal CA1 subregion and was decreased 6 hr to 1 d after ischemia. Correspondingly, these proteins showed a marked increase in the cytosol at the same time points. The increase in cytosolic cytochrome c/Smac could derive from other sources, such as ischemia-induced upregulation or impaired protein import. Endoplasmic reticulum and ribosome are known to be the sites of protein production; however, those organelles were excluded by ultracentrifuge. We cannot exclude the possibility that cytosolic cytochrome c and Smac are partly derived from impaired protein transport, and further investigation is necessary to clarify this point. From the findings of immunohistochemistry and Western blotting, we believe that increased cytosolic cytochromec and Smac are substantially derived from the mitochondria of CA1 neurons.
Apoptotic cell death in vulnerable hippocampal CA1 neurons after transient global ischemia
Neuronal death in the hippocampal CA1 subregion after global ischemia has been shown to occur in a delayed manner (Kirino, 1982), and recent studies demonstrated that these neuronal deaths are caused in part by apoptosis (MacManus et al., 1993; Nitatori et al., 1995). TUNEL staining has been used as a marker to detect “apoptotic cells” after global cerebral ischemia (Nitatori et al., 1995;Sugawara et al., 1999), and internucleosomal DNA fragmentation has also been detected by a DNA laddering pattern (Heron et al., 1993; Sugawara et al., 1999). However, methods to detect TUNEL and DNA laddering are dependent on detection of DNA breaks, and their specificity for apoptosis is still controversial. The method used in the present study to detect ssDNA is supposed to be more specific for apoptotic cells. Formamide-induced DNA denaturation has been reported to be specific for condensed chromatin of apoptotic cells, and the denaturation is likely caused by digestion of histones by caspases (Frankfurt and Krishan, 2001). In this study, ∼85% of the CA1 neurons in Wt animals and 45% of the neurons in Tg animals were morphologically damaged by 3 d after ischemia, and most of these cells were also TUNEL- and ssDNA-positive (Fig. 4). The specificity of TUNEL and ssDNA has to be clarified further. On the other hand, electron microscopic studies showed observations against apoptosis, such as early organelle swelling, disaggregation of polyribosomes, and cell and nuclear membrane breaks in CA1 neurons (Colbourne et al., 1999). However, the protective effects of caspase inhibitors on delayed neuronal death in the present (Fig. 7) and other studies (Chen et al., 1998) strongly suggest that the biochemical caspase cascade plays a major role in neuronal death after global ischemia.
Mitochondrial pathway of apoptotic cell death was partially blocked in Tg animals
One well established pathway of caspase activation in mammalian cells is initiated by the release of cytochrome c from mitochondria. Once cytochrome c is released from mitochondria in the early stages of apoptosis, it interacts with the CED-4 homolog, Apaf-1, and dATP/ATP, leading to the activation of caspase-9 (Li et al., 1997; Zou et al., 1997; Kuida et al., 1998;Yoshida et al., 1998). Caspase-9, which is presumably an initiator of the cytochrome c-dependent caspase cascade, then activates an effector caspase, caspase-3, followed by caspase-2, -6, -8, and -10 activation downstream (Thornberry and Lazebnik, 1998; Slee et al., 1999). The DNA repair enzyme PARP is one of the substrates of caspase-3 (Nicholson et al., 1995), and cleavage of this enzyme induces ATP depletion or Ca2+/Mg2+-dependent endonuclease activation, ultimately leading to cell death (Boulares et al., 1999). In contrast, the IAP family proteins negatively regulate caspase activation. IAPs suppress apoptosis by inhibiting the enzymatic activity of active caspases (Deveraux and Reed, 1999; Miller, 1999). The newly identified apoptosis regulator, Smac, is released from mitochondria into the cytosol concurrently with cytochromec. It eliminates the inhibitory effects of many IAPs, thereby promoting caspase activation (Du et al., 2000; Verhagen et al., 2000).
In the present study, superoxide production peaked 1 hr after ischemia, cytochrome c/Smac release started at 6 hr, caspase-9 cleavage increased at 12 hr, and increased caspase-3 proform/active form and cleaved PARP were evident at 3 d in the Wt animals (Figs.5, 6). With regard to the order in which signaling factors are activated in the rat hippocampal CA1 subregion, these data are compatible with the latest concept of the cytochromec-dependent mitochondrial caspase cascade. Furthermore, exclusive spatial expression of cytochrome c, Smac, and caspase-3 in CA1 neurons also supports this hypothesis. In Tg animals, superoxide production, cytochrome c/Smac release, and caspase-9 activation increase of caspase-3 were observed at the same time as in Wt animals, but active caspase-3 and PARP cleavage were not increased afterward (Figs. 5, 6). These results indicate that oxidative stress to the mitochondria may be an initiator of the cytochromec-dependent caspase cascade and that reduced oxidative stress in Tg animals may not be sufficient to induce complete activation of the cytochrome c-dependent caspase cascade. Furthermore, a greater release of cytochrome c/Smac in Wt animals compared with Tg animals may indicate that their release is a critical step in the pathway. However, there is an unexplainable time lag between caspase-9 activation (12 hr) and caspase-3 activation (3 d) in the Wt animals. Although we provided the supportive findings for our mitochondrial pathway hypothesis, a mitochondrial-independent pathway, such as Fas (APO-1)-mediated caspase cascade, may have played a role in the cell death. Further studies are needed to examine the possible cascade that links active caspase-3 and caspase-9 after global ischemia. We also demonstrated that the caspase-3 inhibitor Z-DEVD-FMK did not alter cytosolic cytochrome c release in either the Wt or Tg animals, but it reduced the proform and active form of caspase-3 and subsequent neuronal damage in the Wt animals. These results confirm the pivotal role of caspase-3 in Wt CA1 neuronal death; however, the inability of Z-DEVD-FMK to prevent cell death in Tg animals and only a partial rescue of Wt CA1 neurons may suggest the existence of other apoptotic or necrotic cell death pathways.
Footnotes
This study was supported by National Institutes of Health Grants NS14543, NS25372, NS36147, NS37530, NS38653, and NO1 NS82386. P.H.C. is a recipient of the Jacob Javits Neuroscience Investigator Award. We thank Cheryl Christensen for editorial assistance and Bernard Calagui, Liza Reola, and Jane O. Kim for technical assistance. We also thank Dr. Xiaodong Wang for providing the antibody against Smac.
Correspondence should be addressed to Dr. Pak H. Chan, Neurosurgical Laboratories, Stanford University, 1201 Welch Road, MSLS #P314, Stanford, CA 94305-5487. E-mail:phchan@leland.stanford.edu.
REFERENCES
- 1.Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A, Kogure K, Itoyama Y. Ischemic delayed neuronal death. A mitochondrial hypothesis. Stroke. 1995;26:1478–1489. doi: 10.1161/01.str.26.8.1478. [DOI] [PubMed] [Google Scholar]
- 2.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- 4.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan PH. Oxygen radicals in focal cerebral ischemia. Brain Pathol. 1994;4:59–65. doi: 10.1111/j.1750-3639.1994.tb00811.x. [DOI] [PubMed] [Google Scholar]
- 6.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 7.Chan PH, Epstein CJ, Kinouchi H, Kamii H, Chen SF, Carlson E, Gafni J, Yang G, Reola L. Neuroprotective role of CuZn-superoxide dismutase in ischemic brain damage. Adv Neurol. 1996;71:271–280. [PubMed] [Google Scholar]
- 8.Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colbourne F, Sutherland GR, Auer RN. Electron microscopic evidence against apoptosis as the mechanism of neuronal death in global ischemia. J Neurosci. 1999;19:4200–4210. doi: 10.1523/JNEUROSCI.19-11-04200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Copin J-C, Gasche Y, Li Y, Chan PH. Prolonged hypoxia during cell development protects mature manganese superoxide dismutase-deficient astrocytes from damage by oxidative stress. FASEB J. 2001;15:525–534. doi: 10.1096/fj.00-0330com. [DOI] [PubMed] [Google Scholar]
- 12.Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 13.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 14.Frankfurt OS, Krishan A. Identification of apoptotic cells by formamide-induced DNA denaturation in condensed chromatin. J Histochem Cytochem. 2001;49:369–378. doi: 10.1177/002215540104900311. [DOI] [PubMed] [Google Scholar]
- 15.Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan PH. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18:1239–1247. doi: 10.1097/00004647-199811000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan PH. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J Neurosci. 2000;20:2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heron A, Pollard H, Dessi F, Moreau J, Lasbennes F, Ben-Ari Y, Charriaut-Marlangue C. Regional variability in DNA fragmentation after global ischemia evidenced by combined histological and gel electrophoresis observations in the rat brain. J Neurochem. 1993;61:1973–1976. doi: 10.1111/j.1471-4159.1993.tb09843.x. [DOI] [PubMed] [Google Scholar]
- 19.Kim GW, Noshita N, Sugawara T, Chan PH. Early decrease in DNA repair proteins, Ku70 and Ku86, and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke. 2001;32:1401–1407. doi: 10.1161/01.str.32.6.1401. [DOI] [PubMed] [Google Scholar]
- 20.Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci USA. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 22.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 23.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 25.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 26.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 27.Miller LK. An exegesis of IAPs: salvation and surprises from BIR motifs. Trends Cell Biol. 1999;9:323–328. doi: 10.1016/s0962-8924(99)01609-8. [DOI] [PubMed] [Google Scholar]
- 28.Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T-T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 30.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. [DOI] [PubMed] [Google Scholar]
- 32.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 33.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith ML, Bendek G, Dahlgren N, Rosen I, Wieloch T, Siesjö BK. Models for studying long-term recovery following forebrain ischemia in the rat. 2. A 2-vessel occlusion model. Acta Neurol Scand. 1984;69:385–401. doi: 10.1111/j.1600-0404.1984.tb07822.x. [DOI] [PubMed] [Google Scholar]
- 35. Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 19 1999. RC39(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugawara T, Kawase M, Lewén A, Noshita N, Gasche Y, Fujimura M, Chan PH. Effect of hypotension severity on hippocampal CA1 neurons in a rat global ischemia model. Brain Res. 2000;877:281–287. doi: 10.1016/s0006-8993(00)02684-6. [DOI] [PubMed] [Google Scholar]
- 37.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 38.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 40.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]