

Abstract
We investigated the neuroprotective properties of two M-type K+ channel blockers, linopirdine and its analog XE991, in rat sympathetic neurons deprived of nerve growth factor (NGF). Linopirdine and XE991 promoted sympathetic neuronal survival 48–72 hr after NGF withdrawal in a concentration-dependent manner. Both drugs prevented neuronal apoptosis by blocking the pathway leading to the release of cytochrome c and development of “competence-to-die” after NGF deprivation. Fura-2 Ca2+ imaging showed no significant difference in intracellular free Ca2+([Ca2+]i) in the presence or absence of NGF; linopirdine and XE991, on the other hand, caused membrane depolarization and increases in [Ca2+]i. Whole-cell recordings showed that linopirdine and XE991 selectively blocked the M current at neuroprotective concentrations, although they additionally inhibited other K+ currents at high concentrations. Membrane depolarization and [Ca2+]i increases induced by linopirdine and XE991 were blocked by the Na+ channel blocker tetrodotoxin (TTX) or by the L-type Ca2+ channel antagonist nifedipine. TTX and nifedipine also prevented the neuroprotection elicited by linopirdine or XE991.
We propose that Na+ channel activation amplifies the membrane depolarization produced by M channel blockade and is essential for subsequent Ca2+ entry via the L-type Ca2+ channel. The interaction of these three classes of ion channels highlights an integrated anti-apoptosis mechanism in sympathetic neurons.
Keywords: apoptosis, calcium, M-type potassium channel, nerve growth factor, sympathetic neuron, cortical neuron, tetrodotoxin, linopirdine, XE991
Apoptosis is an important regulatory process during the development of the nervous system. It also contributes to neuronal loss in stroke, trauma, and some neurodegenerative disorders (Oppenheim, 1991; Choi, 1996; Henderson, 1996). Elevation of extracellular K+([K+]o) blocks apoptosis in neurons from a variety of peripheral and central locations, including sympathetic ganglia, hippocampus, neocortex, cerebellum, and dorsal root ganglia (Gallo et al., 1987; Collins and Lile, 1989; Koike et al., 1989; Collins et al., 1991; Franklin et al., 1995; Galli et al., 1995; Pike et al., 1996; Tong et al., 1997; Yu et al., 1997; Colom et al., 1998). Two different mechanisms have been proposed to explain the anti-apoptotic effect of high [K+]o. In sympathetic neurons, cerebellar granule cells, and some other types of neurons, the anti-apoptotic effect of elevated [K+]o has been shown to be mediated by increased intracellular Ca2+ concentration ([Ca2+]i) as a result of membrane depolarization and activation of the L-type voltage-dependent Ca2+ channels (Gallo et al., 1987; Collins and Lile, 1989; Koike et al., 1989; Franklin et al., 1995; Galli et al., 1995; Tong et al., 1997). In contrast, recent work on central neurons, such as neocortical neurons, and several peripheral cells supports the idea that elevated extracellular K+, or K+channel blockers, such as tetraethylammonium (TEA), suppress apoptosis attributable to prevention of K+efflux and intracellular K+ loss (Yu et al., 1997, 1998, 1999; Colom et al., 1998; Dallaporta et al., 1998;Hughes and Cidlowski, 1999). This protective effect can be independent of changes in [Ca2+]i (Yu et al., 1997, 1998, 1999).
Given these results, we wondered whether newly developed, selective M-type K+ channel blockers would be anti-apoptotic in cultured rat sympathetic neurons after nerve growth factor (NGF) deprivation, and, if so, whether the neuroprotective mechanism would be mediated by elevation of [Ca2+]i or direct inhibition of K+ efflux. Sympathetic neurons undergo apoptosis within 48–72 hr after NGF withdrawal (Edwards et al., 1991; Deckwerth and Johnson, 1993; Deshmukh and Johnson, 1997; Werth et al., 2000). It has been shown that NGF deprivation induces two parallel processes that are sufficient to induce apoptotic death: (1) protein synthesis-dependent, caspase-independent loss of mitochondrial cytochrome c; and (2) the development of “competence-to-die,” which requires no macromolecular synthesis (Deshmukh and Johnson, 1998). This cell death can be inhibited by cycloheximide (CHX), boc-aspartyl(OMe)-fluoromethylketone (BAF), and some other neuroprotective agents (Martin et al., 1988; Rydel and Greene, 1988;Koike et al., 1989; Franklin et al., 1995; Deshmukh et al., 1996;McCarthy et al., 1997).
Linopirdine [3,3-bis(4-pyridinylmethyl)-1-phenylindolin-2-one;DUP996] and its analog XE991 [10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone] are potent blockers of M-type K+ channels in a variety of neurons (Costa and Brown, 1997; Schnee and Brown, 1998; Wang et al., 1998, 2000; Brown and Yu, 2000). They are also representative of a class of cognition-enhancing compounds that increase the release of neurotransmitters (Kristufek et al., 1999). The neuroprotective potential of these compounds, however, has not been investigated previously. The present study demonstrates that M channel blockers are highly neuroprotective against NGF deprivation-induced apoptosis in sympathetic neurons; the protection requires block of the M channel, as well as activation of voltage-gated Ca2+and Na+ channels.
MATERIALS AND METHODS
Sympathetic neuronal cultures. Primary cultures of sympathetic neurons from superior cervical ganglion were prepared by dissecting tissue from rat fetuses on embryonic day 21 as described previously (Johnson and Argiro, 1983; Martin et al., 1988). Briefly, the ganglia were placed in Leibovitz's L-15 medium withl-glutamine (Life Technologies, Gaithersburg, MD), digested with 1 mg/ml collagenase (Worthington, Freehold, NJ) for 30 min at 37°C, followed by another 30 min digestion in trypsin (Worthington), and then resuspended in modified HBSS. The digestion was stopped by AM50, which contained minimum essential medium with Earle's salts (nol-glutamine), 10% fetal calf serum (HyClone, Logan, UT), 2 mm glutamine, 20 mm floxuridine, 20 mmuridine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml mouse 2.5 S NGF (Harlan Sprague Dawley, Indianapolis, IN). Ganglia were then dissociated into a suspension of individual cells and preplated on a 100 mm Falcon or Primaria culture dish (Becton Dickinson, Lincoln Park, NJ). After 2 hr, the medium containing the unattached cells, virtually all neurons, was removed and triturated again.
The cell suspension was plated on 24-well tissue culture plates (Costar, Wilmington, MA), glass-bottomed 35 mm dishes (Corning, Corning, NY), or two-well chamber slides (Nunc, Naperville, IL) that have been coated previously with collagen and air dried. Cells were allowed to attach for 0.5–2 hr. Approximately 1500 cells, or 25% of the cells obtained from a single ganglion, were plated into each well. Cultures were then incubated at 37°C in 5% CO2and 95% air atmosphere.
Neocortical cultures. Mixed cortical cultures (containing neurons and a confluent glia bed) were prepared as described previously (Rose et al., 1993). Dissociated neocortices obtained from fetal mice were plated onto a previously established glial monolayer at a density of 0.35–0.40 hemispheres per milliliter on 24-well plates (Falcon or Primaria), in Eagle's minimal essential medium (Earle's salts) supplemented with 20 mm glucose, 5% fetal bovine serum, and 5% horse serum. Medium was changed after 1 week to MEM containing 20 mm glucose and 10% horse serum, as well as cytosine arabinoside (10 μm) to inhibit cell division. Experiments were performed after 10–12 d in culture.
Neuronal death–survival assay. Sympathetic neurons were plated on 24-well plates as stated above or, alternatively, on two-well chamber slides. The cultured sympathetic neurons could be killed by adding medium (AM0) lacking NGF and containing 0.05% goat anti-NGF. AM0 caused the death of the neurons over a period of 48–72 hr. For experiments with potassium channel blockers, drugs were added at the time when NGF was removed. To quantify neuronal death and survival, the cultures were fixed in 4% paraformaldehyde or 10% formalin in PBS, stained with toluidine blue, and counted using a phase-contrast microscope. Neurons were scored as viable if they had a clear nucleolus and nuclei and were clearly stained with toluidine.
Cortical neuronal cell death induced by staurosporine (0.1 μm, 24 hr) was assessed in 24-well plates by measuring lactate dehydrogenase (LDH) released into the bathing medium (MEM plus 20 mm glucose and 30 mmNaHCO3) using a multiple plate reader (Molecular Devices, Sunnyvale, CA). Neuronal loss is expressed as a percentage of LDH release measured in each experimental condition normalized to negative (sham wash) and positive (complete neuronal death induced by 24 hr exposure to 300 μm NMDA) controls.
Immunohistochemistry. Sympathetic neuronal cultures were immunostained as described previously (Easton et al., 1997; Deshmukh and Johnson, 1998). Briefly, cells were grown on collagen-coated, two-well glass chamber slides. For staining, cultures were washed once with PBS and fixed with 4% paraformaldehyde in PBS for 30 min at 4°C, followed by washing three times with Tris-buffered saline (TBS) (0.9% NaCl and 100 mm Tris-HCl, pH 7.6). After incubation in blocking buffer (5% goat serum and 0.3% Triton X-100 in TBS) for 30 min at room temperature (21 ± 1°C), cultures were exposed to the anti-cytochrome c primary antibody (PharMingen, San Diego, CA) overnight at 4°C. The primary antibody was diluted 1:1000 (final concentration of 0.5 μg/ml) in blocking buffer. Cells were then washed three times with TBS and incubated with an anti-mouse FITC-conjugated secondary antibody (1:300 with a final concentration of 2 μg/ml) (Jackson ImmunoResearch, West Grove, PA) for 2–4 hr at 4°C. Finally, the cells were washed twice in TBS and stained with the nuclear dye bisbenzimide (Hoechst 33258 used at 1 μg/ml; Molecular Probes, Eugene, OR) for 15 min at room temperature. After washing twice with TBS, samples were mounted (50% glycerin and 0.1% paraphenylenediamine in PBS) and examined under fluorescence microscopy.
Cell counts for loss of cytochrome c. After 5–7 d in the NGF-containing medium, cultured neurons were deprived of NGF in the presence or absence of linopirdine or XE991. Parallel control cultures were deprived of NGF in the presence of protein synthesis inhibitor CHX or the caspase inhibitor BAF (Enzyme Systems Products, Livermore, MO). Forty-eight hours after NGF withdrawal, cultures were fixed and immunostained with anti-cytochrome c antibodies as stated above. Sympathetic neurons maintained with NGF exhibited a punctate staining pattern with anti-cytochrome c antibodies, and this staining pattern became very diffuse after NGF deprivation (Deshmukh and Johnson, 1998). For each condition, the number of cells that lost the punctate staining pattern for cytochrome c was counted by a blinded observer, from a random sampling of 100–150 cells.
Microinjections and quantification of cell death.Microinjection of cytochrome c into sympathetic neurons was performed as described previously (Deshmukh and Johnson, 1998). Briefly, sympathetic neuronal cultures were grown in the appropriate medium on collagen-coated, 35 mm dishes and then switched to Leibovitz's L-15 medium containing 100 μg/ml penicillin and 100 μg/ml streptomycin before injection. To identify the injected cells, the injection solution (100 mm KCl and 10 mm Kpi, pH 7.4) contained rhodamine dextran (4 mg/ml). The solution containing rhodamine dextran with or without cytochrome c (15 mg/ml, diluted in water and freshly prepared for each experiment) was injected into the cytoplasm of neurons by using Femtotips needles (Eppendorf Inc., Madison, WI). Immediately after the injections, the number of injected viable cells was determined by counting the number of rhodamine-positive cells that had intact, phase-bright cell bodies. Cultures were then switched to the appropriate medium, and, at various time after injections, the number of remaining viable injected neurons was determined by using the same counting criterion.
Calcium imaging. After 12–16 hr of treatment, we used ratiometric fluorescence imaging with fura-2 AM (Teflabs, Houston, TX) to measure the intracellular free Ca2+concentration, [Ca2+]i, in neuronal cell bodies. Fura-2 AM (5 μm) was bath loaded into neurons at 37°C for 1 hr, followed by another 1 hr of incubation at room temperature. Fluorescent cells were imaged on an inverted microscope (Diaphot; Nikon, Melville, NY), using a 40×, 1.3 numerical aperture fluorite oil immersion objective (Nikon) and a cooled CCD camera (Sensys; Photometrics, Tucson, AZ). A 75 W xenon arc lamp provided fluorescence excitation. Ratio images were obtained by acquiring pairs of images at alternate excitation wavelengths (340 and 380 nm) and filtering the emission at 510 nm. Image acquisition and processing were controlled by a computer connected to the camera and filter wheel (Metafluor; Universal Imaging Corporation, West Chester, PA). A background image for each wavelength was acquired from a field lacking fluorescent neurons and subtracted from each fluorescent image.
The actual [Ca2+]iin a region of interest was calculated from the following formula: [Ca2+]i =KdB(R −Rmin)/(Rmax− R), where Kd is the fura-2 dissociation constant for Ca2+ (224 nm), R is the average ratio of fluorescence intensity at 340 and 380 nm wavelength in the region of interest, Rmax andRmin are the ratios at saturating Ca2+ and zero Ca2+, respectively, and B is the ratio of the fluorescence intensity of the 380 nm wavelength at zero and saturating Ca2+ (Grynkiewicz et al., 1985). Rmin,Rmax, and B for fura-2 on our microscope were determined by imaging a droplet (20 μl) that evenly filled the microscopic field and contained 0 (10 mm EGTA) or 2 mm added Ca2+, 25 μmfura-2/K+, and an artificial intracellular solution. The concentration of fura-2 in the calibration solution was selected to provide similar fluorescence intensity to that of dye-loaded neurons.
Electrophysiology. Whole-cell recording was used to measure the membrane potential and potassium currents in sympathetic neurons. Neurons were cultured for 7–11 d in 35 mm dishes and placed on the stage of an inverted microscope (Diaphot; Nikon) that allowed us to record under direct vision. We used an EPC-7 amplifier (List Electronic, Darmstadt, Germany); patch electrodes had tip resistances between 7 and 10 MΩ (fire polished). The extracellular solution contained (in mm): 115 NaCl, 2.5 KCl, 2.0 MnCl2, 10 HEPES, 0.1 BAPTA, and 10d-glucose. Tetrodotoxin (TTX) (0.1 μm) was added in some of the experiments. Mn2+ was chosen to replace Ca2+ to block Ca2+ channel activation. The electrode solution contained (in mm): 120 KCl, 1.5 MgCl2, 1.0 CaCl2, 2.0 Na2-ATP, 1.0 BAPTA, and 10 HEPES. After forming gigaohm seals, whole-cell recording mode was established by slight suctions. Current and voltage traces were displayed and stored on a computer using the data acquisition–analysis program package PULSE (Heka Electronik, Lambrecht/Pfalz, Germany).
Reagents. All reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated. Linopirdine and XE991 were kindly provided by DuPont Pharmaceuticals (Wilmington, DE).
Statistics. Significant changes were determined if the two-tailed p value was at least <0.05. Multiple comparisons were performed using one-way ANOVA, followed by Tukey's test using commercial InStat (GraphPad Software Inc., San Diego, CA). Data were represented as mean ± SEM.
RESULTS
M channel blockers protected sympathetic neurons from programmed cell death induced by NGF deprivation
The M channel blocker linopirdine or XE991 showed marked neuroprotection against apoptosis induced by NGF withdrawal. Within 48 hr of NGF deprivation, 90% of sympathetic neurons underwent apoptosis as indicated by cell shrinkage, phase darkness, irregular membranes, and neurite fragmentation (Fig.1B). In the presence of linopirdine or XE991, the NGF-deprived neurons maintained their phase-bright appearance and intact neurites and looked similar to neurons in NGF-maintained medium (Fig.1A,C,D). The protective effect of both linopirdine and XE991 was concentration dependent (Fig.2). After 48 hr incubation in NGF-deprived medium, >90% of the neurons survived with 30 μm linopirdine or 50 μmXE991. The EC50 values of linopirdine and XE991 against NGF deprivation-induced cell death were 3.5 and 0.7 μm, respectively (Fig. 2).
Fig. 1.
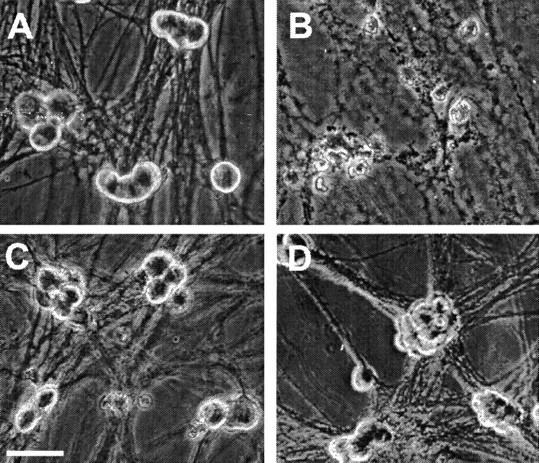
Linopirdine and XE991 promoted survival of sympathetic neurons deprived of NGF. A, Healthy sympathetic neurons maintained in the presence of NGF showed phase-bright appearance in phase-contrast photographs.B, Cells maintained in NGF for 5 d after plating and then deprived of NGF for 2 d had irregular membranes and neurite fragmentation indicative of apoptosis. C,D, Neuronal death was prevented in NGF-deprived sympathetic neurons when 20 μm linopirdine (C) or 5 μm XE991 (D) was added in the medium at the time of NGF withdrawal. Photographs were taken 2 d after NGF deprivation. Scale bar, 30 μm.
Fig. 2.
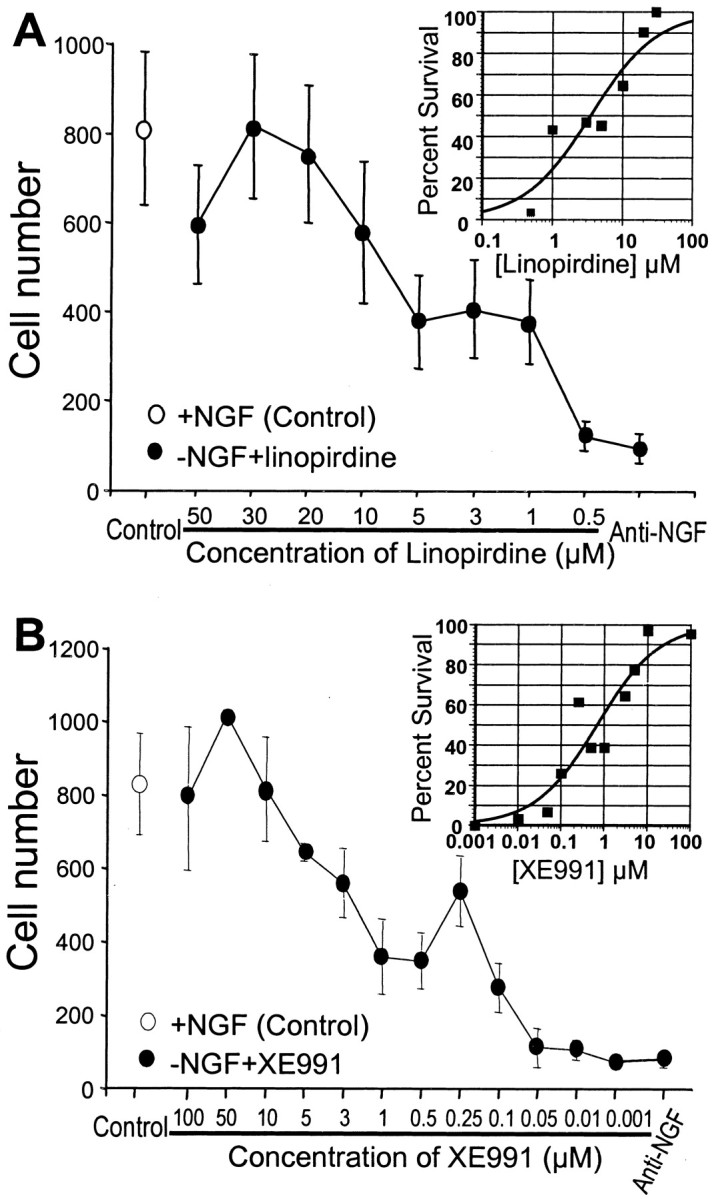
Dose-dependent protective effects of linopirdine and XE991 on sympathetic neuronal apoptosis. Cultures were fixed and stained with toluidine blue, and live cells were counted under a microscope. A, Linopirdine protected sympathetic neurons from apoptosis in a dose-dependant manner. At 30 μm,linopirdine showed the most potent protective effect. Theinset is an exponential curve-fitting plot on a log axis; the fitted curve gives an EC50 of 3.5 μm for the effect of linopirdine on cell survival.B, XE991 at 50 μm completely protected sympathetic neurons from death. The inset of fitted curve yields an EC50 of 0.7 μm for the effect of XE991 on cell survival. Depicted are the mean ± SEM value for each condition.
Mediation of the neuroprotection by M-type potassium channels
The M channel is a non-inactivating K+ channel (Brown and Yu, 2000). Previous work in hippocampal neurons showed that linopirdine blocked the M current with an IC50 of 2.4 μm(Schnee and Brown, 1998). In sympathetic neurons, the M current was blocked by 54 ± 5% by 20 μm linopirdine (n = 10; p < 0.05) and 62 ± 8% by 10 μm XE991 (n = 10;p < 0.05) (Fig. 3). At concentrations that blocked approximately half of cell death, linopirdine (5 μm) and XE991 (3 μm) suppressed 10 ± 1 and 15 ± 1% M current (n = 5 and 7 respectively; p< 0.05); at these concentrations, they showed little inhibitory effect on other potassium currents (Fig. 4). Higher concentrations of linopirdine (20 μm) and XE991 (10 μm), nevertheless, suppressed the outward delayed rectifier K+ currentIK (Fig. 4). Neither linopirdine (20 μm) nor XE991 (10 μm) showed significant inhibitory effect on the A-type K+ current,IA, when evaluated at a membrane potential of −20 mV (Fig. 4). Both drugs did, however, attenuate ∼20–30% of IA current triggered by voltage steps to positive membrane potentials (+20 or +40 mV). The pharmacological profile of linopirdine and XE991 appeared similar in normal sympathetic neurons or neurons deprived of NGF for 7–10 hr (Figs. 3, 4). The selective IA channel blocker 4-aminopyridine (4-AP) (0.1 and 1.0 mm) showed no protective effect against the NGF deprivation-induced apoptosis (Fig. 4), supporting our hypothesis thatIA was not involved in the neuroprotection.
Fig. 3.
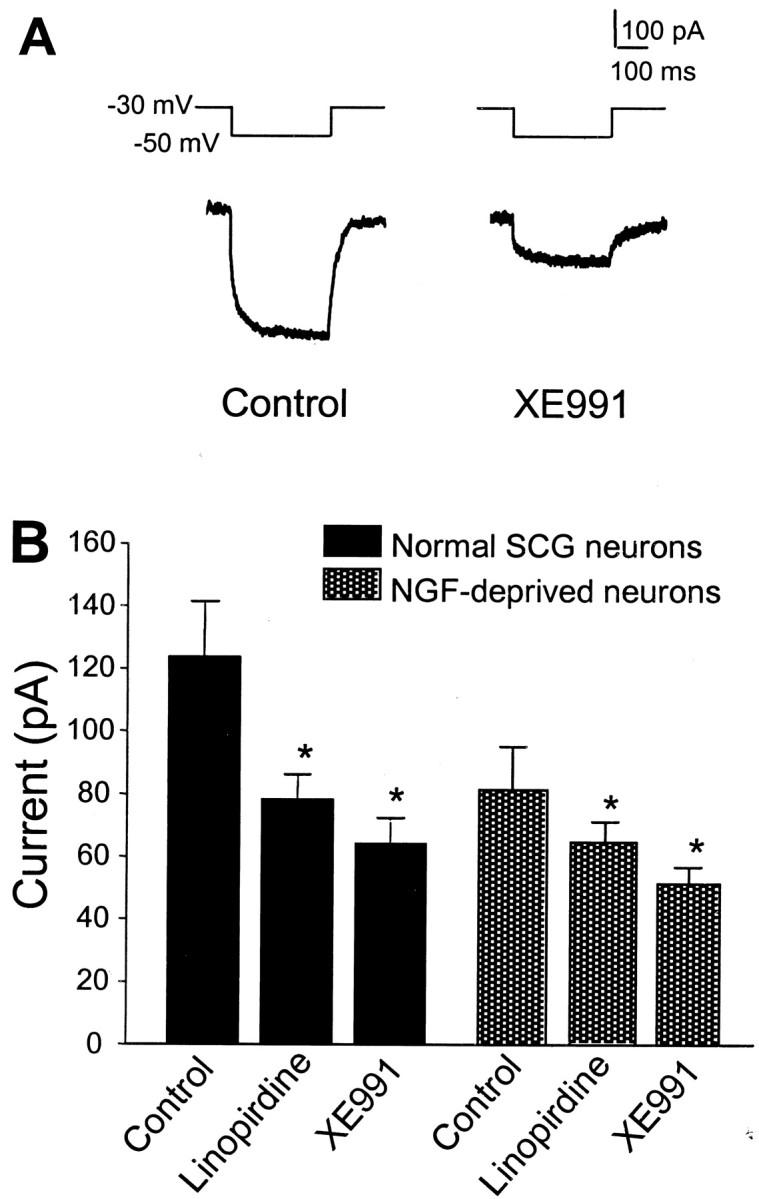
Inhibition of the M current by linopirdine and XE991 in sympathetic neurons. A, In whole-cell recording, the membrane potential was held at −30 mV to allow M channels to stay in an open state; when the membrane was hyperpolarized to −50 mV, a slow inward current was generated, representing the time-dependent closing of M channels; during depolarizing back to −30 mV, an outward current associated with channel reopening appeared. After 5 min application of 10 μm XE991, the M current was substantially suppressed. B, The inhibitory effects of 20 μm linopirdine (n = 7) and 10 μm XE991 (n = 5) on M currents in normal sympathetic neurons or neurons deprived of NGF for 7–10 hr. *p < 0.05 indicates significant difference from the current before drug application (control) (paired ttest).
Fig. 4.
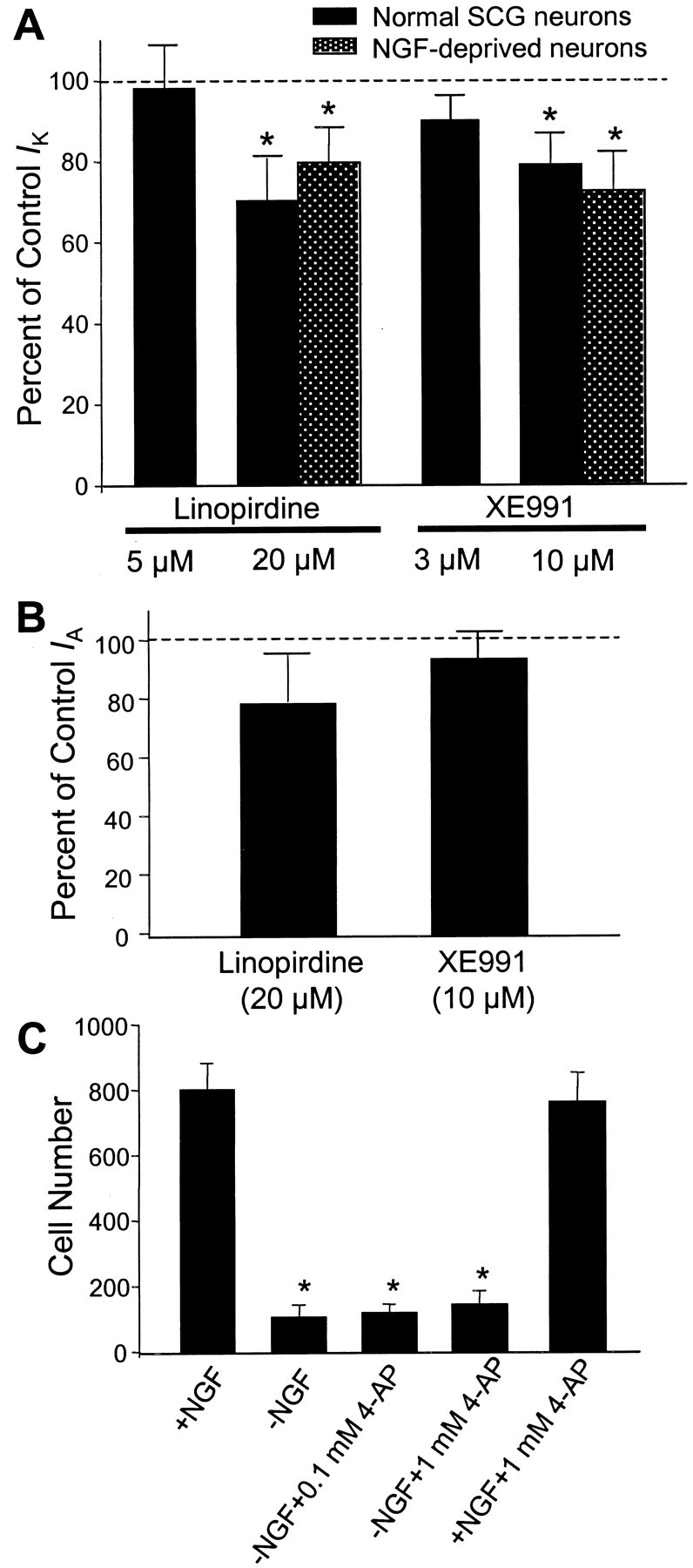
Effects of linopirdine and XE991 onIK and IAcurrents in sympathetic neurons. A, The outward delayed rectifier IK was not affected by 5 μm linopirdine and 3 μm XE991 added into the medium for up to 20 min. IK was partly depressed by 20 μm linopirdine (27 ± 5% inhibition; n = 8; p < 0.05) and by 10 μm XE991 (18 ± 2% block;n = 8; p < 0.05). Similar inhibitory effects were seen in neurons deprived of NGF for 7–10 hr (n = 5 for each test).IK was triggered by a voltage step from the holding potential of −70 to +40 mV for 300 msec; steady-state current was measured for the drug effect. *p < 0.05 indicates significant difference from the control current recorded before drug application. B, Linopirdine (20 μm) and XE991 (10 μm) showed no significant effect on the A-type K+ current triggered by a voltage step from −110 to −20 mV. The IA peak current was measured for the drug effect (n = 8 for each group).C, The A-type K+ channel blocker 4-AP (0.1 and 1.0 mm) showed no protection against the NGF deprivation-induced cell death; 4-AP at tested concentrations was not toxic to SCG neurons. *p < 0.05 indicates significant difference from controls with NGF.
In contrast to sympathetic neurons, cortical neurons in our culture condition often do not have detectable M current (Fig.5). In agreement with this observation, linopirdine (1–10 μm) and XE991 (1–10 μm) showed no neuroprotective effect against apoptosis in cortical neuronal cultures (Fig. 5).
Fig. 5.
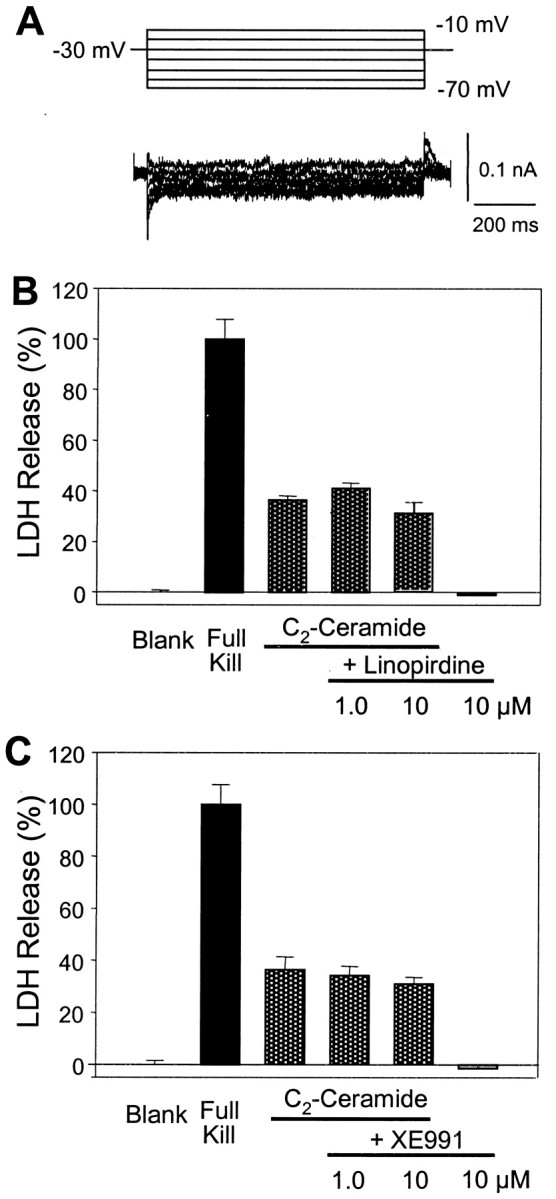
Lack of neuroprotective effects of linopirdine and XE991 in neocortical cultures. A, In cultured cortical neurons, little or small M current was detected, even with voltage steps of wide ranges specific for M channel activation (Brown and Adams, 1980; Yu, 1995). B, The membrane-permeable sphingomyelin metabolite C2-ceramide produces apoptosis in cortical neurons, which can be attenuated by K+channel blockers TEA and clofilium (Yu et al., 1999). Cortical cultures were exposed to 25 μm C2-ceramide alone or coapplied with linopirdine or XE991. Cell death was assayed 48 hr later by LDH release. The C2-ceramide-induced cell death was not affected by linopirdine (1–10 μm); linopirdine alone showed no influence on cell viability (n = 12 cultures for each test). C, XE991 (1–10 μm) showed no protective effect on C2-ceramide-induced apoptosis; XE991 alone was not toxic to cortical cells (n = 12 cultures for each test). MK-801 (1 μm) was added into the medium to prevent NMDA receptor-mediated excitotoxicity. Complete neuronal death was achieved by 300 μm NMDA in the absence of MK-801.
Linopirdine and XE991 prevented cytochrome c translocation from mitochondria to cytosol
Because linopirdine and XE991 protected sympathetic neurons from apoptotic cell death under NGF deprivation, we examined whether these compounds blocked the pathway leading to the release of cytochrome c and/or the development of competence-to-die (Deshmukh and Johnson, 1998). Immunocytochemistry showed that, in NGF-containing medium or in NGF-depleted medium containing CHX, >90% of the neurons possessed intact mitochondrial cytochrome c by exhibiting a punctate staining pattern. Cells deprived of NGF and treated with the caspase inhibitor BAF showed a diffused cytoplasmic pattern (Deshmukh and Johnson, 1998). Cultures treated with linopirdine or XE991 under NGF deprivation retained intact mitochondrial cytochrome c in over 90% of the cells (Fig. 6). Therefore, the K+ channel blockers inhibited neuronal apoptosis by acting at a point before the release of cytochrome c from mitochondria to cytosol.
Fig. 6.

Linopirdine and XE991 prevented cytochrome c release in NGF-deprived sympathetic neurons. Sympathetic neurons were immunostained with anti-cytochrome c antibody, and the neurons that retained a punctate cytochrome c staining pattern (intact cytochrome c) were counted under different conditions. CHX (1 μg/ml) and BAF (50 μm) were used as positive and negative control, respectively. In medium supplemented with linopirdine (20 μm) and XE991 (10 μm), >90% neurons maintained intact cytochrome c.
Linopirdine and XE991 inhibited the competence-to-die of sympathetic neurons
We then examined whether linopirdine or XE991 prevented sympathetic neuronal apoptosis by inhibiting the development of competence-to-die (Deshmukh and Johnson, 1998). Sympathetic neurons were deprived of NGF in the presence of linopirdine or XE991 for 36–48 hr. Parallel control cultures were deprived of NGF in the presence of cycloheximide. To examine whether these neurons had developed competence-to-die, cells were microinjected with mammalian cytochrome c, and their survival was assessed at multiple time points after cytosolic microinjection with mammalian cytochrome c. As reported previously (Deshmukh and Johnson, 1998), the NGF-deprived, cycloheximide-saved neurons developed “competence,” because microinjection of cytochrome c induced rapid cell death in these neurons. In contrast, >80% of the linopirdine- or XE991-treated neurons were alive even 12 hr after cytosolic microinjection of cytochrome c (Fig. 7). The fact that microinjection of cytochrome c did not induce cell death in the presence of linopirdine or XE991 indicates that these neurons, although deprived of NGF, had not developed competence-to-die. Thus, both linopirdine and XE991 appear to block the pathway leading to the development of competence-to-die during NGF deprivation.
Fig. 7.

Linopirdine and XE991 inhibited the development of competence-to-die in sympathetic neurons deprived of NGF. Sympathetic neurons were maintained in NGF for 5 d and then deprived of NGF in the presence of CHX (1 μg/ml; circles), linopirdine (20 μm; triangles), or XE991 (10 μm; squares) for 36–48 hr. Cells were microinjected with 15 mg/ml mammalian cytochrome c. At each time point after the injection, the number of microinjected cells that remained viable was determined and expressed as a percentage of the total number of microinjected cells. Values represent mean ± SEM. More than 80% of the linopirdine-saved (triangles) or XE991-saved (squares) neurons were alive, even 12 hr after cytosolic microinjection of cytochrome c. In contrast, the NGF-deprived, cycloheximide-saved neurons (circles) developed competence as microinjection of cytochrome c induced rapid neuronal death.
Increased intracellular calcium was correlated with sympathetic neuronal survival in NGF deprivation
Intracellular Ca2+ concentration is critical for survival in some cell death paradigms (Gallo et al., 1987;Collins and Lile, 1989; Koike et al., 1989; Collins et al., 1991;Franklin et al., 1995). We, therefore, investigated whether nifedipine could reduce the capacity of M channel blockers to promote neuron survival. Nifedipine dramatically reversed the protective effect of linopirdine and XE991 (Fig. 8). When nifedipine (100 nm) was added together with linopirdine (30 μm) or XE991 (50 μm) in NGF-deficient medium, the protective effect of these K+channel blockers was reduced by ∼80%. Furthermore, nifedipine mostly reduced cell survival promoted by 40 mmK+ in NGF-deprived medium; the survival rate decreased from 80 to 20% in the presence of nifedipine.
Fig. 8.

Nifedipine reversed the protective effect of linopirdine or XE991 on NGF-deprived sympathetic neurons. Withdrawal of NGF from the culture medium for 2 d caused widespread neuronal death. Elevation of extracellular K+, 30 μm linopirdine, or 50 μm XE991 at the time of NGF deprivation all show prominent neuroprotection. The L-type Ca2+ channel antagonist nifedipine (100 nm) was able to primarily reverse the neuroprotective effects induced by these three treatments. Cell survival was assayed 48 hr after incubation, cells were stained with toluidine blue, and live cells were counted. Significant difference was determined with one-way ANOVA, followed by Tukey's test. *p < 0.001 indicates significant difference from the control group with NGF; #p < 0.001 indicates significant difference from the corresponding group of deprived NGF plus the treatment without nifedipine (i.e., the bar on the left).n = 8 cultures for each testing group.
To better understand the role of Ca2+ in neuronal survival of NGF deprivation, we next examined alterations in [Ca2+]i after NGF deprivation. After 12 hr treatment, NGF-deprived neurons showed a statistically insignificant small decline in [Ca2+]i compared with neurons maintained in NGF. However, there was a significant increase in [Ca2+]i in the presence of linopirdine (20 μm) or XE991 (10 μm) over 12 hr in the NGF-deprived medium. The magnitude of the [Ca2+]iincrease produced by linopirdine or XE991 was even greater when NGF was present (Fig. 9). The [Ca2+]i elevation induced by either linopirdine or XE991 was prevented by the L-type Ca2+ channel antagonist nifedipine (100 nm) (Fig. 9). On the other hand, the A-type K+ channel blocker 4-AP (0.1 mm, 10–70 min) did not elevate [Ca2+]i; [Ca2+]i was 20 ± 4 and 9 ± 1 nm in control and 4-AP-treated cells, respectively (n = 55 cells for each group;p < 0.05).
Fig. 9.

Increases in intracellular free Ca2+ by linopirdine and XE991 and prevention by nifedipine. NGF withdrawal did not cause a significant change in [Ca2+]i measured by fura-2 imaging. In NGF-deprived medium, linopirdine (20 μm) or XE991 (10 μm) raised [Ca2+]iapproximately twofold beyond the baseline. In NGF-maintained medium, [Ca2+]i was increased approximately threefold above the baseline by the same concentration of linopirdine or XE991. The increase in [Ca2+]i was eliminated by coapplied nifedipine (100 nm). Data were taken from at least 100 cells. *p < 0.001 indicates significant difference from the basal level of [Ca2+]i; ≠p < 0.001 indicates significant difference from the corresponding group with NGF; #p < 0.001 indicates significant difference from the corresponding group without nifedipine.
Effect of linopirdine and XE991 on membrane potential
To understand the mechanism by which the M channel blockers elevated [Ca2+]i, we determined the effect of linopirdine and XE991 on resting membrane potential. When the membrane potential was measured using whole-cell current clamp in the absence of TTX, frequent action potentials were generated after application of linopirdine and XE991 (data not shown). After 30–60 min exposure, the membrane potential was depolarized from −60.3 ± 2.3 to −47.3 ± 4.7 mV (p< 0.05) by linopirdine (20 μm) and from −64.6 ± 2.3 to −52.7 ± 2.8 mV (p< 0.05) by XE991 (10 μm), respectively (Fig.10). The activity of Na+ channels seemed critical for linopirdine- and XE991-induced depolarization. When TTX (100 nm) was coapplied to block Na+ channels, neither linopirdine nor XE991 altered the resting membrane potential, even after several hours of incubation (Fig. 10). Whereas prolonged incubation of 16–24 hr with XE991 (10 μm) and TTX slightly depolarized the membrane, prolonged incubation with linopirdine (20 μm) failed to induce any depolarization in the presence of TTX (Fig. 10).
Fig. 10.

Effects of linopirdine and XE991 on membrane potential of sympathetic neurons and their dependence on activation of Na+ channels. Sympathetic neurons were patched at multiple time points for recording of membrane potentials during 1–24 hr treatments. A, Neurons were treated with sham wash or exposed to 20 μm linopirdine or 10 μm XE991 for 10–60 min in the absence of TTX. Compared with the membrane potential before drug application or the time-matched controls, linopirdine and XE991 induced mild but significant depolarization after 30–60 min incubation (n = 3–10 cells for each time point). B, When TTX (100 nm) was added into the medium, there was no significant difference of membrane potential between sham wash and linopirdine-treated cells during several hours of incubation. Prolonged incubation of up to 24 hr with XE991 induced a mild depolarization (n = 10–20 cells for each time point). Data shown in plots are from actual time points. *p < 0.05 versus sham washing at same time point; unpaired t test.
To test the contribution of Na+ channel activation to Ca2+ homeostasis, we measured changes in [Ca2+]i after acute application (60 min) of linopirdine (20 μm) or XE991 (10 μm) in the presence or absence of TTX (100 nm). TTX diminished the linopirdine-induced or XE991-induced [Ca2+]ielevations, stabilizing [Ca2+]i at or near normal range during the incubation (Fig.11). Consistent with the [Ca2+]i data, the neuroprotective effects of either linopirdine or XE991 were prevented by coapplied TTX (Fig. 12).
Fig. 11.

Effects of linopirdine and XE991 on [Ca2+]i and their dependence on activation of Na+ channels. Effects of linopirdine and XE991 on [Ca2+]i in sympathetic neurons were tracked by fura-2 imaging in the presence (triangles) and absence (squares) of TTX.A, Linopirdine (20 μm) alone caused a gradual increase in [Ca2+]i(n = 12 cells); the [Ca2+]i increase was prevented by coapplied TTX (100 nm; n = 14).B, [Ca2+]i was raised by XE991 (10 μm; n = 10), and the effect was blocked by TTX (100 nm; n = 7).
Fig. 12.

Requirement of Na+ channel activation in the protective effect of linopirdine and XE991 on sympathetic neurons deprived of NGF. Substantial neuronal death was induced by 2 d NGF withdrawal. In the presence or absence of NGF, TTX (100 nm) alone showed little influence on cell survival. Addition of 30 μm linopirdine or 50 μm XE991 promoted cell survival in the absence of NGF; 100 nm TTX abolished the neuroprotection induced by linopirdine or XE991. Cell survival was assayed 48 hr after incubation, cells were stained with toluidine blue, and live cells were counted.n = 4 cultures for each group. *p < 0.001 indicates significant difference from the control with NGF; ≠p < 0.05 indicates significant difference from NGF deprived cells without antagonist; #p < 0.05 indicates significant difference from the corresponding group without TTX (i.e., the next baron the left).
DISCUSSION
The experiments described above demonstrate that K+ channel blockers targeting the M-type channel strongly promote sympathetic neuronal survival by activating voltage-gated Na+ and Ca2+ channels and increasing [Ca2+]i. We further reveal that interactions of these ion channels contribute to the neuroprotection observed in sympathetic neurons.
Membrane depolarization and activation of voltage-gated channels in the neuroprotection
The membrane depolarization caused by linopirdine and XE991 was magnified by activation of voltage-gated Na+ channels. Although we anticipated that these drugs would block membrane K+channels and depolarize the neurons directly, we saw little evidence for a membrane depolarization induced directly by blocking M channels. The small decrease in membrane potential in the presence of TTX was insufficient to open voltage-gated calcium channels (Franklin et al., 1995). Therefore, it appears that block of M channels by linopirdine and XE991 requires activation of Na+channels to amplify the depolarization and, consequently, to promote Ca2+ entry via the L-type Ca2+ channel and neuron survival. The interdependence between activation of Ca2+and Na+ channels in neuronal death and survival has also been reported after traumatic axonal injury, in which Na+ influx could subsequently trigger an increase in [Ca2+]i via the opening of voltage-gated Ca2+ channels and reversal of the Na+–Ca2+exchanger (Wolf et al., 2001).
Roles of calcium and potassium in the neuroprotection
The precise cellular mechanism(s) used by [Ca2+]i to enhance neuronal survival remain elusive. Translocation of cytochrome c and development of competence-to-die could be directly inhibited by elevated [Ca2+]i(Putcha et al., 1999). We found that both linopirdine and XE991 block the pathways leading to the release of cytochrome c and the development of competence-to-die in sympathetic neurons under NGF deprivation. This is consistent with previous observations that depolarization with elevated extracellular K+ could block the release of cytochrome c and development of competence-to-die (Putcha et al., 1999). On the other hand, we are also intrigued by the observation that [Ca2+]i does not seem to decrease significantly after NGF is withdrawn for 12–16 hr (Franklin et al., 1995) (Fig. 9). This suggests that NGF does not necessarily maintain [Ca2+]i; rather, manipulations that elevate [Ca2+]i above normal levels are capable of substituting for the removal of the NGF. Consequently, two separate trophic pathways may be present in the sympathetic neurons: one requiring NGF and one dependent on elevated [Ca2+]i. Very large elevations in [Ca2+]i are neurotoxic, but these are well above the levels seen in the protected sympathetic neurons (Hyrc et al., 1997).
The K+ hypothesis for apoptosis has been proposed based on observations that an excessive K+ efflux and intracellular K+ depletion are early events in apoptotic cascade and prerequisites for apoptotic shrinkage, caspase-3 cleavage, and endonuclease activation (McCarthy and Cotter, 1997; Yu et al., 1997; Dallaporta et al., 1998; Hughes and Cidlowski, 1999). The K+ mechanism of apoptosis has been implicated in cortical (Yu et al., 1997, 1998, 1999), hippocampal (Nadeau et al., 2000), and basal forebrain cholinergic neurons (Colom et al., 1998), as well as in peripheral cells such as lymphocytes (Dallaporta et al., 1998; Hughes and Cidlowski, 1999). In the present study, although nifedipine maintained [Ca2+]i at the resting level, it did not completely eliminate the neuroprotection by linopirdine and XE991. It is possible that the residual ±20% neuronal survival is attributable to attenuated K+efflux. This might also explain the report that Na+ channel activation delays sympathetic neuronal death induced by NGF deprivation in the absence of calcium entry (Tanaka and Koike, 1997). In this situation, Na+ entry might enhance the activity of the Na+, K+-ATPase and preserve levels of intracellular K+.
Based on available evidence, we currently believe that intracellular Ca2+ and K+both contribute to regulation of neuronal apoptosis; the dominant mechanism, however, may be different depending on the cell types and apoptotic pathways involved (Yu et al., 2001). Specifically, the Ca2+-dependent mechanism is likely the principal mechanism for the protection against NGF deprivation-induced apoptosis in sympathetic neurons.
M-type potassium channel block and anti-apoptotic neuroprotection
The M channel is a G-protein-coupled K+ channel inhibited by muscarinic cholinergic agonists, originally described in bullfrog sympathetic neurons (Brown and Adams, 1980). It is a voltage- and time-dependent, low-threshold, non-inactivating channel, and the primary K+ channel activated near the threshold for Na+ channel activation and generation of action potentials. The M channel, thus, plays important roles in determining the membrane potential and membrane excitability. Our study is the first report of an anti-apoptotic effect associated with antagonism of this K+ channel. We conclude that the neuroprotection achieved by linopirdine and XE991 is mainly attributable to inhibition of the M channel based on the following observations: (1) the half effective concentrations for M current block and neuroprotection are both in low micromolar range; (2) at concentrations that prevented ∼50% neuronal death, linopirdine and XE991 show no significant effect on other K+ currents; and (3) linopirdine and XE991 have little anti-apoptotic effect in cultured cortical neurons that have most major K+ currents but lack the M current. On the other hand, because these two compounds inhibitIK at high concentrations, their powerful neuroprotection at these concentrations could involve block ofIK and even other K+ currents (Schnee and Brown, 1998). We did not test the effects of low concentrations of linopirdine and XE991 on cytochrome c release and [Ca2+]i increase; however, based on their concentration-dependent neuroprotective effects against apoptosis, it is reasonable to predict that these two cellular events are likely affected by linopirdine and XE991 in concentration-dependent manners.
Final remarks
The complicated interaction of several voltage-gated channels leading to neuroprotection in these experiments was a surprise to us, knowing that other investigators described previously exacerbation of neuronal death by activation of voltage-gated Na+ channels (Koike et al., 2000). Our results illustrate the complex interrelationship between ion channel activities and suggest that synchronized manipulation of K+, Ca2+, and Na+ channel activities may be necessary for a neuroprotective reagent in a specific paradigm. Given the potency and specificity of the two M-type K+channel antagonists used in the present study, we predict that these or similar drugs may offer a more practical approach to K+ channel blockade as a neuroprotective strategy than elevation of extracellular K+ or administration of lower potency and less selective K+ channel antagonists.
Footnotes
This work was supported by National Science Foundation Grant IBN-9817151 (S.P.Y.), American Heart Association Grant 0170064N (S.P.Y.), and National Institutes of Health Grants NS37773 (S.M.R.) and NS38651 (E.M.J.).
Correspondence should be addressed to Shan Ping Yu, Department of Neurology and Center for the Study of Nervous System Injury, Box 8111, Washington School of Medicine, St. Louis, MO 63110. E-mail:yus@neuro.wustl.edu.
M. Deshmukh's present address: Neuroscience Center, University of North Carolina, Chapel Hill, NC 27599.
REFERENCES
- 1.Brown BS, Yu SP. Modulation and genetic identification of the M channel. Prog Biophys Mol Biol. 2000;73:135–166. doi: 10.1016/s0079-6107(00)00004-3. [DOI] [PubMed] [Google Scholar]
- 2.Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- 3.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 4.Collins F, Lile JD. The role of dihydropyridine-sensitive voltage-gated calcium channels in potassium-mediated neuronal survival. Brain Res. 1989;502:99–108. doi: 10.1016/0006-8993(89)90465-4. [DOI] [PubMed] [Google Scholar]
- 5.Collins F, Schmidt MF, Guthrie PB, Kater SB. Sustained increase in intracellular calcium promotes neuronal survival. J Neurosci. 1991;11:2582–2587. doi: 10.1523/JNEUROSCI.11-08-02582.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colom LV, Diaz ME, Beers DR, Neely A, Xie WJ, Appel SH. Role of potassium channels in amyloid-induced cell death. J Neurochem. 1998;70:1925–1934. doi: 10.1046/j.1471-4159.1998.70051925.x. [DOI] [PubMed] [Google Scholar]
- 7.Costa AM, Brown BS. Inhibition of M-current in cultured rat superior cervical ganglia by linopirdine: mechanism of action studies. Neuropharmacology. 1997;36:1747–1753. doi: 10.1016/s0028-3908(97)00155-x. [DOI] [PubMed] [Google Scholar]
- 8.Dallaporta B, Hirsch T, Susin SA, Zamzami N, Larochette N, Brenner C, Marzo I, Kroemer G. Potassium leakage during the apoptotic degradation phase. J Immunol. 1998;160:5605–5615. [PubMed] [Google Scholar]
- 9.Deckwerth TL, Johnson EM. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deshmukh M, Johnson EM. Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- 11.Deshmukh M, Johnson EM. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 12.Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM. Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards SN, Buckmaster AE, Tolkovsky AM. The death programme in cultured sympathetic neurones can be suppressed at the posttranslational level by nerve growth factor, cyclic AMP, and depolarization. J Neurochem. 1991;57:2140–2143. doi: 10.1111/j.1471-4159.1991.tb06434.x. [DOI] [PubMed] [Google Scholar]
- 15.Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM. Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+ influx but not Trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallo V, Kingsbury A, Balazs R, Jorgensen OS. The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci. 1987;7:2203–2213. doi: 10.1523/JNEUROSCI.07-07-02203.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 19.Henderson CE. Programmed cell death in the developing nervous system. Neuron. 1996;17:579–585. doi: 10.1016/s0896-6273(00)80191-9. [DOI] [PubMed] [Google Scholar]
- 20.Hughes FM, Cidlowski JA. Potassium is a critical regulator of apoptotic enzymes in vitro and in vivo. Adv Enzyme Regul. 1999;39:157–171. doi: 10.1016/s0065-2571(98)00010-7. [DOI] [PubMed] [Google Scholar]
- 21.Hyrc K, Handran SD, Rothman SM, Goldberg MP. Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J Neurosci. 1997;17:6669–6677. doi: 10.1523/JNEUROSCI.17-17-06669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson MI, Argiro V. Techniques in the tissue culture of rat sympathetic neurons. Methods Enzymol. 1983;103:334–347. doi: 10.1016/s0076-6879(83)03022-0. [DOI] [PubMed] [Google Scholar]
- 23.Koike T, Martin DP, Johnson EM. Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic-factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc Natl Acad Sci USA. 1989;86:6421–6425. doi: 10.1073/pnas.86.16.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koike T, Tanaka S, Oda1 T, Ninomiya T. Sodium overload through voltage-dependent Na+ channels induces necrosis and apoptosis of rat superior cervical ganglion cells in vitro. Brain Res Bull. 2000;51:345–355. doi: 10.1016/s0361-9230(99)00246-4. [DOI] [PubMed] [Google Scholar]
- 25.Kristufek D, Koth G, Motejlek A, Schwarz K, Huck S, Boehm S. Modulation of spontaneous and stimulation-evoked transmitter release from rat sympathetic neurons by the cognition enhancer linopirdine: insights into its mechanisms of action. J Neurochem. 1999;72:2083–2091. doi: 10.1046/j.1471-4159.1999.0722083.x. [DOI] [PubMed] [Google Scholar]
- 26.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarthy JV, Cotter TG. Cell shrinkage and apoptosis: a role for potassium and sodium ion efflux. Cell Death Differ. 1997;4:756–770. doi: 10.1038/sj.cdd.4400296. [DOI] [PubMed] [Google Scholar]
- 28.McCarthy MJ, Rubin LL, Philpott KL. Involvement of caspases in sympathetic neuron apoptosis. J Cell Sci. 1997;110:2165–2173. doi: 10.1242/jcs.110.18.2165. [DOI] [PubMed] [Google Scholar]
- 29.Nadeau H, McKinney S, Anderson DJ, Lester HA. ROMK1 (Kir1.1) causes apoptosis and chronic silencing of hippocampal neurons. J Neurophysiol. 2000;84:1062–1075. doi: 10.1152/jn.2000.84.2.1062. [DOI] [PubMed] [Google Scholar]
- 30.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 31.Pike CJ, Balazs R, Cotman CW. Attenuation of beta-amyloid neurotoxicity in vitro by potassium-induced depolarization. J Neurochem. 1996;67:1774–1777. doi: 10.1046/j.1471-4159.1996.67041774.x. [DOI] [PubMed] [Google Scholar]
- 32.Putcha GV, Deshmukh M, Johnson EM. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rose K, Goldberg MP, Choi DW. Methods in toxicology, Vol 1, Pt A, In vitro biological methods. Academic; San Diego: 1993. Cytotoxicity in murine cortical cell culture. pp. 46–60. [Google Scholar]
- 34.Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnee ME, Brown BS. Selectivity of linopirdine (DuP 996), a neurotransmitter release enhancer, in blocking voltage-dependent and calcium-activated potassium currents in hippocampal neurons. J Pharmacol Exp Ther. 1998;286:709–717. [PubMed] [Google Scholar]
- 36.Tanaka S, Koike T. Veratridine delays apoptotic neuronal death induced by NGF deprivation through a Na+-dependent mechanism in cultured rat sympathetic neurons. Int J Dev Neurosci. 1997;15:15–27. doi: 10.1016/s0736-5748(96)00082-2. [DOI] [PubMed] [Google Scholar]
- 37.Tong JX, Vogelbaum MA, Drzymala RE, Rich KM. Radiation-induced apoptosis in dorsal root ganglion neurons. J Neurocytol. 1997;26:771–777. doi: 10.1023/a:1018566431912. [DOI] [PubMed] [Google Scholar]
- 38.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 39.Wang HS, Brown BS, McKinnon D, Cohen IS. Molecular basis for differential sensitivity of KCNQ and IKs channels to the cognitive enhancer XE991. Mol Pharmacol. 2000;57:1218–1223. [PubMed] [Google Scholar]
- 40.Werth JL, Deshmukh M, Cocabo J, Johnson EM, Rothman SM. Reversible physiological alterations in sympathetic neurons deprived of NGF but protected from apoptosis by caspase inhibition or Bax deletion. Exp Neurol. 2000;161:203–211. doi: 10.1006/exnr.1999.7241. [DOI] [PubMed] [Google Scholar]
- 41.Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu SP. Roles of arachidonic acid, lipoxygenases and phosphatases in calcium-dependent modulation of M-current in bullfrog sympathetic neurons. J Physiol (Lond) 1995;487:797–811. doi: 10.1113/jphysiol.1995.sp020919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 44.Yu SP, Farhangrazi ZS, Ying HS, Yeh CH, Choi DW. Enhancement of outward potassium current may participate in beta-amyloid peptide-induced cortical neuronal death. Neurobiol Dis. 1998;5:81–88. doi: 10.1006/nbdi.1998.0186. [DOI] [PubMed] [Google Scholar]
- 45.Yu SP, Yeh CH, Gottron F, Wang X, Grabb MC, Choi DW. Role of the outward delayed rectifier K+ current in ceramide-induced caspase activation and apoptosis in cultured cortical neurons. J Neurochem. 1999;73:933–941. doi: 10.1046/j.1471-4159.1999.0730933.x. [DOI] [PubMed] [Google Scholar]
- 46.Yu SP, Canzoniero LMT, Choi DW. Ion homeostasis and apoptosis. Cur Opin Cell Biol. 2001;13:405–411. doi: 10.1016/s0955-0674(00)00228-3. [DOI] [PubMed] [Google Scholar]