

Abstract
Despite the characterization of neuroprotection by transforming growth factor-β1 (TGF-β1), the signaling pathway mediating its protective effect is unclear. Bad is a proapoptotic member of the Bcl-2 family and is inactivated on phosphorylation via mitogen-activated protein kinase (MAPK). This study attempted to address whether MAPK signaling and Bad phosphorylation were influenced by TGF-β1 and, furthermore, whether these two events were involved in the antiapoptotic effect of TGF-β1. We found a gradual activation of extracellular signal-regulated kinase 1/2 (Erk1/2) and MAPK-activated protein kinase-1 (also called Rsk1) and a concomitant increase in Bad phosphorylation at Ser112 in mouse brains after adenovirus-mediated TGF-β1 transduction under nonischemic and ischemic conditions induced by transient middle cerebral artery occlusion. Consistent with these effects, the ischemia-induced increase in Bad protein level and caspase-3 activation were suppressed in TGF-β1-transduced brain. Consequently, DNA fragmentation, ischemic lesions, and neurological deficiency were significantly reduced. In cultured rat hippocampal cells, TGF-β1 inhibited the increase in Bad expression caused by staurosporine. TGF-β1 concentration- and time-dependently activated Erk1/2 and Rsk1 accompanied by an increase in Bad phosphorylation. These effects were blocked by U0126, a mitogen-activated protein kinase/Erk kinase 1/2 inhibitor, suggesting an association between Bad phosphorylation and MAPK activation. Notably, U0126 and a Rsk1 inhibitor (Ro318220) abolished the neuroprotective activity of TGF-β1 in staurosporine-induced apoptosis, indicating that activation of MAPK is necessary for the antiapoptotic effect of TGF-β1 in cultured hippocampal cells. Together, we demonstrate that TGF-β1 suppresses Bad expression under lesion conditions, increases Bad phosphorylation, and activates the MAPK/Erk pathway, which may contribute to its neuroprotective activity.
Keywords: TGF-β1, neuroprotection, MAPK/Erk signaling, Bad phosphorylation, cerebral ischemia, rat hippocampal cells
Transforming growth factor-β1 (TGF-β1) is a multifunctional cytokine capable of regulating diverse cellular processes. The neuroprotective effect of TGF-β1 in vitro (Krieglstein and Krieglstein, 1997) has been related to its ability to maintain the mitochondrial membrane potential, to stabilize Ca2+ homeostasis, to increase the expression of the anti-apoptotic proteins Bcl-2 and Bcl-xl (Prehn et al., 1994), to inhibit caspase-3 activation (Zhu et al., 2001), and to induce plasminogen activator inhibitor-1 (Buisson et al., 1998). TGF-β1 has also been shown to reduce brain damage after either ischemic (Henrich-Noack et al., 1996; Flanders et al., 1998; Pang et al., 2001) or excitotoxic injury (Ruocco et al., 1999). However, the signaling pathway underlying the neuroprotective effect of TGF-β1 remains unclear.
TGF-β1 has been suggested to mediate signaling from the cell membrane to the nucleus through the activation of TGF-β type II receptors and subsequent activation of type I receptors, which in turn phosphorylates the Smad protein family (Massagué, 2000). However, recent data indicated the involvement of mitogen-activated protein kinase (MAPK) in TGF-β1 signaling. Activation of the Ras–MAPK cascade was shown to be necessary for multiple functions of TGF-β1 in kidney and epithelial cells (Chio, 2000; Mulder, 2000). Chin et al. (1999)reported that TGF-β1 rescued macrophages from apoptosis via the MAPK/extracellular signal-regulated kinase (Erk) pathway. Thus, it is of high interest to investigate whether MAPK/Erk signaling can be activated by TGF-β1 in brain tissue and in cultured neurons and whether MAPK activation contributes to the neuroprotective effect of TGF-β1.
Bad, a proapoptotic member of the Bcl-2 family, is inactivated on phosphorylation, which creates consensus sites for Bad to interact with 14-3-3 proteins instead of Bcl-xl and Bcl-2, resulting in the liberation of these antiapoptotic proteins (Yang et al., 1995; Zha et al., 1996; Scheid et al., 1999). So far, four sites on Bad, Ser112, Ser136, Ser155, and Ser170, have been reported to be phosphorylated in response to different stimuli (Lizcano et al., 2000,Dramsi et al., 2002). In particular, Ser112 could be phosphorylated on activation of MAPK/Erk signaling (Fang et al., 1999; Shimamura et al., 2000). Rsk1 is an immediate downstream target of Erk1/2 and phosphorylates Bad. Moreover, Bad phosphorylation is required for the cell survival-promoting effect mediated by Rsk1 (Bonni et al., 1999;Fang et al., 1999; Shimamura et al., 2000). Interestingly, the survival-promoting effects of epidermal growth factor (EGF), BDNF, and interleukin-3 (IL-3) are associated with phosphorylation of Bad at Ser112 via activation of MAPK/Erk1/2 (Downward, 1999).
The present study aims to investigate whether TGF-β1 can activate the MAPK/Erk1/2 cascade and, if so, whether Bad is subsequently phosphorylated and, furthermore, whether these events contribute to the neuroprotective effect of TGF-β1.
MATERIALS AND METHODS
Vector construction and intracerebral injection. The construction of a human adenovirus serotype 5-derived adenoviral vector that transduces Escherichia coli β-galactosidase (AdRSVlacZ) was performed as described previously (Pang et al., 2001). A recombinant replication-defective (E1, E3 deleted) adenoviral serotype 5 vector (AdRSV) was constructed for the overexpression of active human TGF-β1 (ahTGF-β1). The human TGF-β1 cDNA was generated as described previously (Arrick et al., 1992), and both 5′ and 3′ ends of cDNA were modified by using NotI linkers. The plasmid AdRSVahTGF-β1 (pAdRSVahTGF-β1) was then used to produce recombinant adenoviral clones. Individual clones of pAdRSVahTGF-β1 were isolated and purified by CsCl banding and desalting through Sephadex G50. High-titer stocks were stored at −20°C in PBS containing 5% glycerol and diluted with PBS immediately before use. Mature male CD-1 mice (Charles River Laboratories, Wilmington, MA) weighing 30–35 gm were anesthetized with 4% chloral hydrate (400 mg/kg, i.p.), and then placed in a stereotactic frame with a mouse holder (model 921; David Kopf Instruments, Tujunga, CA). A burr hole was drilled in the right pericranium 1 mm lateral to the sagittal suture and 1 mm posterior to the coronal suture. A 28 gauge needle affixed to a Hamilton (Reno, NV) syringe was slowly inserted into the right lateral ventricle (3.0 mm deep from dura). An adenoviral suspension (1 μl) containing 1 × 1012 virus particles/ml was injected into the lateral ventricle at a rate of 0.2 μl/min. Control animals received the same amount of saline or AdRSVlacZ at the same injection rate. The needle was then withdrawn slowly. The hole was sealed with bone wax, and the wound was closed with a suture. The animals were allowed to recover in their cages.
TGF-β1 ELISA. Mice were anesthetized as described above, and their brains were removed from nontransduced control mice and from TGF-β1-transducd mice at defined time points. Blood plasma samples were collected at the same time from each animal. These brain and blood samples were quickly frozen in liquid nitrogen until performance of ELISA. After sonication and centrifugation of probes at 30,000 ×g for 30 min, the supernatants were collected for measurement of TGF-β1 concentration by using a TGF-β1 ELISA kit (R & D Systems, Minneapolis, MN). The detection was done under acidified conditions according to the manufacturer's protocol, thereby representing the total levels of TGF-β1. Absorbance of the peroxidase reaction product was measured at 450 nm excitation wavelength. Recombinant human TGF-β1 was used for establishing the standard curve.
Transient middle cerebral artery occlusion and infarct volume assay. On the fifth day after either AdRSVahTGF-β1, AdRSVlacZ, or saline injection, the mice were anesthetized with 1.5% isoflurane in 70/30% N2O/O2. A PE-10 catheter was introduced into the left femoral artery for continuous monitoring of arterial blood pressure, sampling of blood gases, and pH analysis before and during ischemia. Rectal temperature was controlled at 37.0 ± 0.5°C with a regulated heating pad (model 73ATD indicating controller; Yellow Springs Instrument Co., Yellow Springs, OH). The internal carotid artery was isolated, and the pterygopalatine artery was ligated. A 2 cm length of a 5-0 rounded nylon suture with a slightly larger tip was gently advanced from the external carotid artery to the beginning of the middle cerebral artery for a distance of 10.0 ± 0.5 mm. After 30 min of occlusion, the suture was partially withdrawn from the internal carotid artery to the common carotid artery. Cerebral ischemia was confirmed by monitoring the surface cerebral blood flow using a laser Doppler flow meter (BPM2 system; Vasamedics Inc., St. Paul, MN) as described previously (Yang et al., 1994).
Mouse brains were removed 1 d after transient middle cerebral artery occlusion and frozen immediately in 2-methylbutane at 42°C for 5 min. Cryostat sections (20 μm thickness) were cut and then stained with cresyl violet. Using NIH Image 1.62 software, the lesioned area was measured and calculated as the difference in area of the contralateral (nonischemic) hemisphere and the normal area of the ipsilateral (ischemic) hemisphere. The ischemic lesion was obtained from multiplying the lesioned areas by the thickness of the sections.
Evaluation of neurological deficiency (beam test). Fine motor coordination was evaluated according to the protocol described byAronowski et al. (1996) with some modifications. The mouse was placed on one end of a narrow wooden beam (8 mm in width and 600 mm in length) which was fixed 300 mm above a 60-mm-thick foam pad. The time of foot faults for the right hindlimb over 50 steps was counted in either direction on the beam. A baseline level of competence at this task was established before surgery with an acceptance level of <10 faults per 50 steps. Mice were examined before adenoviral vector injections before and 24 hr after transient middle cerebral artery occlusion (tMCAO).
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling. The mice were anesthetized 1 d after tMCAO and transcardially perfused with 4% paraformaldehyde in PBS. The brains were removed, and coronal paraffin sections (5 μm thickness) were cut from the striatum for the evaluation of cellular DNA fragmentationin situ. Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling staining was performed using an ApoTag kit (Oncor, Gaithersburg, MD) according to the previous protocol (Zhu et al., 1998). Sections were then analyzed using a confocal laser scanning microscope (Zeiss, Jena, Germany). The numbers of TUNEL-positive cells were counted in four different fields with the size of 326 × 326 μm in the ipsilateral side of each animal. Three mice were used in each group.
Immunocytochemistry. Coronal paraffin sections were prepared 8 hr after tMCAO as described above. After deparaffinization and permeabilization, sections were incubated with blocking buffer containing 2% bovine serum albumin (BSA) and 10% normal goat serum for 1 hr at 37°C. Rabbit anti-TGF-β1 (1:20; Santa Cruz Biotechnology, Heidelberg, Germany), rabbit anti-phospho-Erk1/2 (P-Erk1/2, 1:200; Cell Signaling Technology, Frankfurt, Germany), mouse anti-Bad (phospho-independent, 1:300; Transduction Laboratories, Heidelberg, Germany), and rabbit anti-active caspase-3 antibody (1:400; Transduction Laboratories) were applied to the sections overnight at 4°C. As negative controls, sections were incubated with an identical concentration of nonspecific mouse or rabbit IgG1 instead of primary antibody. After washing the sections with washing buffer [1% BSA in Tris-buffered saline (TBS)], biotin-conjugated anti-mouse or anti-rabbit IgG was added to the sections, followed by incubation with fluorescein-avidin D. For double staining, the sections were incubated with mouse anti-Bad and rabbit anti-neurofilament 200 (1:200; Sigma, Deisenhofen, Germany) antibodies, mouse anti-Bad and rabbit anti-caspase-3 antibodies, rabbit anti-TGF-β1 and mouse anti-Bad antibodies, rabbit anti-P-Erk1/2 and mouse anti-neuron-specific enolase (NSE) antibodies (1:100; Capricorn, Scarborough, ME), or rabbit anti-P-Erk1/2 and mouse anti-glia fibrillary acidic protein (GFAP) antibodies (1:100; Santa Cruz Biotechnology, Santa Cruz, CA). The sections were subsequently incubated with biotin-conjugated anti-mouse or anti-rabbit IgG followed by the fluorescein-avidin D reaction. Thereafter, a rhodamine-conjugated anti-rabbit or anti-mouse IgG was applied to the sections. Finally, the sections were mounted with a mounting medium (Dako, Hamburg, Germany) and analyzed by confocal laser scanning microscopy.
Western blotting. The mice were deeply anesthetized, and the striatum was quickly removed. The brain tissue was homogenized by sonication in a lysis buffer containing 10% glycerol, 3% sodium lauryl sulfate, 0.5 m Tris, 1 mmphenylmethylsulfonyl fluoride, 1 μm calpain inhibitor I, and 7 μg/ml trypsin inhibitor. Lysates were centrifuged at 20,000 × g (4°C) for 20 min, and the supernatants were collected for protein concentration assay using a BCA kit (Pierce, Rockford, IL). Samples containing an equal amount of total protein were loaded on 12.5 or 15% SDS-polyacrylamide gels. After electrophoresis, the proteins were transferred onto a nitrocellulose membrane. The protein transfer was controlled by staining with Ponceau S. Unspecific binding was blocked by a buffer containing 0.1% Tween 20, 2% BSA, and 5% nonfat dry milk in TBS. The blots were then incubated with primary antibodies diluted in blocking buffer overnight at 4°C. The following antibodies were used for Western blotting: phospho-specific anti-rabbit Erk1/2, phospho-specific anti-rabbit Rsk1 (Thr360/Ser364), phospho-specific anti-rabbit Bad (Ser112, 1:1000; Cell Signaling Technology), anti-mouse Bad (1:300), anti-rabbit antibody against both pro- and active caspase-3 (1:400; Santa Cruz Biotechnology), and anti-mouse α-tubulin (1:2000; Sigma). After washing the membranes with 0.1% Tween 20 in TBS, the blots were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (1:2500) at room temperature for 1 hr. Peroxidase activity was detected by the Amersham Biosciences (Arlington Heights, IL) ECL detection system. To control the amount of protein loaded in the each lane, α-tubulin was detected in parallel. The integrated optical density (IOD) of the signals was semiquantified and expressed as the ratio of IOD from the tested proteins to IOD from α-tubulin. Data were given as means ± SD of the percentage ratio of the control.
Primary rat hippocampal cultures. Mixed primary rat hippocampal cultures were prepared as described previously (Zhu et al., 2001). Briefly, the hippocampi from neonatal Fischer 344 rats (postnatal days 1–2) were dissected and incubated for 20 min in Leibovitz's L15 medium supplemented with 1 mg/ml papain and 0.2 mg/ml BSA. Thereafter, the cell suspension was layered onto growth medium containing 1% trypsin inhibitor and 10% BSA. After centrifugation at 200 × g for 10 min, the pellet was resuspended and seeded into poly-l-lysine-coated Petri dishes with a density of 2–3 × 104cells/cm2. The cells were cultured in neurobasal medium with 0.5 mm glutamine, B27 supplement, and antibiotics for 8 d.
Drug treatment. TGF-β1 (R & D Systems, Wiesbaden, Germany) was administered to the cells on the seventh day of the culture. To induce apoptosis, staurosporine was added to the cells at a final concentration of 100 nm in the absence of TGF-β1 or 24 hr after the onset of TGF-β1 treatment. In some experiments, U0126 at a final concentration of 10 or 20 μm (Cell Signaling Technology) or Ro318220 at the final concentration of 5 or 10 μm (Cell Signaling Technology) was added to the cells 2 hr before the TGF-β1 treatment to block the activation of MAPK. Control cultures received vehicle only. There were four series of experiments designed for cultured hippocampal cells: (1) the concentration-dependent effect of TGF-β1 on the activation of MAPK and on the phosphorylation of Bad was established, in which cultured cells received 0.1, 1.0, and 10 ng/ml TGF-β1 and were harvested 8 hr after the onset of the treatment for Western blot analysis; (2) to evaluate the time-dependent effect of TGF-β1 on the activation of MAPK and phosphorylation of Bad, the cells were treated with 10 ng/ml TGF-β1 and collected 15 and 30 min and 1, 8, and 24 hr after the treatment; (3) to find out whether TGF-β1 modulates Bad protein expression, cells were harvested at 6 and 24 hr after the exposure to staurosporine in the absence or presence of TGF-β1 (1 and 10 ng/ml) for Western blot analysis; and (4) to reveal the role of MAPK activation in the antiapoptotic effect of TGF-β, apoptosis was evaluated by the nuclear staining 24 hr after the exposure of staurosporine in the absence and in the presence of TGF-β1 (10 ng/ml). Using the same treatment protocol, the cells were harvested for Western blot analysis to study the activation of MAPK and Bad phosphorylation in parallel.
Nuclear staining. Cells were fixed with methanol and incubated with Hoechst 33258 (10 μg/ml; Sigma) at 37°C for 10 min, followed by washing with methanol and PBS. Thereafter, the nuclear morphology was analyzed under the fluorescence microscope. Cells showing condensed chromatin or fragmented nuclei were counted as apoptotic cells, and the data were given as the means ± SD of the percentage of apoptotic cells.
Statistics. All data were expressed as means ± SD. ANOVA followed by Scheffé's test was used in Figures1A, 3, B and C, and 9,A and C. Student's t test was used in Figures 2D, c, 4B, 5B,6B, 7, A and B, and 8,B and D.
Fig. 1.
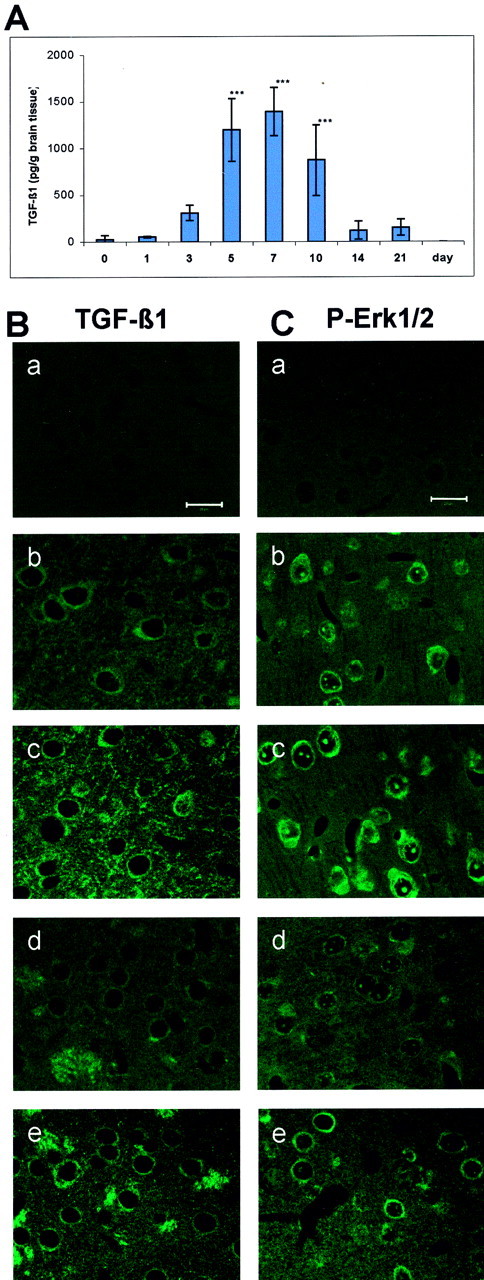
Adenovirus transduction causes an increase in TGF-β1 and P-Erk1/2 levels in mouse brain tissue. A, Time course of TGF-β1 concentration detected in mouse brain tissue. Adenovirus carrying the human active TGF-β1 gene was injected into the lateral ventricle of the mice. The brains were removed immediately (0 d) and 1, 3, 5, 7, 10, 14, and 21 d after adenoviral injection. TGF-β1 concentration was measured by ELISA under acidified conditions, thereby representing the total level of TGF-β1. ***p < 0.001 compared with 0 d. Data are expressed as means ± SD from five mice for each time point. B, Immunostaining of TGF-β1 in the cortex (b, c) and the striatum (d, e) 5 d after lacZ (b, d) or TGF-β1 (c, e) transduction. C, Immunostaining of P-Erk1/2 in the cortex (b, c) and the striatum (d, e) 5 d after lacZ (b, d) or TGF-β1 (c, e) transduction. Negative controls of TGF-β1 and P-Erk1/2 immunostaining are shown in B, a, and C, a, respectively. Scale bars, 20 μm.
Fig. 3.

Overexpression of TGF-β1 reduces the ischemic lesion and improves the neurological outcome of ischemic mice. The brains were removed from the saline-treated and lacZ- and TGF-β1-transduced mice. The cryostat sections were cut continuously and stained with cresyl violet. A, Photographs of the ischemic lesion 1 d after tMCAO. B, Quantitative analysis of the ischemic lesion 1 d after tMCAO. Data are expressed as means ± SD of 10 animals for each group. There was no difference between saline-treated and lacZ-transduced mice. ***p < 0.001 compared with lacZ-transduced mice.C, Improvement of the neurological outcome in TGF-β1-transduced mice after ischemia. The neurological deficiency was evaluated by a beam test before cerebral injection on the fifth day after cerebral injection without ischemia and 1 d after tMCAO. Intracerebral injection did not cause neurological deficiency in nonischemic mice (shaded columns) of all three groups in comparison with those before injection (open columns). Thirty minutes of MCAO led to a severe neurological deficiency in saline-treated or lacZ-transduced mice at 1 d after ischemia but to a lesser extent in TGF-β1-transduced mice as evidenced by the significant decrease in the falls from the beam (on the sixth day after cerebral injection; filled columns). Data are expressed as means ± SD of 13–20 animals for each group. There was no difference between saline-treated and lacZ-transduced mice. *p < 0.05 compared with lacZ-transduced mice.
Fig. 9.

MEK/Erk1/2 and Rsk inhibitors block the antiapoptotic effect of TGF-β1 and inhibit TGF-β1-mediated phosphorylation of Erk1/2, Rsk1 and Bad in cultured rat hippocampal cells. TGF-β1 (10 ng/ml) was administered to the hippocampal cells on the seventh day of culture. Staurosporine (Stau, 100 nm) was added to the medium 24 hr after onset of TGF-β1 treatment. To block the activation of MEK/Erk1/2 or Rsk1, U0126 (20 μm) or Ro318220 (5 μm) was added to the cells 2 hr before exposure of TGF-β1, respectively. Controls received vehicle only. A, C, U0126 and Ro318220 abolished the antiapoptotic effect of TGF-β1. The nuclear morphology was analyzed under a fluorescence microscope after nuclear staining with Hoechst 33258. The cells showing condensed chromatin, and fragmented nuclei were counted as apoptotic cells. The data are expressed as the percentage of means ± SD ###p < 0.001 compared with vehicle-treated control; **p < 0.01; ***p < 0.001 compared with cultures treated with staurosporine alone; ++p < 0.01;+++p < 0.001, compared with cultures treated with staurosporine plus TGF-β1. The experiments were representative of three independent cultures. B, D, TGF-β1-mediated activation of Erk1/2, Rsk1 and increase in phosphorylation of Bad were inhibited by U0126 (B) and Ro318220 (D). The cells were harvested in lysis buffer 24 hr after adding staurosporine. Thirty micrograms of total protein from each group were loaded to SDS-polyacrylamide gels, and P-Erk1/2, P-Rsk1 (Thr360/Ser364) and P-Bad (Ser112) proteins were analyzed using specific antibodies. α-Tubulin blots were used for controlling the amount of protein loaded in each lane. The experiments were representative of three independent experiments.
Fig. 2.
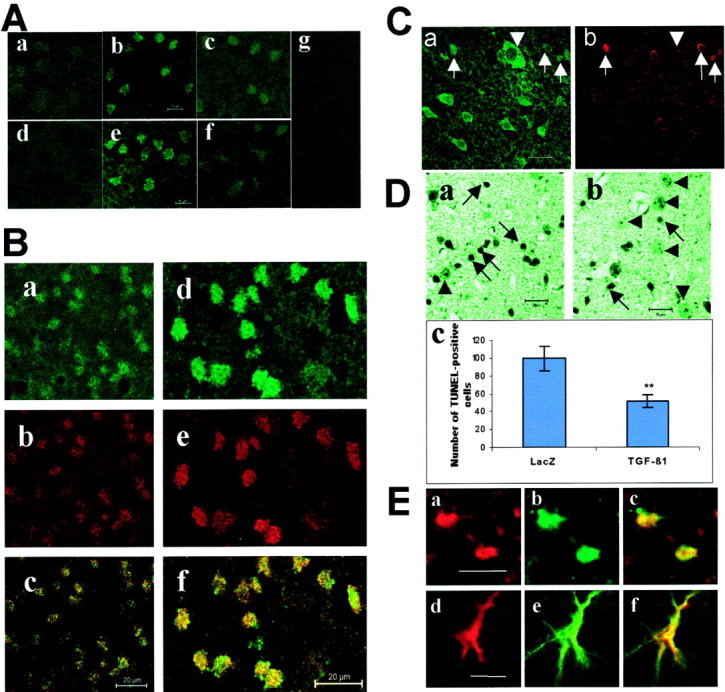
Overexpression of TGF-β1 inhibits the expression of Bad, the activation of caspase-3, and DNA fragmentation in ischemic brain after 30 min of MCAO. A, Immunostaining of Bad (a–c) and active caspase-3 (d–f) in striatal sections prepared 8 hr after reperfusion. Only weak Bad immunoreactivity was detected in the contralateral side (a), whereas it markedly increased in the ipsilateral side (b) of control mice, which were subjected to intracerebral lacZ injection. In comparison, activation of caspase-3 was not detected in the contralateral side (d) but was clearly visible in the ipsilateral side (e) of control mice. TGF-β1 transduction inhibited both Bad expression (c) and caspase-3 activation (f) in the ipsilateral side. Negative controls did not show detectable immunofluorescence (g). Scale bar, 10 μm. B, Double staining of a striatum section prepared 8 hr after cerebral ischemia.a–c, Double staining of Bad and neurofilament 200. Bad immunoreactivity (a) was colocalized with the neuronal marker neurofilament 200 (b), as evidenced by the overlap image (c).d–f, Double staining of Bad and active caspase-3. Bad immunoreactivity (d) was colocalized with active caspase-3 (e), as revealed by the overlap image (f). The immunostaining was reproduced in three mice per group. Scale bar, 20 μm. C, Double staining of TGF-β1 (a) and Bad (b) in the penumbra of a lesioned area 8 hr after tMCAO. TGF-β1 immunoreactivity was detected in the extracellular matrix and in the cytoplasma of cells with normal morphology and very low Bad immunoreactivity (arrowhead). Arrowsindicate dying cells, which showed increased Bad immunoreactivity. Scale bar, 20 μm. D, TUNEL staining. The number of TUNEL-positive cells was counted in four different fields with a size of 326 × 326 μm in the ipsilateral side of each animal. Three mice were used in each group. A high number of TUNEL-positive cells was detected 1 d after tMCAO in the striatum of lacZ-transduced mice (a). These TUNEL-positive cells showed apoptotic features, including shrunken cell bodies and condensed nuclei, as indicated by arrows. The number of TUNEL-positive cells was reduced in TGF-β1-transduced mice (b).Arrowheads indicate TUNEL-negative cells with normal morphology. Scale bar, 20 μm. Quantitative analysis indicated a significant reduction of the number of TUNEL-positive cells in the ipsilateral side of TGF-β1-transduced mice (c). **p < 0.01, compared with lacZ control.E, Double staining of P-Erk1/2 and neuronal marker NSE (a–c) or astrocyte marker GFAP (d–f) in the ischemic core of TGF-β1-transduced mice 48 hr after tMCAO. P-Erk1/2 (a, c) was colabeled with NSE (b) and GFAP (e). The overlap of P-Erk1/2-NSE and P-Erk1/2-GFAP is shown in c and f, respectively. Scale bar, 20 μm.
Fig. 4.
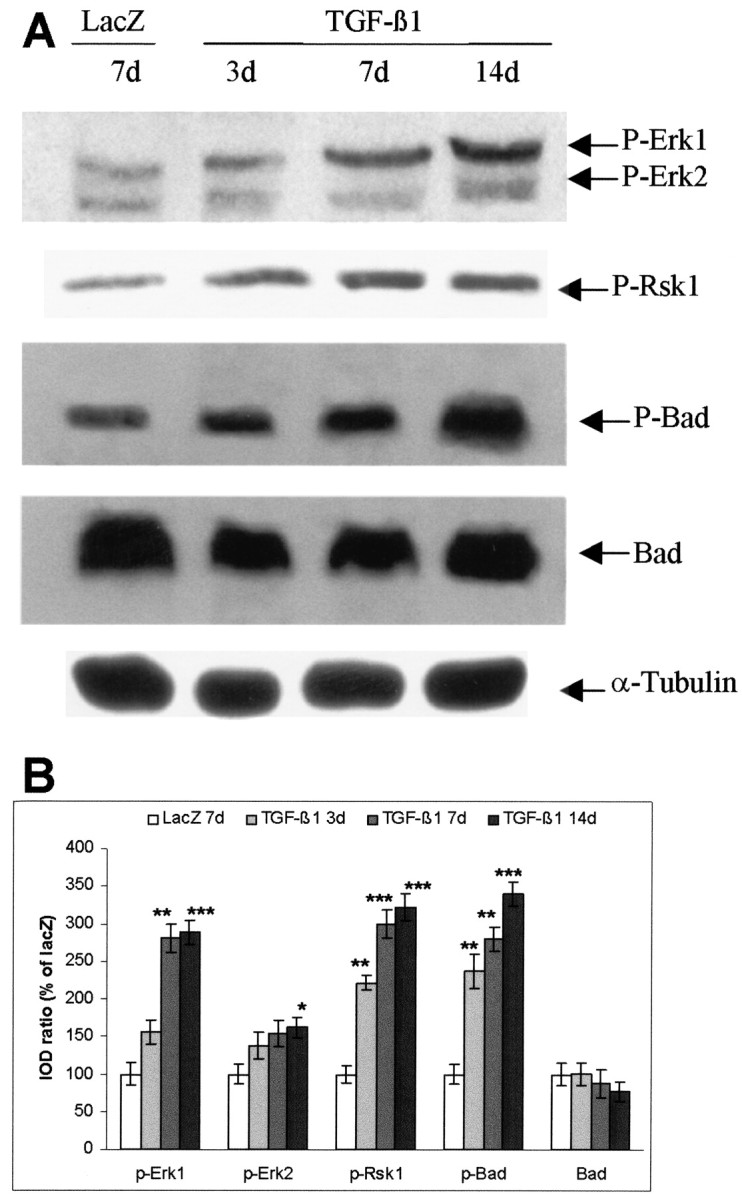
Adenovirus-mediated TGF-β1 overexpression activates Erk1/2 and Rsk1 and increases phosphorylation of Bad in the striatum of nonischemic mice. A, Representative blots for P-Erk1/2, P-Rsk1 (Thr360/Ser364), P-Bad (Ser112), Bad, and α-tubulin. Mice were subjected to intracerebral injection of either AdRSVlacZ or AdRSVahTGF-β1. The striatum tissues were isolated, and proteins were extracted at different time points after injection. Sixty micrograms of total protein were loaded to each lane for Western blot. Blots were representative of three animals at each time point. B. Semiquantification of blots. The ratio of the IOD of P-Erk1, 2, P-Rsk1, P-Bad, and Bad signals to α-tubulin signals was calculated. The ratios obtained from LacZ-injected mice were normalized to 100%, and the data were given as means ± SD of the percentage of lacZ-transduced controls. *p < 0.05; **p < 0.01; ***p < 0.001 compared with lacZ-transduced mice.
Fig. 5.
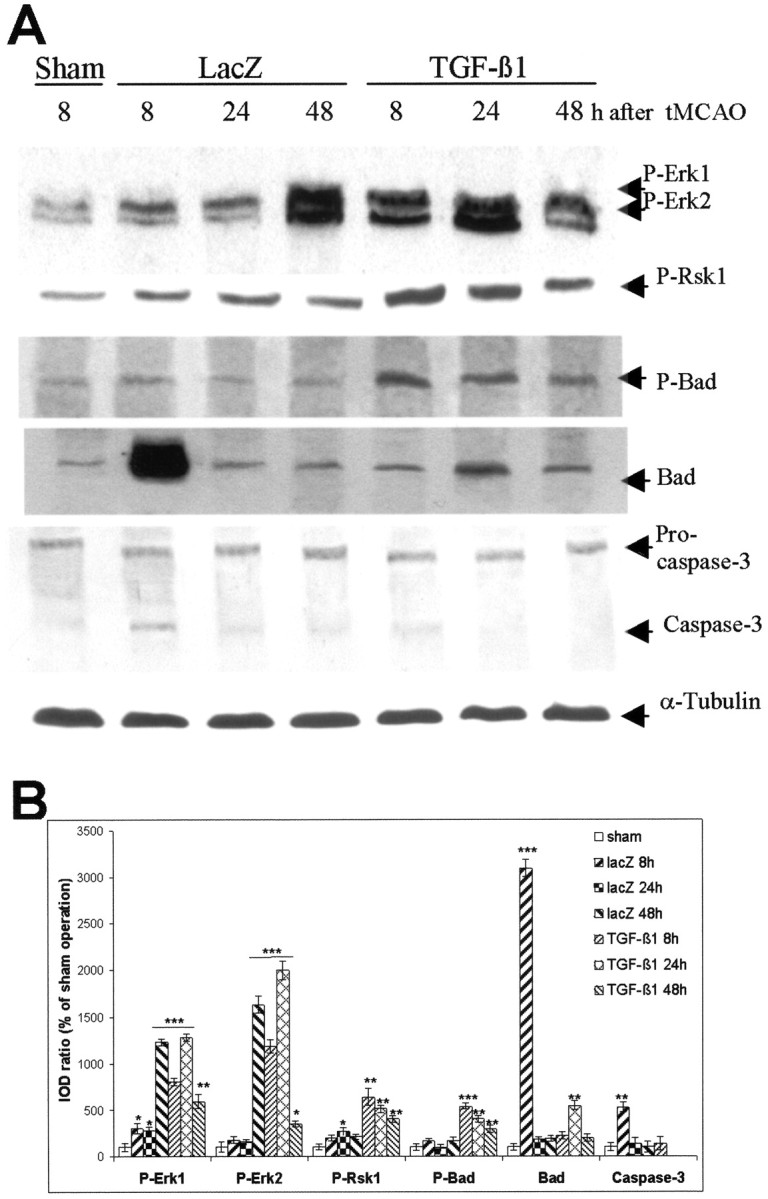
Adenovirus-mediated TGF-β1 overexpression activates Erk1/2 and Rsk1 and interrupts apoptotic cascades through modulation of Bad expression and inhibition of caspase-3 activation in ischemic mice. A, Representative blots of P-Erk1/2, P-Rsk1 (Thr360/Ser364), P-Bad (Ser112), Bad, caspase-3, and α-tubulin. The mice were subjected to intracerebral injection of either AdRSVlacZ or AdRSVahTGF-β1. Thirty minutes of MCAO or sham operation was performed on the fifth day after the injection. The striatal tissues were isolated, and proteins were extracted at the defined time points after ischemia. Sixty micrograms of total protein were loaded to each lane for Western blot. Blots were representative for three animals for each time point. B, Semiquantification of blots. The ratio of IOD from P-Erk1/2, P-Rsk1, P-Bad, Bad, and caspase-3 signals to α-tubulin signals was calculated. The ratios obtained from sham-operated mice were normalized to 100%, and the data were given as means ± SD of the percentage of controls. Caspase-3 activation was not detectable in TGF-β1-transduced mice at 24 and 48 hr after tMCAO. *p < 0.05; **p< 0.01; ***p < 0.001 compared with sham-operated mice.
Fig. 6.

TGF-β1 concentration-dependently activates Erk1/2 and Rsk1 and increases phosphorylation of Bad in cultured rat hippocampal cells. A, Representative blots of P-Erk1/2, P-Rsk1 (Thr360/Ser364), P-Bad (Ser112), Bad, and α-tubulin. TGF-β1 (0.1, 1.0, and 10 ng/ml) was administered to the hippocampal cells on the seventh day of culture. Controls received vehicle only. The cells were harvested in a lysis buffer 8 hr after adding TGF-β1 or vehicle. Thirty micrograms of total protein from each group were loaded to SDS-polyacrylamide gel. Blots were representative of three independent experiments. B, Semiquantification of the blots. We calculated the ratio of IOD from the signals of P-Erk1/2, P-Rsk1, and P-Bad to α-tubulin, and the ratios obtained from vehicle-treated controls (0 ng/ml) were normalized to 100%. The data were given as means ± SD of the percentage of controls. *p< 0.05; ***p < 0.001 compared with vehicle treated-control.
Fig. 7.

Time course of activation of Erk1/2 and Rsk1 as well as phosphorylation of Bad after administration of TGF-β1 in the cultured rat hippocampal cells. A, B, Representative blots of P-Erk1/2, P-Rsk1 (Thr360/Ser364), P-Bad (Ser112), Bad and α-tubulin and the semiquantification of the blots. TGF-β1 (10 ng/ml) was added to the hippocampal cells on the seventh day of culture. To block MAPK signaling, U0126 (20 μm) was added 2 hr before the exposure of TGF-β1. Controls received vehicle only. The cells were harvested in a lysis buffer at 15 and 30 min (A) as well as 1, 8, and 24 hr (B) after adding TGF-β1. Thirty micrograms of total protein from each group were loaded to SDS-polyacrylamide gel. Blots were representative of three independent cultures. The ratio of IOD from P-Erk1/2, P-Rsk1, and P-Bad signals to α-tubulin signals was calculated. The ratios obtained from vehicle-treated controls (either 30 min or 24 hr) were normalized to 100%, and the data were given as means ± SD of the percentage of the corresponding control. *p < 0.05; **p < 0.01; ***p < 0.001, compared with vehicle-treated control (30 min);#p < 0.05;##p < 0.01;###p < 0.001 compared with vehicle-treated control (24 hr).
Fig. 8.

TGF-β1 suppresses the increase in Bad protein level in cultured rat hippocampal cells induced by staurosporine.A, Representative blots of Bad and α-tubulin 6 and 24 hr after the treatment of TGF-β1 (10 ng/ml) in the absence of staurosporine. B, Semiquantification of the blots presented in A. The ratio of IOD from Bad signals to α-tubulin signals was calculated. The ratios obtained from vehicle-treated controls (either 6 or 24 hr) were normalized to 100%, and the data were given as means ± SD of the percentage of controls. C, Representative blots of Bad and α-tubulin in the presence of staurosporine. TGF-β1 (1 and 10 ng/ml) was added to the hippocampal cells 24 hr before the onset of staurosporine (Stau, 100 nm) treatment. Cells were harvested at 6 and 24 hr after the exposure to staurosporine. Controls received vehicle only. Blots were representative for three independent experiments. D, Semiquantification of the blots presented in C. *p < 0.05 compared with the vehicle-control; #p < 0.05 compared with staurosporine-treated cells.
RESULTS
Adenovirus-mediated TGF-β1 transduction increased TGF-β1 level in mouse brain
The levels of TGF-β1 protein in the brain and in the blood plasma were measured in normal mice (nontransduced) and immediately (0 d) and 1, 3, 5, 7, 10, 14, and 21 d after cerebral TGF-β1 transduction. TGF-β1 levels were very low in brain tissue of nontransduced mice and immediately after transduction (0 d, ∼23 pg/gm of brain tissue). TGF-β1 levels gradually increased from day 3 to day 7 after transduction. The level of TGF-β1 in mouse brain tissue peaked at 7 d after transduction (1395 ± 258 pg/gm of brain tissue) and was maintained at higher levels up to 10 d after transduction. It was then declined nearly to the basal level 14 d after adenoviral injection (Fig.1A). The basal concentration of TGF-β1 in blood plasma of nontransduced mice was 1683 ± 611 pg/ml, which was much higher than that in brain tissue of nontransduced mice. There were no significant changes in plasma levels of TGF-β1 up to 21 d after TGF-β1 transduction, indicating that cerebral transduction of TGF-β1 did not influence the level of TGF-β1 in the blood (data not shown).
We further studied the distribution of TGF-β1 in brain tissue 5 d after either lacZ or TGF-β1 transduction by immunostaining. A negative control did not show detectable immunofluorescence (Fig.1B, a). TGF-β1 immunoreactivity was considerably increased in both the extracellular matrix and neuronal cytoplasm of cortical (Fig. 1B, c) and striatal (Fig.1B, e) regions of TGF-β1-transduced mice in comparison with lacZ-transduced mice (Fig. 1B, b, d). The TGF-β1 level seemed to be particularly high in the cytoplasm and in the striatal fibers. In addition, a high level of TGF-β1 protein was detected in the region surrounding cerebral vessels in TGF-β1-transduced brain.
Overexpression of TGF-β1 interfered with apoptotic cascades, attenuated brain damage, and improved the neurological outcome after tMCAO
Physiological parameters including the animal's body temperature, blood pressure, blood PO2, PCO2, and pH were controlled at normal levels during and after tMCAO. These parameters and surface cerebral blood flow did not differ among TGF-β1-transduced-, lacZ-, and saline-injected mice (data not shown).
To determine whether ischemia-activated apoptotic cascades could be suppressed by TGF-β1 transduction, immunostaining of Bad and active caspase-3 was performed in brain sections 8 hr after tMCAO. There was no visible fluorescence in negative control sections (Fig.2A, g). Enhanced Bad immunoreactivity was detected in the ipsilateral striatum (Fig.2A, b) accompanied by a significant activation of caspase-3 (Fig. 2A, e), whereas Bad immunoreactivity was only weakly visible (Fig. 2A, a), and no active caspase-3 (Fig. 2A, d) was seen in the contralateral side. Double staining indicated colocalization of Bad with the neuronal marker neurofilament 200 (Fig. 2B, a–c), as well as colocalization of Bad with active caspase-3 (Fig. 2B, d–f). The ischemia-induced increase in Bad immunoreactivity and activation of caspase-3 were inhibited in TGF-β1-transduced mice, as shown in Figure 2A, cand f, respectively. Furthermore, it was noted that cells in the ischemic penumbra highly expressing TGF-β1 showed normal morphology (Fig. 2C, a, arrowhead) where Bad immunoreactivity in these cells appeared considerably weak (Fig.2C, b, arrowhead). In contrast, increased Bad immunoreactivity was seen in the damaged cells, which showed reduced expression of TGF-β1 (Fig. 2C, arrows). These findings were in agreement with the results obtained from Western blot analysis (see Fig. 5A, fourth and fifth panels).
To confirm the antiapoptotic effect of TGF-β1 in vivo, TUNEL staining was performed 1 d after tMCAO. A high number of TUNEL-positive cells with shrunken nuclei and condensed chromatin, as indicated by arrows, was detected in the lesioned striatum of lacZ-injected control mice (Fig. 2D, a), whereas the number of TUNEL-positive cells was much less in the striatum of TGF-β1-transduced mice (Fig. 2D, b). Quantitative analysis revealed an ∼50% reduction of the number of TUNEL-positive cells in TGF-β1-transduced brains (Fig. 2D, c).
We further tested whether inhibition of apoptotic cascades by TGF-β1 contributed to neuroprotection and improvement of the neurological outcome after ischemia. As shown in Figure3A, the striatal region was severely lesioned in saline and lacZ controls, but to a much less extent in TGF-β1-transduced mice. Quantitative analysis revealed that the ischemic lesion in TGF-β1-transduced mice was reduced by 40% (p < 0.001) 1 d after tMCAO (Fig.3B) in comparison with that in saline and lacZ controls. Evaluation of neurological outcome by the beam test showed that cerebral injection of either saline or AdRSVlacZ or AdRSVTGF-β1 to nonischemic mice did not cause any neurological deficiency. Thirty minutes of cerebral ischemia markedly increased the number of falls from the beam in mice subjected to either saline or AdRSVlacZ injection but to a lesser extent in TGF-β1-transduced mice (Fig.3C). The reduction of the ischemic lesion and the functional improvement of neurological outcome indicated a neuroprotective potency of TGF-β1 in vivo.
Overexpression of TGF-β1 increased phosphorylation of Erk1/2, Rsk1, and Bad in the striatum of nonischemic mice
To investigate whether TGF-β1 transduction was able to activate the MAPK cascade and to phosphorylate Bad, the levels of P-Erk1/2, phospho-Rsk1 (P-Rsk1), and phospho-Ser112-Bad (P-Bad) were detected by Western blot analysis of striatal tissue, the most vulnerable region to tMCAO. As shown in Figure4A, the levels of P-Erk1/2 and P-Rsk1 increased in a time-dependent manner after TGF-β1 transduction. The P-Bad level was enhanced concomitantly in the striatum of TGF-β1-transduced mice. Semiquantification of the blots revealed an approximately threefold increase in the levels of P-Erk1, P-Rsk1, and P-Bad from days 7 to 14 after TGF-β1 transduction in comparison with the lacZ control (Fig. 4B). The level of Bad protein in these nonischemic mouse brains was not changed at any detected time point after adenoviral transduction. The results suggest a correlation between activation of MAPK signaling and phosphorylation of Bad at Ser112 mediated by TGF-β1.
The levels of P-Erk1/2 were also immunohistochemically studied 5 d after either lacZ or TGF-β1 transduction in mouse brain. The negative control did not show detectable immunofluorescence (Fig. 1C, a). Immunoreactivity of P-Erk1/2 was weakly detected in the cytoplasma of neurons in cortex (Fig. 1C, b) and striatum (Fig. 1C, d) of lacZ-transduced mice, whereas enhanced P-Erk1/2 expression was observed in both the extracellular space and the cytoplasma of cortical (Fig. 1C, c) and striatal (Fig.1C, e) neurons in TGF-β1-transduced mice. It was of note that P-Erk1/2 was also highly expressed in the region surrounding vessels, similar to that observed in TGF-β1 immunostaining (Fig.1B, c, e), indicating a concomitant increase in TGF-β1 expression and activation of Erk1/2.
Overexpression of TGF-β1 increased phosphorylation of Erk1/2, Rsk1, and Bad, and suppressed expression of death protein Bad as well as caspase-3 activation in brain tissue after tMCAO
Next, we investigated how TGF-β1 transduction in mouse brain influenced the activation of MAPK cascades after cerebral ischemia. The level of P-Erk1/2 moderately increased 8 and 24 hr after and was markedly enhanced (12- and 16-fold increases in P-Erk1 and P-Erk2, respectively) 48 hr after tMCAO in the striatum of lacZ-transduced mice in comparison with lacZ-injected, sham-operated mice (Fig.5A, first panel, B). Phosphorylation of Rsk1, a downstream kinase of Erk1/2, increased moderately in lacZ-transduced mice after tMCAO (Fig. 5A, second panel, B), whereas the level of P-Bad was slightly elevated (Fig.5A, third panel, B). Notably, ischemia led to a 30-fold increase in Bad expression 8 hr after reperfusion (Fig. 5A, fourth panel, B), accompanied by a fivefold activation of caspase-3 (Fig. 5A, fifth panel, B). TGF-β1 transduction resulted in 4- and 15-fold increases in P-Erk1 and P-Erk2 levels, respectively, at 8 hr after tMCAO, and they further increased 24 hr after tMCAO compared with the lacZ controls (Fig. 5A, first panel, B). A 3.5-fold increase in the P-Rsk1 level was observed in TGF-β1-transducd mice at 8 hr after tMCAO compared with lacZ-transduced controls (Fig. 5A, second panel, B), whereas P-Bad was increased approximately fourfold (Fig. 5A, third panel, B). P-Bad was maintained at this level up to 24 hr in TGF-β1-transduced brain after ischemia and then declined 48 hr after tMCAO, but it was still twofold higher than in lacZ-transduced controls. Overexpression of TGF-β1 significantly reduced the level of Bad protein (Fig. 5A, fourth panel, B) and the activation of caspase-3 8 hr after ischemia. The latter was no longer detectable in TGF-β1-transduced mice 24 and 48 hr after tMCAO (Fig. 5A, fifth panel, B). These results were consistent with the findings revealed by immunostaining of Bad and active caspase-3 (Fig.2A).
To further identify the cell phenotypes that accounted for the persistent elevation of the P-Erk1/2 level in the TGF-β1-transduced brains at the late stage of reperfusion, double staining of P-Erk1/2-NSE and P-Erk1/2-GFAP was performed on the brain sections prepared 48 hr after tMCAO. As shown in Figure 2E, P-Erk1/2 (Fig. 2E, a, d) was colabeled with the neuronal marker NSE (Fig. 2E, b, c) as well as with the astrocyte marker GFAP (Fig. 2E, e, f), indicating that both neurons and astrocytes were involved in the expression of P-Erk1/2.
TGF-β1 activated Erk1/2 and Rsk1 and enhanced phosphorylation of Bad in cultured hippocampal cells, which was blocked by U0126
The effect of TGF-β1 on activation of MAPK cascades and on phosphorylation of Bad was further characterized in neuronal cultures. Cultured rat hippocampal cells were treated with different concentrations of TGF-β1, and cells were harvested at different time points after the onset of the treatment. As shown in Figure6A, the levels of P-Erk1/2, P-Rsk1, and P-Bad concomitantly increased in a concentration-dependent manner. Notably, TGF-β1 (10 ng/ml) caused a fivefold increase in the level of P-Bad, as revealed by semiquantitative analysis of the blots (Fig. 6B).
Activation of Erk1/2 and Rsk1 as well as an increase in Bad phosphorylation were detected in cultured hippocampal cells as early as 15 min after adding 10 ng/ml TGF-β1 (Fig.7A). The levels of P-Erk1/2, P-Rsk1, and P-Bad were continuously elevated 1 and 8 hr after the onset of TGF-β1 treatment. The semiquantitative analysis of the blots indicated a fivefold increase in P-Bad level 8 hr after the onset of TGF-β1 treatment. They declined 24 hr after the administration of TGF-β1 but were still higher than those detected in the controls (Fig. 7B). To get evidence that activation of MAPK by TGF-β1 mediated the increase in Bad phosphorylation, U0126, a specific MEK and Erk1/2 inhibitor, was added to the cells 2 hr before the exposure of TGF-β1. U0126 completely blocked the phosphorylation of Erk1/2, Rsk1, and Bad, suggesting that TGF-β1-mediated phosphorylation of Bad is mediated by the MAPK/Erk1/2 cascade (Fig.7A).
TGF-β1 suppressed the level of Bad protein in staurosporine-treated hippocampal cells
To confirm the inhibitory effect of TGF-β1 on the lesion-induced increase in Bad protein expression, we detected Bad protein expression in staurosporine-treated hippocampal cells in the presence and absence of TGF-β1. The level of Bad protein increased 1.8- and 1.6-fold 6 and 24 hr after the exposure to staurosporine, respectively, compared with the corresponding vehicle-treated controls. TGF-β1 at the concentration of 1 ng/ml effectively diminished the elevation of Bad protein level caused by staurosporine, whereas 10 ng/ml TGF-β1 further reduced Bad expression by 50 and 30% at 6 and 24 hr, respectively, after the treatment of staurosporine in comparison with controls (Fig. 8C,D). However, TGF-β1 did not influence Bad protein levels in the absence of staurosporine (Fig. 8A,B).
U0126 and Ro318220 abolished the TGF-β1-mediated increase in phosphorylation of Bad and the antiapoptotic effect of TGF-β1 in cultured hippocampal cells
To clarify whether the antiapoptotic effect of TGF-β1 was mediated by activation of MAPK signaling, U0126 and Ro318220 were added to cells 2 hr before the exposure of TGF-β1 (10 ng/ml). Apoptosis was induced by 100 nm staurosporine, which was given to the cells 24 hr after the onset of TGF-β1 treatment. As revealed by nuclear staining with Hoechst 33258, TGF-β1 significantly reduced the percentage of apoptotic neurons, which was in agreement with our previous report (Zhu et al., 2001). This effect was completely abolished by U0126 (20 μm) (Fig.9A) and Ro318220 (5 μm) (Fig. 9C). U0126 at the concentration of 10 μm only partially blocked the neuroprotection by TGF-β1, and Ro318220 at the concentration of 10 μm was toxic (data not shown).
Using the same treatment protocol, it was further demonstrated that U0126 completely blocked the basal phosphorylation of Erk1/2 and suppressed the basal phosphorylation of Rsk1 and Bad under control conditions. The latter implies that Erk1/2 is not the sole kinase for activation of Rsk1. Staurosporine alone did not affect the level of P-Erk1/2 and P-Rsk1, supporting the finding that Erk was a staurosporine-insensitive kinase (McCubrey et al., 2000). However, increased phosphorylation of Erk1/2, Rsk1, and Bad was clearly detected when TGF-β1 was added to the cells 24 hr before the exposure of staurosporine, and this increase was no longer seen in the presence of U0126 (Fig. 9B). To support these findings, another MAPK pathway inhibitor, Ro318220, was used. Ro318220 nearly abolished both the basal and TGF-β1-mediated phosphorylation of Rsk1, whereas the phosphorylation of Erk1/2 was not influenced, suggesting that Rsk1 acted downstream of Erk1/2. Importantly, Ro318220 markedly suppressed the basal level of P-Bad as well as the TGF-β1-mediated increase in Bad phosphorylation, indicating the requirement of Rsk1 activation for the phosphorylation of Bad (Fig. 9D).
DISCUSSION
Most cells, including those in the CNS, are capable of expressing TGF-β1 and TGF-β receptors. Accordingly, the spectrum of cellular activities of TGF-β1 is extremely diverse (Unsicker and Strelau, 2000). The role of TGF-β1 in apoptosis varies depending on the cell type and their developmental stage as well as on the specific stimulus (De Luca et al., 1996; Krieglstein and Krieglstein, 1997; Krieglstein et al., 2000). Indeed, little is known about the signal transduction pathways mediating its role in apoptosis. In the present study we demonstrated the following: (1) Adenovirus-mediated overexpression of TGF-β1 in mouse brain interfered with the apoptotic process after tMCAO through suppressing Bad protein expression, increasing Bad phosphorylation, and inhibiting caspase-3 activation. Consequently, ischemia-induced DNA degradation and cerebral lesions were decreased, and the neurological outcome was improved in TGF-β1-transduced mice. The inhibitory effect of TGF-β1 on Bad expression was also found in staurosporine-treated hippocampal cells. (2) TGF-β1 was able to activate the MAPK/Erk1/2 pathway under nonischemic and ischemic conditions, which was concomitantly related to an increase in phosphorylation of Bad in mouse brain. Similarly, TGF-β1 activated MAPK/Erk1/2 signaling and increased phosphorylation of Bad in a concentration- and time-dependent manner in cultured rat hippocampal cells. Both events were completely blocked by U0126, suggesting a link between MAPK/Erk1/2 and phosphorylation of Bad caused by TGF-β1. (3) TGF-β1 protected cultured rat hippocampal cells from staurosporine-induced apoptosis, and this effect was abolished by either U0126 or Ro318220, indicating a requirement of Erk1/2 and Rsk1 activation for the antiapoptotic activity of TGF-β1.
We used adenovirus-mediated TGF-β1-transduced mice in the present study because this model has advantages for studying the role of TGF-β1 in vivo. First, a desired TGF-β1 level (1–1.5 ng/gm of brain tissue), which was almost the same as the neuroprotective concentration of TGF-β1 identified in our previous study (Henrich-Noack et al., 1996), was achieved in brain tissue from the 5th to 10th day after a single injection of AdRSVahTGF-β1 (Fig.1) without an influence of the level of TGF-β1 in the blood plasma. This is usually difficult to achieve by the single or multiple intracerebral injection of exogenous TGF-β1 because of its short half-life (Wakefield et al., 1990). Second, adenovirus transduction of TGF-β1 did not influence the cerebral blood flow; thus we can exclude the possibility that an increase in cerebral blood flow contributed to the neuroprotective effect of TGF-β1.
Bcl-2 and caspase family proteins are known to play a central role in the regulation of apoptosis. Activation of caspase-3, a critical member of the caspase family, accelerates apoptosis and thus has been suggested as an apoptotic marker (Chen et al., 1998; Lee et al., 1999;Schulz et al., 1999). Bad is a Bcl-2 homology domain-only proapoptotic member of the Bcl-2 family, and a massive increase in Bad expression has been found to kill cells. Furthermore, dephosphorylated Bad forms a complex with the antiapoptotic proteins Bcl-xl or Bcl-2, thereby displacing the apoptotic promoter Bax. Phosphorylated Bad can dissociate from this complex, which in turn results in the liberation of Bcl-xl and Bcl-2 (Hsu et al., 1997; Scheid et al., 1999). Thus, phosphorylation of Bad has been suggested to be essential for inactivation of its proapoptotic activity (Lizcano et al., 2000; Virdee et al., 2000; Zhou et al., 2000; Dramsi et al., 2002). Phosphorylation of Bad at Ser112 in response to MAPK activation was absolutely required for dissociation of Bad from Bcl-xl (Bonni et al., 1999; Fang et al., 1999; Scheid et al., 1999). In light of these findings, we propose that modulation of Bad expression by TGF-β1 would interfere with the apoptotic cascade in the CNS. Here we provide evidence that TGF-β1 increased Bad phosphorylation at Ser112 in striatal tissue under both nonischemic and ischemic conditions, as well as in cultured hippocampal cells in the presence or absence of apoptotic stimuli. This event is tightly associated with activation of the MAPK/Erk1/2 pathway. In addition, overexpression of TGF-β1 inhibited Bad expression, which had been dramatically increased after tMCAO. These effects may crucially contribute to the antiapoptotic effect of TGF-β1, i.e., inhibition of caspase-3 activation, prevention of DNA fragmentation, reduction of ischemic lesion, and the final improvement of neurological function after cerebral ischemia. Interestingly, the inhibitory effect of TGF-β1 on Bad expression was observed only under damaging conditions in both in vivo and in vitroexperiments. Most likely, TGF-β1 interferes with a mechanism that upregulates Bad in apoptosis and that is yet to be defined.
An important link between kinase signaling pathways and the cellular apoptotic machinery has been established after the discovery that Bad is phosphorylated in response to activation of MEK/Erk1/2, phosphatidylinositol 3-kinase/Akt, and cAMP-dependent protein kinase. In particular, MAPK/Erk1/2 has been suggested to be important for intracellular signaling mediating cell survival-promoting effects of growth factors such as glial cell line-derived neurotrophic factor (Nicole et al., 2001), BDNF (Hetman et al., 1999; Han and Holtzman, 2000), basic fibroblast growth factor (Gardner and Johnson, 1996; Abe et al., 2001), EGF (Gleichmann et al., 2000), and insulin-like growth factor-1 (Parrizas et al., 1997) and the cytokine IL-3 (Shimamura et al., 2000). More recently, evidence for either positive (Reimann et al., 1997; Chin et al., 1999; Chio, 2000; Lhuillier and Dryer, 2000; Mulder, 2000) or negative (Chang, 2000) regulatory effects of TGF-β1 on MAPK activation has been shown, depending on the cell type and experimental conditions. However, little is known about the role of TGF-β1 in MAPK activation in the nervous system, and to our knowledge, data dealing with TGF-β1-mediated MAPK activation and Bad phosphorylation are not yet available. In this study, we demonstrated that TGF-β1 concomitantly increased phosphorylation of Erk1/2, Rsk1, and Bad (Ser112) both in mouse brain and in cultured rat hippocampal cells. Blocking of Bad phosphorylation by the MEK/Erk1/2 inhibitor U0126 in cultured rat hippocampal cells indicated that the MAPK pathway was required for the phosphorylation of Bad at Ser112.
The MAPK/Erk signaling pathway has been intensively studied in different types of cultured cells, including cultured neurons (Anderson and Tolkovsky, 1999; Hetman et al., 1999; Abe et al., 2001;Chang and Karin, 2001). However, its role in the brain and in setting CNS disease is not clear. The activation of MAPK/Erk signaling in neurons plays a central role in controlling synaptic plasticity and memory (Sweatt, 2001) and is implicated in the pathological processes after cerebral ischemia (Sugino et al., 2000; Wu et al., 2000).Alessandrini et al. (1999) reported a reduction of brain damage by the MEK1 inhibitor PD98059 after focal cerebral ischemia. The same group also recently showed the neuroprotective effect of the MEK/Erk1/2 inhibitor U0126 after either global or focal cerebral ischemia (Namura et al., 2001). However, others found that MAPK/Erk1/2 inhibitors did not reduce brain damage after global ischemia (Sugino et al., 2000) or after hypoxia (Han and Holtzman, 2000). Furthermore, it has been documented that activation of MAPK/Erk1/2 was involved in the mechanisms of ischemic tolerance (Gonzalez-Zulueta et al., 2000) and mediated the neuroprotective effect of BDNF after either hypoxic–ischemic injury in neonatal rats (Han and Holtzman, 2000) or traumatic CNS injury in adult rats (Klöcker et al., 2000). In this study, we showed that activation of MAPK/Erk1/2 signaling was consistently accompanied by an increase in Bad phosphorylation in either nonischemic or ischemic mouse brain after adenovirus-mediated TGF-β1 transduction. Moreover, overexpression of TGF-β1 in mouse brain resulted in a neuroprotective effect after tMCAO, as evidenced by attenuation of DNA degradation, reduction of the brain infarction, and improvement of the neurological outcome, in which MAPK/Erk1/2 was consistently activated. These findings suggest a beneficial contribution of the MAPK pathway to the neuroprotection by TGF-β1. To support this hypothesis, we showed that U0126 and Ro318220 completely abolished the protective effect of TGF-β1 in cultured hippocampal cells, indicating that activation of Erk1/2-Rsk1 signaling was necessary for the antiapoptotic effect of TGF-β1. Consistent with the blockade of the antiapoptotic activity of TGF-β1, the phosphorylation of Erk1/2, Rsk1, and Bad mediated by TGF-β1 was diminished by both MAPK inhibitors. These results obtained from the in vitro experiments could to some extent support our findings observed in ischemic brain tissue. However, it is realized that staurosporine-induced apoptosis may have less ischemic relevance, although it is a widely used model for study of apoptosis. Therefore, it is worthwhile to confirm these findings using a more ischemic relevant in vitro model, for instance, hypoxia. Furthermore, application of an MAPK/Erk1/2 inhibitor in vivo would directly reveal the role of MAPK activation in the neuroprotective effect of TGF-β1 after cerebral ischemia.
The major findings of the present study are schematically summarized in Figure 10. Considering the importance of sustaining survival signaling and, in addition, suppressing death promoters in the injured brain, we suggest that the inhibition of Bad protein expression under lesion conditions and the increase in Bad phosphorylation through the MAPK/Erk1/2 pathway by TGF-β1 may represent a pivotal step in the neuroprotective effect of this cytokine.
Fig. 10.

Scheme summarizing the major findings of this study. Either adenovirus-mediated overexpression of TGF-β1 in mouse brain or administration of TGF-β1 to the cultured hippocampal cells caused an activation of Erk1/2 and Rsk1, consistently accompanied by an increase in phosphorylation of Bad (Ser112). TGF-β1-mediated activation of MAPK, phosphorylation of Bad, and an antiapoptotic effect in cultured hippocampal cells were abolished by MEK/Erk1/2 and Rsk1 inhibitors, indicating a tight association between MAPK activation and Bad phosphorylation, as well as a requirement of MAPK pathway for the antiapoptotic activity of TGF-β1. It is proposed that the increase in phosphorylation of Bad may lead to a dissociation of Bcl-2 and Bcl-xl from the complex of Bad/Bcl-xl or Bad/Bcl-2, which in turn interrupts multiple apoptotic cascades, including inhibition of caspase-3 activation. These effects, together with the inhibitory effect of TGF-β1 on the lesion-mediated increase in Bad level, may crucially contribute to the neuroprotective activity of TGF-β1, as evidenced by decrease in DNA degradation and ischemic lesions, as well as the improvement of the neurological outcome after cerebral ischemia.
Footnotes
This study was supported by Deutsche Forschungsgemeinschaft Grant Kr 359/16-3,4 and National Institutes of Health Grant R01 NS-35089. We thank Michaela Stumpf and Sandra Engel for skillful technical assistance.
Correspondence should be addressed to Dr. Yuan Zhu, Institut für Pharmakologie und Toxikologie, Philipps-Universität Marburg, Ketzerbach 63, D-35032 Marburg, Germany. E-mail:zhu@mailer.uni-marburg.de.
REFERENCES
- 1.Abe K, Aoyagi A, Saito H. Sustained phosphorylation of mitogen-activated protein kinase is required for basic fibroblast growth factor-mediated axonal branch formation in cultured rat hippocampal neurons. Neurochem Int. 2001;38:309–315. doi: 10.1016/s0197-0186(00)00093-0. [DOI] [PubMed] [Google Scholar]
- 2.Alessandrini A, Namura S, Moskowitz MA, Bonventre J. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson CNG, Tolkovsky AM. A role for MAPK/ERK in sympathetic neuron survival: protection against a p53-dependent, JUK-independent induction of apoptosis by cytosine arabinoside. J Neurosci. 1999;19:664–673. doi: 10.1523/JNEUROSCI.19-02-00664.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aronowski J, Samways E, Strong R, Rhoades HM, Grotta JC. An alternative method for the quantitation of neuronal damage after experimental middle cerebral artery occlusion in rats: analysis of behavioral deficit. J Cereb Blood Flow Metab. 1996;16:705–713. doi: 10.1097/00004647-199607000-00022. [DOI] [PubMed] [Google Scholar]
- 5.Arrick BA, Lopez AR, Elfman R, Damsky CH, Derynck R. Altered metabolic and adhesive properties and increased tumorigenesis associated with increased expression of transforming growth factor beta 1. J Cell Biol. 1992;118:715–726. doi: 10.1083/jcb.118.3.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signalling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 7.Buisson A, Nicole O, Docagne F, Sartelet H, MacKenzie ET, Vivien D. Up-regulation of a serine-protease inhibitor (PAI-1) in astrocytes mediates the neuroprotective activity of transforming growth factor-β1 (TGF-β1). FASEB J. 1998;12:1683–1691. [PubMed] [Google Scholar]
- 8.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 9.Chang NS. TGF-β-induced matrix proteins inhibit p42/44 MAPK and JUK activation and suppress TNF-mediated IκBα degradation and NF-κB nuclear transduction in L929 fibroblasts. Biochem Biophys Res Commun. 2000;267:194–200. doi: 10.1006/bbrc.1999.1909. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Nagayama T, Jin K, Statler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3 like protease may mediate delayed neuronal death in the hippocampus after transient forebrain ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin BY, Petrache I, Chio AMK, Chio ME. Transforming growth factor-beta1 rescues serum deprivation-induced apoptosis via MAPK pathway in macrophages. J Biol Chem. 1999;274:11362–11368. doi: 10.1074/jbc.274.16.11362. [DOI] [PubMed] [Google Scholar]
- 12.Chio ME. Mechanism of transforming growth factor-beta1 signalling: role of the mitogen-activated protein kinase. Kidney Int. 2000;58:53–58. [Google Scholar]
- 13.De Luca A, Weller M, Fontana A. TGF-β1-induced apoptosis of cerebellar granule neurons is prevented by depolarization. J Neurosci. 1996;16:4174–4185. doi: 10.1523/JNEUROSCI.16-13-04174.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Downward J. How Bad phosphorylation is good for survival. Nature. 1999;1:33–35. doi: 10.1038/10026. [DOI] [PubMed] [Google Scholar]
- 15.Dramsi S, Scheid MP, Maiti A, Hojabrpour P, Chen X, Schubert K, Goodlett DR, Aebersold R, Duronio V. Identification of a novel phosphorylation site, Ser170, as a regulator of Bad pro-apoptotic activity. J Biol Chem. 2002;277:6399–6405. doi: 10.1074/jbc.M109990200. [DOI] [PubMed] [Google Scholar]
- 16.Fang X, Yu S, Eder A, Mao M, Bast RC, Boyd D, Mills GB. Regulation of Bad phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncology. 1999;18:6635–6640. doi: 10.1038/sj.onc.1203076. [DOI] [PubMed] [Google Scholar]
- 17.Flanders KC, Ren RF, Lippa CF. Transforming growth factor-betas in neurodegenerative diseases. Prog Neurobiol. 1998;54:71–85. doi: 10.1016/s0301-0082(97)00066-x. [DOI] [PubMed] [Google Scholar]
- 18.Gardner AM, Johnson GL. Fibroblast growth factor-2 suppression of tumor necrosis factor alpha-mediated apoptosis requires Ras and the activation of mitogen-activated protein kinase. J Biol Chem. 1996;271:14560–14566. doi: 10.1074/jbc.271.24.14560. [DOI] [PubMed] [Google Scholar]
- 19.Gleichmann M, Weller M, Schulz JB. Insulin-like growth factor-mediated protection from neuronal apoptosis is linked to phosphorylation of the pro-apoptotic protein Bad but not to inhibition of cytochrome c translocation in rat cerebellar neurons. Neurosci Lett. 2000;282:69–72. doi: 10.1016/s0304-3940(00)00857-0. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Requirement for nitric oxide activation of p21ras/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci USA. 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the Erk pathway. J Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henrich-Noack P, Prehn JHM, Krieglstein J. TGF-β1 protects hippocampal neurons against degeneration caused by transient global ischemia: dose-response relationship and potential neuroprotective mechanisms. Stroke. 1996;27:1609–1615. doi: 10.1161/01.str.27.9.1609. [DOI] [PubMed] [Google Scholar]
- 23.Hetman M, Kanning AK, Cavanaugh JE, Xia ZG. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 24.Hsu SY, Kaipia A, Zhu L, Hsueh AJW. Interference of Bad (Bcl-xl/Bcl-2-associated death promoter)-induced apoptosis in mammalian cells by 14–3-3 isoforms and p11. Mol Endocrinol. 1997;11:1858–1867. doi: 10.1210/mend.11.12.0023. [DOI] [PubMed] [Google Scholar]
- 25.Klöcker N, Kermer P, Weishaupt JH, Labes M, Ankerhold R, Bähr M. Brain-derived neurotrophic factor-mediated neuroprotection of adult rat retinal ganglion cells in vivo does not exclusively depend on phosphatidyl-inositol-3′-kinase/protein kinase B signalling. J Neurosci. 2000;20:6962–6967. doi: 10.1523/JNEUROSCI.20-18-06962.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krieglstein K, Krieglstein J. Transforming growth factor-β signalling and neuroprotection. In: Mattson MP, editor. Neuroprotective signal transduction. Humana; Totawa, NJ: 1997. pp. 119–144. [Google Scholar]
- 27.Krieglstein K, Richter S, Farkas L, Schuster N, Dünker N, Oppenheim R, Unsicker K. Reduction of endogenous transforming growth factors β prevents ontogenetic neuron death. Nat Neurosci. 2000;3:1085–1090. doi: 10.1038/80598. [DOI] [PubMed] [Google Scholar]
- 28.Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischemic brain injury mechanisms. Nat Neurosci Rev. 1999;399:7–14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 29.Lhuillier L, Dryer SE. Developmental regulation of neuronal KCa channels by TGF-β1: transcriptional and posttranscriptional effects mediated by Erk MAP kinase. J Neurosci. 2000;20:5616–5622. doi: 10.1523/JNEUROSCI.20-15-05616.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lizcano JM, Morrice N, Cohen P. Regulation of Bad by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Massagué J. How cells read TGF-β signals. Nature Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 32.McCubrey J, May WS, Duronio V, Mufson A. Serine/threonine phosphorylation in cytokine signal transduction. Leukemia. 2000;14:9–21. doi: 10.1038/sj.leu.2401657. [DOI] [PubMed] [Google Scholar]
- 33.Mulder KM. Role of Ras and MAPKs in TGF-β signalling. Cytokine Growth Factor Rev. 2000;11:23–35. doi: 10.1016/s1359-6101(99)00026-x. [DOI] [PubMed] [Google Scholar]
- 34.Namura S, Lihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicole O, Ali C, Docagne F, Plawinski L, MacKenzie ET, Vivien D, Buisson A. Neuroprotection mediated by glial cell line-derived neurotrophic factor: Involvement of a reduction of NMDA-induced calcium influx by the mitogen-activated protein kinase pathway. J Neurosci. 2001;21:3024–3033. doi: 10.1523/JNEUROSCI.21-09-03024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pang L, Wen Y, Che XM, Roessler BJ, Betz AL, Yang GY. Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-β1 expression. Stroke. 2001;32:544–552. doi: 10.1161/01.str.32.2.544. [DOI] [PubMed] [Google Scholar]
- 37.Parrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J Biol Chem. 1997;272:154–161. doi: 10.1074/jbc.272.1.154. [DOI] [PubMed] [Google Scholar]
- 38.Prehn JHM, Bindokas VP, Marcuccilli CJ, Krajewski D, Reed JC, Miller RJ. Regulation of neuronal Bcl-2 protein expression and calcium homeostasis by transforming growth factor type β confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA. 1994;91:12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reimann T, Hempel U, Krautwald S, Axmann A, Scheibe R, Seidel D, Wenzel KW. Transforming growth factor-beta1 induces activation of Ras, Raf-1, MEK and MAPK in rat hepatic stellate cells. FEBS Lett. 1997;403:57–60. doi: 10.1016/s0014-5793(97)00024-0. [DOI] [PubMed] [Google Scholar]
- 40.Ruocco A, Nicole O, Docagne F, Ali C, Chazalviel L, Komesli S, Yablonsky F, Roussel S, MacKenzie ET, Vivien D, Buisson A. A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J Cereb Blood Flow Metab. 1999;19:1345–1353. doi: 10.1097/00004647-199912000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Scheid MP, Schubert KM, Duronio V. Regulation of Bad phosphorylation and association with Bcl-xl by the MAPK/Erk kinase. J Biol Chem. 1999;274:31108–31113. doi: 10.1074/jbc.274.43.31108. [DOI] [PubMed] [Google Scholar]
- 42.Schulz JB, Weller M, Moskowitz MA. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 43.Shimamura A, Ballif BA, Richards SA, Blenis J. Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr Biol. 2000;10:127–135. doi: 10.1016/s0960-9822(00)00310-9. [DOI] [PubMed] [Google Scholar]
- 44.Sugino T, Nozaki K, Takagi Y, Hattori I, Hashimoto N, Moriguchi T, Nishida E. Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J Neurosci. 2000;20:4506–4514. doi: 10.1523/JNEUROSCI.20-12-04506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 46.Unsicker K, Strelau J. Functions of transforming growth factor-β isoforms in the nervous system. Eur J Biochem. 2000;267:6972–6975. doi: 10.1046/j.1432-1327.2000.01824.x. [DOI] [PubMed] [Google Scholar]
- 47.Virdee K, Parone PA, Tolkovsky AM. Phosphorylation of the pro-apoptotic protein Bad on serine 155, a novel site, contributes to cell survival. Curr Biol. 2000;10:1151–1154. doi: 10.1016/s0960-9822(00)00702-8. [DOI] [PubMed] [Google Scholar]
- 48.Wakefield LM, Winokur TS, Hollands RS, Christopherson K, Levinson AD, Sporn MB. Recombinant latent transforming growth factor beta 1 has a longer plasma half-life in rats than active transforming growth factor beta 1, and a different tissue distribution. J Clin Invest. 1990;86:1976–1984. doi: 10.1172/JCI114932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu DC, Ye W, Che XM, Yang GY. Activation of mitogen-activated protein kinase after permanent cerebral artery occlusion in mouse brain. J Cereb Blood Flow Metab. 2000;20:1320–1330. doi: 10.1097/00004647-200009000-00007. [DOI] [PubMed] [Google Scholar]
- 50.Yang GY, Betz AL, Chenevert TL, Brunberg JA, Hoff JT. Experimental intracerebral hemorrhage: relationship between brain edema, blood flow and blood brain barrier permeability in rats. J Neurosurg. 1994;81:93–102. doi: 10.3171/jns.1994.81.1.0093. [DOI] [PubMed] [Google Scholar]
- 51.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xl and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 52.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist Bad in response to survival factor results in binding to 14–3-3 not Bcl-xl. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 53.Zhou XM, Liu Y, Payne G, Lutz RJ, Chittenden T. Growth factors inactivate the cell death protein Bad by phosphorylation of its BH3 domain on Ser155. J Biol Chem. 2000;275:25046–25051. doi: 10.1074/jbc.M002526200. [DOI] [PubMed] [Google Scholar]
- 54.Zhu Y, Culmsee C, Semkova I, Krieglstein J. Stimulation of β2-adrenoceptors inhibits apoptosis in rat brain after transient forebrain ischemia. J Cereb Blood Flow Metab. 1998;18:1032–1039. doi: 10.1097/00004647-199809000-00013. [DOI] [PubMed] [Google Scholar]
- 55.Zhu Y, Ahlemeyer B, Bauerbach E, Krieglstein J. TGF-β1 prevents neuronal apoptosis in rat hippocampal cultures involving inhibition of caspase-3 activation. Neurochem Int. 2001;38:227–235. doi: 10.1016/s0197-0186(00)00084-x. [DOI] [PubMed] [Google Scholar]