

Abstract
Drosophila Slowpoke (dSlo) calcium-dependent potassium channels bind directly to the catalytic subunit of cAMP-dependent protein kinase (PKAc). We demonstrate here that coexpression of PKAc with dSlo in mammalian cells results in a dramatic decrease of dSlo channel activity. This modulation requires catalytically active PKAc but is not mediated by phosphorylation of S942, the only PKA consensus site in the dSlo C-terminal domain. dSlo binds to free PKAc but not to the PKA holoenzyme that includes regulatory subunits and is inactive. Activators of endogenous PKA that stimulate dSlo phosphorylation, but do not produce detectable PKAc binding to dSlo, do not modulate channel function. Furthermore, the catalytically inactive PKAc mutant does bind to dSlo but does not modulate channel activity. These results are consistent with the hypothesis that both binding of active PKAc to dSlo and phosphorylation of dSlo or some other protein are necessary for channel modulation.
Keywords: Ca2+-dependent K+channel, dSlo, cAMP-dependent protein kinase, protein–protein interaction, phosphorylation, whole-cell recording, modulation
It is well established that ion channels are modulated by protein kinases, phosphatases, and other signaling proteins (Levitan, 1999; Catterall, 2000). In some cases, modulation may involve the intimate association of the ion channel with one or more of these regulatory proteins. Such regulatory protein complexes are formed either through direct binding or via scaffold adaptor and anchoring proteins and are presumed to target modulatory enzymes to sites where they can be accessed optimally by activators and also affect particular substrates (Tavalin et al., 1999; Colledge et al., 2000). One important type of ion channel that is modulated by associated regulatory proteins is the large conductance calcium-dependent potassium (KCa) channel (Atkinson et al., 1991; Adelman et al., 1992). This unique class of ion channel responds to both the membrane potential and intracellular calcium. Because KCa channels are expressed ubiquitously in both excitable and nonexcitable tissues, modulation of these channels can influence many important physiological functions, including neurotransmitter release, hormone secretion, and muscle contraction (Vergara et al., 1998; Shao et al., 1999; Brenner et al., 2000).
KCa channels in native tissues can be modulated by a number of different protein kinases (White et al., 1991; Tian et al., 1998; Schopf et al., 1999). We and others have shown previously that KCa channels reconstituted in lipid bilayers or isolated membrane patches can be modulated by the addition of ATP alone, without an exogenous protein kinase, suggesting that these channels might be intimately associated with protein kinase activities (Chung et al., 1991; Bielefeldt and Jackson, 1994; Reinhart and Levitan, 1995). After the cloning of the first KCa channel from the Drosophilaslowpoke locus (dSlo), genetic and biochemical studies have provided more direct evidence of such regulatory complexes involving KCa channels and other proteins (Schopperle et al., 1998; Xia et al., 1998; Zhou et al., 1999). In previous studies, we found that endogenous protein kinase activity co-immunoprecipitates with both native and recombinant dSlo calcium-dependent potassium channels. We also showed that the dSlo channel can bind simultaneously to two different protein kinases, one the catalytic subunit of the cAMP-dependent protein kinase (PKAc) and the other the Src tyrosine kinase (Wang et al., 1999). Additionally, overlay experiments demonstrated that PKAc can bind directly to a 172 amino acid region in the C-terminal domain of dSlo, indicating that PKAc interacts directly with the dSlo channel (Wang et al., 1999) rather than via regulatory subunits or anchoring proteins (Colledge et al., 2000).
In this study, we investigated the effect of PKAc binding on dSlo channel function. We show here that dSlo channel activity, measured using the whole-cell voltage-clamp technique, is downregulated upon coexpression of PKAc. Although modulation of dSlo activity requires catalytically active PKAc, it is not mediated by phosphorylation of serine 942, the only consensus PKA substrate site in the dSlo C-terminal domain. We also show that dSlo binds to free PKAc but not to the holoenzyme in which PKAc is bound to its regulatory subunit. In addition, we provide evidence that direct binding of active PKAc may be necessary for the profound modulation of dSlo channel activity. Taken together with our previous observations, these results suggest that the dSlo channel itself can serve as a scaffold for modulatory proteins that influence channel function.
MATERIALS AND METHODS
Antibodies. The polyclonal anti-dSlo antibody was generated and purified with fusion proteins containing the C-terminal 58 amino acids of dSlo (splice variant, A1C2E1G3I0) (Adelman et al., 1992). The sequence- and phosphorylation-specific antibody anti-pS942 was generated and purified as described previously (Wang et al., 1999). Monoclonal anti-PKA catalytic and regulatory subunits antibodies were purchased from Transduction Laboratories (Lexington, KY).
cDNA constructs and mutagenesis. cDNAs encoding mouse PKAc and PKA regulatory subunits RIα and RIβ (kindly provided by Dr. G. Stanley McKnight, University of Washington, Seattle, WA) were subcloned into the mammalian expression vector pcDNA3. Construction of the glutathione S-transferase (GST) fusion proteins GST-Cter and GST-A was as reported previously (Wang et al., 1999). Site-directed mutagenesis of the PKAc gene was performed using the Quickchange strategy (Stratagene), with appropriate primers to introduce single mutations at K72, H87, R133, or W196. The same approach was used to generate mutations in dSlo at R939, R940, and S942. A deletion mutant of dSlo (Δ922–956) was constructed using a standard PCR approach.
Transfection and immunoprecipitation. tsA201 cells were maintained in DMEM supplemented with 10% fetal bovine serum. A calcium phosphate transfection protocol was used to introduce cDNAs into the cells. Forty-eight hours after transfections, cells were lysed in lysis buffer containing 1% CHAPS, 20 mmTris-HCl, pH 7.5, 10 mm EDTA, 120 mm NaCl, 50 mm KCl, 2 mm DTT, and protease inhibitors [1 mm PMSF, 1 μg/ml each aprotonin, leupeptin, and pepstatin A (Sigma, St. Louis, MO)]. After a centrifugation to remove insoluble debris from the lysate, the supernatant was precleared with 20 μl/ml protein A-agarose (Santa Cruz Biotechnology, Santa Cruz, CA). dSlo was immunoprecipitated by incubation with anti-dSlo antibodies (2 μg/ml) for 2 hr at 4°C, followed by incubation with 20 μl/ml protein A-agarose. The immunoprecipitates were then washed with lysis buffer five times.
Western blot. Proteins in the cell lysates or immunoprecipitates were separated on polyacrylamide gels and transferred to nitrocellulose membranes. After blocking with 5% nonfat milk in TBST (0.1% Tween 20 in TBS), the blots were probed with appropriate primary antibodies in blocking buffer at 4°C overnight. The membranes were then washed with TBST and incubated with a 1:3000 dilution of horseradish peroxidase-conjugated donkey anti-rabbit or sheep anti-mouse IgG (Amersham Biosciences, Arlington Heights, IL) for 1 hr at room temperature. After three washes of the membrane with TBST and one wash with TBS, protein complexes were visualized using the enhanced chemiluminescence system (Amersham).
Protein kinase assay. The enzymatic activity of PKAc was measured in vitro using the SignalTECH PKA assay system (Promega, Madison, WI). The reaction mixture contained PKA assay buffer, 100 μmol biotinylated Kemptide, 0.5 mm[γ-32P]ATP mix at 10 μCi/μl, 0.25 pmol PKAc, and varying amounts of GST fusion proteins or PKA RII subunit. The reaction mix was pre-equilibrated at room temperature for 5 min, and the reaction was initiated by adding the enzyme. After a 5 min incubation at 30°C, the reaction was terminated by adding the termination buffer, and spotted onto a SAM2 Biotin Capture Membrane square. After washing eight times with 2 m NaCl and twice with deionized water, the membrane was dried. Radioactivity was measured by liquid scintillation spectrometry.
Whole-cell recording. dSlo channel activity was recorded in the whole-cell configuration from tsA201 cells expressing dSlo, alone or together with PKAc. The Fugene 6 transfection reagent (Roche, Indianapolis, IN) was used to transfect cells with these cDNAs along with the pEGFP-N1 vector cDNA (Clontech) in a 9:1 ratio. Recordings were done 1–2 d after transfection on an Axiovert 25 inverted fluorescence microscope (Zeiss). Transfected cells bearing GFP fluorescence were identified with an FITC filter set. Patch electrodes with resistances of 1–3 MΩ were pulled from borosilicate glass and fire polished. The bath solution contained (in mm): 2 KCl, 148 NaCl, 2 MgCl2, 1 EGTA, and 10 HEPES, pH 7.2. The pipette solution contained (in mm): 150 KCl, 2 MgCl2, 0.5 BAPTA, and 10 HEPES, pH 7.2. Sufficient CaCl2 was added to the pipette solution to bring the free calcium concentration to 10 μm, determined with a calcium electrode as described previously (Schopperle et al., 1998). Whole-cell currents were filtered at 1 kHz and digitized at 20 kHz with an Axopatch 200A amplifier. Data acquisition and voltage clamp were performed with pCLAMP7 software.
RESULTS
dSlo channel activity is downregulated by coexpression of PKAc
We have shown previously that the dSlo calcium-dependent potassium channel can bind directly to PKAc. To study the effect of the bound kinase on dSlo channel activity, we measured dSlo current with the whole-cell voltage-clamp configuration in a heterologous expression system. In previous experiments we reported that cotransfection of both Src and PKAc together does not influence dSlo channel activity (Wang et al., 1999). Because of the possibility that this result arose from opposing actions of the two kinases, in this study we focused solely on the effect of PKAc.
In tsA201 cells transfected with dSlo cDNA, whole-cell K+ currents were elicited by depolarizing voltage steps from a holding potential of −80 mV, with 10 μm intracellular free Ca2+(Fig. 1A). Like macroscopic dSlo K+ currents recorded in inside-out membrane patches, the peak current and the rate of activation of whole-cell dSlo currents are dependent on both the membrane potential and the intracellular Ca2+ concentration (Adelman et al., 1992;DiChiara and Reinhart, 1995). When PKAc is coexpressed with dSlo, peak dSlo channel current is decreased dramatically (Fig.1B,C). As shown in Figure1D, the activation of whole-cell dSlo current in response to a depolarizing voltage step to +165 mV is also much slower in PKAc cotransfected cells. The time constant for activation (τ) of dSlo current is 22.5 ± 1.98 msec in the absence and 43.7 ± 3.89 msec in the presence of PKAc (Fig. 1D). In addition, the peak amplitude of dSlo current is significantly lower in cells cotransfected with PKAc. The dSlo current density at +165 mV is 1807 ± 191 pA/pF in control and 708 ± 99.2 pA/pF in PKAc cotransfected cells (Fig. 1C). A similar slowdown of activation kinetics and a reduction of current amplitude were observed at other voltages examined (Fig.1A,B).
Fig. 1.

Decrease of dSlo channel activity upon coexpression of PKAc. dSlo currents were evoked by 300 msec depolarizing voltage steps from −80 mV in 15 mV increments, in the whole-cell voltage-clamp configuration, with 10 μmintracellular free Ca2+. Currents were recorded from tsA201 cells transfected with either dSlo alone (A) or dSlo and PKAc (B).C, The peak dSlo current density at +165 mV (mean ± SEM), normalized to membrane capacitance (I/Cm), is significantly lower in cells cotransfected with PKAc (p < 0.001). D, The activation time constant (τ) (mean ± SEM), from single exponential fits of dSlo currents evoked at +165 mV, is significantly increased with cotransfection of PKAc (p < 0.001).
Modulation of dSlo activity requires catalytically active PKAc
Coexpression of PKAc results in a large increase in the phosphorylation of dSlo channels (Wang et al., 1999). To examine separately the effects of PKAc binding and phosphorylation, we constructed a mutant form of PKAc (K72E). Residue K72 of PKAc has long been recognized as being essential for its maximal enzyme activity, and such a mutation results in much reduced catalytic activities for PKAc and other kinases (Hanks et al., 1988; Zhong et al., 1997; Cauthron et al., 1998). To determine the effect of the K72E mutation on the ability of PKAc to phosphorylate and to interact with dSlo, wild-type or K72E PKAc was coexpressed with dSlo in tsA201 cells. dSlo was immunoprecipitated from cotransfected cells, and the immunoprecipitate was probed for bound PKAc with anti-PKAc antibody and for dSlo phosphorylation with a sequence- and phosphorylation-specific antibody, anti-pS942 (Wang et al., 1999). As shown in Figure2 (middle panel), K72E PKAc is much less effective than wild-type PKAc in phosphorylating S942. On the other hand, a similar amount of PKAc co-immunoprecipitates with dSlo from wild-type and K72E PKAc cotransfected cells (Fig. 2,top panel). These results demonstrate that K72E PKAc is catalytically inactive but can still bind to dSlo.
Fig. 2.

dSlo binds to a catalytically inactive PKAc mutant (K72E). tsA201 cells were transfected with dSlo alone (lane 1), dSlo and K72E PKAc (lane 2), or dSlo and wild-type PKAc (WT; lane 3). dSlo immunoprecipitates were probed on parallel Western blots for PKAc with anti-PKAc antibody (top panel) or for dSlo phosphorylation with anti-pS942 antibody (middle panel). The expression of PKAc is similar for wild-type and mutant PKAc under these transfection conditions (bottom panel).
We then recorded whole-cell dSlo currents in K72E PKAc cotransfected cells. As shown in Figure 3, Aand B, dSlo currents are not modulated by cotransfection of K72E PKAc. Note that there is no significant difference in either the peak amplitude (Fig. 3C) or activation kinetics (Fig.3D) of dSlo currents. At +150 mV, the time constant for activation of dSlo current is 24.7 ± 2.14 msec in the absence and 25.8 ± 2.6 msec in the presence of K72E PKAc (Fig.3D), and the dSlo current density is 1613 ± 139 pA/pF in control and 1662 ± 167 pA/pF in K72E PKAc cotransfected cells (Fig. 3C). This indicates that binding of catalytically inactive PKAc to dSlo is not sufficient to modulate dSlo channel activity.
Fig. 3.

K72E PKAc does not modulate dSlo channel activity. Whole-cell dSlo currents were elicited by 400 msec depolarizing voltage steps from −80 mV in 20 mV increments. Currents were recorded from tsA201 cells transfected with either dSlo alone (A) or dSlo and K72E PKAc (B). There is no significant difference in the peak current density (mean ± SEM) of dSlo currents recorded at +150 mV from cells transfected with dSlo alone or from cells transfected with dSlo and K72E PKAc (C) (p > 0.8). The activation time constant (mean ± SEM) of dSlo currents evoked at +150 mV is also not significantly changed with cotransfection of K72E PKAc (D) (p > 0.8).
Modulation of dSlo activity is not mediated by phosphorylation of S942 in dSlo
There is a consensus PKA substrate site (Kemp and Pearson, 1990) at serine residue 942 (S942) in the C-terminal domain of dSlo (Wang et al., 1999). To investigate whether modulation of dSlo activity on cotransfection of PKAc is mediated by the increase of phosphorylation of dSlo S942, we recorded whole-cell currents from cells transfected with a dSlo mutant, in which S942 is mutated to an alanine (S942A dSlo). Similar to what has been reported previously (Bowlby and Levitan, 1996; Silberberg et al., 1996), there is no difference in current amplitude or activation kinetics between wild-type and S942A dSlo transfected cells. The time constants for activation of dSlo currents at 165 mV are 22.5 ± 1.98 msec for wild-type dSlo (Fig.1D) and 23.4 ± 2.87 msec for S942A dSlo (Fig.4D), and the dSlo peak current amplitudes are 1807 ± 191 pA/pF in wild-type dSlo (Fig.1C) and 1778 ± 130 pA/pF in S942A dSlo (Fig.4C) transfected cells. Furthermore, S942A dSlo is modulated normally in cells cotransfected with PKAc (Fig.4A,B). The extent of reduction of current amplitude and the slowdown of activation by cotransfection of PKAc are not significantly different between wild-type and S942A dSlo. With cotransfection of PKAc, the activation time constants of dSlo currents at 165 mV are 43.7 ± 3.89 msec for wild-type dSlo (Fig.1D) and 39.3 ± 3.52 msec for S942A dSlo (Fig.4D), and the peak amplitudes are 708 ± 99.2 pA/pF in wild-type dSlo (Fig. 1C) and 793 ± 126 pA/pF in S942A dSlo (Fig. 4C) transfected cells. This shows that the modulation of dSlo is not mediated by phosphorylation of dSlo at S942.
Fig. 4.

Modulation of S942A dSlo by PKAc. Whole-cell dSlo currents were recorded from cells transfected with a dSlo mutant (S942A) alone (A) or S942A dSlo together with PKAc (B), using recording conditions identical to those described in Figure 1. Cotransfection of PKAc significantly decreases the peak S942A dSlo current density (mean ± SEM) (C) (p < 0.01) and increases the activation time constant (mean ± SEM) (D) (p < 0.01) at 165 mV.
To determine whether there are PKA phosphorylation sites other than S942 in the dSlo protein, we performed an in vitrophosphorylation assay. Because most of the intracellular portion of the dSlo sequence is the C-terminal domain, we constructed a GST fusion protein (GST-Cter) that contains the entire C-terminal domain of dSlo, amino acid residues 337–1164. We also constructed a smaller GST-fusion protein (GST-A), which contains amino acid residues 821–993 (Fig.5A). As shown in Figure 5,B and C, both GST fusion proteins are readily phosphorylated in vitro by PKAc. When S942 was replaced with alanine in the fusion proteins, no phosphorylation by PKAc was detected in either GST fusion protein in our in vitro kinase assay (Fig. 5B,C). These results show that S942 is the only residue that can be phosphorylated in vitro by PKAc in the entire C-terminal domain of dSlo.
Fig. 5.

S942 is the only target in the dSlo C-terminal domain of in vitro PKA phosphorylation.A, Schematic diagram of the dSlo channel protein shows the two regions in the dSlo C-terminal domain used to generate GST-fusion proteins. At the bottom is the amino acid sequence in the region from residues 922–956 that is critical for dSlo binding to PKAc. B, C, In vitro, PKAc phosphorylates both wild-type GST-A and GST-Cter (B,C, left lanes), but does not phosphorylate either S942A GST-A (B, right lane) or S942A GST-Cter (C, right lane). Equal amounts of wild-type and mutant GST-fusion proteins were used in these experiments.
dSlo binds to free PKAc, but not to the PKA holoenzyme
The PKA holoenzyme contains two catalytic subunits and a regulatory subunit dimer. Using a protein overlay assay, we have shown previously that PKAc binds directly to the dSlo fusion protein, GST-A, suggesting a direct interaction between dSlo and PKAc (Wang et al., 1999). However, interactions between other ion channels and the PKA holoenzyme are often mediated by PKA anchoring proteins (AKAPS), which bind to both the regulatory subunit and the channel protein (Tavalin et al., 1999; Dodge and Scott, 2000). To characterize more fully the protein complex containing dSlo and PKAc, we examined the interaction between dSlo and PKAc in the presence of the regulatory subunits, RIα or RIβ. We found that dSlo does not bind to either of the PKA regulatory subunits (data not shown). When RIα or RIβ is coexpressed with dSlo and PKAc, the interaction between dSlo and PKAc is nearly abolished, as assayed by co-immunoprecipitation using anti-dSlo and anti-PKAc antibodies (Fig.6). These data suggest that the regulatory subunits might compete with dSlo for binding to PKAc. To confirm this, we constructed a PKAc mutant (H87Q/W196R) that no longer associates with the regulatory subunits in cells (Orellana and McKnight, 1992; Gibson and Taylor, 1997). As shown in Figure 6, dSlo binds well to H87Q/W196R PKAc, and neither RIα nor RIβ inhibits this binding.
Fig. 6.

dSlo binds to free PKAc but not to the holoenzyme. dSlo immunoprecipitates were probed on Western blots for PKAc (top panel). Cells were transfected with dSlo alone (lane 1), dSlo and wild-type (WT) PKAc, together with either control vector (lane 2), PKA regulatory subunit RIα (lane 3), or RIβ (lane 4) or dSlo and H87Q/W196R (MT) PKAc, together with RIα (lane 5), RIβ (lane 6), or control vector (lane 7). Cotransfection of RIα or RIβ abolishes the interaction between dSlo and WT PKAc but has no effect on binding of dSlo to MT PKAc (top panel). Expression of both WT and MT PKAc is similar under all transfection conditions (middle panel). Lysates were probed with anti-RI antibody for expression of RIα or RIβ (bottom panel).
PKAc binds directly to the region where S942 is located
We examined further the region of dSlo that is important for PKAc binding. On the basis of our previous finding that PKAc binds to a GST-fusion protein (GST-A), which contains amino acid residues 821–993 of the dSlo C-terminal domain (Wang et al., 1999), we made a mutant dSlo channel in which amino acids 922–956 were deleted (dSloΔ1). As shown in Figure 7A, deletion of these 35 amino acids results in the loss of binding between dSlo and PKAc. Interestingly, this stretch of amino acids in dSlo contains the consensus PKA substrate site, S942 (Fig. 5A). Accordingly, we asked whether the dSlo–PKAc binding can be competed by the heat stable inhibitor peptide, PKI, which contains a pseudosubstrate site (Wen and Taylor, 1994; Poteet-Smith et al., 1997). After incubating dSlo and PKAc cotransfected cells with a membrane-permeable form of PKI, much less PKAc was detected in anti-dSlo immunoprecipitates, indicating that PKI inhibits binding between dSlo and PKAc (Fig.7B). In addition, we used site-directed mutagenesis to assess the role that residues in the dSlo substrate site might play in binding to PKAc. As shown in Figure 7A, mutations of dSlo at serine 942 (S942A), or at arginine 939 and arginine 940 (R939A/R940A), do not prevent dSlo from binding to PKAc. These results show that the interaction between PKAc and dSlo is not simply an enzyme-substrate association and that residues separate from the substrate site are critical in achieving the high-affinity binding to PKAc.
Fig. 7.
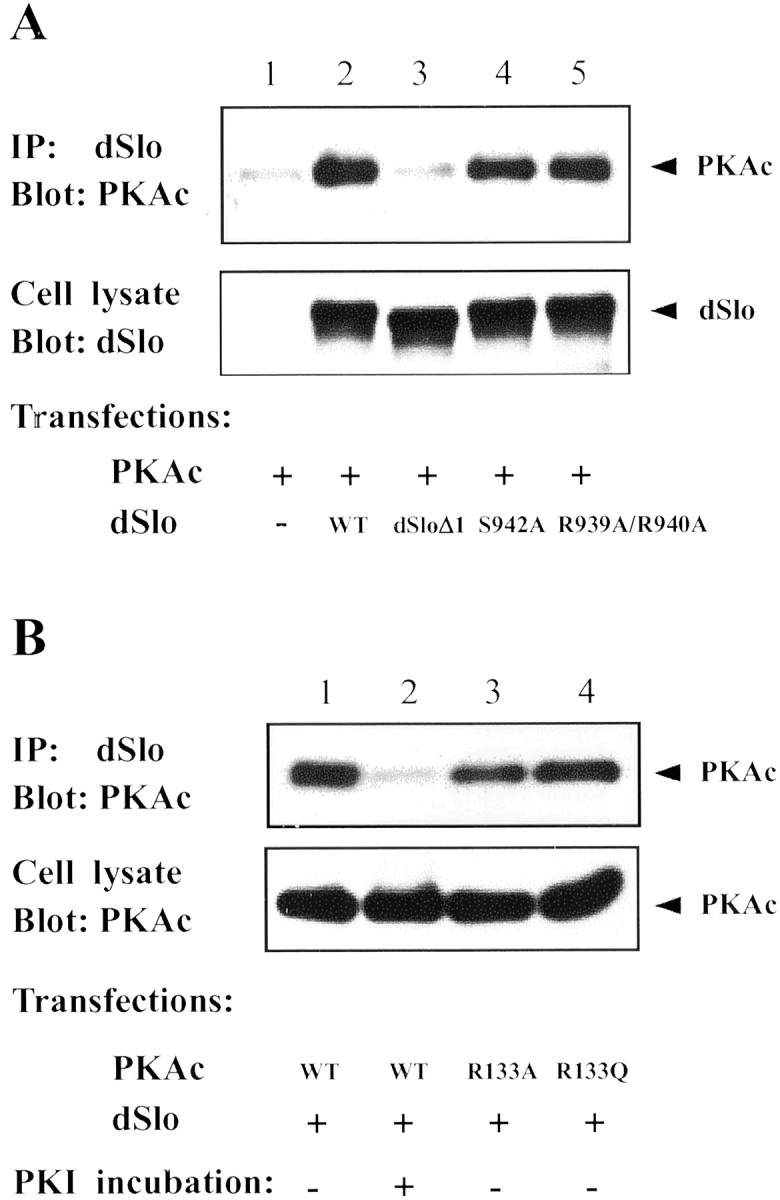
Mutational analysis of the dSlo-PKAc interaction. dSlo immunoprecipitates were probed on Western blot for PKAc.A, Cells were transfected with PKAc, together with control vector (lane 1), wild-type (WT) dSlo (lane 2), dSlo deletion mutant (dSloΔ1, lane 3), S942A dSlo (lane 4), or R939A/R940A dSlo (lane 5). Deletion of 35 amino acids in dSlo abolishes its binding to PKAc (lane 3, top panel), but the dSlo mutants bind well to PKAc (lanes 4, 5, top panel). Expression of WT and mutant dSlo channels is similar under all transfection conditions (bottom panel). B, dSlo was cotransfected with wild-type (WT) PKAc (lanes 1,2), R133A PKAc (lane 3), or R133Q PKAc (lane 4). Less PKAc was detected in dSlo immunoprecipitates after incubation of cells with the membrane-permeant PKI (lane 2, top panel). Neither R133 mutation in PKAc affects its binding to dSlo (lanes 3, 4, top panel). Expression of wild-type and mutant PKAc is similar under all transfection conditions (bottom panel).
To determine the regions or amino acid residues in PKAc that are involved in its binding to dSlo, we examined the binding between dSlo and PKAc mutants that have been shown to have decreased affinity for PKI or the regulatory subunits. Two such mutants, R133A and R133Q PKAc, have a decreased affinity for binding to both RI and PKI but maintain their affinity for RII. Another PKAc mutant (H87Q/W196R) exhibits altered interaction with both RI and RII (Orellana and McKnight, 1992;Poteet-Smith et al., 1997; Cheng et al., 2001). We found that dSlo binds as well to all of these PKAc mutants as to wild-type PKAc (Figs.6, 7B), suggesting that PKAc binds to dSlo via a region different from that necessary for its binding to PKI or the regulatory subunits.
PKAc maintains its enzymatic activity when it binds to dSlo
The catalytic activity of PKAc is inhibited after binding to the regulatory subunits or PKI. It has also been reported that PKAc is maintained in an inactive state through association with Iκ B-α or IκB-β in an NF-κB–IκB–PKAc complex (Zhong et al., 1997; Doucas et al., 2000). To determine the effect of dSlo binding on the catalytic activity of PKAc, we performed a standardin vitro protein phosphorylation assay using biotinylated Kemptide as substrate. As shown in Figure8A, PKAc activity is not significantly changed after addition of up to 500 ng of the GST fusion protein (GST-A) that binds to PKAc (Wang et al., 1999). We also tested whether GST-A affects the inhibition of PKAc activity by the regulatory subunit. In the absence of cAMP, 5 ng RII subunit results in a >50% reduction of PKAc activity, and addition of 8.5 μg GST-A fusion protein does not significantly change the RII-induced inhibition (Fig. 8B). This suggests a relatively lower binding affinity of PKAc for dSlo compared with that for the RII subunit.
Fig. 8.

dSlo-bound PKAc is catalytically active in vitro. Activity of PKAc was measured using the biotinylated Kemptide assay system. A, PKAc activity is not significantly different in reactions that contain PKAc alone (left), PKAc together with 5, 50, or 500 ng of either GST (right bars, n = 3) or the GST-dSlo fusion proteins (GST-A, middle bars, n = 3). B, PKAc activity is dramatically decreased in the presence of 5 ng of PKA regulatory subunit (RII, n = 3). Addition of 8.5 μg of either GST (RII + GST,n = 3) or GST-A fusion proteins (RII + GST-A, n = 3) does not significantly alter the inhibition of PKAc activity by RII.
K72E PKAc prevents phosphorylation of dSlo by endogenous PKA
Activation of the PKA holoenzyme is induced by binding of cAMP to the regulatory subunits, which leads to the dissociation of active PKAc. Hence a rise in intracellular cAMP leads to a significant increase of phosphorylation of cellular proteins, including ion channels and receptors (Swope et al., 1999; Catterall, 2000), by PKA. We examined the phosphorylation of dSlo at S942 and the binding of endogenous PKAc to dSlo induced by the PKA activators, forskolin and cBIMPS (Blackstone et al., 1994; Gjertsen et al., 1995; Cantrell et al., 1997). After incubating dSlo-transfected cells with 30 μm forskolin or 50 μm cBIMPS for 20 min, lysates were subjected to immunoprecipitation with anti-dSlo antibodies. Immunoprecipitates were analyzed by Western blotting using anti-pS942 antibody to assess the phosphorylation of dSlo S942 and anti-PKAc to probe the bound PKAc. As shown in Figure9A (bottom panel), incubation with either forskolin or cBIMPS results in a robust increase of phosphorylation of dSlo at S942, comparable to that seen with cotransfection of PKAc. However, no detectable PKAc is found in dSlo immunoprecipitates after either forskolin or cBIMPS treatment (Fig. 9A, top panel).
Fig. 9.
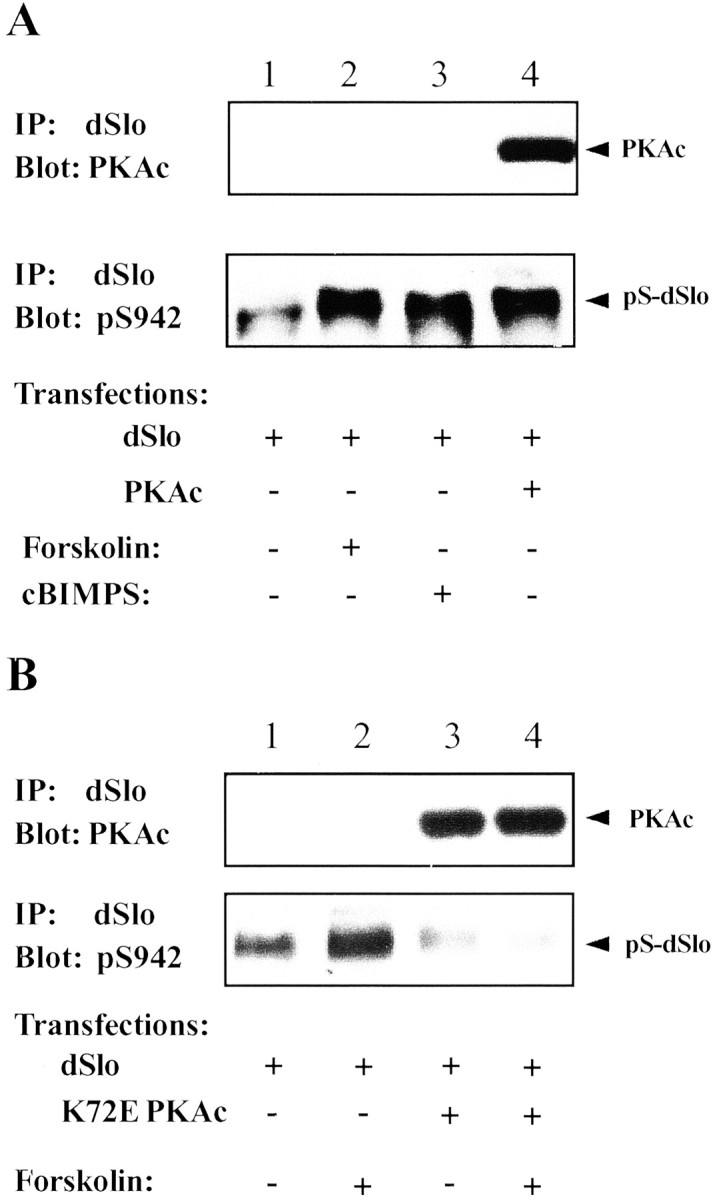
Phosphorylation of dSlo by PKA activators. dSlo immunoprecipitates were probed on parallel Western blots for PKAc with anti-PKAc antibody (top panels) or for dSlo phosphorylation at S942 with anti-pS942 antibody (bottom panels). A, When cells were transfected with dSlo alone (lanes 1–3), no PKAc was detected in dSlo immunoprecipitates in control (lane 1, top panel) or after incubation of cells for 20 min with either 30 μm forskolin (lane 2, top panel) or 50 μm cBIMPS (lane 3, top panel). However, PKAc was present in dSlo immunoprecipitates when dSlo was cotransfected with PKAc (lane 4, top panel). dSlo phosphorylation at S942 is increased by treatment with either forskolin (lane 2, bottom panel) or cBIMPS (lane 3, bottom panel) as well as by cotransfection of PKAc (lane 4, bottom panel). B, When K72E PKAc is cotransfected with dSlo, forskolin treatment does not increase dSlo phosphorylation (lanes 3, 4,bottom panel). However, K72E PKAc is present in dSlo immunoprecipitates in dSlo and K72E PKAc cotransfected cells (lanes 3, 4, top panel).
Because co-immunoprecipitation failed to detect binding between dSlo and endogenous PKAc, an alternative approach was used to test whether phosphorylation of dSlo by endogenous PKA activity requires a binding interaction between dSlo and endogenous PKAc. As shown in Figure9B (bottom panel), when cells are transfected with the catalytically inactive PKAc mutant, K72E PKAc, 20 min incubation of transfected cells with 30 μmforskolin no longer induces dSlo phosphorylation at S942. This result indicates that production of free endogenous PKAc is not sufficient to phosphorylate dSlo. It suggests that both endogenous and exogenous PKAc bind to dSlo at the same site, and an excess of the mutant PKAc can function as a dominant negative to prevent endogenous PKAc from binding to and phosphorylating dSlo.
dSlo is not modulated by cAMP-activated endogenous PKA activity
In addition, we investigated whether dSlo channel activity can be modulated by forskolin or cBIMPS, both of which stimulate phosphorylation of dSlo at the consensus substrate site but do not produce detectable binding between dSlo and endogenous PKAc. The experiments were done in two ways. The first approach was to apply forskolin directly into the bath chamber after establishing a stable whole-cell dSlo recording. In these experiments, 5 mmMg-ATP was included in the pipette solution. Figure10 shows the results of a typical experiment in which we measured the peak current amplitude and activation time constant of dSlo currents evoked by a depolarizing voltage step to +100 mV from a holding potential of −80 mV. We found no significant difference in current amplitude (Fig.10A) or activation kinetics (Fig.10B) between control cells and cells treated with forskolin at a concentration of 50 μm. As shown in Figure 10B, the activation of dSlo current becomes faster over time both in the absence and in the presence of forskolin. This may be contrasted with the dramatic slowing of activation of dSlo current in cells cotransfected with PKAc (Fig. 1D). Similar results were obtained with cBIMPS (data not shown). In addition, we measured whole-cell dSlo currents after preincubation with either forskolin or cBIMPS, under conditions similar to those used for the biochemical studies. Again, the peak current amplitude and activation kinetics of dSlo currents recorded from forskolin- or cBIMPS-treated cells are not different from those in control cells (data not shown). Thus, although dSlo modulation requires active PKAc (Fig. 3), these results suggest that phosphorylation alone may not be sufficient to produce the modulation.
Fig. 10.

dSlo is not modulated by activation of endogenous PKA. Whole-cell dSlo currents were elicited by 300 msec test pulses to +100 mV from a holding potential of −80 mV. Forskolin was applied directly to the bath solution at the arrows, after a stable recording of dSlo current was established. The normalized peak current and the activation time constant are plotted as a function of time, with (▪) or without (■) the addition of forskolin. There was no apparent difference in either the peak currents (A) or activation time constant (B) in the absence and presence of 50 μm forskolin.
DISCUSSION
The initial evidence on modulation of calcium-dependent potassium channels by closely associated protein kinases or phosphatases came from experiments in which KCa channels reconstituted from rat brain were activated in an artificial lipid bilayer by the addition of ATP (Chung et al., 1991; Reinhart and Levitan, 1995). We subsequently identified PKAc as one of several protein kinases closely associated with dSlo channels (Wang et al., 1999). To determine whether the bound PKAc modulates channel function, we recorded dSlo currents in the whole-cell voltage-clamp configuration. We show here that there is a dramatic downregulation of dSlo channel functional activity with cotransfection of PKAc. Although we reported previously that channel function is not affected by coexpression with both PKAc and Src (Wang et al., 1999), it is now known that Src enhances Slo channel activity (Ling et al., 2000), and this upregulation by Src may have masked the PKAc downregulation that we describe here.
The reduction of whole-cell dSlo in PKAc-cotransfected cells represents a change in channel functional activity. Although it is difficult to obtain an accurate conductance–voltage relation because of the large current amplitude in the whole-cell configuration, both the lower current amplitude and slower activation kinetics of dSlo currents in PKAc-cotransfected cells are consistent with a decreased sensitivity of the dSlo channel to voltage and calcium. The changes in current do not appear to result from alterations in channel expression or membrane targeting, because we found that the expression level of dSlo protein measured by quantitative Western blot is actually several-fold higher, and its surface localization measured by immunocytochemistry is unchanged, in PKAc-cotransfected cells (data not shown). In any event, it is difficult to explain the change in channel kinetics that we demonstrate here in terms of protein expression level.
Although questions still remain about the precise molecular mechanism underlying the profound modulation of dSlo channel activity by cotransfected PKAc, our studies suggest strongly that it requires the association between active PKAc and the dSlo channel. However, dSlo channel activity is not altered by cotransfection of a catalytically inactive PKAc (K72E), although this mutant PKAc is capable of binding to dSlo. This demonstrates that the modulation of dSlo activity is not simply the result of binding of PKAc per se, but also requires phosphorylation. This is also consistent with other reports of modulation of native and expressed Slo family channels by PKAc-dependent phosphorylation (White et al., 1991; Hall and Armstrong, 2000; Tian et al., 2001). On the other hand, our results with forskolin suggest that phosphorylation, although necessary, is not by itself sufficient to produce modulation. Accordingly, we favor the hypothesis that modulation requires phosphorylation, of either dSlo or some other protein, by active PKAc bound to the channel.
To determine the potential molecular target the phosphorylation of which might mediate the modulation of dSlo activity, we examined the role of serine 942 in the dSlo C-terminal domain. This residue has long been recognized as a consensus PKA substrate (Hanks and Hunter, 1995), and it is readily phosphorylated by PKA both in vitro andin vivo (Wang et al., 1999). Interestingly, we found that the mutation of serine 942 to alanine does not affect dSlo binding to or modulation by PKAc. This is consistent with a previous report showing that S942A dSlo is not different from the wild-type channel in its kinetic variability when expressed in Xenopus oocytes (Bowlby and Levitan, 1996; Silberberg et al., 1996). Although it is convenient to use the anti-pS942 antibody, as we do here, to measure channel phosphorylation, we do not mean to imply that S942 participates in dSlo modulation by PKAc.
In addition to S942, other serine and threonine residues are thought to be exposed to the intracellular milieu. To identify other PKA substrate sites on dSlo, we performed an in vitro phosphorylation assay using a recombinant fusion protein containing the entire dSlo C-terminal domain. This domain is considerably longer than many other voltage-dependent potassium channels and constitutes approximately two-thirds of the total dSlo channel protein (Atkinson et al., 1991). Somewhat surprisingly, we found that other than S942, no additional amino acid in the entire dSlo C-terminal domain is phosphorylated by PKAc in vitro. However, we have not yet examined PKA phosphorylation of several serine or threonine residues in the intracellular loops between transmembrane domains. On the other hand, it is equally plausible that PKAc phosphorylates other proteins that may themselves or through some signaling pathway modulate dSlo channel activity. Studies by us and others have shown that dSlo is indeed regulated by several closely associated proteins (Schopperle et al., 1998; Xia et al., 1998). At least one dSlo-interacting protein, 14-3-3, interacts with dSlo via the adaptor protein Slob in a phosphorylation-dependent manner, and formation of this protein complex results in inhibition of channel activity (Zhou et al., 1999). A recent report also showed that a Slo channel associating protein, cPLA2-α, is a target for phosphorylation. Phosphorylation of cPLA2-α results in activation of the channel, whereas phosphorylation of other regulatory elements causes channel inactivation (Denson et al., 2000). Finally, we show here that dSlo channel activity is not modulated by activators of endogenous PKA, including forskolin and cBIMPS, which do not enhance binding of PKAc to dSlo. Although these results do not lend themselves readily to unequivocal interpretation, they are consistent with the hypothesis that whatever the actual substrates of PKA are, targeting of sufficient PKAc via binding to dSlo is also a requirement for channel modulation.
It has been well documented that PKA forms regulatory complexes with some ion channels and ligand-gated receptors. PKA targeting is often achieved through AKAPs, proteins that bind to the PKA regulatory subunit as well as the substrate (Glantz et al., 1992; McCartney et al., 1995). However, our previous overlay experiment showed a direct binding between dSlo and PKAc, suggesting a novel channel–PKA protein complex. The present studies support this hypothesis and provide additional molecular details about this regulatory complex. Using co-immunoprecipitation approaches, we showed that dSlo binds only to free PKAc but not to the PKA holoenzyme, and that both PKA regulatory subunit and PKI inhibit the association between dSlo and PKAc. We also identified a 35 amino acid region in the dSlo C-terminal domain that is essential for PKAc binding. Interestingly, this region includes the consensus PKA substrate site, S942, although residues within the substrate site itself (RRXS) do not appear to be critical for binding to PKAc. This demonstrates that the dSlo–PKAc association is not simply an enzyme-substrate complex. It is also consistent with previous studies on interactions between PKAc and regulatory subunits or PKI that show that residues separate from the pseudosubstrate site of the regulatory subunits or PKI are important in mediating the high-affinity binding to PKAc (Gibson and Taylor, 1997; Cheng et al., 2001). Moreover, our in vitro phosphorylation results suggest that binding of dSlo to PKAc does not prevent the enzyme from phosphorylating its substrates. It remains to be determined how the activity of dSlo-bound PKAc is regulated in cells.
It is noteworthy that channels of the Slo family can physically associate with other protein kinases, including the Src tyrosine kinase and type I cGMP-dependent protein kinase (Wang et al., 1999; Ling et al., 2000; Swayze and Braun, 2001). This suggests that modulation of channel activity may involve multiple regulatory mechanisms. Further biochemical and electrophysiological studies to dissect these regulatory pathways will undoubtedly give us important insights into the relationship between various signal transduction pathways and neuronal physiology.
Footnotes
This work was supported by a grant from the National Institutes of Health to I.B.L. We are grateful to G. Stanley McKnight for providing us with PKAR constructs. We also thank Leslie Griffith and former and present members of the Levitan laboratory for helpful discussions and critical comments on this manuscript.
Correspondence should be addressed to Dr. Irwin B. Levitan, Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, PA 19104. E-mail: levitani@mail.med.upenn.edu.
J. Wang's present address: Caliper Technologies Corp., 605 Fairchild Drive, Mountain View, CA 94043.
REFERENCES
- 1.Adelman JP, Shen K-Z, Kavanaugh MP, Warren RA, Wu Y-N, Lagrutta A, Bond CT, North RA. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- 3.Bielefeldt K, Jackson MB. Phosphorylation and dephosphorylation modulate a Ca2+-activated K+ channel in rat peptidergic nerve terminals. J Physiol (Lond) 1994;475:241–254. doi: 10.1113/jphysiol.1994.sp020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackstone C, Murphy TH, Moss SJ, Baraban JM, Huganir RL. Cyclic AMP and synaptic activity-dependent phosphorylation of AMPA-preferring glutamate receptors. J Neurosci. 1994;14:7585–7593. doi: 10.1523/JNEUROSCI.14-12-07585.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowlby MR, Levitan IB. Kinetic variability and modulation of dSlo, a cloned calcium-dependent potassium channel. Neuropharmacology. 1996;35:867–875. doi: 10.1016/0028-3908(96)00090-1. [DOI] [PubMed] [Google Scholar]
- 6.Brenner R, Yu JY, Srinivasan K, Brewer L, Larimer JL, Wilbur JL, Atkinson NS. Complementation of physiological and behavioral defects by a slowpoke Ca2+-activated K+ channel transgene. J Neurochem. 2000;75:1310–1319. doi: 10.1046/j.1471-4159.2000.751310.x. [DOI] [PubMed] [Google Scholar]
- 7.Cantrell AR, Smith RD, Goldin AL, Scheuer T, Catterall WA. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel alpha subunit. J Neurosci. 1997;17:7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 9.Cauthron RD, Carter KB, Liauw S, Steinberg RA. Physiological phosphorylation of protein kinase A at Thr-197 is by a protein kinase A kinase. Mol Cell Biol. 1998;18:1416–1423. doi: 10.1128/mcb.18.3.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng X, Phelps C, Taylor SS. Differential binding of cAMP-dependent protein kinase regulatory subunit isoforms I and II to the catalytic subunit. J Biol Chem. 2001;276:4102–4108. doi: 10.1074/jbc.M006447200. [DOI] [PubMed] [Google Scholar]
- 11.Chung SK, Reinhart PH, Martin BL, Brautigan D, Levitan IB. Protein kinase activity closely associated with a reconstituted calcium-activated potassium channel. Science. 1991;253:560–562. doi: 10.1126/science.1857986. [DOI] [PubMed] [Google Scholar]
- 12.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 13.Denson DD, Wang X, Worrell RT, AIKalili O, Eaton DC. Cytosolic phospholipase A2 is required for optimal ATP activation of BK channels in GH(3) cells. J Biol Chem. 2000;276:7136–7142. doi: 10.1074/jbc.M009566200. [DOI] [PubMed] [Google Scholar]
- 14.DiChiara TJ, Reinhart PH. Distinct effects of Ca2+ and voltage on the activation and deactivation of cloned Ca2+-activated K+ channels. J Physiol (Lond) 1995;489:403–418. doi: 10.1113/jphysiol.1995.sp021061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dodge K, Scott JD. AKAP79 and the evolution of the AKAP model. FEBS Lett. 2000;476:58–61. doi: 10.1016/s0014-5793(00)01671-9. [DOI] [PubMed] [Google Scholar]
- 16.Doucas V, Shi Y, Miyamoto S, West A, Verma I, Evans RM. Cytoplasmic catalytic subunit of protein kinase A mediates cross-repression by NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA. 2000;97:11893–11898. doi: 10.1073/pnas.220413297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson RM, Taylor SS. Dissecting the cooperative reassociation of the regulatory and catalytic subunits of cAMP-dependent protein kinase. Role of Trp-196 in the catalytic subunit. J Biol Chem. 1997;272:31998–32005. doi: 10.1074/jbc.272.51.31998. [DOI] [PubMed] [Google Scholar]
- 18.Gjertsen BT, Mellgren G, Otten A, Maronde E, Genieser HG, Jastorff B, Vintermyr OK, McKnight GS, Doskeland SO. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J Biol Chem. 1995;270:20599–20607. doi: 10.1074/jbc.270.35.20599. [DOI] [PubMed] [Google Scholar]
- 19.Glantz SB, Amat JA, Rubin CS. cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol Biol Cell. 1992;3:1215–1228. doi: 10.1091/mbc.3.11.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall SK, Armstrong DL. Conditional and unconditional inhibition of calcium-activated potassium channels by reversible protein phosphorylation. J Biol Chem. 2000;275:3749–3754. doi: 10.1074/jbc.275.6.3749. [DOI] [PubMed] [Google Scholar]
- 21.Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 22.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 23.Kemp BE, Pearson RB. Protein kinase recognition sequence motifs. Trends Biol Sci. 1990;15:342–346. doi: 10.1016/0968-0004(90)90073-k. [DOI] [PubMed] [Google Scholar]
- 24.Levitan IB. Modulation of ion channels by protein phosphorylation. How the brain works. Adv Second Messenger Phosphoprotein Res. 1999;33:3–22. doi: 10.1016/s1040-7952(99)80003-2. [DOI] [PubMed] [Google Scholar]
- 25.Ling S, Woronuk G, Sy L, Lev S, Braun AP. Enhanced activity of a large conductance, calcium-sensitive K+ channel in the presence of Src tyrosine kinase. J Biol Chem. 2000;275:30683–30689. doi: 10.1074/jbc.M004292200. [DOI] [PubMed] [Google Scholar]
- 26.McCartney S, Little BM, Scott JD. Analysis of a novel A-kinase anchoring protein 100, (AKAP 100). Biochem Soc Trans. 1995;23:268S. doi: 10.1042/bst023268s. [DOI] [PubMed] [Google Scholar]
- 27.Orellana GA, McKnight GS. Mutations in the catalytic subunit of cAMP-dependent protein kinase result in unregulated biological activity. Proc Natl Acad Sci USA. 1992;89:4726–4730. doi: 10.1073/pnas.89.10.4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poteet-Smith CE, Shabb JB, Francis SH, Corbin JD. Identification of critical determinants for autoinhibition in the pseudosubstrate region of type I cAMP-dependent protein kinase. J Biol Chem. 1997;272:379–388. doi: 10.1074/jbc.272.1.379. [DOI] [PubMed] [Google Scholar]
- 29.Reinhart PH, Levitan IB. Kinase and phosphatase activities intimately associated with a reconstituted calcium-dependent potassium channel. J Neurosci. 1995;15:4572–4579. doi: 10.1523/JNEUROSCI.15-06-04572.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schopf S, Bringmann A, Reichenbach A. Protein kinases A and C are opponents in modulating glial Ca2+-activated K+ channels. NeuroReport. 1999;10:1323–1327. doi: 10.1097/00001756-199904260-00031. [DOI] [PubMed] [Google Scholar]
- 31.Schopperle WM, Holmqvist MH, Zhou Y, Wang J, Wang Z, Griffith LC, Keselman I, Kusinitz F, Dagan D, Levitan IB. Slob, a novel protein that interacts with the slowpoke calcium-dependent potassium channel. Neuron. 1998;20:565–573. doi: 10.1016/s0896-6273(00)80995-2. [DOI] [PubMed] [Google Scholar]
- 32.Shao LR, Halvorsrud R, Borg-Graham L, Storm JF. The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol (Lond) 1999;521:135–146. doi: 10.1111/j.1469-7793.1999.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silberberg SD, Lagrutta A, Adelman JP, Magleby KL. Wanderlust kinetics and variable Ca2+-sensitivity of Drosophila, a large conductance Ca2+-activated K+ channel, expressed in oocytes. Biophys J. 1996;70:2640–2651. doi: 10.1016/S0006-3495(96)79833-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swayze RD, Braun AP. A catalytically inactive mutant of type I cGMP-dependent protein kinase prevents enhancement of large conductance, calcium-sensitive K+ channels by sodium nitroprusside and cyclic GMP. J Biol Chem. 2001;276:19729–19737. doi: 10.1074/jbc.M005711200. [DOI] [PubMed] [Google Scholar]
- 35.Swope SL, Moss SI, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res. 1999;33:49–78. doi: 10.1016/s1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- 36.Tavalin SJ, Westphal M, Colledge LK, Scott JD. The molecular architecture of neuronal kinase/phosphatase signalling complexes. Biochem Soc Trans. 1999;27:539–542. doi: 10.1042/bst0270539. [DOI] [PubMed] [Google Scholar]
- 37.Tian L, Knaus H-G, Shipston MJ. Glucocorticoid regulation of calcium-activated potassium channels mediated by serine/threonine protein phosphatase. J Biol Chem. 1998;273:13531–13536. doi: 10.1074/jbc.273.22.13531. [DOI] [PubMed] [Google Scholar]
- 38.Tian L, Duncan RR, Hammond MS, Coghill LS, Wen H, Rusinova R, Clark AG, Levitan IB, Shipston MJ. Alternative splicing switches potassium channel sensitivity to protein phosphorylation. J Biol Chem. 2001;276:7717–7720. doi: 10.1074/jbc.C000741200. [DOI] [PubMed] [Google Scholar]
- 39.Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 40. Wang J, Zhou Y, Wen H, Levitan IB. Simultaneous binding of two protein kinases to a calcium-dependent potassium channel. J Neurosci 19 1999. RC4(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wen W, Taylor SS. High affinity binding of the heat-stable protein kinase inhibitor to the catalytic subunit of cAMP-dependent protein kinase is selectively abolished by mutation of Arg133. J Biol Chem. 1994;269:8423–8430. [PubMed] [Google Scholar]
- 42.White RE, Schonbrunn A, Armstrong DL. Somatostatin stimulates Ca2+-activated K+ channels through protein dephosphorylation. Nature. 1991;351:570–573. doi: 10.1038/351570a0. [DOI] [PubMed] [Google Scholar]
- 43.Xia X-M, Hirschberg B, Smolik S, Forte M, Adelman JP. dSlo interacting protein 1, a novel protein that interacts with large-conductance calcium-activated potassium channels. J Neurosci. 1998;18:2360–2369. doi: 10.1523/JNEUROSCI.18-07-02360.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kB is regulated by the IkB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 45.Zhou Y, Schopperle WM, Murrey H, Jaramillo A, Dagan D, Griffith LC, Levitan IB. A dynamically regulated 14–3-3, Slob, and Slowpoke potassium channel complex in Drosophila presynaptic nerve terminals. Neuron. 1999;22:809–818. doi: 10.1016/s0896-6273(00)80739-4. [DOI] [PubMed] [Google Scholar]