

Abstract
Ischemic proliferative retinopathy (e.g., diabetes mellitus, retinopathy of prematurity, or retinal vein occlusion) is a major cause of blindness worldwide. Apart from neovascularization, ischemic proliferative retinopathy leads to retinal degeneration. Apoptosis has been ascribed to be the leading mechanism in ischemic retinal degeneration. We showed recently that inducible nitric oxide synthase (iNOS) is expressed in the avascular retina in proliferative retinopathy in vivo and that iNOS expression in retinal glial cells is responsible for retinal neuronal cell death in vitro. Here we show that retinal apoptosis and subsequent degeneration occur in the murine model of ischemic proliferative retinopathy. Furthermore, because NO can have beneficial or detrimental effects in the retina, we analyzed the role of iNOS on retinal apoptosis in ischemic proliferative retinopathy. Using iNOS knock-out mice and iNOS inhibitor 1400W, we demonstrate in vivothat iNOS expression induces apoptosis locally in the inner nuclear layer of the avascular retina and that protein nitration may be involved in this process.
These findings are the first evidence for retinal apoptosis in an animal model of ischemic proliferative retinopathy, demonstrating that iNOS plays a crucial role not only in retinal neovascular disease but also in retinal degeneration. We show that it is an ideal target to protect the hypoxic retina from degeneration and to improve its vascularization.
Keywords: inducible nitric oxide synthase, apoptosis, retina, knock-out mice, ROP, ischemia, peroxynitrite, neovascularization
Ischemic proliferative retinopathy, as in diabetes mellitus, retinopathy of prematurity, or retinal vein occlusion, is a major cause of blindness worldwide. The vascular changes and molecular mechanisms of vitreal neovascularization have been the focus of intense research for many years (D'Amore, 1994;Adamis et al., 1999). Independently of vitreal neovascularization, pathological changes occur in retinal neurons in the ischemic retina (Barber et al., 1998). Atrophy in the inner nuclear layer (INL) and the ganglion cell layer (GCL) in diabetic retinopathy (Wolter, 1961; Bek, 1994) and structural changes in a model of retinopathy of prematurity (Lachapelle et al., 1999; Dembinska et al., 2001) have been described. Theses changes clinically manifest as progressive loss in visual function in diabetic retinopathy independent of the occurrence of neovascularization (Collier et al., 1985; Palmowski et al., 1997), and functional differences in retinal activity can be found years after the resolution of neovascular complications in retinopathy of prematurity (Fulton and Hansen, 1996; Reisner et al., 1997; Fulton et al., 2001). Very little is known about the mechanisms inducing neuronal apoptosis in theses diseases, and none of the treatments proposed for ischemic proliferative retinopathy seem to alter the course of the degeneration in the retina.
Nitric oxide (NO) is an important signaling molecule that mediates a variety of essential physiological processes, including neurotransmission, vasodilatation, and host cell defense (Christopherson and Bredt, 1997; MacMicking et al., 1997; Nathan, 1997). NO is synthesized from l-arginine by NO synthase (NOS). The constitutive NOS isoforms, originally described in endothelial cells and in neurons, produce small amounts of NO in response to appropriate signals (Christopherson and Bredt, 1997). The cytokine-inducible NO synthase (iNOS), whose expression requires protein synthesis, has been demonstrated and cloned in a wide variety of mammalian cells (Nathan, 1997). NO is known to influence apoptosis in a variety of models, and its effect can be pro-apoptotic or anti-apoptotic (Brune et al., 1998).
Because we demonstrated previously that iNOS is expressed in the INL of the ischemic retina in a murine model of ischemic proliferative retinopathy in vivo (Sennlaub et al., 2001) and that the iNOS-related NO release from stimulated retinal Müller glial cells (RMG) induces neuronal cell death in vitro (Goureau et al., 1999), we here investigate the influence of iNOS on retinal apoptosis in vivo.
MATERIALS AND METHODS
Animals. C57BL/6×129SvEv mice with a targeted disruption of the iNOS gene (knock-out iNOS), generated as described previously (MacMicking et al., 1995), were generously provided by Drs J. D. MacMicking, C. Nathan (Cornell University Medical College, Ithaca, NY) and J. S. Mudgett (Merck Research Laboratories, Rahway, NJ). iNOS-deficient mice were mated with C57BL/6×129SvEv wild-type (+/+) mice to produce heterozygous (+/−) iNOS-deficient mice. Heterozygote (+/−) mice were then mated with (+/+) mice for eight subsequent generations. Heterozygote (+/−) mice of the ninth generation were mated with another to provide iNOS-deficient, mice iNOS(−/−), and wild-type littermates (+/+) with the same genetic background. The animals used in the experiment were between generations 10 and 15, bred continuously from these iNOS knock-out and wild-type littermates. Genotyping to verify the absence or presence of the iNOS gene, or of the targeting vector, was accomplished by PCR of DNA from tail biopsies. The animals were given food and water ad libitum and maintained under pathogen-free conditions of a 12 hr light/dark cycle.
Murine model of oxygen-induced retinopathy of prematurity.All procedures were conducted in accordance with the Use of Animals in Ophthalmic and Vision Research statement of the Association for Research in Vision and Ophthalmology. Mice at postnatal day 7 (P7) were exposed, with their mothers, for 5 d to hyperoxic conditions (75%), inducing vaso-obliteration and subsequent capillary loss of the central retinal vasculature (Smith et al., 1994). At P12, the mice were returned to room-air conditions, and extensive vitreal neovascularization occurred in 100% of iNOS(+/+) mice.
Intravitreal injections of iNOS inhibitor 1400W in oxygen-induced retinopathy. The right eye of oxygen-incubated C57BL/6 iNOS(+/+) mice were intravitreally injected at P13 and P15, with 2 μl of 15 mg/ml 1400W (Calbiochem, France Biochem, Meudon, France), a highly specific inhibitor of iNOS (Garvey et al., 1997) in vehicle (n = 8) or vehicle only [50% polyethylenglycol (Sigma-Aldrich, St. Quentin Fallavier, France), 40% PBS, and 10% ethanol] (n = 8). At P17, eyes were enucleated. Four treated eyes were subjected to retinal whole-mount histochemistry, and four treated eyes were subjected to intravitreal neovascularization quantification and retinal layer thickness measurements (see below). The left eyes served as untreated controls.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. P14 iNOS(+/+)and iNOS(−/−) mice eyes were enucleated, immediately frozen in OCT (Tissue Tek, Puteaux, France), and sectioned (10 μm). iNOS(−/−) and iNOS(+/+) sections crossing the optic nerve were collected on the same glass slides, thus undergoing the same procedures. Before terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), they were fixed in 70% ethanol and 30% acidic acid for 5 min at −20°C. Then sections were washed three times in double-distilled H2O and then incubated for 60 min at 37°C with 0.25 U/μl terminal deoxynucleotidyl transferase (Boehringer Mannheim, Meylan, France) in the supplied reaction buffer containing 1 mmCoCl2 and 5 nmol/ml biotin 16-dUTP. After six washes in PBS, sections were incubated in a 1:100 Extravidin-tetramethylrhodamine isothiocyanate (TRITC) (Sigma-Aldrich) and 1:1000 Toto-3 iodide (Molecular Probes, Leiden, The Netherlands) PBS solution for 60 min, washed three times, mounted, viewed, and photographed with a Zeiss (Oberkochen, Germany) LSM 510 LASER scanning microscope. Experiments using the Apoptag TUNEL detection kit (Intergen, Purchase, NY) gave similar results. The experiment was performed on a minimum of three independent samples of each group.
Retinal layer thickness measurements. The eyes were enucleated at P17, fixed in Bouin's fixative for 24 hr, and embedded in paraffin. Sections (7 μm) were cut sagittally parallel to the optic nerve and stained with periodic acid Schiff (PAS) and hemalun. Retinal layer thickness was measured on four sections containing the optic nerve, 14 μm apart from one another using digitalized images and Visilog image analysis system (Noesis, Les Ullis, France) using a fluorescence microscope (Aristoplan; Leitz, Jena, Germany) and a Spot RT digital camera (Spot Diagnostic Instruments, Burroughs, MI). Ganglion cell density was quantified on sections by counting ganglion cell nuclei on a defined length of retina and are expressed in ganglion cells per 100 μm. The data were averaged for each eye, and the mean values from the individual eyes were statistically analyzed. Investigators performed measurements unaware of the provenance of the samples.
Immunohistochemical analysis. P14 iNOS(+/+) and iNOS(−/−) mice eyes were enucleated, immediately frozen in OCT (Tissue Tek), and sectioned (10 μm). iNOS(−/−) and iNOS(+/+) sections crossing the optic nerve were collected on the same glass slides, thus undergoing the same immunohistochemical procedures. Before immunohistochemistry, they were fixed in 4% paraformaldehyde (PAF) for 5 min at 4°C. The slides were incubated overnight at 4°C with polyclonal anti-nitrotyrosine antibody (Upstate Biotechnology, Euromedex, Strasbourg, France) and TRITC-conjugated lectin Griffonia simplicifolia(Sigma-Aldrich) diluted at 1:50 and 1:100, respectively, in 0.1% PBS Triton X-100 (w/v). Sections for glial fibrillary acidic protein (GFAP) histochemistry were incubated overnight at 4°C with polyclonal anti-GFAP antibody (Dako, Trappes, France) diluted at 1:100 in 0.1% PBS Triton X-100 (w/v). After washing, all sections were incubated in a solution of 1:100 of secondary FITC-conjugated goat anti-rabbit antibody (Biosys, Compiegne, France) for 60 min. The slides were then washed, mounted, viewed with a fluorescence microscope (Leitz Aristoplan), and photographed with a Spot RT digital camera (Spot Diagnostic Instruments). Control experiments omitting the first antibody gave no staining (data not shown). The experiment was performed on four independent samples.
Retinal whole-mount histochemistry. Eyes were enucleated and fixed in 4% PAF for 15 min at room temperature. Retinas were dissected and post-fixed in methanol for 10 min at −20°C. The retinas were incubated overnight with TRITC-conjugated lectin Griffonia simplicifolia (Sigma-Aldrich) diluted at 1:100 in 1% PBS Triton X-100. After washing, the retinas were mounted, viewed, and photographed with a fluorescence microscope (see above), and the total surface and the surface of the capillary-free area were measured using a computerized image-analysis system (NIH Image; Scion, Frederick, MD). Control experiments omitting the lectin gave no staining (data not shown).
Quantification of vitreal neovascularization. Briefly, the eyes were enucleated at different time points, fixed in Bouin's fixative for 24 hr, and embedded in paraffin. Serial sections (7 μm) were cut sagittally parallel to the optic nerve and stained with PAS and hemalun. Vascular cell nuclei found on the vitreal side of the inner limiting membrane were then counted on four sections 20 μm apart from one another on each side of the optic nerve (Smith et al., 1994). Investigators performed counting unaware of the provenance of the samples. No vascular cell nucleus anterior to the internal limiting membrane was found in room-air-raised animals.
Statistical analysis. Results were expressed as mean ± SEM. Statistical analyses were performed using the Mann–Whitney test.
RESULTS
Apoptosis in P14 retina (ischemic phase of the experiment) detected by TUNEL
Because we hypothesized that NO production induces neuronal apoptotic cell death in vivo, we analyzed retinal apoptosis in proliferative ischemic retinopathy at P14, when iNOS induction had been detected. TUNEL-positive cells were found in the central INL of post-oxygen-incubated iNOS(+/+) sections (Fig. 1A), whereas room-air-raised mice did not show any TUNEL-positive cells in this area (Fig. 1C). The TUNEL-positive cells in the INL of post-oxygen-incubated iNOS(+/+) eyes additionally exhibited signs of chromatin condensation and pycnic nuclei, indicating the apoptotic nature of the DNA strand breaks detected by TUNEL technique (Fig. 1A,inset). Furthermore, in post-oxygen-exposed iNOS(−/−) mice, only very few TUNEL-positive cells were observed in the avascular retina (Fig.1B), suggesting that NO release was responsible for the observed apoptosis in iNOS(+/+) mice. Apoptotic cells in the GCL or outer nuclear layer (ONL) could not be detected in any samples at P14. The signal on sections of post-oxygen-incubated iNOS(+/+) mice was restricted to the INL of the central retina (Fig.1D), corresponding to the avascular area as seen by endothelial cell staining on adjacent sections (Fig.1E), in which iNOS expression had been detected previously (Sennlaub et al., 2001). RMG cell activation was detected by GFAP staining in the same pattern (Fig. 1F).
Fig. 1.
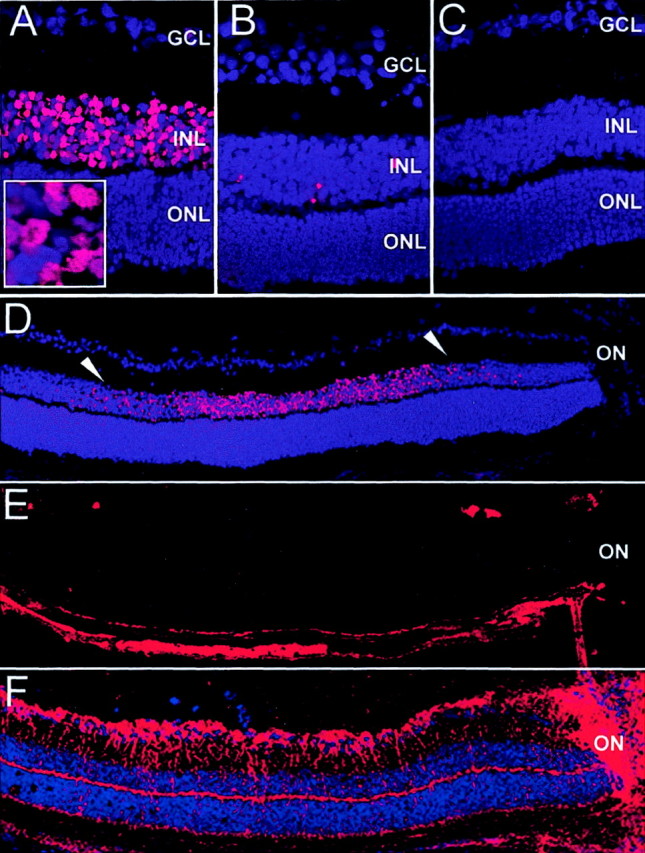
Apoptosis in the central P14 retina detected by TUNEL staining. A–D, TUNEL staining; E, lectin Griffonia simplicifolia; F, GFAP, immunohistochemistry. A, D–F, Eyes of iNOS(+/+) mice of the ischemic phase of the experiment. B, Eyes of iNOS(−/−)mice of the ischemic phase of the experiment. C, Room-air-raised iNOS(+/+) control mice. TUNEL-positive cells were found in the central inner nuclear layer of ischemic iNOS(+/+) sections (A). Room-air-raised iNOS(+/+)mice (C) do not show any TUNEL-positive cells, and ischemic iNOS(−/−) eyes show only very few positive cells (B). The TUNEL-positive cells in the INL of post-oxygen-incubated iNOS(+/+) eyes also exhibited signs of chromatin condensation and pyknotic nuclei (inset in A). The signal on sections of ischemic iNOS(+/+) mice is restricted to the INL of the central retina (D, betweenarrowheads). This area corresponds to the capillary-free retina, as demonstrated by endothelial cell staining (E) in which RMG cell activation can be visualized by GFAP immunohistochemistry (F) (adjacent sections). Experiment represents one of three independent experiments that gave similar results. ON, Optic nerve. Panel height: A–C, 240 μm; inset inA, 20 μm; D–F, 370 μm.
Retinal layer thickness in P17 retina
A decrease of the INL thickness in the ischemic retina can be expected after disappearance of the apoptotic cells. We therefore analyzed the INL thickness in paraffin-embedded sections at P17, 3 d after the apoptosis had occurred.
The central INL and inner plexiform layer (IPL) of post-oxygen-incubated iNOS(+/+) sections (Fig. 2A) were found to be severely reduced in thickness compared with room-air-raised mice (Fig. 2C), sometimes to as little as two to three rows of nuclei in the INL. The nerve fiber layer (NFL), GCL, outer plexiform layer (OPL), and ONL seemed to be relatively unchanged in post-oxygen-incubated iNOS(+/+) animals compared with room-air-raised mice. The changes in the IPL and INL of post-oxygen-exposed iNOS(−/−) mice (Fig.2B), although thinned compared with room-air-raised mice, were much less pronounced compared with post-oxygen-incubated iNOS(+/+) mice, whereas the morphology of the remaining retinal layers seemed comparable with sections from room-air-raised animals. The reduction in post-oxygen-exposed iNOS(+/+) retinal layer thickness was mainly found in the central area, 450 to 750 μm from the optic nerve (Fig. 2D, between arrowheads), in the same distribution than iNOS expression (Sennlaub et al., 2001) and the TUNEL-positive cells (Fig. 1). Quantification of the retinal layers at P17 revealed no differences in retinal layer thickness between room-air-raised iNOS(+/+) and iNOS(−/−) animals (data not shown). In the model of ischemic proliferative retinopathy, we observed a generalized thinning of the IPL in post-oxygen-exposed mice, iNOS(+/+) and iNOS(−/−), compared with room-air-raised mice (Fig. 3A). This difference, found throughout the entire retina, might be ascribed to disseminated apoptosis detected in the INL of oxygen-incubated iNOS(+/+) and iNOS(−/−) mice during the hyperoxic phase of the model at P12 (data not shown).
Fig. 2.
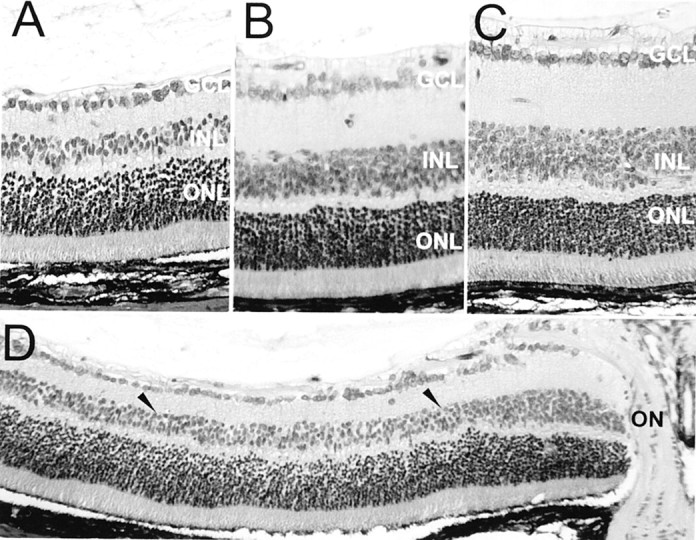
Histology of P17 retina. Periodic acid Schiff and hemalun staining. A, D, Eyes of iNOS(+/+) mice of the ischemic phase of the experiment. B, Eyes of iNOS(−/−)mice of the ischemic phase of the experiment. C, Room-air-raised control mice. The central INL and IPL of eyes of iNOS(+/+) mice of the ischemic phase of the experiment (A) were found to be severely reduced in thickness compared with room-air-raised mice (C). Changes in the IPL and INL of eyes of iNOS(−/−) mice of the ischemic phase of the experiment (B), although thinned compared with room-air-raised mice, were much less pronounced compared with ischemic iNOS(+/+) mice. The reduction in post-oxygen-exposed iNOS(+/+) retinal layer thickness was mainly found in the central area, 450–750 μm from the optic nerve (D, between arrowheads).ON, Optic nerve. Panel height: A–C, 240 μm; D, 370 μm.
Fig. 3.
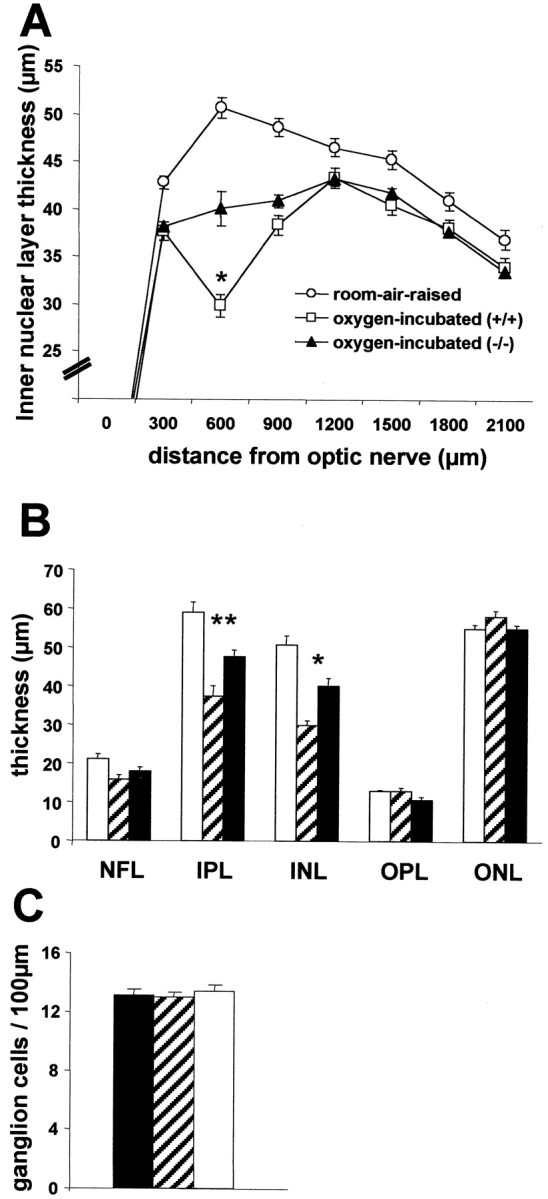
Retinal layer thickness and ganglion cell density at P17 (n = 6 per group). A, INL thickness of the retina. B, Differential retinal layer thickness analysis of the central retina (450–750 μm).C, Ganglion cell density in the central retina (450–750 μm). Room-air-raised control mice, White circles(A) and white columns(B, C); eyes of iNOS(+/+) mice of the ischemic phase of the experiment, white squares (A) andhatched columns (B, C); eyes of iNOS(−/−) mice of the ischemic phase of the experiment, black triangles(A) and black columns(B, C). A, Quantification of the INL at P17 revealed a generalized thinning of the INL in eyes of iNOS(+/+) and iNOS(−/−) mice of the ischemic phase of the experiment compared with room-air-raised control mice. More striking and highly significant differences in the thickness of the INL were observed in the central area (600 μm distance from nerve head), which corresponds to the ischemic area at P14 in post-oxygen-incubated animals. For A andB, * indicates significant differences by the Mann–Whitney test: p = 0.0001, post-oxygen-incubated iNOS(+/+) mice compared with room-air-raised animals; p = 0.0079, post-oxygen-incubated iNOS(−/−) mice compared with room-air-raised animals; p = 0.0016, post-oxygen-incubated iNOS(+/+) mice compared with post-oxygen-incubated iNOS(−/−) mice. B, In the central area (450–750 μm), additionally to the INL thinning, a significant thinning of the IPL can be observed. ** indicates significant differences by the Mann–Whitney test: p = 0.0020, post-oxygen-incubated iNOS(+/+) mice compared with room-air-raised animals; p = 0.0089, post-oxygen-incubated iNOS(−/−) mice compared with room-air-raised animals; p = 0.0074, post-oxygen-incubated iNOS(+/+) mice compared with post-oxygen-incubated iNOS(−/−) mice.C, Analysis of ganglion cell density showed no alteration in any of the groups.
More striking and highly significant differences in the thickness of the INL were observed in the central area, which corresponds to the ischemic area at P14 in post-oxygen-incubated animals. In this area, the INL of post-oxygen-incubated iNOS(+/+)mice is thinned to 29.81 ± 1.34 μm compared with 50.7 ± 2.49 μm in room-air-raised animals (p = 0.0001). The INL of post-oxygen-incubated iNOS(−/−) mice (40.07 ± 2.14 μm) is thinned compared with room-air-raised animals (p = 0.0079) but less so than in post-oxygen-incubated iNOS(+/+) mice. The difference in INL thickness of the central area between post-oxygen-incubated iNOS(+/+) and iNOS(−/−) mice observed are highly significant (p = 0.0016).
A more detailed analysis of the central retina (450–750 μm) revealed similar differences in the IPL layer (Fig. 3B), but no significant differences were observed in the thickness of any of the other retinal layers (Fig. 3B). Furthermore, analysis of ganglion cell density showed no alteration in any of the groups (Fig.3C).
Intravitreal injections of 1400W in proliferative retinopathy
We showed previously that the effects of iNOS expression in ischemic proliferative retinopathy could be partially prevented by systemic (subcutaneous) administration of 1400W, a highly specific iNOS inhibitor (Sennlaub et al., 2001). We here tested the effect of this highly specific inhibitor on retinal cell death in ischemic proliferative retinopathy. 1400W was administered by two intravitreal injections during the hypoxic phase (P13 and P15) to achieve a more complete iNOS inhibition in the ischemic retina compared with subcutaneous administration. At P17, the intravitreal neovascularization, the avascular area, and the thickness of the central INL (450–750 μm distance from the optic nerve) were quantified.
Intravitreal 1400W injections significantly inhibited pathological intravitreal neovascularization compared with vehicle (Fig.4A) (p = 0.028). Note that there was also an important nonspecific inhibitory effect of vehicle injection (p = 0.0003), which has been reported by others (Penn et al., 2001). Furthermore, revascularization of the ischemic retina was significantly enhanced in the 1400W-treated group compared with vehicle (Fig. 4B) (p = 0.023). Interestingly the vehicle effect on intraretinal revascularization was not significant (p = 0.079).
Fig. 4.

Intravitreal injections of 1400W (a specific iNOS inhibitor) in proliferative retinopathy (n = 4 per group). Mice of the same litter were injected at P13 and P15 with 2 μl of vehicle or 15 mg/ml 1400W. A, Intravitreal neovascularization at P17 (intravitreal endothelial nuclei per section). B, Avascular area at P17 (avascular area expressed as percentage of total retinal surface). C, INL thickness in the central retina (450–750 μm). Untreated mice,White columns; vehicle-injected mice, hatched columns; 1400W-injected mice, black columns.A, Intravitreal 1400W injections significantly inhibited pathological intravitreal neovascularization compared with vehicle. *p = 0.028; Mann–Whitney test. Note that there was also an important nonspecific inhibitory effect of vehicle injection (p = 0.0003). B, Revascularization of the ischemic retina was significantly enhanced in the 1400W-treated group compared with vehicle. **p= 0.023; Mann–Whitney test. The vehicle effect on intraretinal revascularization was not significant (p = 0.079). C, Analysis of the INL revealed a significant protection from thinning by 1400W compared with vehicle, confirming the role of iNOS in retinal apoptosis in ischemic proliferative retinopathy. ***p = 0.0034; Mann–Whitney test.
Analysis of the INL revealed a significant protection from thinning by 1400W compared with vehicle (Fig. 4C) (p = 0.0034), confirming the role of iNOS in retinal apoptosis in ischemic proliferative retinopathy. The differences in INL thickness between vehicle-treated eyes and untreated ones were not significant (p = 0.078).
Nitrotyrosine immunohistochemistry at P14
The nitration of free tyrosine or protein tyrosine residues generates 3-nitrotyrosine, the detection of which has been used as a footprint for the in vivo formation of peroxynitrite and other reactive nitrogen species (Greenacre and Ischiropoulos, 2001). We examined sections of room-air-raised and post-oxygen-incubated iNOS(+/+) and iNOS(−/−) mice at P14 immunocytochemically for the presence of nitrotyrosine, using an anti-nitrotyrosine-specific antibody.
Post-oxygen-incubated iNOS(+/+) sections revealed nitrotyrosine-positive cells at the edge of the central ischemic retina in proximity to blood vessels, in a “RMG cell”-like distribution (Fig. 5A). No positive cells were observed in the central retina in post-oxygen-exposed iNOS(−/−) (Fig.5B), demonstrating the role of iNOS in protein nitration in this model. Control, room-air-raised iNOS(+/+) mice revealed no positive cells in the central retina at P14 (Fig. 5C). Control experiments on post-oxygen-incubated iNOS(+/+) mice, omitting the primary antibody, revealed no staining (data not shown).
Fig. 5.

Protein nitration detected by nitrotyrosine immunostaining at P14. A, Eyes of iNOS(+/+) mice of the ischemic phase of the experiment. B, Eyes of iNOS(−/−)mice of the ischemic phase of the experiment. C, Room-air-raised iNOS(+/+) control mice. Nitrotyrosine-positive cells were found at the edge of the capillary-free, central retina of ischemic iNOS(+/+)sections in an RMG cell-like distribution in proximity to blood vessels (A). Ischemic iNOS(−/−) mice (B) and room-air-raised iNOS(+/+) mice (C) did not show any positive cells. Panel height: A–C, 240 μm.
DISCUSSION
These data demonstrate for the first time that neuronal apoptosis occurs in a model of ischemic proliferative retinopathy. Therefore, this model, apart from developing the same type of neovascularization, displays the feature of retinal apoptotic cell death described in diabetic retinopathy (Barber et al., 1998).
TUNEL-positive cells were detected in the INL of the avascular retina of post-oxygen-incubated animals during the ischemic phase at P14. Additionally, the TUNEL-positive cells exhibited morphological signs of apoptosis, such as chromatin condensation and pyknotic nuclei, pointing toward an apoptotic nature of the DNA strand breaks detected by the TUNEL technique. Furthermore, this apoptosis led to a significant thinning of the INL in this area 3 d later at P17. Moreover, a significant thinning of the IPL, in which the synapses between bipolar, amacrine, and ganglion cells are established, was observed.
The INL consists of the nuclei of amacrine, bipolar, horizontal, and Müller cells. However, we were not able to further characterize the nature of the apoptotic cells because of the lack of specific nuclear markers of the different cell types. Histological sections stained with specific antibodies to specific cytoplasmic proteins of the different cell types were not conclusive because they did not allow differential cell–nuclei counts in the INL, and cell counts on the basis of morphological nuclear differences did not seem to be reliable. Judging from the extent of cell death, sometimes to as little as two rows of cells in the INL, it seems likely that all prevalent cell types contribute to the programmed cell death observed.
In the model of retinal ischemia–reperfusion, apoptosis and subsequent thinning in the inner retina have well been documented (Geyer et al., 1995; Lam et al., 1999), and thinning of the IPL and INL was also reported in a rat model of diabetic retinopathy (Barber et al., 1998). In contrast to our observations in proliferative retinopathy, ischemia–reperfusion and diabetic retinopathy lead to ganglion cell rarefaction additionally to the thinning of the inner retinal layers (INL and IPL). The ganglion cells of the ischemic retina in our experiments did not undergo cell death at any rate. It might be that ganglion cells in newborn mice have better protection against ischemia compared with adult animals used in the other models. On the other hand, ganglion cell loss in the ischemia–reperfusion model might mainly be attributable to mechanisms of reperfusion, which do not occur in our model in that way. In a model of ischemic proliferative retinopathy in the rat, Lachapelle et al. (1999)described previously histological and electrophysiological changes. This group described a significant thinning of the OPL and a rarefaction of horizontal cells of the INL that were ascribed to oxygen toxicity. We did not detect thinning of the OPL in the mouse model at P17, and we were not able to perform differential horizontal cell counts on morphological features in the mouse. We cannot exclude that horizontal cell death might contribute to the thinning observed in our model, and OPL thinning might occur later in the murine model. It is, however, difficult to compare the rat and mouse model of ischemic proliferative retinopathy. In the rat model, oxygen exposures consist of high oxygen levels interrupted by multiple daily low (or normoxic) oxygen level periods. Because high oxygen exposure leads to vasoconstriction and obliteration (Alon et al., 1995), it can be assumed that the daily low (normoxic) periods represent periods of relative hypoxia for the retina. It is therefore more difficult in the rat model to differentiate between hyperoxic and hypoxic effects and the differences described by Lachapelle et al. (1999), notably that the horizontal cell loss might paradoxically be attributable to hypoxia rather than oxygen toxicity.
With regard to the mechanism inducing apoptotic cell death in the central INL, we analyzed the role of iNOS. We described previously that iNOS mRNA is expressed in this model, with a peak at P14. We localized iNOS mRNA and iNOS-related NADPH-diaphorase activity to the cytoplasm of cells of the INL of the central ischemic retina at P14 (Sennlaub et al., 2001). This corresponds precisely to the area in which apoptotic cells in the INL were found.
Using mice lacking the iNOS gene, we evaluated the role of NO released by iNOS on the apoptosis in the central INL in ischemic proliferative retinopathy. Apoptotic cells in the INL of the central retinal area at P14 were considerably more numerous in iNOS(+/+) than in iNOS(−/−) animals. At P17, a significant thinning in this area was observed in the iNOS-expressing animals compared with iNOS(−/−) mice. These differences between iNOS(+/+) and iNOS(−/−) mice could only be detected in the central area, corresponding exactly to the capillary-free area in which iNOS is expressed at P14 in iNOS(+/+) mice. Differences in the detected apoptosis could not be attributable to differences of the ischemic state in iNOS(+/+) and iNOS(−/−) mice, because the size of the capillary-free area or in the intravitreal neovascular response in iNOS(+/+) and iNOS(−/−) mice at P14 are equivalent (Sennlaub et al., 2001). Furthermore, intravitreal injections of the potent iNOS inhibitor 1400W to post-oxygen-incubated iNOS(+/+) mice in the ischemic phase, when iNOS is expressed, significantly inhibited central INL thinning compared with vehicle-injected mice. The efficiency of iNOS inhibition by intravitreal administration could also be seen in a marked improvement of intraretinal revascularization and an inhibition in pathological intravitreal neovascularization compared with control. Together, this is good evidence that iNOS activity induces apoptotic cell death and retinal degeneration in the central capillary-free retina in ischemic proliferative retinopathy.
Regarding the mechanism involved in NO induced-apoptosis, nitrosative stress, leading to protein nitration, is thought to be one of the major mechanisms responsible for NO-mediated neurotoxicity. Indeed, the nitration of proteins can lead to the loss of function of proteins, particularly mitochondrial and antioxidant proteins (Yamakura et al., 1998; MacMillan-Crow and Thompson, 1999; Aulak et al., 2001), which can ultimately lead to impairment of the mitochondrial respiratory chain and result in apoptosis or necrosis depending on the energy status of the cell (Bolanos et al., 1997). In our model, tyrosine nitration was detected in cells of the INL at the edge of the ischemic, central retina of post-oxygen-incubated iNOS(+/+). The absence of nitrotyrosine staining in post-oxygen-incubated iNOS(−/−) and room-air-raised iNOS(+/+) mice demonstrated that protein nitration was subsequent to iNOS activity. NO does not directly nitrate tyrosine but provides the biological precursor for nitrating agents that perform this modification in vivo. NO can form nitrating agents in a number of ways, including reaction with superoxide to make peroxynitrite and through enzymatic oxidation of nitrite to generate NO2° (Beckman and Koppenol, 1996; Halliwell et al., 1999). We therefore propose that protein nitration, leading to protein inactivation, in cells of the INL could participate in the induction of apoptosis in our model after iNOS induction, as we demonstrated previously in vitro for NO-induced neuronal cell death (Goureau et al., 1999).
The distribution of the anti-nitrotyrosine staining in an RMG cell-like pattern in post-oxygen-incubated iNOS(+/+) mice and the activation of RMG cells in the ischemic area seen by GFAP immunohistochemistry, as well as our previous in vitro studies showing the ability of RMG cells to express iNOS (Goureau et al., 1999), are additional indications that these cells might be a major source of iNOS expression in this model.
As concerns other molecular mechanisms of NO-induced retinal cell death, we analyzed Fas and tumor necrosis factor-α expression, two factors that have been described to mediate NO-related apoptosis (Sun et al., 1998; Garban and Bonavida, 2001). Western blot and immunohistochemical analysis revealed no significant induction of theses proteins (data not shown). Involvement of cGMP, the poly(ADP-ribose) polymerase pathway, and caspases, although not involved in retinal cell death induced by NO in vitro(Goureau et al., 1999), are currently under investigation.
Recently, El-Asra et al. (2001) reported that iNOS is expressed in human diabetic retinopathy and in ocular ischemic syndrome. They identified the iNOS-expressing cells to be Müller cells of the INL, supporting the hypothesis that mechanisms similar to the ones we found in the mouse model are involved in human ischemic retinopathy. Our data are the first evidence that iNOS plays a crucial role in retinal apoptosis in ischemic proliferative retinopathy. Current treatments proposed for ischemic proliferative retinopathy do not alter the course of the degeneration in the ischemic retina. Selective iNOS inhibition is able to improve its vascularization and to inhibit vitreal neovascularization (Sennlaub et al., 2001) and to protect the hypoxic retina from degeneration, confirming that iNOS is an ideal target for the treatment of ischemic proliferative retinopathy.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale and by grants from the Association pour la Recherche sur le Cancer and the Deutscher Akademischer Austauschdienst. We gratefully acknowledge Drs. J. S. MacMicking, C. Nathan, and J. Mudgett for providing the mice. We thank Christophe Klein, Sylvie Thomasseau, and Laurent Jonet for technical assistance and Jean Claude Jeanny.
Correspondence should be addressed to Florian Sennlaub, Développement, Vieillissement et Pathologie de la Rétine, Institut National de la Santé et de la Recherche Médicale U450, 15, rue de l'École de Médecine, 75270 Paris Cedex 06, France. E-mail: fsennlau@infobiogen.fr.
REFERENCES
- 1.Adamis AP, Aiello LP, D'Amato RA. Angiogenesis and ophthalmic disease. Angiogenesis. 1999;3:9–14. doi: 10.1023/a:1009071601454. [DOI] [PubMed] [Google Scholar]
- 2.Alon T, Hemo I, Itin A, Pe'er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- 3.Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW, Stuehr DJ. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci USA. 2001;98:12056–12061. doi: 10.1073/pnas.221269198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 6.Bek T. Transretinal histopathological changes in capillary-free areas of diabetic retinopathy. Acta Ophthalmol. 1994;72:409–415. doi: 10.1111/j.1755-3768.1994.tb02787.x. [DOI] [PubMed] [Google Scholar]
- 7.Bolanos JP, Almeida A, Fernandez E, Medina JM, Land JM, Clark JB, Heales SJ. Potential mechanisms for nitric oxide-mediated impairment of brain mitochondrial energy metabolism. Biochem Soc Trans. 1997;25:944–949. doi: 10.1042/bst0250944. [DOI] [PubMed] [Google Scholar]
- 8.Brune B, von Knethen A, Sandau KB. Nitric oxide and its role in apoptosis. Eur J Pharmacol. 1998;351:261–272. doi: 10.1016/s0014-2999(98)00274-x. [DOI] [PubMed] [Google Scholar]
- 9.Christopherson KS, Bredt DS. Nitric oxide in excitable tissues: physiological roles and disease. J Clin Invest. 1997;100:2424–2429. doi: 10.1172/JCI119783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collier A, Mitchell JD, Clarke BF. Visual evoked potential and contrast sensitivity function in diabetic retinopathy. Br Med J Clin Res. 1985;291:248. doi: 10.1136/bmj.291.6490.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Amore PA. Mechanisms of retinal and choroidal neovascularization. Invest Ophthalmol Vis Sci. 1994;35:3974–3979. [PubMed] [Google Scholar]
- 12.Dembinska O, Rojas LM, Varma DR, Chemtob S, Lachapelle P. Graded contribution of retinal maturation to the development of oxygen-induced retinopathy in rats. Invest Ophthalmol Vis Sci. 2001;42:1111–1118. [PubMed] [Google Scholar]
- 13.El-Asrar AM, Desmet S, Meersschaert A, Dralands L, Missotten L, Geboes K. Expression of the inducible isoform of nitric oxide synthase in the retinas of human subjects with diabetes mellitus. Am J Ophthalmol. 2001;132:551–556. doi: 10.1016/s0002-9394(01)01127-8. [DOI] [PubMed] [Google Scholar]
- 14.Fulton AB, Hansen RM. Photoreceptor function in infants and children with a history of mild retinopathy of prematurity. J Opt Soc Am A. 1996;13:566–571. doi: 10.1364/josaa.13.000566. [DOI] [PubMed] [Google Scholar]
- 15.Fulton AB, Hansen RM, Petersen RA, Vanderveen DK. The rod photoreceptors in retinopathy of prematurity: an electroretinographic study. Arch Ophthalmol. 2001;119:499–505. doi: 10.1001/archopht.119.4.499. [DOI] [PubMed] [Google Scholar]
- 16.Garban HJ, Bonavida B. Nitric oxide inhibits the transcription repressor yin-yang 1 binding activity at the silencer region of the fas promoter: a pivotal role for nitric oxide in the up-regulation of fas gene expression in human tumor cells. J Immunol. 2001;167:75–81. doi: 10.4049/jimmunol.167.1.75. [DOI] [PubMed] [Google Scholar]
- 17.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 18.Geyer O, Almog J, Lupu-Meiri M, Lazar M, Oron Y. Nitric oxide synthase inhibitors protect rat retina against ischemic injury. FEBS Lett. 1995;374:399–402. doi: 10.1016/0014-5793(95)01147-7. [DOI] [PubMed] [Google Scholar]
- 19.Goureau O, Regnier-Ricard F, Courtois Y. Requirement for nitric oxide in retinal neuronal cell death induced by activated Muller glial cells. J Neurochem. 1999;72:2506–2515. doi: 10.1046/j.1471-4159.1999.0722506.x. [DOI] [PubMed] [Google Scholar]
- 20.Greenacre SA, Ischiropoulos H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res. 2001;34:541–581. doi: 10.1080/10715760100300471. [DOI] [PubMed] [Google Scholar]
- 21.Halliwell B, Zhao K, Whiteman M. Nitric oxide and peroxynitrite. The ugly, the uglier and the not so good: a personal view of recent controversies. Free Radic Res. 1999;31:651–669. doi: 10.1080/10715769900301221. [DOI] [PubMed] [Google Scholar]
- 22.Lachapelle P, Dembinska O, Rojas LM, Benoit J, Almazan G, Chemtob S. Persistent functional and structural retinal anomalies in newborn rats exposed to hyperoxia. Can J Physiol Pharmacol. 1999;77:48–55. doi: 10.1139/cjpp-77-1-48. [DOI] [PubMed] [Google Scholar]
- 23.Lam TT, Abler AS, Tso MO. Apoptosis and caspases after ischemia-reperfusion injury in rat retina. Invest Ophthalmol Vis Sci. 1999;40:967–975. [PubMed] [Google Scholar]
- 24. MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 81 1995. 641 650 [Erratum (1995) 81:1170] [DOI] [PubMed] [Google Scholar]
- 25.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 26.MacMillan-Crow LA, Thompson JA. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Arch Biochem Biophys. 1999;366:82–88. doi: 10.1006/abbi.1999.1202. [DOI] [PubMed] [Google Scholar]
- 27.Nathan C. Inducible nitric oxide synthase: what difference does it make? J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmowski AM, Sutter EE, Bearse MA, Jr, Fung W. Mapping of retinal function in diabetic retinopathy using the multifocal electroretinogram. Invest Ophthalmol Vis Sci. 1997;38:2586–2596. [PubMed] [Google Scholar]
- 29.Penn JS, Rajaratnam VS, Collier RJ, Clark AF. The effect of an angiostatic steroid on neovascularization in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2001;42:283–290. [PubMed] [Google Scholar]
- 30.Reisner DS, Hansen RM, Findl O, Petersen RA, Fulton AB. Dark-adapted thresholds in children with histories of mild retinopathy of prematurity. Invest Ophthalmol Vis Sci. 1997;38:1175–1183. [PubMed] [Google Scholar]
- 31.Sennlaub F, Courtois Y, Goureau O. Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J Clin Invest. 2001;107:717–725. doi: 10.1172/JCI10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith LE, Wesolowski E, McLellan A, Kostyk SK, D'Amato R, Sullivan R, D'Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 33.Sun D, Coleclough C, Cao L, Hu X, Sun S, Whitaker JN. Reciprocal stimulation between TNF-alpha and nitric oxide may exacerbate CNS inflammation in experimental autoimmune encephalomyelitis. J Neuroimmunol. 1998;89:122–130. doi: 10.1016/s0165-5728(98)00124-6. [DOI] [PubMed] [Google Scholar]
- 34.Wolter JR. Diabetic retinopathy. Am J Ophthalmol. 1961;51:1123–1139. doi: 10.1016/0002-9394(61)91802-5. [DOI] [PubMed] [Google Scholar]
- 35.Yamakura F, Taka H, Fujimura T, Murayama K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]