

Abstract
We have shown that cortical neurons challenged with toxic concentrations of β-amyloid peptide (βAP) enter the S phase of the cell cycle before apoptotic death. Searching for a signaling molecule that lies at the border between cell proliferation and apoptotic death, we focused on the disialoganglioside GD3. Exposure of rat cultured cortical neurons to 25 μm βAP(25–35) induced a substantial increase in the intracellular levels of GD3 after 4 hr, a time that precedes neuronal entry into S phase. GD3 levels decreased but still remained higher than in the control cultures after 16 hr of exposure to βAP(25–35). Confocal microscopy analysis showed that the GD3 synthesized in response to βAP colocalized with nuclear chromatin. The increase in GD3 was associated with a reduction of sphingomyelin (the main source of the ganglioside precursor ceramide) and with the induction of α-2,8-sialyltransferase (GD3 synthase), the enzyme that forms GD3 from the monosialoganglioside GM3. A causal relationship between GD3, cell-cycle activation, and apoptosis was demonstrated by treating the cultures with antisense oligonucleotides directed against GD3 synthase. This treatment, which reduced βAP(25–35)-stimulated GD3 formation by ∼50%, abolished the neuronal entry into the S phase and was protective against βAP(25–35)-induced apoptosis.
Keywords: Alzheimer's disease, β-amyloid, cell cycle, ganglioside GD3, apoptosis, neurodegeneration
It is currently believed that neuronal degeneration in Alzheimer's disease (AD) is caused by extracellular β-amyloid peptide (βAP) (for review, see Selkoe, 2001). Cultured neurons exposed to βAP predominantly show an apoptotic phenotype (Forloni et al., 1993; Loo et al., 1993), although neuronal apoptosis by β-amyloid is not the only factor that contributes to the pathophysiology of AD (Behl, 2000; Mattson, 2000;Joseph et al., 2001; Roth, 2001; Small et al., 2001). Molecular determinants of βAP-induced neuronal death have been investigated extensively, but they are still poorly defined. In vivo andin vitro studies have shown that an untimely activation of a cell cycle in terminally differentiated neurons may be a requisite antecedent to neuronal apoptosis in AD (Herrup and Busser, 1995;Vincent et al., 1996; Arendt et al., 1998; Busser et al., 1998; Nagy et al., 1998; Copani et al., 1999; Giovanni et al., 1999, 2000; McShea et al., 1999; Yang et al., 2001). We have shown that full-length βAP (fragment 1–42) and its active fragments βAP(1–40) and βAP(25–35) promote the activation of a cell cycle in differentiated cultured cortical neurons. In particular, βAP-treated cortical neurons express the repertoire of proteins necessary to exit quiescence and eventually enter S phase. These neurons undergo apoptosis before entering the G2/M phase (Copani et al., 1999).
Because βAP-induced activation of this “neuronal cycle” seems to be critical for the development of apoptosis, it becomes important to disclose the signaling pathway(s) leading to reactivation of the cell cycle in neurons.
Gangliosides, sialic acid-containing glycosphingolipids, constitute a signaling system involved in the modulation of processes of neuronal proliferation and differentiation. GD3 is highly expressed in the embryonic nervous system, particularly in neuroprogenitor cells. GD3 levels are low in the adult brain (Percy et al., 1991; Svennerholm et al., 1991; Goldman and Reynolds, 1996; Kawai et al., 1998), although they increase in the brains of patients with AD (Kalanj et al., 1991) and Creutzfeldt–Jakob disease (Ando et al., 1984). Interestingly, endogenous GD3 neosynthesis is associated with the appearance of a tumor phenotype in melanocytes (Birkle et al., 2000), and overexpression of GD3 synthase (the α-2,8-sialyltransferase that generates GD3 from GM3) enhances the proliferation rate of both rat C6 glioma (Sottocornola et al., 1998) and PC12 pheochromocytoma cell lines (Fukumoto et al., 2000). Thus, it appears that GD3 is able to modify the cell proliferation status under physiological and pathological conditions.
In the present study we show that mature rat cortical neurons in culture, which respond to βAP by re-entering the cell cycle, show an early increase in the intracellular levels of GD3. GD3 synthesis in βAP-treated neurons is required for their entrance into the S phase and contributes to the development of apoptosis.
MATERIALS AND METHODS
Pure neuronal culture. Cultures of pure cortical neurons were obtained from rats at embryonic day 15 as described previously (Copani et al., 1999). Briefly, dissociated cortical cells were plated in a medium consisting of DMEM/Ham's F12 (1:1) supplemented with the following components: 10 mg/ml bovine serum albumin, 10 μg/ml insulin, 100 μg/ml transferrin, 100 μm putrescine, 20 nm progesterone, 30 nm selenium, 2 mm glutamine, 6 mg/ml glucose, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cortical cells were plated at a density of 2 × 106cells/dish on 35 mm Nunc (Roskilde, Denmark) dishes precoated with 0.1 mg/ml poly-d-lysine. Cytosine-β-d-arabinofuranoside (10 μm) was added to the cultures 18 hr after plating to avoid the proliferation of non-neuronal elements and was kept for 3 d before medium replacement. This method yields >99% pure neuronal cultures, as judged by immunocytochemistry for glial fibrillary acidic protein and neuron-specific microtubule-associated protein 2 (Copani et al., 1999). βAP has always been applied to mature cultures at 8 din vitro (DIV).
Handling of β-amyloid peptide. βAP(25–35) and the reverse peptide βAP(35–25) were purchased from Bachem AG (Bubendorf, Switzerland). Different lots of peptides were used. βAP(25–35) and βAP(35–25) were solubilized in sterile, doubly distilled water at an initial concentration of 2.5 mm and stored frozen at −20°C. They were used at a final concentration of 25 μm in the presence of the glutamate receptor antagonists MK-801 (10 μm) and DNQX (30 μm) to prevent the excitotoxicity mediated by endogenous glutamate (Copani et al., 1999).
Addition of antisense oligonucleotides to the cultures.Cultures were also treated with the following “end-capped” phosphorothioate antisense oligonucleotides directed against the enzyme α-2,8-sialyltransferase (GD3 synthase): 5′-CAGTACAGCCATGGCCCCTCT-3′. A scrambled oligonucleotide was used as a control: 5′-CGACCTACCTATGCGCT-ACCG-3′. Oligonucleotides (3 μm) were applied to the cultures 16 hr before the addition of βAP(25–35).
Fluorescence-activated cell sorter analysis.Fluorescence-activated cell sorter analysis was performed as described previously (Copani et al., 1999). Cells were harvested by incubation with 0.25% trypsin for 3 min, and the suspension was centrifuged at low speed after addition of 50% fetal calf serum. Each pellet was washed with PBS and finally fixed in 70% ethanol. Before staining with propidium iodide (50 μg/ml in the dark for 30 min), suspended cells were treated for 1 hr at 37°C with RNase (100 μg/ml). The DNA content and ploidy were assessed using a Coulter Elite flow cytometer (Beckman, Fullerton, CA). The Multicycle AV software program (Phoenix Flow Systems, San Diego, CA) was used to analyze cell-cycle distribution profiles.
Evaluation of sphingomyelin hydrolysis. Cortical neurons were incubated in the presence of [3H]serine (specific activity, 26 Ci mmol−1; Amersham Biosciences, Milan, Italy) for 72 hr before exposure to βAP(25–35) for 4 hr. The reaction was stopped by adding methanol:chloroform:HCl (100:100:1, v/v/v) and a balanced salt solution containing 10 mm EDTA; the aqueous and lipid phases were separated by centrifugation. Glycerophospholipids present in the lipid phase were saponified in methanolic KOH (0.1 mfor 1 hr at 37°C) before resolution of sphingomyelin by sequential one-dimensional TLC, using chloroform:benzene:ethanol (80:40:75, v/v/v) followed by chloroform:methanol:28% ammonia (65:25:5, v/v/v) as solvents. Plates were analyzed using a digital autoradiographer (Berthold, Bad Wildbad, Germany).
Assessment of intracellular GD3 levels. Cultures were washed twice with ice-cold PBS, pH 7.4, and cells were scraped from the dishes and homogenized. Gangliosides were extracted according to the method ofSvennerholm and Fredman (1980) as described previously (Dotta et al., 1998) and analyzed by high-performance TLC (HPTLC) using analytical precoated Silica gel 60 HPTLC plates (Merck, Darmstadt, Germany). All plates were first activated by heating to 100°C for 30 min. Samples were spotted onto plates with a Hamilton syringe in chloroform:methanol:0.25% KCl (5:4:1, v/v/v). Authentic GD3 (provided by Fidia S.p.A., Abano Terme, Italy) was used as standard. GD3 was immunodetected by using the R24 anti-GD3 monoclonal antibody (1:100). The plates were incubated for 1 hr at room temperature with the primary antibody, washed twice with PBS–Tween 20, and then incubated for 45 min at room temperature with a horseradish peroxidase-conjugated rabbit anti-mouse antibody (1:200; Sigma, St. Louis, MO). Detection was performed by ECL (Amersham Biosciences). The bands were quantified by scanning densitometric analysis.
Immunofluorescence analysis of GD3. Cultures were fixed with 2% paraformaldehyde. After incubation with 3% nonimmunized mouse serum in PBS, the R24 anti-GD3 monoclonal antibody (1:100) was applied at 4°C for 72 hr. Cells were washed three times and FITC-conjugated anti-mouse Ig (1:200; Cappel, ICN Biomedicals, Milan, Italy) was applied for 1 hr at room temperature to visualize the labeled sites. Nuclei were stained with propidium iodide (50 μg/ml) in PBS. Fluorescence was detected by a Zeiss (Oberkochen, Germany) LSM510 laser scanner microscope.
Reverse transcriptase-PCR analysis of GD3 synthase. Total RNA was extracted from cultures of primary cortical neurons essentially as described previously (Auffray and Rougeon, 1980), except that cells were washed twice with ice-cold PBS and then scraped in 2 ml of cold 3m LiCl/6 m urea and the procedure was scaled down appropriately. Finally, total RNA was subjected to DNase I treatment (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer's instructions. Two micrograms of total RNA were then used for cDNA synthesis, using Superscript II (Invitrogen, San Diego, CA) and an oligo(dT) primer according to the manufacturer's instructions. The reverse transcriptase (RT) product was diluted to 100 μl with sterile, distilled water, and 1 μl of cDNA was used in each subsequent PCR amplification. Amplification of GD3 synthase cDNA was performed using the following primers: forward (5′-CCAGCATAATTCGCCAGAGA-3′) and reverse (5′-TTGCATGTTCACGGAGAAGG-3′). For β-actin cDNA amplification, the primers were those described by Roelen et al. (1994), which span an intron and yield products of different sizes depending on whether cDNA or genomic DNA is used as a template (400 bp for a cDNA-derived product and 600 bp for a genomic DNA-derived amplification). Reaction conditions included an initial denaturation step (94°C for 3 min) followed by 45 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec. A final extension step (72°C for 10 min) concluded the reaction. PCR products (one-third of the reaction) were analyzed electrophoretically on 2% agarose gels poured and run in 1× Tris acetate-EDTA.
RESULTS
We have shown previously that βAP(1–42) or its active fragments βAP(1–40) and βAP(25–35) reactivate the cell cycle and induce apoptotic death in pure cultures of cortical neurons (Copani et al., 1999). Because identical effects were seen with the three peptides, we used βAP(25–35) in the present study. βAP(25–35) (25 μm) was applied to mature cultures (8–9 DIV) in the presence of a mixture of ionotropic glutamate receptor antagonists (10 μm MK-801 plus 10 μmDNQX) to avoid any endogenous excitotoxic component (Copani et al., 1999). As expected, ∼8–10% of cultured neurons were found in S phase 16 hr after the addition of βAP(25–35), whereas no S phase was seen at earlier times (4 or 8 hr). The number of apoptotic neurons increased progressively after 16 hr, reaching >50% of the cell population at 20 hr (see also Copani et al., 1999).
TLC analysis combined with immunodetection showed a large increase in GD3 content 4 hr after the addition of βAP(25–35) (i.e., at a time that precedes both neuronal entry into S phase and apoptotic death) (Fig. 1). GD3 levels decreased, but were still higher than in control cultures, 16 hr after the addition of βAP(25–35) (Fig. 1A,B).
Fig. 1.

Intracellular GD3 levels in cultured cortical neurons exposed to βAP(25–35) for 4 or 16 hr. A, A representative TLC stained with anti-GD3 antibodies. B, A densitometric analysis of three independent experiments. Four culture dishes (2 × 106 neurons per dish) were pooled for each condition in any experiment. Values are expressed as percentages of controls and represent means ± SEM. *p < 0.05 (one-way ANOVA plus Fisher's least significant difference) versus controls.
In Figure 2A,B,D, double-fluorescence analysis of GD3 (green) and nuclear chromatin (red) showed few neurons expressing GD3 in control cultures or in cultures exposed to the reverse peptide βAP(35–25). Nearly all neurons became immunopositive for GD3 in neurons exposed to βAP(25–35). Immunoreactivity was mostly detected in cell nuclei (yellow) after the addition of βAP(25–35), although it was also found outside the nuclear region in late degenerating neurons (Fig. 2C,D).
Fig. 2.
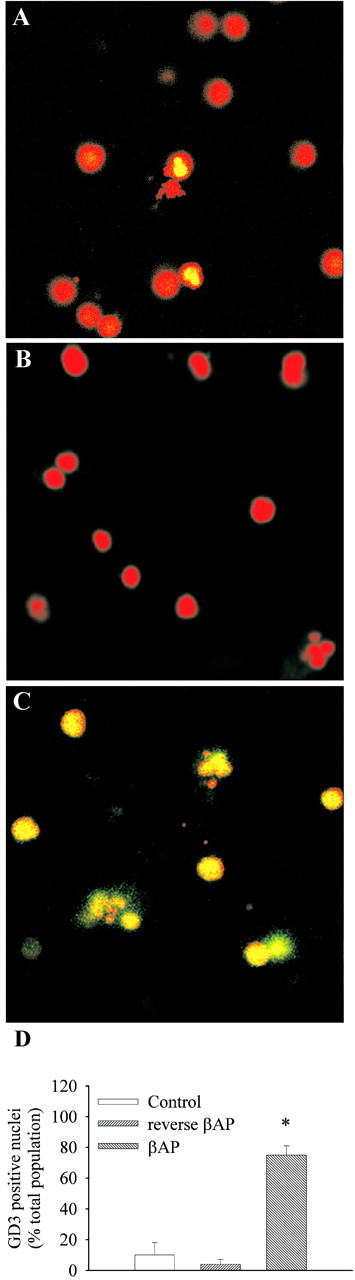
Immunofluorescence analysis of GD3 in control cultures (A) and in cultures exposed to reverse βAP(35–25) (B) or to βAP(25–35) (C) for 16 hr. GD3 immunofluorescence is ingreen. Fluorescence staining of DNA with propidium iodide is in red. Colocalization between GD3 and DNA is in yellow. The count of GD3-positive nuclei is shown inD. Values were calculated by a blinded observer from three random fields per culture dish for a total of four to six culture dishes per condition in two to three independent experiments. Each single dish has been considered as an individual value in the statistical analysis (i.e., n = 4–6). Values are means ± SEM. *p < 0.05 (one-way ANOVA plus Fisher's least significant difference) versus controls or reverse βAP(35–25). Reverse βAP(35–25) was not toxic in these experiments.
The early increase in GD3 expression paralleled a reduction of the sphingomyelin content in neurons exposed to βAP(25–35) for 4 hr (Fig. 3A), suggesting that GD3 is synthesized after βAP(25–35)-induced sphingomyelin hydrolysis. RT-PCR analysis showed that βAP(25–35) induced the expression of GD3 synthase mRNA after 2, 8, and 16 hr (Fig. 3B).
Fig. 3.

A, Exposure of cultured cortical neurons to βAP(25–35) for 4 hr reduces sphingomyelin (SM) levels. Values are means ± SEM of five individual determinations. *p < 0.01 (Student'st test) compared with control cultures.B, Expression of GD3 synthase in primary cultures of rat cortical neurons exposed to βAP(25–35) for the indicated times. The results of two representative experiments are shown.CTRL, Control cultures. Amplification of β-actin cDNA was performed to confirm the integrity of the cDNA preparations and to control for genomic DNA contamination. The 600 bp of β-actin was not detected, thus excluding any genomic contamination.
To examine whether the increase in GD3 levels was causally related to the reactivation of the cell cycle and apoptotic death induced by βAP, we treated the cultures with a 3 μm concentration of end-capped antisense oligonucleotides directed against GD3 synthase for 16 hr before the addition of βAP(25–35). GD3 synthase antisense oligonucleotides substantially reduced the rise of neuronal GD3 content induced by βAP(25–35) at 4 hr, whereas a scrambled oligonucleotide had a smaller effect (Fig.4A). Cytofluorometric analysis showed that GD3 synthase antisense oligonucleotides abolished the neuronal re-entry into the S phase of the cell cycle in response to βAP(25–35) (Fig. 4B) and substantially protected against βAP(25–35)-induced apoptosis (Fig. 4C). Scrambled oligonucleotides could also reduce βAP(25–35)-induced S phase and apoptosis, but their effect was much smaller and was significantly different from that produced by GD3 synthase antisense oligonucleotides (Fig. 4B,C).
Fig. 4.
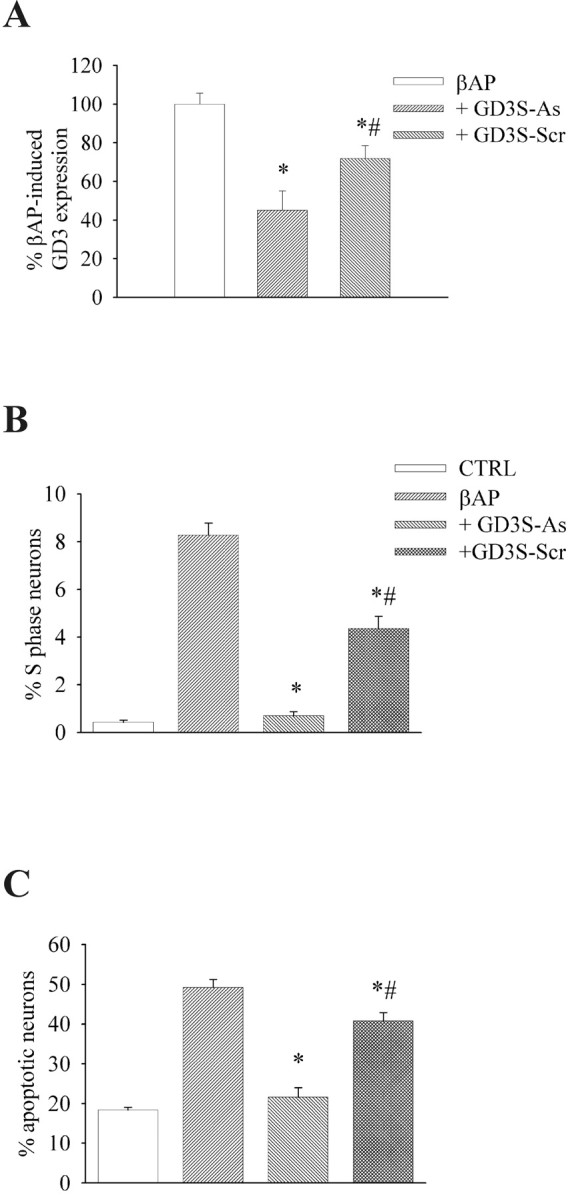
Intracellular GD3 levels (A), percentage of neurons in the S phase of the cell cycle (B), and percentage of apoptotic neurons (C) in cultured cortical neurons pretreated for 16 hr with GD3 synthase antisense oligonucleotides (GD3S-As, 3 μm) or a scrambled oligonucleotide (GD3S-Scr, 3 μm), and then exposed to βAP(25–25) for 4 hr (A) or 20 hr (B, C). Densitometric analysis of GD3 levels from three independent experiments is shown in A. Four culture dishes (2 × 106 neurons per dish) were pooled for each condition in any experiment. Values in B andC were calculated from eight individual culture dishes from three independent experiments. Each single dish has been considered as an individual value in the statistical analysis (i.e.,n = 8). p < 0.05 (one-way ANOVA plus Fisher's least significant difference) if compared with cultures treated with βAP(25–35) alone (∗) or with βAP plus GD3S-As (#).
Together, these results indicate that βAP(25–35)-induced GD3 synthesis is antecedent and causally related to neuronal cell-cycle reactivation and apoptosis.
DISCUSSION
After the induction of apoptosis, GD3 accumulates in non-neuronal cells, where it contributes to the death pathway by a dual mechanism that involves the opening of mitochondrial permeability transition pores and the suppression of the nuclear factor-κB-dependent survival pathway (De Maria et al., 1997; Kristal and Brown, 1999; Malisan and Testi, 1999; Scorrano et al., 1999; Rippo et al., 2000; Colell et al., 2001). In neurons, a number of studies have been performed with exogenously added gangliosides (Favaron et al., 1988; Manev et al., 1990; Saito et al., 1998, 1999; Ryu et al., 1999), but the functional role of endogenously generated gangliosides in neurodegenerative processes has never been investigated.
In this study, we show that GD3 accumulates in rat cortical neurons that have been exposed to βAP(25–35). We have demonstrated previously that mature neurons must re-enter the cell cycle and cross the G1/S transition before undergoing βAP-induced apoptosis (Copani et al., 1999). βAP-treated cortical neurons express a battery of proteins that are typically expressed by proliferating cells during G1/S phases, such as cyclin D1, phosphoretinoblastoma, cyclin E, and cyclin A. Neurons eventually enter the S phase, as shown by quantitative cytofluorometric analysis, and then die by apoptosis before crossing the border between the S and G2 phases (Copani et al., 1999). The use of a dexamethasone-inducible cyclin D1 mRNA antisense, a negative dominant mutant of cyclin-dependent kinase type 2 (CDK2) or chemical CDK inhibitors has shown that the unscheduled cell cycle is causally related to apoptotic death in neurons exposed to βAP (Copani et al., 1999, 2001). These in vitro studies have their in vivo counterpart in the AD brain, in which Yang et al. (2001)provided evidence for chromosomal replication in “at-risk” neurons. The present data indicate that GD3 is an essential component of the signaling pathway(s) leading to the reactivation of the cell cycle in βAP-treated neurons (Fig. 5). This evidence is consistent with the regulatory functions of GD3 in cellular proliferation and differentiation processes. PC12 cells overexpressing the GD3 synthase gene showed an enhanced rate of proliferation attributable to a sustained activation of the Ras/mitogen-activated protein–extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase pathway and failed to differentiate in response to NGF (Fukumoto et al., 2000). In malignant cells, GD3 induces the suppression of differentiation phenotypes and promotes proliferation (Sottocornola et al., 1998;Birkle et al., 2000).
Fig. 5.

Hypothetical model of the role of GD3 in βAP-induced cell-cycle activation and apoptosis. SM, Sphingomyelin.
In neurons that degenerate in response to βAP, neosynthesized GD3 accumulated inside the nuclear region and colocalized with nuclear chromatin. In other cell types, in which apoptosis is not associated with a reactivation of the cell cycle, GD3 is instead consistently found outside the nucleus (Malisan and Testi, 1999). To date, only gangliosides of the “a” series have been described in the cell nucleus. GM1 is found in the nuclear membrane, where its expression is upregulated during axonogenesis (Wu et al., 1995, 2001;Kozireski-Chuback et al., 1999). Interestingly, GM1 inhibits DNA synthesis and the activity of DNA polymerase α in isolated nuclei of HeLa cells (Ohsawa et al., 1988). An attractive hypothesis is that GD3 acts as a functional counterpart of GM1 by increasing cell proliferation via a nuclear mechanism. Accordingly, in neurons exposed to βAP, GD3 might provide a nuclear signal for the aberrant DNA replication. The evidence that the antisense-induced decrease in GD3 levels was highly effective in preventing the unscheduled S phase, and the ensuing apoptotic phenotype strengthens the hypothesis of a causal relationship among GD3 formation, cell-cycle activation, and neuronal death. However, we cannot exclude the possibility that GD3 contributes to βAP-induced apoptosis through additional mechanisms (Fig. 5), for example by targeting the mitochondrial pathway of cell death.
Ceramide released from sphingomyelin hydrolysis is a likely source for GD3 synthesis (De Maria et al., 1998). Sphingomyelin hydrolysis might follow the interaction of βAP aggregates with a membrane receptor, the identity of which is unknown. The p75 low-affinity neurotrophin receptor is a possible candidate (Yaar et al., 1997), because this receptor has been shown to transduce the extracellular signal via the activation of acidic sphingomyelinase (Brann et al., 1999; Bilderback et al., 2001). The finding of an early induction of GD3 synthase in response to βAP is particularly interesting because it provides the first evidence that this enzyme is upregulated in response to a death signal. This suggests a novel strategy against βAP-induced neurotoxicity based on the pharmacological regulation of GD3 synthase expression.
Footnotes
This work was supported by Ministero dell' Istruzione, dell' Universitá e della Ricerca cofin 2000 (M.A.S.), by Ricerca Finalizzata 2000 (F.N.), and by Alzheimer's Association Grant IIRG-01-2824 (A.C.).
Correspondence should be addressed to Dr. Agata Copani, Department of Pharmaceutical Sciences, University of Catania, Viale A. Doria 6, 95125 Catania, Italy. E-mail: acopani@katamail.com.
REFERENCES
- 1.Ando S, Toyoda Y, Nagai Y, Ikuta F. Alterations in brain gangliosides and other lipids of patients with Creutzfeldt–Jakob disease and subacute sclerosing panencephalitis (SSPE). Jpn J Exp Med. 1984;54:229–234. [PubMed] [Google Scholar]
- 2.Arendt T, Holzer M, Gartner U, Bruckner MK. Aberrancies in signal transduction and cell cycle related events in Alzheimer's disease. J Neural Transm. 1998;54:147–158. doi: 10.1007/978-3-7091-7508-8_14. [DOI] [PubMed] [Google Scholar]
- 3.Auffray C, Rougeon F. Purification of mouse immunoglobulin heavy-chain messenger RNAs from total myeloma tumor RNA. Eur J Biochem. 1980;107:303–314. doi: 10.1111/j.1432-1033.1980.tb06030.x. [DOI] [PubMed] [Google Scholar]
- 4.Behl C. Apoptosis in Alzheimer's disease. J Neurol Transm. 2000;107:1325–1344. doi: 10.1007/s007020070021. [DOI] [PubMed] [Google Scholar]
- 5.Bilderback TR, Gazula VR, Dobrowsky R. Phosphoinositide 3-kinase regulates crosstalk between Trk A tyrosine kinase and p75(NTR)-dependent sphingolipid signaling pathways. J Neurochem. 2001;76:1540–1551. doi: 10.1046/j.1471-4159.2001.00171.x. [DOI] [PubMed] [Google Scholar]
- 6.Birkle S, Gao L, Zeng G, Yu RK. Down regulation of GD3 ganglioside and its O-acetylated derivative by stable transfection with antisense vector against GD3-synthase gene expression in hamster melanoma cells: effects on cellular growth, melanogenesis, and dendricity. J Neurochem. 2000;75:547–554. doi: 10.1046/j.1471-4159.2000.740547.x. [DOI] [PubMed] [Google Scholar]
- 7.Brann AB, Scott R, Neuberger Y, Abulafia D, Boldin S, Fainzilber M, Futerman AH. Ceramide signaling downstream of the p75 neurotrophin receptor mediates the effects of nerve growth factor on outgrowth of cultured hippocampal neurons. J Neurosci. 1999;19:8199–8206. doi: 10.1523/JNEUROSCI.19-19-08199.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colell A, Garcia-Ruiz C, Roman J, Ballesta A, Fernandez-Checa JC. Ganglioside GD3 enhances apoptosis by suppressing the nuclear factor-κ B-dependent survival pathway. FASEB J. 2001;15:1068–1070. doi: 10.1096/fj.00-0574fje. [DOI] [PubMed] [Google Scholar]
- 10.Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F, Sortino MA. Mitotic signaling by β-amyloid causes neuronal death. FASEB J. 1999;13:2225–2234. [PubMed] [Google Scholar]
- 11.Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Memo M. Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci. 2001;24:25–31. doi: 10.1016/s0166-2236(00)01663-5. [DOI] [PubMed] [Google Scholar]
- 12.De Maria R, Lenti L, Malisan F, d'Agostino F, Tomassini B, Zeuner A, Rippo MR, Testi R. Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science. 1997;277:1652–1655. doi: 10.1126/science.277.5332.1652. [DOI] [PubMed] [Google Scholar]
- 13.De Maria R, Rippo MR, Schuchman EH, Testi R. Acidic sphingomyelinase (ASM) is necessary for fas-induced GD3 ganglioside accumulation and efficient apoptosis in lymphoid cells. J Exp Med. 1998;187:897–902. doi: 10.1084/jem.187.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dotta F, Previti M, Neerman-Arbez M, Dionisi S, Cucinotta D, Lenti L, Di Mario U, Halban PA. The GM2–1 ganglioside islet autoantigen in insulin-dependent diabetes mellitus is expressed in secretory granules and is not beta-cell specific. Endocrinology. 1998;139:316–319. doi: 10.1210/endo.139.1.5708. [DOI] [PubMed] [Google Scholar]
- 15.Favaron M, Manev H, Alho H, Bertolino M, Ferret B, Guidotti A, Costa E. Gangliosides prevent glutamate and kainate neurotoxicity in primary neuronal cultures of neonatal rat cerebellum and cortex. Proc Natl Acad Sci USA. 1988;85:7351–7355. doi: 10.1073/pnas.85.19.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forloni G, Chiesa R, Smiroldo S, Verga L, Salmona M, Tagliavini F, Angeretti N. Apoptosis mediated neurotoxicity induced by chronic application of β-amyloid 25–35. NeuroReport. 1993;4:523–526. doi: 10.1097/00001756-199305000-00015. [DOI] [PubMed] [Google Scholar]
- 17.Fukumoto S, Mutoh T, Hasegawa T, Miyazaki H, Okada M, Goto J, Furukawa K, Urano T. GD3 synthase gene expression in PC12 cells results in the continuous activation of TrkA and ERK1/2 and enhanced proliferation. J Biol Chem. 2000;275:5832–5838. doi: 10.1074/jbc.275.8.5832. [DOI] [PubMed] [Google Scholar]
- 18.Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F × DP, in β-amyloid-induced neuronal death. J Biol Chem. 1999;274:19011–19016. doi: 10.1074/jbc.274.27.19011. [DOI] [PubMed] [Google Scholar]
- 19.Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N, Robertson GS, Slack RS, Park DS. E2F1 mediates death of β-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem. 2000;275:11553–11560. doi: 10.1074/jbc.275.16.11553. [DOI] [PubMed] [Google Scholar]
- 20.Goldman JE, Reynolds R. A reappraisal of ganglioside GD3 expression in the CNS. Glia. 1996;16:291–295. doi: 10.1002/(SICI)1098-1136(199604)16:4<291::AID-GLIA1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 21.Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- 22.Joseph J, Shukitt-Hale B, Denisova NA, Martin A, Perry G, Smith MA. Copernicus revisited: amyloid beta in Alzheimer's disease. Neurobiol Aging. 2001;22:131–146. doi: 10.1016/s0197-4580(00)00211-6. [DOI] [PubMed] [Google Scholar]
- 23.Kalanj S, Kracun I, Rosner H, Cosovic C. Regional distribution of brain gangliosides in Alzheimer's disease. Neurol Croat. 1991;40:269–281. [PubMed] [Google Scholar]
- 24.Kawai H, Sango K, Mullin KA, Proia RL. Embryonic stem cells with a disrupted GD3 synthase gene undergo neuronal differentiation in the absence of b-series gangliosides. J Biol Chem. 1998;273:19634–19638. doi: 10.1074/jbc.273.31.19634. [DOI] [PubMed] [Google Scholar]
- 25.Kozireski-Chuback D, Wu G, Ledeen RM. Axogenesis in neuro-2a cells correlates with GM1 upregulation in the nuclear and plasma membranes. J Neurosci Res. 1999;57:541–550. [PubMed] [Google Scholar]
- 26.Kristal BS, Brown AM. Apoptogenic GD3 directly induces the mitochondrial permeability transition. J Biol Chem. 1999;274:23169–23175. doi: 10.1074/jbc.274.33.23169. [DOI] [PubMed] [Google Scholar]
- 27.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malisan F, Testi R. Lipid signaling in CD95-mediated apoptosis. FEBS Lett. 1999;452:100–103. doi: 10.1016/s0014-5793(99)00543-8. [DOI] [PubMed] [Google Scholar]
- 29.Manev H, Favaron M, Vicini S, Guidotti A, Costa E. Glutamate-induced neuronal death in primary cultures of cerebellar granule cells: protection by synthetic derivatives of endogenous sphingolipids. J Pharmacol Exp Ther. 1990;252:419–427. [PubMed] [Google Scholar]
- 30.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 31.McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Med Hypotheses. 1999;52:525–527. doi: 10.1054/mehy.1997.0680. [DOI] [PubMed] [Google Scholar]
- 32.Nagy Z, Esiri MM, Smith AD. The cell division cycle and the pathophysiology of Alzheimer's disease. Neuroscience. 1998;87:731–739. doi: 10.1016/s0306-4522(98)00293-0. [DOI] [PubMed] [Google Scholar]
- 33.Ohsawa T, Ikeda H, Senshu T. Effects of ganglioside GM1 on DNA synthesis in isolated nuclei and on the activity of DNA polymerase alpha derived from S-phase HeLa cells. Biochim Biophys Acta. 1988;949:305–310. doi: 10.1016/0167-4781(88)90156-x. [DOI] [PubMed] [Google Scholar]
- 34.Percy AK, Gottfries J, Vilbergsson G, Mansson JE, Svennerholm L. Glycosphingolipid glycosyltransferases in human fetal brain. J Neurochem. 1991;56:1461–1465. doi: 10.1111/j.1471-4159.1991.tb02038.x. [DOI] [PubMed] [Google Scholar]
- 35.Rippo MR, Malisan F, Ravagnan L, Tomassini B, Condo I, Costantini P, Susin SA, Rufini A, Todaro M, Kroemer G, Testi R. GD3 ganglioside directly targets mitochondria in a bcl-2-controlled fashion. FASEB J. 2000;14:2047–2054. doi: 10.1096/fj.99-1028com. [DOI] [PubMed] [Google Scholar]
- 36.Roelen BA, Lin HY, Knezevic V, Freund E, Mummery CL. Expression of TGF-βs and their receptors during implantation and organogenesis of the mouse embryo. Dev Biol. 1994;166:716–728. doi: 10.1006/dbio.1994.1350. [DOI] [PubMed] [Google Scholar]
- 37.Roth KA. Caspases, apoptosis, and Alzheimer disease: causation, correlation and confusion. J Neuropathol Exp Neurol. 2001;60:829–838. doi: 10.1093/jnen/60.9.829. [DOI] [PubMed] [Google Scholar]
- 38.Ryu BR, Choi DW, Hartley DM, Costa E, Jou I, Gwag BJ. Attenuation of cortical neuronal apoptosis by gangliosides. J Pharmacol Exp Ther. 1999;290:811–816. [PubMed] [Google Scholar]
- 39.Saito M, Guidotti A, Berg MJ, Marks N. The semisynthetic ganglioside LIGA20 potently protects neurons against apoptosis. Ann NY Acad Sci. 1998;845:253–262. doi: 10.1111/j.1749-6632.1998.tb09678.x. [DOI] [PubMed] [Google Scholar]
- 40.Saito M, Berg MJ, Guidotti A, Marks N. Gangliosides attenuate ethanol-induced apoptosis in rat cerebellar granule neurons. Neurochem Res. 1999;24:1107–1115. doi: 10.1023/a:1020704218574. [DOI] [PubMed] [Google Scholar]
- 41.Scorrano L, Petronilli V, Di Lisa F, Bernardi P. Commitment to apoptosis by GD3 ganglioside depends on opening of the mitochondrial permeability transition pore. J Biol Chem. 1999;274:22582–22585. doi: 10.1074/jbc.274.32.22581. [DOI] [PubMed] [Google Scholar]
- 42.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 43.Small DH, Mok SS, Bornstein JC. Alzheimer's disease and Abeta toxicity: from top to bottom. Nat Rev Neurosci. 2001;2:595–598. doi: 10.1038/35086072. [DOI] [PubMed] [Google Scholar]
- 44.Sottocornola E, Colombo I, Vergani V, Taraboletti G, Berra B. Increased tumorigenicity and invasiveness of C6 rat glioma cells transfected with the human α-2,8 sialyltransferase cDNA. Invasion Metastasis. 1998;18:142–154. doi: 10.1159/000024507. [DOI] [PubMed] [Google Scholar]
- 45.Svennerholm L, Fredman P. A procedure for the quantitative isolation of brain gangliosides. Biochim Biophys Acta. 1980;617:97–109. doi: 10.1016/0005-2760(80)90227-1. [DOI] [PubMed] [Google Scholar]
- 46.Svennerholm L, Rynmark BM, Vilbergsson G, Fredman P, Gottfries J, Mansson JE, Percy A. Gangliosides in human fetal brain. J Neurochem. 1991;56:1763–1768. doi: 10.1111/j.1471-4159.1991.tb02078.x. [DOI] [PubMed] [Google Scholar]
- 47.Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu G, Lu ZH, Ledeen RW. GM1 ganglioside in the nuclear membrane modulates nuclear calcium homeostasis during neurite outgrowth. J Neurochem. 1995;65:1419–1422. doi: 10.1046/j.1471-4159.1995.65031419.x. [DOI] [PubMed] [Google Scholar]
- 49.Wu G, Lu ZH, Xie X, Ledeen R. Comparison of ganglioside profiles in nuclei and whole cells of NG-108 and NG-CR72 lines: changes in response to different neuritogenic stimuli. Brain Res Dev Brain Res. 2001;126:183–190. doi: 10.1016/s0165-3806(00)00150-4. [DOI] [PubMed] [Google Scholar]
- 50.Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA. Binding of β-amyloid to the p75 neurotrophin receptor induces apoptosis: a possible substrate for Alzheimer's disease. J Clin Invest. 1997;100:2333–2340. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]