

Abstract
Chronic opiate exposure induces numerous neurochemical adaptations in the noradrenergic system, including upregulation of the cAMP-signaling pathway and increased expression of tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine biosynthesis. These adaptations are thought to compensate for opiate-mediated neuronal inhibition but also contribute to physical dependence, including withdrawal after abrupt cessation of drug exposure. Little is known about molecules that regulate the noradrenergic response to opiates. Here we report that noradrenergic locus ceruleus (LC) neurons of mice with a conditional deletion of BDNF in postnatal brain respond to chronic morphine treatment with a paradoxical downregulation of cAMP-mediated excitation and lack of dynamic regulation of TH expression. This was accompanied by a threefold reduction in opiate withdrawal symptoms despite normal antinociceptive tolerance in the BDNF-deficient mice. Although expression of TrkB, the receptor for BDNF, was high in the LC, endogenous BDNF expression was absent there and in the large majority of other noradrenergic neurons. Therefore, a BDNF-signaling pathway originating from non-noradrenergic sources is essential for opiate-induced molecular adaptations of the noradrenergic system.
Keywords: norepinephrine, locus coeruleus, Cre recombinase, loxP, BDNF, opiates, plasticity, cAMP
The noradrenergic system plays a central role in the neurobiology of several major psychiatric diseases, including mood disorders (Aston-Jones et al., 1999), chronic pain conditions, and opiate dependence (Maldonado, 1997). Chronic opiate exposure induces numerous neurochemical adaptations in various neurotransmitter systems (for review, see Williams et al., 2001), including the noradrenergic system. These include increased adenylyl cyclase (AC) expression and upregulation of the cAMP pathway, increased expression of tyrosine hydroxylase (TH), the rate-limiting enzyme in norepinephrine biosynthesis (for review, see Nestler and Aghajanian, 1997), altered neuropeptide expression (Zachariou et al., 2000), and opiate receptor desensitization (for review, see Tsao et al., 2001). These adaptive responses are thought to counterbalance the inhibitory effects of activated opiate receptors and therefore restore the baseline activity of noradrenergic neurons during opiate exposure. However, they also contribute to the development of physical dependence: when drug exposure is stopped abruptly, these adaptations, in conjunction with an increased excitatory afferent input, cause a disinhibition of the noradrenergic system and a withdrawal reaction (Maldonado, 1997; Nestler and Aghajanian, 1997; Saiepour et al., 2001;Williams et al., 2001). Because the α2-adrenoreceptor agonist clonidine and other drugs that downregulate noradrenergic neuron activity are very effective for the treatment of opiate withdrawal (Washton and Resnick, 1983; Maldonado, 1997), it is clear that the disinhibition of noradrenergic neurons is a key factor in the neurobiology of opiate withdrawal.
Very little is known about endogenous factors that regulate opiate-induced adaptations of noradrenergic neurons. Drug-induced adaptations in reward and addiction circuitry could be viewed as a form of neuronal plasticity (for review, see Koob et al., 1998) and thus may depend on similar molecular mechanisms that operate in learning and memory centers such as the hippocampus. For example, neurotrophin-signaling pathways that use molecules from the nerve growth factor family such as BDNF play a key role for some forms of long-term potentiation at hippocampal synapses [Kang and Schuman, 1995; Kang et al., 1997; Minichiello et al., 1999; Patterson et al., 2001 (and references therein)].
Neurons of the locus ceruleus (LC), which is the largest noradrenergic cell cluster in the brainstem, express very high levels of TrkB, the high-affinity receptor tyrosine kinase for BDNF (Numan et al., 1998;King et al., 1999; Yamuy et al., 2000). This may raise the question that BDNF, which is expressed at high levels in adult brain (Maisonpierre et al., 1990), is an important regulator of noradrenergic neuron function. In vitro studies with fetal tissue demonstrated that addition of BDNF to the cell culture medium increases noradrenergic neuron survival (Friedman et al., 1993) and upregulates the expression of noradrenergic markers (Sklair-Tavron and Nestler, 1995). However, in vivo evidence for a role of BDNF in noradrenergic neuron function and plasticity is still lacking. Studies with BDNF-deficient mice would be ideal for these purposes but until recently were hampered by the fact that conventional BDNFnull-mutant mice do not survive into adulthood (Ernfors et al., 1994;Jones et al., 1994). Using a conditional knock-out strategy with Cre/loxP-mediated BDNF gene deletion in postnatal mouse brain (Rios et al., 2001), we analyzed the physiological adaptations to chronic opiate exposure in the absence of BDNF. We report that LC neurons of BDNF-deficient mice had a highly abnormal response to morphine treatment, as evidenced by a profound dysregulation of cAMP-mediated cellular signaling and a failure to upregulate TH expression. These abnormalities occurred in conjunction with a strongly attenuated opiate withdrawal reaction and provide, for the first time, evidence that BDNF is essential for drug-related plasticity in the noradrenergic system.
MATERIALS AND METHODS
Conditional deletion of BDNF in postnatal brain. Details of the generation of the BDNF conditional allele (BDNF2lox) were reported previously (Rios et al., 2001). Briefly, in this line of mice, exon 5 of the BDNF gene was flanked by loxP sites. Exon 5 is the single coding exon ofBDNF, and thus its Cre-mediated deletion results in a nullBDNF allele (Rios et al., 2001). To achieve BDNF gene deletion in postnatal CNS neurons, we used the calmodulin-kinase II (CamK)-cre transgenic mice in which cre expression is under the control of the neuronal CamK promoter (Chen et al., 2001; Fan et al., 2001). Regional and cellular expression patterns were monitored by crossing the CamK-cre transgenic line to a lacZ reporter line for cre/loxP-mediated recombination (Soriano, 1999). In all experiments, 12- to 16-week-old conditional mutants and wild-type (wt) littermate controls were used, in accordance with an approved animal protocol. The genetic backgrounds of all mutant and control mice were mixed but were predominantly 129/Sv. Breeding strategies were chosen to obtain littermates with two functional BDNF alleles (wt/wt or wt/2lox) as control for the BDNF conditional mutants, to minimize the variability in genetic background.
Behavioral studies. For the behavioral studies, a minimum of 10 animals per genotype per test were used. For somatic withdrawal ratings, opiate dependence was induced by a progressive intraperitoneal treatment schedule of morphine every 6–10 hr (day 1, 20–40–60 mg/kg; day 2, 80–100–100 mg/kg; day 3, 100 mg/kg). Withdrawal was precipitated 4–5 hr after the last morphine injection with a subcutaneous injection of naloxone HCl (1 mg/kg). The overall withdrawal score was determined by the total number of checked signs (diarrhea, rhinorhea, lacrimation, abnormal posture, ptosis) and the frequency of each counted sign (wet dog shakes, freezing, grooming, rear, dig, jumping, tremors) over a period of 20 min by a trained observer, who was blind to experimental treatment and genotype.
Acute and chronic opiate tolerance were assessed by measuring nociceptive hot plate latency (56°C) as described by Bohn et al. (2000). In the acute tolerance paradigm, mice were treated with morphine (100 mg/kg, s.c.) or with saline. Twenty-four hours later, the mice were challenged with low-dose morphine (10 mg/kg, s.c.). Hot plate latencies were measured at baseline under drug-naïve conditions and 30 min after the low-dose morphine. To prevent burn injuries and tissue damage, plate exposure was kept to a 30 sec artificial maximum time. Data were recorded as the “maximum possible effect” (%MPE): 100% × [(drug response time − basal response time)/(30 sec − basal response time)]. In the chronic tolerance paradigm, mice were treated daily with 10 mg/kg morphine subcutaneously for 5 d. Each day, antinociception was measured 30 min after the morphine injection, using the hot plate assay as described above. Statistical differences between mutants and controls were calculated by using two-tailed t tests.
Electrophysiology. A minimum of 12 animals was used for each genotype and animal treatment (saline or chronic morphine). Morphine dependence was induced by a subcutaneous implantation of a single 75 mg morphine pellet (Bohn et al., 2000), and recordings were conducted 65 hr after implantation. In pilot experiments, we compared electrophysiological adaptations of pellet-treated animals with those treated with repeated daily injections of morphine (total morphine dose 500 mg/kg). Results were indistinguishable between these two morphine administration protocols, and here we report the data using the pellet protocol.
Electrophysiological recordings from LC neurons were performed as described previously (Lane-Ladd et al., 1997). Brainstem slices were transferred onto the stage of a gas–liquid interface brain slice chamber under constant flow of humidified 95% O2/5% CO2 and physiological buffer. The A6 (LC) were visually identified, and single-unit extracellular potentials were recorded with glass microelectrodes filled with 2 m NaCl. Recordings began not earlier than 90 min after setting up the slice preparation. This waiting period ensured complete wash-out of morphine in the drug-treated animals, as described elsewhere (Kogan et al., 1992). Transient inhibition of spontaneous firing after brief application of the short-lived, endogenous opiate receptor agonist enkephalin (100 μm) was used to confirm that recordings were done from LC neurons. Consecutive cells were sampled by multiple electrode tracks randomly positioned within the A6 and recorded for a minimum of 3–5 min to ensure that the firing rates were stable. Firing rates of LC neurons were recorded before and during bath application (for at least 10 min) of forskolin (10 μm). At this forskolin concentration, maximum electrophysiological responses from A6 neurons are elicited. Additional recordings were conducted in the presence or absence of the opiate receptor antagonist naloxone (1 μmol). Furthermore, μ-(opioid) receptor inhibition was examined by dose–response curves with the synthetic agonist [d-Ala2, NMePhe4, Gly-ol]-enkephalin (DAMGO). For statistical analysis, the firing rates of 10–14 neurons from a single LC were averaged and considered as a single data point. Statistical significance of differences between mutants and controls was then tested by simple, two-tailed t tests.
cAMP ELISA. Mice were treated with saline or with morphine intraperitoneally three times per day (day 1, 20–40–60 mg/kg; day 2, 80–100–100 mg/kg; day 3, 100 mg/kg) with a total dose of 500 mg/kg during 65 hr. Animals were killed, and tissue from the dorsal pons in the area of the LC was homogenized by polytron in 10 mm Tris, pH 7.4, 5 mm EDTA buffer and pooled (six animals/experimental group and treatment), and membrane extracts were prepared by centrifugation at 20,000 ×g for 15 min at 4°C. Membranes were washed repeatedly in this buffer, and then adenylyl cyclase activity was assessed using a protocol that is similar to the one described by Hausdorff et al. (1990). First, membranes were resuspended in 2× assay buffer (75 mm Tris, pH 7.4, 15 mmMgCl2, 2 mm EDTA, 500 μmIBMX). Membrane protein aliquots (10 μg per tube) were then assayed for forskolin-stimulated adenylyl cyclase activity for 10 min at 25οC in 1× assay buffer containing 50 μm GTP, 100 μm ATP, 2.7 mm phosphoenolpyruvate, 20 U/ml myokinase (Sigma), 4 U/ml pyruvate kinase, and 10 μmforskolin (Calbiochem) in a final volume of 50 μl. Samples were frozen at −20°C for a period of 12 hr, and then the concentration of cAMP was quantified using the BIOTRAK EIA system (RPN 225; Amersham Biosciences) according to the manufacturer's instructions (Protocol I: non-acetylation EIA procedure; see RPN 225 manual). Assays were conducted in triplicate, and animals of each genotype and treatment were assayed in parallel experiments. Statistical significance of differences was calculated using two-tailed t tests.
Western blots. A minimum of five animals per genotype per treatment were used. Mice were anesthetized with isofluorane on days 1 and 3, morphine pellets (25 mg; NIDA) were implanted subcutaneously, and the incision was closed with wound clips. Sham animals were anesthetized similarly and incised but did not receive pellets. Mice were decapitated on day 5, and 14 gauge needle punch samples of the locus ceruleus were obtained from chilled 1 mm coronal slabs of brain.
Relative tyrosine hydroxylase protein levels were determined by Western blot analysis. Briefly, samples were solubilized by sonication in buffer containing 0.5% SDS, lima bean trypsin inhibitor (10 μg/ml), leupeptin (10 μg/ml), phenylmethylsulfonyl fluoride (15 μg/ml),N-p-Tosyl-l-phenylalanine chloromethyl ketone (15 μg/ml), Nα-p-Tosyl-lysine chloromethyl ketone (15 μg/ml), and MG-132 (10 μm). Protein concentrations were determined by the method of Lowry with bovine serum albumin as standard, and for each sample 20 μg of protein was electrophoresed on a denaturing 7% polyacrylamide gel and transferred to nitrocellulose. Subsequently, nitrocellulose membranes were blocked in PBS containing 3% non-fat milk and 0.1% Tween 20. TH was detected by incubating overnight at 4°C in rabbit polyclonal anti-tyrosine hydroxylase antiserum (1:2500; Chemicon, Temecula, CA), washing repeatedly with PBS/01.% Tween 20 (PBS-T), and incubating in horse radish peroxidase-conjugated goat anti-rabbit (1:5000; Vector Labs, Burlingame, CA) for 30 min at room temperature. Immunoreactive bands were detected by enhanced chemiluminescence and quantitated densitometrically using NIH image software. Subtracted background densities were obtained immediately below each band. Relative levels of protein were analyzed by ANOVA followed by post hoc analysis with Tukey's multiple comparison test.
To control for sample loading, membranes were reprobed with a rabbit polyclonal antibody recognizing catalytic subunits α and β of protein kinase A (PKA-c) (1:1000; Santa Cruz Biotechnology), using a procedure similar to the one described for TH immunolabeling. We choose PKA-c immunolabeling as a control because we observed that in mice, PKA-c levels are not differentially regulated between drug-naïve and morphine-treated animals.
Immunohistochemistry. For immunohistochemical experiments, drug-naïve mutant and control mice were compared (minimum of five animals per genotype). All mutant and controls brains were perfusion-fixed with phosphate-buffered 4% paraformaldehyde, postfixed in the same fixative for 2 hr, then cryoprotected in phosphate-buffered 30% sucrose and frozen in dry ice. Blocks containing pons and medulla were cut in series of 15-μm-thick coronal sections, which were permeabilized with 0.3% Triton X-100 in 0.1 mPO4, pH 7.4. Free-floating sections were incubated at room temperature for 12–24 hr, with a mouse monoclonal antibody against tyrosine hydroxylase (1:1000; Incstar), in combination with rabbit antiserum against phospho-cAMP-responsive DNA element binding protein (CREB) (1:500; Upstate Biotechnology). Sections were washed repeatedly, incubated at room temperature for 60 min with two secondary antibodies (FITC-coupled horse anti-mouse antibodies and Texas Red-coupled goat anti-rabbit; 1:200; both from Vector Labs), and then washed repeatedly, mounted on slides, and visualized with a confocal microscope (Zeiss) and LSM (Zeiss) software.
Additional immunohistochemical experiments were conducted in mice in which the single coding exon of BDNF had been replaced with alacZ reporter coding sequence (Farinas et al., 2001). Coronal sections from the area of the pons and of the medulla of adult BDNF/lacZ reporter mice brains were prepared as described above, processed for immunohistochemistry using the anti-TH antibody (1:1000; Incstar) and a rabbit anti-lacZ antiserum (1:500; Molecular Probes), and then further processed with secondary antibodies using a protocol similar to the one described above.
Cell counting studies. Drug-naïve mutant and control animals were used for cell counting studies (minimum of five animals for each genotype). Brains were processed as described above, and for each brain the entire pons and medulla were cut into series of 15-μm-thick free-floating sections and processed for TH immunohistochemistry, mounted on slides, and counterstained with 4′,6-diamidino-2-phenylindole for labeling of nuclei. Every second section was used for counting. Because both A6 and A2 showed considerable variability in neuronal distribution and density along the rostrocaudal axis in all mutants and controls, optical dissector and volume-based cell counting methods that rely on constant cell densities in a given volume were inaccurate for the present study. Instead, TH-immunoreactive neurons cut through the level of the nucleus were counted in each section, and total neuronal numbers were calculated by adding counts of single sections. The noradrenergic cell group A2 was localized using the detailed cytoarchitectonic descriptions of Kalia et al. (1985).
In situ hybridization. Brains of wild-type mice were perfusion fixed with phosphate-buffered 4% paraformaldehyde, incubated overnight in 10% phosphate buffered formalin, cut into blocks, and embedded in paraffin. Coronal sections (8 μm thick) were obtained from blocks containing the dorsal pons, mounted on Fisher Superfrost slides, and processed for in situ hybridization histochemistry as described by Tessarollo and Parada (1995). Briefly, slides were deparaffinized in xylene and rehydrated in graded (100–30%) ethanol solutions. After fixing in 4% paraformaldehyde, the tissues were pretreated with proteinase K (20 μg/ml) (Boehringer Mannheim), refixed, immersed in triethanolamine buffer containing acetic anhydrate, and dehydrated. Sections were hybridized with antisense cRNA probes (5 × 105 cpm) in a buffer containing 50% formamide, 0.3 m NaCl, 20 mm Tris-Cl, pH 7.4, 1× Denhardt's solution, 0.5 mg/ml yeast tRNA, and 10 mm DTT at 50°C for 20 hr. After hybridization, washes were performed in 4× SSC and 10 mm DTT at 50°C. The slides were then incubated for 30 min at 37°C with RNase A (20 μg/ml) and RNase T1 (2 μg/ml) followed by a 30 min incubation at 55°C in 50% formamide, 0.2× SSC, 10 mm DTT, washed twice for 30 min in 0.2× SSC, 1% NaPyrophosphate (w/v), 10 mm DTT, and dehydrated. The slides were dipped in Kodak emulsion NTB-2 and exposed for 7 and 14 d at 4°C for the TrkB and BDNF probes, respectively. The slides were then developed in Kodak D-19, fixed as recommended by the manufacturer. Sense and antisense probes labeled with 35S were prepared by standard procedures by using UTP as the labeled nucleotide. The BDNF probe was synthesized from a full-length BDNF-specific rat cDNA. The TrkB probe was derived from a 450 bp fragment from the extracellular domain, as described in detail by Klein et al. (1989), that recognizes the active kinase receptor and also truncated forms (Middlemas et al., 1991). Because it has been shown recently that there appears to be signaling going through the truncated forms of the Trk B receptor (Yacoubian and Lo, 2000), it is important to look at expression localization of all forms of the receptor.
RESULTS
Conditional deletion of BDNF
The spatial and temporal distribution pattern of CamK-cre-mediated recombination was examined in mice carrying the CamK-cre transgene and a lacZ reporter for Cre/loxP-mediated recombination (Soriano, 1999). In 2.5-week-old transgenic mice, the CamK-cre-mediated gene deletion was widespread in the forebrain, midbrain, and hindbrain, with the exception of the cerebellar cortex (Fig.1A). As described previously (Chen et al., 1991), the lacZ+, blue cells were morphologically CNS neurons, confirming the neuronal specificity of the CamK-cre transgene. Cre-mediated recombination was also detected in several noradrenergic nuclei, including the locus ceruleus (A6) in the dorsal pons (Fig. 1B, LC) and the A2/C2 group in the dorsal medulla.
Fig. 1.

Regional pattern of CamIIK-cre transgene expression. A, Representative parasagittal sections of P18 (left) and E19 animal (bottom right) showing widespread activation of the Rosa-26 Cre/lox lacZreporter by the CamIIK-cre transgene in P18 but not in E19 animal.B, Representative coronal section of dorsal pons of adult mouse, showing activation of the Rosa-26 Cre/lox lacZreporter in the locus ceruleus (LC) and surrounding structures by the CamIIK-cre transgene. Notice that the CamIIK-cre transgene is not active in mesencephalic trigeminal tract nucleus and cerebellar cortex. CB, Cerebellar cortex;CC, cerebral cortex; LC, locus ceruleus;MeV, mesencephalic trigeminal tract nucleus;OB, olfactory bulb; R, rostral;V, ventral; asterisk, IVth ventricle. Scale bars: A, 2.5 mm; B, 0.25 mm.
In perinatal brain, few blue cells were observed (Fig.1A), confirming previous observations that the bulk of recombination in this CamK-cre transgenic line occurs after birth (Chen et al., 2001; Fan et al., 2001). The spatial and temporal distribution pattern of the lacZ-positive cells in theCamK-cre;lacZ reporter transgenic mice is also in accordance with Southern blot analysis for the deletion of the conditional BDNF allele in various brain regions ofCamK-cre;BDNF conditional mutants (Rios et al., 2001). Adult conditional BDNF mutants were recovered at the expected Mendelian ratio, indicating that BDNF deficiency in postnatal CNS neurons did not affect animal viability.
Attenuated opiate withdrawal in BDNF mutant mice
We examined opiate withdrawal behavior in mutant and control mice. A withdrawal reaction was induced in 12- to 16-week-old mice after 65 hr of a progressive treatment of morphine (total dose 500 mg/kg) with a subcutaneous injection of naloxone (1 mg/kg) 4 hr after the last morphine injection. Wild-type mice showed symptoms characteristic of opiate withdrawal, including signs of autonomic hyperactivity, wet dog shakes, forepaw tremors, digging, and other withdrawal-related behaviors. The total withdrawal score for the mutant mice was threefold lower in comparison to age-matched littermate controls [total score mutant 18.3 ± 4.0 (mean ± SE); controls 53.7 ± 3.2;p < 0.01] (Fig. 2A). As shown in Figure 2B, the BDNF conditional mutant mice scored, in comparison to wild-type mice, 40% lower on ratings for autonomic hyperactivity. Furthermore, mutants had a threefold decrease in various behavioral symptoms, including tremors (Fig. 2C) and wet dog shakes (Fig.2D). Similar results were obtained when withdrawal was induced 65 hr after implantation of a single, 75 mg morphine pellet (data not shown).
Fig. 2.
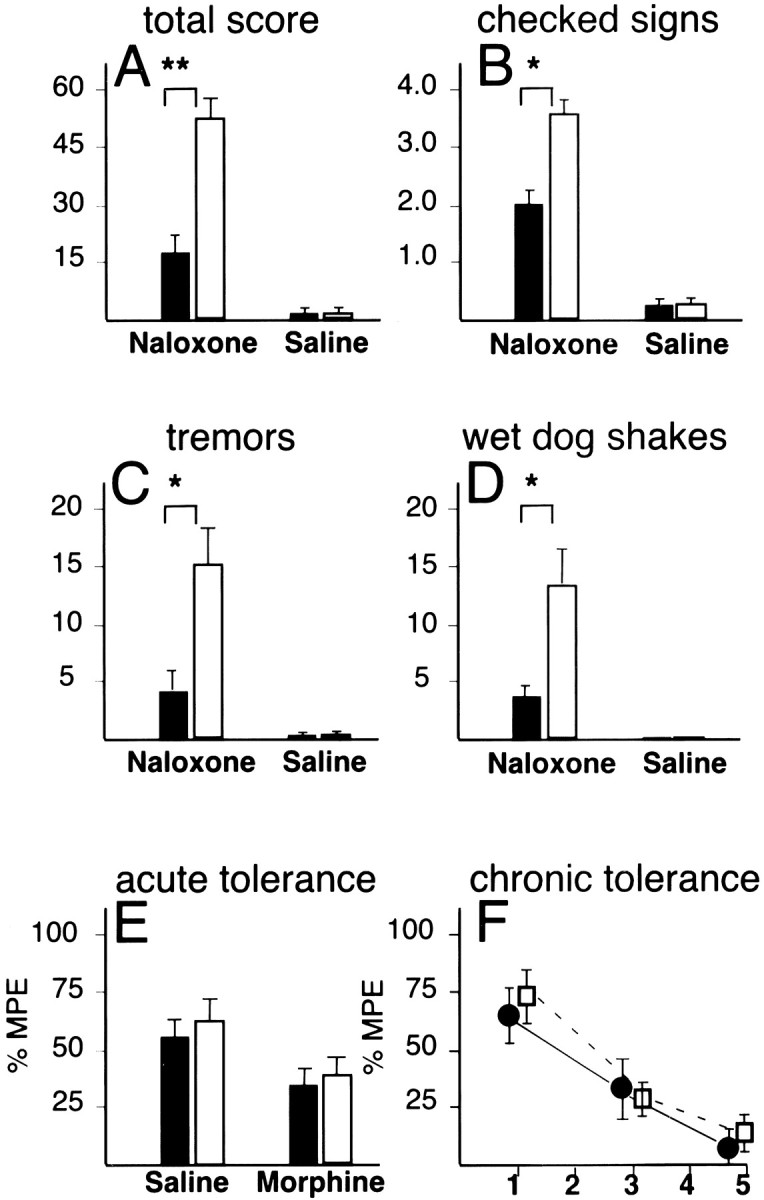
Attenuated opiate withdrawal reactions inBDNF mutant mice. A–D summarize the observer-based ratings of naloxone-precipitated withdrawal inBDNF conditional mutant mice (black bars) in comparison to littermate wild-type controls (open bars) after chronic morphine administration (500 mg/kg, total dose).A, Total overall withdrawal score; B, total number of checked signs (diarrhea, rhinorhea, lacrimation, ptosis, abnormal posture); C, tremors; D, wet dog shakes; over a 20 min period. Differences between naloxone-treated mutants and controls in A–D are significant (t tests; *p < 0.05, **p < 0.01). E, Acute antinociceptive tolerance in BDNF conditional mutants and controls, measured as maximum possible effect (%MPE) of hot plate latencies 30 min after administration of morphine (10 mg/kg, i.p.) and 24 hr after a one-time challenge with saline or morphine (100 mg/kg). F, Chronic antinociceptive tolerance inBDNF conditional mutants and controls, measured as %MPE in hot plate assay 30 min after administration of morphine (10 mg · kg−1 · d−1) over the course of 5 d. Filled bars, Mutants;open bars, controls. Data are expressed as mean ± SE.
This attenuated withdrawal reaction in the mutant mice could be caused by either a generalized failure to develop tolerance or a dysfunction in the neural circuitry mediating withdrawal, or both. To distinguish between these two possibilities, we examined morphine tolerance to a single low dose (10 mg/kg) after a single high-dose challenge (100 mg/kg) administered 24 hr previously. Both mutant and control mice exhibited a 40% decrease in antinociception after a single high-dose challenge (Fig. 2E). Next, we monitored the development of tolerance over the course of several days, by subjecting mice to daily injections of 10 mg/kg morphine and measuring antinociception with the hot plate assay. Both mutant and control mice developed a greater than threefold decline in morphine responsiveness over the course of 5 d (Fig. 2F). Therefore, the attenuated withdrawal observed in the BDNF-deficient mice is not caused by a generalized failure to develop tolerance.
BDNF deficiency results in a paradoxical downregulation of cAMP signaling after chronic opiate exposure
The noradrenergic system plays a key role in the neurobiology of opiate withdrawal, and therefore a dysfunction of noradrenergic signaling in BDNF-deficient mice could contribute to the observed withdrawal deficits. To examine whether genetic BDNF ablation alters LC noradrenergic neuron function, we conducted single-unit electrophysiological recordings from brainstem slice preparations. We first examined the function of G-protein-coupled opiate receptors that mediate inhibition of noradrenergic neurons. Activation of the μ-(opioid) receptor is well known to decrease the firing rate of LC neurons (Williams et al., 1988). When we studied the suppression of spontaneous neuronal firing by successively increased concentrations of the synthetic μ-(opioid) receptor agonist DAMGO in LC neurons, no reduction in response was found in drug-naïve mutants as compared with controls (Fig. 3). In addition, application of 100 μm enkephalin to the perfusate resulted in complete cessation of neuronal firing in all mutants and control neurons (data not shown). We conclude that the mechanisms of opiate-mediated neuronal inhibition are preserved in the LC of BDNF-deficient mice.
Fig. 3.

Opiate receptor-mediated inhibition is preserved in mutant LC. Dose–response curve for μ-(opioid) receptor agonist DAMGO, measured as percentage suppression of baseline firing rate (y-axis) after successive increase in DAMGO concentration in perfusate (x-axis) in mutant (●) and control (▪) mice. Note similar dose–response curves in mutant and control. Data shown as mean ± SE.
Superactivation of the cAMP pathway after chronic morphine treatment has been studied as a cellular hallmark of opiate withdrawal in animals (Nestler and Aghajanian, 1997; Chieng and Williams, 1998) and in cell culture [Finn and Whistler, 2001 (and references therein)]. We hypothesized that the attenuated withdrawal reaction of the BDNF-deficient mice could be caused by altered cAMP signaling in noradrenergic neurons. To examine this, we recorded from single units in saline- and morphine-treated animals in the absence and presence of forskolin (10 μm) in the perfusate. Forskolin activates adenylyl cyclases that generate cAMP, resulting in activation of nonselective ion channels and neuronal excitation (Nestler and Aghajanian, 1997).
Representative tracings from saline-treated wild-type and mutant LC neurons are shown in Figure 4,A and B, respectively. Baseline LC neuron firing rates of wild-type and mutant mice were not significantly different [controls 0.52 ± 0.14 (SE) Hz or 5.2 ± 1.4 spikes/10 sec vs mutants 0.42 ± 0.11 Hz) (Fig.4E,F). When forskolin was added to the slice perfusate, neuronal firing in saline-treated wild-type and saline-treated mutant LC neurons was increased more than threefold (Fig.4A,B,E,F). Therefore, the cAMP-signaling pathway in mutant LC neurons appeared to be normal when studied under drug-naïve conditions.
Fig. 4.

Morphine downregulates cAMP signaling in BDNF-deficient mice. A–D, Representative traces of single-unit recordings of LC neurons in wild-type animals (A, C) and BDNF conditional mutant mice (B, D) treated with saline (A, B) or chronic morphine (C, D). The y-axis shows firing rate (spikes/10 sec); the horizontal axis represents running time (calibration: 5 min); arrows mark time period when forskolin (10 μm) was added to the perfusate. Notice decrease in firing in conditional mutant (D).Bar graph in E summarizes recording data for saline- and morphine-treated controls, and bar graphin F summarizes recording data for saline- and morphine-treated BDNF mutants. Open bars inE and F show neuronal firing rate in the absence of forskolin; black bars indicate neuronal firing rate with forskolin (Fo) added to the perfusate.G summarizes forskolin-stimulated AC enzymatic activity (picomoles per milligram of tissue per minute) in saline- and morphine-treated conditional mutants (hatched bars) and controls (diagonal line bars). Sal., Saline; Morph., morphine. Data shown as mean ± SE. *p < 0.05; **p < 0.01 (t tests).
To examine the superactivation of the cAMP pathway, we next recorded from LC neurons of mice that were treated for 72 hr with morphine pellets. Representative tracings from morphine-treated wild-type and mutant neurons are shown in Figure 4, C and D, respectively. The firing rate of wild-type LC neurons in the absence of forskolin in the perfusate was 0.72 ± 0.21 Hz (Fig.4C,E). Addition of forskolin to the perfusate caused a robust increase in neuronal firing in morphine-treated wild-type mice that was, on average, stronger than the forskolin-induced neuronal firing in saline-treated wild-type mice (2.55 ± 0.46 vs 1.63 ± 0.20 Hz) (Fig.4E). This increase was significant (p < 0.05). This upregulation of the cAMP pathway in morphine-treated wild-type mice is consistent with previous results in rats (Kogan et al., 1992; Osborne and Williams, 1996;Lane-Ladd et al., 1997) and in mice (Akbarian et al., 2001). Unexpectedly and in striking contrast to morphine-treated controls, LC neurons of morphine-treated conditional mutants had a very low spike rate at baseline (0.23 ± 0.06 Hz) (Fig.4D,F). After addition of forskolin to the perfusate, the absolute spike rate of morphine-treated mutant LC neurons was 0.90 ± 0.11 Hz (Fig.4D,F), which represents a twofold decrease in comparison to saline-treated mutants (Fig. 4, compareB,D,F) and a threefold decrease in comparison to morphine-treated controls (Fig.4E). These differences were significant (p < 0.05–0.01).
Thus, morphine treatment in wild-type animals induced an expected significant increase in cAMP-mediated excitation but induced a paradoxical significant decrease in cAMP-mediated excitation in the BDNF-deficient mice. The dramatic decrease in neuronal firing in the mutant LC is not explained by an abnormal activation of inhibitory opiate receptors because all slices were subjected to a 90 min wash-out period to remove residual morphine. Furthermore, we repeated the electrophysiological recordings after adding 1 μmnaloxone, an opioid receptor antagonist, to the perfusate. We found that naloxone did not increase the firing rate in slices from morphine-treated mutant or control mice (data not shown), in agreement with previous studies on morphine-treated rats (Kogan et al., 1992). Therefore, we conclude that chronic morphine administration results in failure to upregulate the cAMP signal transduction pathway in the BDNF mutant mice.
Opiate-induced superactivation of the cAMP pathway is caused, in part, by increased expression and activity of several ACs (for review, seeNestler and Aghajanian, 1997). To examine whether a dysregulation in AC enzymatic activity contributes to the paradoxical downregulation of cAMP-mediated excitation in the BDNF mutant mice, we measured AC activity in tissue extracts of the dorsal pons at the level of the LC. Tissue from wild-type mice showed a 1.7-fold increase in AC activity after chronic morphine treatment, but this upregulation was completely absent in morphine-treated mutants (Fig. 4G). Therefore, lack of AC upregulation contributes to the neuronal hypoactivity and the altered cAMP signaling in the morphine-treated mutant mice.
BDNF is required for opiate-induced upregulation of tyrosine hydroxylase
TH is the rate-limiting enzyme in norepinephrine biosynthesis. During chronic morphine exposure, TH expression is upregulated in noradrenergic neurons of rats and mice, presumably as a compensatory mechanism for increased neuronal inhibition (Boundy et al., 1998). To examine whether BDNF is required for this dynamic regulation of TH, we compared TH expression in LC of wild-type and BDNF-deficient mice. Morphine pellets (25 mg, s.c.) were administered on days 1 and 3 to mutant and control mice that were killed later on day 5. Changes in TH levels in microdissected LC were examined and compared with those of drug-naïve, sham-operated animals using Western blot analysis. At baseline, under drug-naïve conditions, TH immunoreactivity was similar in wild-type and mutant LC (Fig.5A,B). Morphine-treated wild-type mice showed a robust increase in TH expression in comparison to drug-naïve wild-type mice (Fig.5A). This on average 2.5-fold increase in TH protein content was significant (p < 0.05) (Fig.5B). In contrast, TH immunolabeling in morphine-treated conditional BDNF mutant mice was similar to the labeling observed in drug-naïve mutant and control mice (Fig. 5A), and no significant differences in TH immunolabeling between drug-naïve and morphine-treated mutant mice were observed (Fig. 5B). We conclude that opiate-induced upregulation of TH requires BDNF.
Fig. 5.

Noradenergic neurons of BDNF-deficient mice lack dynamic regulation of TH. A, Representative Western blot showing TH immunolabeling and PKA (catalytic subunit) immunolabeling as loading control in LC of wild-type and BDNF-deficient mutant mice chronically treated with morphine (+) or sham (−) surgeries. Notice increased TH immunolabeling in morphine-treated wild type but not in morphine-treated mutant. B, Relative TH-immunoreactivity (y-axis) as determined densitometrically in mutant and wild-type mice. Bars with diagonal lines indicate drug-naïve animals (sham surgery), andhatched bars indicate morphine-treated mice. Data are expressed as mean ± SE. *p < 0.05.
BDNF-deficient mice maintain normal numbers of noradrenergic neurons
In cell culture studies, BDNF has been implicated in the survival of noradrenergic neurons (Friedman et al., 1993). To determine whether increased death of noradrenergic neurons contributes to the attenuated opiate withdrawal reaction in the BDNF-deficient mice, we analyzed total neuronal numbers in noradrenergic cell groups of adult mutant and controls. Coronal sections from the brainstem of BDNFconditional mutant mice (Fig.6A) and controls (Fig.6D) were immunostained for TH. Total numbers of LC (A6) neurons in the dorsal pons and of A2 neurons in the medial solitary tract nucleus, as outlined by Kalia et al. (1985), were counted. There was no significant difference between mutants and controls in the number of A6 neurons (controls, 933 ± 76; mutants, 866 ± 57) and A2 neurons (controls, 265 ± 91; mutants, 313 ± 110) (Fig. 6F). We conclude that BDNF is not essential for survival of noradrenergic neurons in adult brain.
Fig. 6.

Total numbers of noradrenergic neurons are maintained in BDNF-deficient mice. A–E, Representative examples of coronal sections, double stained for TH and for CREB immunoreactivity. LC of BDNF mutant (A) and littermate control (D) shows Texas Red-labeled TH immunoreactive neurons that show robust expression of FITC-labeled phospho-CREB (B, E). Note robust expression of CREB in nuclei of mutant (C). F, Total numbers of TH+ neurons (y-axis) in unilateral LC (A6) and A2 noradrenergic cell groups. Filled bars, BDNF mutant mice;open bars, wild-type littermate controls. Images were taken at 10 × 20 magnification (A, B, D, E) and at 100 × 10 magnification (C).
CREB is a transcription factor known to be expressed by noradrenergic neurons (Widnell et al., 1994) and many other CNS neurons (Takeda et al., 1991; Konradi and Heckers, 1995). In peripheral sensory and sympathetic neurons, normal levels of neurotrophins are required to maintain phosphorylated CREB, which promotes gene expression (Riccio et al., 1999; Watson et al., 1999). To examine whether BDNF deficiency alters CREB expression in central noradrenergic neurons, we conducted immunohistochemical studies using an antibody that specifically recognized phosphorylated CREB. Immunoreactivity was fully preserved in LC (Fig. 6B,C), A2, and all other noradrenergic cell groups of the BDNF mutant mice. No detectable differences in immunohistochemical staining were found in comparison to controls (Fig. 6E), suggesting that central noradrenergic neurons do not require normal levels of neurotrophins to maintain CREB phosphorylation.
Regional distribution pattern of BDNF expression implies target-derived supply for noradrenergic neurons
Our studies suggest that BDNF is essential for drug-induced molecular adaptations of the noradrenergic system. Noradrenergic neurons are known to have widespread projections throughout the CNS and thus could derive BDNF from non-noradrenergic neuronal populations or express BDNF themselves, or both. To examine these possibilities, we analyzed by in situ hybridization the mRNA expression pattern for BDNF and TrkB (the BDNF high-affinity receptor) in the area of the wild-type LC. In emulsion-dipped sections from wild-type pons hybridized with radiolabeled TrkB antisense RNA, the strongest labeling was observed over the full dorsoventral extension of the locus ceruleus (Fig. 7A,arrows), whereas labeling in other areas of the dorsal pons and in the overlying cerebellar cortex was less intense. In sections from wild-type pons hybridized with radiolabeled BDNF antisense RNA, the strongest labeling was observed in the granule cell layer of the cerebellar cortex, whereas labeling in the dorsal pons was more scattered and included a loose cluster of labeled cells in the vicinity of the ventral border of the locus ceruleus (Fig.7B,C, arrows). These results are in accordance with previous studies describing BDNF mRNA-expressing cells in the dorsal pons by means of film and emulsion autoradiography (Castren et al., 1995; Conner et al., 1997; Numan et al., 1998).
Fig. 7.

Expression pattern of BDNF and TrkB in dorsal pons. A–C, Representative examples of emulsion-dipped coronal sections of adult wild-type mice hybridized with TrkB cRNA (A) and BDNF cRNA (B, C). Notice very strong TrkB mRNA expression over the locus ceruleus (A, arrows) and BDNF mRNA-expressing cells in ventrolateral position to IVth ventricle (B, C, arrows). D–F show coronal sections of mice carrying a lacZ reporter knock-in into the endogenous BDNF locus (Farinas et al., 2001). Sections were double stained with Texas Red-labeled lacZ and FITC-labeled TH immunoreactivity.D, Dorsal pons in area of the LC; E, higher magnification of D;arrow inD and E points to the same neuron.F, Dorsal medulla in the area of A2.Arrow in F points to double-labeled neuron (TH+, lacZ+). Notice lack of double-labeled neurons in the LC (D, E).CBL, Cerebellum; asterisk, IVth ventricle. Images taken at 2 × 10 magnification inA; 4 × 10 magnification in B, C; 10 × 10 magnification in D; 100 × 10 magnification in E, F.
To examine whether the BDNF-expressing cells in the dorsal pons overlap with the population of noradrenergic LC neurons, we used mice that express the lacZ reporter coding sequence under the control of the endogenous BDNF promoter (Farinas et al., 2001). Sections that were processed for lacZ immunoreactivity containedlacZ+ cells in an area medial to the LC (Fig. 7D) that corresponded to the area that contains BDNF mRNA-expressing cells in wild-type mice (Fig.7B,C, arrows). In sections that were double labeled for lacZ and TH immunoreactivity, lacZ+ cells were found in close spatial proximity medial to TH+ neurons, but double-labeled cells were not observed in the area of the LC (Fig.7D,E). A similar analysis in sections obtained from the dorsal medulla revealed that a small portion (<15%) of catecholaminergic cells in the area of A2 werelacZ+ positive (Fig. 7F). We conclude that most of the noradrenergic neurons, including the entire population of LC neurons, do not express BDNF. However, gene expression for TrkB, the BDNF high-affinity receptor, is comparatively high in the LC.
DISCUSSION
We report genetic evidence for a role of BDNF in noradrenergic neuron function and opiate withdrawal. Loss of BDNF expression in postnatal brain dramatically alters the response of the noradrenergic system to chronic opiate exposure, as evidenced by an abnormal decrease in neuronal firing rates, a paradoxical downregulation of cAMP-mediated excitation, and a failure to upregulate levels of enzymes that are important for noradrenergic signal transduction, including adenylyl cyclase and tyrosine hydroxylase, the rate-limiting enzyme for norepinephrine biosynthesis. In contrast to the profound abnormalities after chronic opiate exposure, no abnormalities were found in LC neurons of drug-naïve BDNF-deficient mice. Notably, wild-type LC neurons do not express BDNF, but they express high levels of TrkB, the high-affinity receptor for BDNF. These results suggest that opiate-induced adaptations of the noradrenergic system depend on a BDNF-signaling pathway that emanates from non-noradrenergic target cell populations or afferent projections.
BDNF deficiency causes an abnormal cellular adaptation to opiate exposure
Upregulated cAMP signaling in noradrenergic neurons restores neuronal activity and compensates for cellular inhibition caused by activated opiate receptors but also contributes to noradrenergic disinhibition and withdrawal behavior when opiate exposure is stopped abruptly (for review, see Nestler and Aghajanian, 1997). Drugs disrupting cAMP-mediated signaling, such as the PKA inhibitor Rp-cAMPS, when infused directly into the LC, attenuate opiate withdrawal reactions (Punch et al., 1997) and suppress the withdrawal hyperactivity of LC neurons (Ivanov and Aston-Jones, 2001). Conversely, drugs that activate the cAMP pathway, such as the PKA activator Sp-cAMPS, induce withdrawal-like behaviors when infused into the LC (Punch et al., 1997). Therefore, we predicted that the threefold reduction in opiate withdrawal behaviors in the BDNF-deficient mice could be caused by a dysregulation of cAMP-mediated excitation in the mutant noradrenergic neurons. This is what we observed. Chronic opiate exposure resulted in a significant reduction in neuronal firing, a paradoxical downregulation of cAMP-mediated excitation, and a failure to upregulate AC enzyme activity in mutant LC neurons.
Activated opiate receptors inhibit the cAMP pathway by the release of the GTP-bound Gαi subunits, blocking AC enzyme activity. The resulting decrease in cAMP levels then inhibits nonselective cation channels, causing a downward shift of the intracellular potential (for review, see Williams et al., 2001) (Fig.8). Interestingly, the reduction in LC neuron firing in the drug-treated mutant mice persisted even after a complete wash-out of morphine from the slice preparation. This finding suggests that chronic opiate exposure resulted in other maladaptations in the mutant LC neurons, in addition to the dysregulation of AC levels. Abnormal activation of potassium channels or inhibition of voltage-sensitive Ca2+ channels, which are regulated by Gβ/γ subunits (North et al., 1987; Williams et al., 2001), and altered endocytosis (Tsao et al., 2001) or constitutive activation of opiate receptors (Liu and Prather, 2001) may contribute to the observed inhibition of mutant LC neurons after chronic morphine treatment. Given the fact that the recordings were conducted on coronal slice preparations that disrupt afferent inputs of the LC, it is unlikely that the observed decrease in LC neuronal firing in the mutants is caused by an imbalance between GABAergic and glutamatergic excitatory signal transmission (Van Bockstaele et al., 2000) to the LC. At present, it is unclear whether a defect in LC neuron synchronization (Zhu and Zhou, 2001) contributes to the observed alterations in the mutant LC.
Fig. 8.

A model whereby neurotrophins regulate opiate-induced adaptations in noradrenergic neurons. Ligand-activated opiate receptors, through GαI subunits, inhibit adenylyl cyclase (AC) activity, and this results in decreased depolarization currents through nonselective cation channels (NSC). Opiate receptors reduce neuronal activity further through Gβ/γ subunits that activate potassium channels and inhibit voltage-sensitive calcium channels (Williams et al., 2001). In the drug-adapted state, BDNF offsets opiate-mediated neuronal inhibition by increasing levels of ACs and by restoring neuronal firing rates. NT-3, which plays a less dominant role in the noradrenergic system, appears to have a weak inhibitory effect on the cAMP pathway after chronic opiate exposure (Akbarian et al., 2001). Both BDNF and NT-3 increase TH expression in the drug-adapted state. It is yet unclear whether the neurotrophins regulate TH and AC expression at the level of gene transcription and whether mitogen-activated protein kinase (MAPK) pathways are involved. Both BDNF and NT-3 are released by non-noradrenergic neurons and activate TrkB (BDNF) and TrkC (NT-3) receptors by dimerization and phosphorylation either at the somal membrane or in distal processes of noradrenergic neurons.
Chronic morphine administration has been shown to increase TH expression and catalytic activity (Guitart et al., 1990; Lane-Ladd et al., 1997; Boundy et al., 1998) in the LC. This upregulation of TH would be expected to increase the capacity of noradrenergic neurons to synthesize norepinephrine, which then could contribute to the dramatic increase in norepinephrine release and to associated behavioral changes described during opiate withdrawal (Koob et al., 1992; Grasing et al., 1997). Therefore, one would predict that the lack of TH upregulation in the LC of BDNF-deficient mice would contribute to an attenuated opiate withdrawal reaction, which is what we observed. It is yet unclear how the disruption of the BDNF-signaling pathway abolishes the dynamic regulation of TH expression during opiate exposure. It has been suggested that TH expression is controlled by transcriptional regulation (Boundy et al., 1998). Interestingly, cAMP-dependent transcription factors and repressors appear to modulate the activity of the TH promoter (Tinti et al., 1996). When levels of CREB in the LC are decreased by infusion of antisense oligonucleotides, TH levels also decrease (Lane-Ladd et al., 1997). However, levels of phosphorylated CREB, indicative of its active state, appeared to be preserved in the nuclei of LC neurons of the BDNF-deficient mice, suggesting that in addition to CREB, other factors are required for the dynamic regulation of TH expression.
Notably, no abnormalities were found in LC neurons of drug-naïve BDNF-deficient mice, and LC neuronal inhibition after acute opiate exposure was fully preserved in the mutants. Therefore, the paradoxical downregulation of the cAMP-signaling pathway and the lack of TH upregulation in chronic morphine-treated mutants most likely reflects an alteration in drug-induced plasticity. The precise timing of these opiate-induced adaptations in the LC, including any effects of exogenous BDNF, remains a subject for future studies.
The neurochemical plasticity of noradrenergic neurons depends on BDNF derived from non-noradrenergic sources
During withdrawal, an increased excitatory afferent input to the LC and other noradrenergic cell groups contributes to the disinhibition of noradrenergic neurons (Akaoka and Aston-Jones, 1991; Aghajanian et al., 1994; Williams et al., 2001). In addition to modulating noradrenergic neuron function in acute withdrawal, our results indicate that non-noradrenergic systems, through a BDNF-signaling pathway, are also essential for the adaptations of noradrenergic neurons during chronic opiate exposure. Noradrenergic neurons have widespread projections to many areas of the adult forebrain, midbrain, and hindbrain that express BDNF (Maisonpierre et al., 1990), suggesting that target-derived BDNF is a key factor for opiate-induced plasticity of noradrenergic neurons. Therefore, one would expect that a widespread genetic ablation of BDNF in the CNS results in functional abnormalities of LC neurons after chronic morphine treatment, which is what we observed. BDNF immunoreactivity is more readily detectable in noradrenergic fibers than in perikarya of noradrenergic neurons (Fawcett et al., 1998), which may be expected if BDNF is taken up but not expressed by noradrenergic neurons. In analogy to the peripheral nervous system, perhaps BDNF is transported back to noradrenergic somata as phosphorylated Trk-ligand complex (Bhattacharyya et al., 1997;Watson et al., 1999). Furthermore, BDNF is also subject to anterograde axonal transport in the CNS (Altar et al., 1997;Conner et al., 1998; Kohara et al., 2001), and therefore afferent projections could serve as an additional source of BDNF for noradrenergic neurons.
Notably, two separate neurotrophin-signaling pathways regulate the neurochemical adaptations of the noradrenergic system in response to opiate exposure (Fig. 8): a BDNF/TrkB-related signal, as shown in this study, and a neurotrophin-3 (NT-3)/TrkC-related signal, as reported previously (Akbarian et al., 2001). The observed LC neuron abnormalities in morphine-treated NT-3-deficient mice appear to be less severe and in part opposite to the observed alterations in the BDNF-deficient mice, perhaps reflecting the fact that NT-3 expression in adult brain is much lower in comparison to BDNF (Maisonpierre et al., 1990). However, in close analogy to the BDNF/TrkB-signaling pathway, the modulation of noradrenergic neuron function by NT-3 also emanates from non-noradrenergic systems (Akbarian et al., 2001). By using these neurotrophin-signaling pathways in variable combinations and intensities, neuronal circuits that are interconnected with noradrenergic neurons could adjust and fine-tune opiate-related adaptations of the noradrenergic system, which may greatly increase the response repertoire of the brain when it is exposed to opiates and other psychoactive drugs.
The LC as a model system for opiate withdrawal
Pharmacological inhibition of noradrenergic neurons is a very effective treatment for opiate withdrawal (Washton and Resnick, 1983;Maldonado, 1997), and this observation has spurred considerable interest in the role of the noradrenergic system for opiate dependence and withdrawal. Traditionally, the LC as the largest noradrenergic cell cluster in the brain has served as a model system for the pharmacology and cellular basis of opiate tolerance, dependence, and withdrawal (Nestler and Aghajanian, 1997). The LC was also the focus of our study. However, noradrenergic neurons are organized in multiple clusters in the pons and medulla, which may explain the observation that lesions of individual clusters and fiber pathways, including those emanating from the LC, do not abolish opiate withdrawal reactions (Christie et al., 1997; Caille et al., 1999; Delfs et al., 2000).
Conclusion
Little is known about molecules that are key regulators of noradrenergic neuron function in adult brain. Here we show that BDNF is essential for adaptations of the noradrenergic system in response to chronic opiate exposure, including superactivation of the cAMP pathway, upregulation of tyrosine hydroxylase, and restoration of neuronal firing. It remains to be elucidated whether BDNF mediates neuronal plasticity in the noradrenergic system beyond these opiate-induced adaptations and whether the molecular mechanisms resemble those that operate, for example, in the Schaffer collateral–CA1 synapse in the hippocampus, where the intracellular signaling cascades of BDNF-dependent synaptic plasticity (Kang and Schuman, 1995) become increasingly recognized (Kang and Schuman, 2000; Patterson et al., 2001).
S.A. and M.R. contributed equally to this work.
This work was supported by the National Alliance for Research on Depression and Schizophrenia, by National Institute of Drug Abuse Grant 1-K08-DA00479 to S.A., and by National Cancer Institute Grant 5-R35-CA44339 to R.J. We thank Ruth Flannery, Jeanne Reis, Jeannie Smith, and Cathy Steffens for technical support and Dr. Haley Melikian for helpful discussions and comments.
Correspondence should be addressed to Dr. Schahram Akbarian, Brudnick Neuropsychiatric Research Institute, 303 Belmont Street, Worcester, MA 01613-2795. E-mail:schahram.akbarian@umassmed.edu.
REFERENCES
- 1.Aghajanian GK, Kogan JH, Moghaddam B. Opiate withdrawal increases glutamate and aspartate efflux in the locus coeruleus: an in vivo microdialysis study. Brain Res. 1994;636:126–130. doi: 10.1016/0006-8993(94)90186-4. [DOI] [PubMed] [Google Scholar]
- 2.Akaoka H, Aston-Jones G. Opiate withdrawal-induced hyperactivity of locus ceruleus neurons is substantially mediated by augmented excitatory amino acid input. J Neurosci. 1991;11:3830–3839. doi: 10.1523/JNEUROSCI.11-12-03830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akbarian S, Bates B, Liu RJ, Skirboll SL, Pejchal T, Coppola V, Sun LD, Fan G, Kucera J, Wilson MA, Tessarollo L, Kosofsky BE, Taylor JR, Bothwell M, Nestler EJ, Aghajanian GK, Jaenisch R. Neurotrophin-3 modulates noradrenergic neuron function and opiate withdrawal. Mol Psychiatry. 2001;6:593–604. doi: 10.1038/sj.mp.4000897. [DOI] [PubMed] [Google Scholar]
- 4.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 5.Aston-Jones G, Rajkowski J, Cohen J. Role of locus coeruleus in attention and behavioral flexibility. Biol Psychiatry. 1999;46:1309–1320. doi: 10.1016/s0006-3223(99)00140-7. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharyya A, Watson FL, Bradlee TA, Pomeroy SL, Stiles CD, Segal RA. Trk receptors function as rapid retrograde signal carriers in the adult nervous system. J Neurosci. 1997;17:7007–7016. doi: 10.1523/JNEUROSCI.17-18-07007.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 8.Boundy VA, Gold SJ, Messer CJ, Chen J, Son JH, Joh TH, Nestler EJ. Regulation of tyrosine hydroxylase promoter activity by chronic morphine in TH9.0-LacZ transgenic mice. J Neurosci. 1998;18:9989–9995. doi: 10.1523/JNEUROSCI.18-23-09989.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caille S, Espejo EF, Reneric JP, Cador M, Koob GF, Stinus L. Total neurochemical lesion of noradrenergic neurons of the locus coeruleus does not alter either naloxone-precipiated or spontaneous opiate withdrawal nor does it influence ability of clonidine to reverse opiate withdrawal. J Pharmacol Exp Ther. 1999;290:881–892. [PubMed] [Google Scholar]
- 10.Castren E, Thoenen H, Lindholm D. Brain-derived neurotrophic factor messenger RNA is expressed in the septum, hypothalamus and in adrenergic brain stem nuclei of adult rat brain and is increased by osmotic stimulation in the paraventricular nucleus. Neuroscience. 1995;64:71–80. doi: 10.1016/0306-4522(94)00386-j. [DOI] [PubMed] [Google Scholar]
- 11.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 12.Chieng B, Williams JT. Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci. 1998;18:7033–7039. doi: 10.1523/JNEUROSCI.18-17-07033.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christie MJ, Williams JT, Osborne PB, Bellchambers CE. Where is the locus in opioid withdrawal? Trends Pharmacol Sci. 1997;18:134–140. doi: 10.1016/s0165-6147(97)01045-6. [DOI] [PubMed] [Google Scholar]
- 14.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–3213. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conner JM, Lauterborn JC, Gall CM. Anterograde transport of neurotrophin proteins in the CNS: a reassessment of the neurotrophic hypothesis. Rev Neurosci. 1998;1998:91–103. doi: 10.1515/revneuro.1998.9.2.91. [DOI] [PubMed] [Google Scholar]
- 16.Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- 17.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 18.Fan G, Beard C, Chen RZ, Csankovszki G, Sun Y, Siniaia M, Biniszkiewicz D, Bates B, Lee PP, Kuhn R, Trumpp A, Poon C, Wilson CB, Jaenisch R. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farinas I, Jones KR, Tessarollo L, Vigers AJ, Huang E, Kirstein M, de Caprona DC, Coppola V, Backus C, Reichardt LF, Fritzsch B. Spatial shaping of cochlear innervation by temporally regulated neurotrophin expression. J Neurosci. 2001;21:6170–6180. doi: 10.1523/JNEUROSCI.21-16-06170.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, McLean JH, Miller FD. Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci. 1998;18:2808–2821. doi: 10.1523/JNEUROSCI.18-08-02808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829–839. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 22.Friedman WJ, Ibanez CF, Hallbook F, Persson H, Cain LD, Dreyfus CF, Black IB. Differential actions of neurotrophins in the locus coeruleus and basal forebrain. Exp Neurol. 1993;119:72–78. doi: 10.1006/exnr.1993.1007. [DOI] [PubMed] [Google Scholar]
- 23.Grasing K, Bills D, Ghosh S, Schlussman SD, Patel AH, Woodward JJ. Opiate modulation of striatal dopamine and hippocampal norepinephrine release following morphine withdrawal. Neurochem Res. 1997;22:239–248. doi: 10.1023/a:1022474318541. [DOI] [PubMed] [Google Scholar]
- 24.Guitart X, Hayward M, Nisenbaum LK, Beitner-Johnson DB, Haycock JW, Nestler EJ. Identification of MARPP-58, a morphine- and cyclic AMP-regulated phosphoprotein of 58 kDa, as tyrosine hydroxylase: evidence for regulation of its expression by chronic morphine in the rat locus coeruleus. J Neurosci. 1990;10:2649–2659. doi: 10.1523/JNEUROSCI.10-08-02649.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hausdorff WP, Hnatowich M, O'Dowd BF, Caron MG, Lefkowitz RJ. A mutation of the beta 2-adrenergic receptor impairs agonist activation of adenylyl cyclase without affecting high affinity agonist binding. Distinct molecular determinants of the receptor are involved in physical coupling to and functional activation of Gs. J Biol Chem. 1990;265:1388–1393. [PubMed] [Google Scholar]
- 26.Ivanov A, Aston-Jones G. Local opiate withdrawal in locus coeruleus neurons in vitro. J Neurophysiol. 2001;85:2388–2397. doi: 10.1152/jn.2001.85.6.2388. [DOI] [PubMed] [Google Scholar]
- 27.Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalia M, Fuxe K, Goldstein M. II. Rat medulla oblongata. Dopaminergic, noradrenergic (A1 and A2) and adrenergic neurons, nerve fibers, and presumptive terminal processes. J Comp Neurol. 1985;233:308–332. doi: 10.1002/cne.902330303. [DOI] [PubMed] [Google Scholar]
- 29.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 30.Kang H, Schuman EM. Intracellular Ca(2+) signaling is required for neurotrophin-induced potentiation in the adult rat hippocampus. Neurosci Lett. 2000;282:141–144. doi: 10.1016/s0304-3940(00)00893-4. [DOI] [PubMed] [Google Scholar]
- 31.Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 32.King VR, Michael GJ, Joshi RK, Priestley JV. trkA, trkB, and trkC messenger RNA expression by bulbospinal cells of the rat. Neuroscience. 1999;92:935–944. doi: 10.1016/s0306-4522(99)00072-x. [DOI] [PubMed] [Google Scholar]
- 33.Klein R, Parada LF, Coulier F, Barbacid M. TrkB, a novel tyrosine protein kinase receptor expressed during mouse neural development. EMBO J. 1989;8:3701–3709. doi: 10.1002/j.1460-2075.1989.tb08545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kogan JH, Nestler EJ, Aghajanian GK. Elevated basal firing rates and enhanced responses to 8-Br-cAMP in locus coeruleus neurons in brain slices from opiate-dependent rats. Eur J Pharmacol. 1992;211:47–53. doi: 10.1016/0014-2999(92)90261-2. [DOI] [PubMed] [Google Scholar]
- 35.Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- 36.Konradi C, Heckers S. Haloperidol-induced Fos expression in striatum is dependent upon transcription factor cyclic AMP response element binding protein. Neuroscience. 1995;65:1051–1061. doi: 10.1016/0306-4522(94)00546-h. [DOI] [PubMed] [Google Scholar]
- 37.Koob GF, Maldonado R, Stinus L. Neural substrates of opiate withdrawal. Trends Neurosci. 1992;15:186–191. doi: 10.1016/0166-2236(92)90171-4. [DOI] [PubMed] [Google Scholar]
- 38.Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- 39.Lane-Ladd SB, Pineda J, Boundy V, Pfeuffer T, Krupinski J, Aghajanian GK, Nestler EJ. CREB in the locus coeruleus: biochemical, physiological, and behavioral evidence for a role in opiate dependence. J Neurosci. 1997;17:7890–7901. doi: 10.1523/JNEUROSCI.17-20-07890.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J-G, Prather PL. Chronic exposure to μ-opioid agonists produces constitutive activation of μ-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol Pharmacol. 2001;60:53–62. doi: 10.1124/mol.60.1.53. [DOI] [PubMed] [Google Scholar]
- 41.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 42.Maldonado R. Participation of noradrenergic pathways in the expression of opiate withdrawal: biochemical and pharmacological evidence. Neurosci Biobehav Rev. 1997;21:91–104. doi: 10.1016/0149-7634(95)00061-5. [DOI] [PubMed] [Google Scholar]
- 43.Middlemas DS, Lindberg RA, Hunter T. trkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 45.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 46.North RA, Williams JT, Surprenant A, Christie MJ. Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc Natl Acad Sci USA. 1987;84:5487–5491. doi: 10.1073/pnas.84.15.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Numan S, Lane-Ladd SB, Zhang L, Lundgren KH, Russell DS, Seroogy KB, Nestler EJ. Differential regulation of neurotrophin and trk receptor mRNAs in catecholaminergic nuclei during chronic opiate treatment and withdrawal. J Neurosci. 1998;18:10700–10708. doi: 10.1523/JNEUROSCI.18-24-10700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osborne PB, Williams JT. Forskolin enhancement of opioid currents in rat locus coeruleus neurons. J Neurophysiol. 1996;76:1559–1565. doi: 10.1152/jn.1996.76.3.1559. [DOI] [PubMed] [Google Scholar]
- 49.Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- 50.Punch LJ, Self DW, Nestler EJ, Taylor JR. Opposite modulation of opiate withdrawal behaviors on microinfusion of a protein kinase A inhibitor versus activator into the locus coeruleus or periaqueductal gray. J Neurosci. 1997;17:8520–8527. doi: 10.1523/JNEUROSCI.17-21-08520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 52.Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan R, Jaenisch R. Conditional deletion of BDNF in the postnatal brain results in obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- 53.Saiepour MH, Semnanian S, Fathollahi Y. Occurrence of morphine tolerance and dependence in the nucleus paragigantocellularis neurons. Eur J Pharmacol. 2001;411:85–92. doi: 10.1016/s0014-2999(00)00862-1. [DOI] [PubMed] [Google Scholar]
- 54.Sklair-Tavron L, Nestler EJ. Opposing effects of morphine and the neurotrophins, NT-3, NT-4, and BDNF, on locus coeruleus neurons in vitro. Brain Res. 1995;702:117–125. doi: 10.1016/0006-8993(95)01029-8. [DOI] [PubMed] [Google Scholar]
- 55.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 56.Takeda J, Maekawa T, Sudo T, Seino Y, Imura H, Saito N, Tanaka C, Ishii S. Expression of the CRE-BP1 transcriptional regulator binding to the cyclic AMP response element in central nervous system, regenerating liver, and human tumors. Oncogene. 1991;6:1009–1014. [PubMed] [Google Scholar]
- 57.Tessarollo L, Parada LF. In situ hybridization. Methods Enzymol. 1995;254:419–430. doi: 10.1016/0076-6879(95)54028-8. [DOI] [PubMed] [Google Scholar]
- 58.Tinti C, Conti B, Cubells JF, Kim KS, Baker H, Joh TH. Inducible cAMP early repressor can modulate tyrosine hydroxylase gene expression after stimulation of cAMP synthesis. J Biol Chem. 1996;271:25375–25381. doi: 10.1074/jbc.271.41.25375. [DOI] [PubMed] [Google Scholar]
- 59.Tsao P, Cao T, von Zastrow M. Role of endocytosis in mediating downregulation of G-protein-coupled receptors. Trends Pharmacol Sci. 2001;22:91–96. doi: 10.1016/s0165-6147(00)01620-5. [DOI] [PubMed] [Google Scholar]
- 60.Van Bockstaele EJ, Saunders A, Commons KG, Liu XB, Peoples J. Evidence for coexistence of enkephalin and glutamate in axon terminals and cellular sites for functional interactions of their receptors in the rat locus coeruleus. J Comp Neurol. 2000;417:103–114. doi: 10.1002/(sici)1096-9861(20000131)417:1<103::aid-cne8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 61.Washton AM, Resnick RB. Recent advances in opiate detoxification: clonidine and lofexidine. NIDA Res Monogr. 1983;43:44–50. [PubMed] [Google Scholar]
- 62.Watson FL, Heerssen HM, Moheban DB, Lin MZ, Sauvageot CM, Bhattacharyya A, Pomeroy SL, Segal RA. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J Neurosci. 1999;19:7889–7900. doi: 10.1523/JNEUROSCI.19-18-07889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Widnell KL, Russell DS, Nestler EJ. Regulation of expression of cAMP response element-binding protein in the locus coeruleus in vivo and in a locus coeruleus-like cell line in vitro. Proc Natl Acad Sci USA. 1994;91:10947–10951. doi: 10.1073/pnas.91.23.10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williams JT, North RA, Tokimasa T. Inward rectification of resting and opiate-activated potassium currents in rat locus coeruleus neurons. J Neurosci. 1988;8:4299–4306. doi: 10.1523/JNEUROSCI.08-11-04299.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 66.Yacoubian TA, Lo DC. Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat Neurosci. 2000;3:342–349. doi: 10.1038/73911. [DOI] [PubMed] [Google Scholar]
- 67.Yamuy J, Sampogna S, Chase MH. Neurotrophin-receptor immunoreactive neurons in mesopontine regions involved in the control of behavioral states. Brain Res. 2000;866:1–14. doi: 10.1016/s0006-8993(00)02204-6. [DOI] [PubMed] [Google Scholar]
- 68.Zachariou V, Thome J, Parikh K, Picciotto MR. Upregulation of galanin binding sites and GalR1 mRNA levels in the mouse locus coeruleus following chronic morphine treatments and precipitated morphine withdrawal. Neuropsychopharmacology. 2000;23:127–137. doi: 10.1016/S0893-133X(00)00094-4. [DOI] [PubMed] [Google Scholar]
- 69. Zhu H, Zhou W. Morphine induces synchronous oscillatory discharges in the rat locus coeruleus. J Neurosci 21 2001. RC179(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]