

Abstract
Recent studies have clarified that endogenous cannabinoids (endocannabinoids) are released from depolarized postsynaptic neurons in a Ca2+-dependent manner and act retrogradely on presynaptic cannabinoid receptors to suppress inhibitory or excitatory neurotransmitter release. This type of modulation has been found in the hippocampus and cerebellum and was called depolarization-induced suppression of inhibition (DSI) or excitation (DSE). In this study, we quantitatively examined the effects of postsynaptic depolarization and a cannabinoid agonist on excitatory and inhibitory synapses in rat hippocampal slices and cultures. We found that both DSE and DSI can be induced, but DSE was much less prominent than DSI. For the induction of DSE, the necessary duration of depolarization was longer than for DSI. The magnitude of DSE was much smaller than that of DSI. To explore the reasons for these differences, we tested the sensitivity of EPSCs and IPSCs to a cannabinoid agonist, WIN55,212-2, in hippocampal cultures. IPSCs were dichotomized into two distinct populations, one with a high sensitivity to WIN55,212-2 (50% block at 2 nm) and the other with no sensitivity. In contrast, EPSCs were homogeneous and exhibited a low sensitivity to WIN55,212-2 (50% block at 60 nm). We estimated that the 5 sec depolarization elevated the local endocannabinoid concentration to a level equivalent to several nanomoles of WIN55,212-2. Using CB1 knock-out mice, we verified that both DSI and DSE were mediated by the cannabinoid CB1 receptor. These results indicate that presynaptic cannabinoid sensitivity is a major factor that determines the extent of DSI and DSE.
Keywords: excitatory transmission, inhibitory transmission, hippocampus, retrograde signal, synaptic modulation, cannabinoid receptor
Marijuana influences various neural functions, with consequences including analgesia, modulation of locomotor control, and impairment of cognition and memory (Deadwyler et al., 1990; Heyser et al., 1993; Howlett, 1995). These effects are thought to be mediated through the interaction of Δ9-tetrahydrocannabinol, the psychoactive component of marijuana, with specific cannabinoid receptors. These receptors belong to the seven transmembrane domain family of G-protein-coupled receptors and consist of type 1 (CB1) and type 2 (CB2) receptors (Matsuda et al., 1990; Munro et al., 1993). Cannabinoid binding sites in the CNS (Herkenham et al., 1991) that correspond to the distribution of CB1 receptors are heterogeneous, with high levels in some regions, including the hippocampus (Matsuda et al., 1993; Tsou et al., 1998; Egertova and Elphick, 2000). Thus, marijuana may act on hippocampal CB1 receptors and interfere with actions of their endogenous ligands. This would disrupt normal information processing in the hippocampus and thereby cause memory impairment.
Two putative endogenous ligands for cannabinoid receptors, anandamide (Devane et al., 1992) and 2-arachidonylglycerol (2-AG) (Mechoulam et al., 1995; Sugiura et al., 1995, 1999), have been identified. These molecules are produced and released from neurons in a Ca2+-dependent manner (Di Marzo et al., 1998; Mechoulam et al., 1998; Piomelli et al., 2000). Activation of cannabinoid receptors exerts variable effects, including inhibition of voltage-gated Ca2+ channels, activation of inwardly rectifying K+ channels, and suppression of neurotransmitter release (Di Marzo et al., 1998; Felder and Glass, 1998). Therefore, the endogenous cannabinoid (endocannabinoid) system is likely to play an important role in controlling neuronal excitability and synaptic transmission.
It was revealed recently that endocannabinoids mediate a form of activity-dependent modulation of synaptic transmission. Depolarization of a neuron induces a transient suppression of inhibitory input, a phenomenon called depolarization-induced suppression of inhibition (DSI) (Llano et al., 1991; Pitler and Alger, 1992; Ohno-Shosaku et al., 1998). DSI is initiated postsynaptically by an elevation of cytoplasmic Ca2+ concentration ([Ca2+]i) and is expressed presynaptically as a suppression of the transmitter release. Therefore, since the discovery of DSI, it has been thought that some retrograde signal must exist from the depolarized postsynaptic neurons to the presynaptic terminals (Alger and Pitler, 1995). Recent studies have demonstrated that endocannabinoids mediate such retrograde signals at inhibitory synapses in both the hippocampus (Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Wilson et al., 2001) and the cerebellum (Kreitzer and Regehr, 2001b; Diana et al., 2002; Yoshida et al., 2002). In addition, a phenomenon similar to DSI was found to occur at excitatory synapses [depolarization-induced suppression of excitation (DSE)] in the cerebellum that is also mediated by endocannabinoids (Kreitzer and Regehr, 2001a; Maejima et al., 2001).
In the present study, we report that DSE can be induced in the hippocampus. Although both DSE and DSI are mediated by CB1 receptors, DSE was less prominent and required longer depolarization for the induction compared with the DSI. Our results suggest that presynaptic cannabinoid sensitivity is a major factor that determines the extent of depolarization-induced retrograde suppression.
MATERIALS AND METHODS
Slices. All experiments were performed according to the guidelines laid down by the animal welfare committees of Kanazawa University and the National Institute for Physiological Sciences. Hippocampal slices were prepared as described previously (Tsubokawa and Ross, 1997; Tsubokawa et al., 2000). Young (10- to 12-d-old) rats were deeply anesthetized with ether and decapitated. The brains were quickly removed and hemisected on filter paper moistened with cutting solution of the following composition (in mm): 120 choline-Cl, 3 KCl, 8 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 20 glucose, equilibrated with 95% O2–5% CO2. Brain tissues containing the hippocampi on each side were dissected out and put into a cutting chamber filled with ice-cold cutting solution. These two blocks were sliced into 300 μm sections transversely to their longitudinal axis with a microtome (Vibroslicer; Campden Instruments, Lafayette, IN). The slices were immediately placed into a reservoir chamber filled with normal solution, incubated at 35°C for ∼30 min, and then maintained at room temperature. In some experiments, hippocampal slices were prepared from the CB1 knock-out mice and wild-type mice by the same procedure as described above. The normal solution was composed of (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 20 glucose, bubbled with a mixture of 95% O2 and 5% CO2, with a final pH of 7.4. For recording, a single slice was transferred to a submerged chamber mounted on the stage of an upright microscope (BX50WI; Olympus Optical, Tokyo, Japan). The slice was superfused continuously with the normal solution at room temperature (for experiments shown in Figs. 2 and 3) or regulated at 30–32°C (Fig.1).
Fig. 2.

Blockade of DSE by a cannabinoid antagonist, SR141716A. Averaged time courses of the changes in EPSC amplitudes induced by depolarization to 0 mV for 10 sec before (A) and after (B) treatment with 5 μm SR141716A are shown. The data shown inB were acquired at least 30 min after the bath application of the antagonist. EPSCs were evoked at 1 Hz. In each experiment, mean amplitudes of five consecutive EPSCs were calculated. Each point is the average of the mean EPSC amplitudes for the 5 sec period from five different cells. EPSC traces acquired before and after depolarization (Depo) were superimposed (right). Each trace is the average of eight consecutive EPSCs.
Fig. 3.

DSE is less prominent than DSI in CA1 pyramidal cells. Averaged time courses of the changes in amplitude of IPSCs (A) and EPSCs (B) are shown. IPSCs and EPSCs were evoked at 0.33 and 1 Hz, respectively. Pyramidal neurons were depolarized to 0 mV for 5 or 10 sec at the times indicated by arrows. In each experiment, mean amplitudes of five consecutive IPSCs or EPSCs were calculated. Eachpoint is the average of the mean amplitudes from five different cells.
Fig. 1.

Transient suppression of EPSCs induced by depolarization of a CA1 pyramidal neuron in the hippocampal slice.A, Example of EPSC traces acquired at the time points indicated in B. Each trace is the average of three consecutive EPSCs. B, Time course of the change in EPSC amplitudes. EPSCs were evoked at 1 Hz. The pyramidal neuron was depolarized to −30 mV or to 0 mV for 7 sec at the times indicated byarrows.
Electrical recordings were made from CA1 pyramidal cell somata in slices using patch pipettes pulled from 1.5-mm-outer diameter (o.d.), thick-walled glass tubing (1511-M; Friedrich & Dimmock, Melville, NJ). The pipette solution contained (in mm): 115 K-gluconate, 10 KCl, 10 NaCl, 10 HEPES, 2 Mg-ATP, and 0.3 GTP, pH adjusted to 7.3 with KOH. The open resistance of the pipettes was 5–7 MΩ. Whole-cell tight seals (>5 GΩ) were made on the soma under visual control with a 40× water-immersion lens (Edwards et al., 1989). Capacitance was fully compensated by a patch-clamp amplifier (Axopatch 1D; Axon Instruments, Foster City, CA). The range of series resistance we accepted was 10–15 MΩ. Bipolar stimulation electrodes constructed from Teflon-coated thin tungsten wire (50 μm o.d.) were placed on the stratum radiatum to generate EPSCs and IPSCs. The membrane potential of neurons was held at −70 mV. The bath solution was supplemented with 10 μm SR95531 or 10 μm bicuculline methiodide for recording EPSCs and with 10 μm6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 50 μmdl-2-amino-5-phosphonovaleric acid (APV) for recording IPSCs. Cells were identified as pyramidal neurons according to both electrical and anatomic criteria.
Cultures. Hippocampal cells were enzymatically (with trypsin) or mechanically dissociated from the hippocampus of newborn rats (1–2 d of age) and were plated onto culture dishes pretreated with poly-l-ornithine (0.01%). The cells were incubated in DMEM/F-12 medium (Invitrogen, San Diego, CA) supplemented with putrescine (0.1 mm), sodium selenite (30 nm),l-glutamine (1.4 mm), gentamycin (10 μg/ml), insulin (5 μg/ml), and fetal calf serum (10%). Cultures were maintained at 36°C in 5% CO2 for 10–14 d. For the experiments shown in Figure 8, hippocampal neurons were cultured from the CB1 knock-out mice and wild-type mice by the same procedure. All experiments were performed at room temperature. The external solution contained (in mm): 140 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, and 10 glucose, pH 7.3 adjusted with NaOH. The bath was perfused with the external solution with or without drugs at a flow rate of 1–3 ml/min. The internal solution contained (in mm): 120 K-gluconate, 15 KCl, 6 MgCl2, 0.2 EGTA, 10 HEPES, 10 KOH, and 5 Na2ATP, pH 7.3 adjusted with KOH. In some of the experiments in which the sensitivity of synaptic currents to a cannabinoid agonist, WIN55,212-2, was examined, the internal solution containing 5 mm EGTA was used. Because the sensitivity to WIN55,212-2 was not different between 0.2 and 5 mm EGTA in the internal solution, data acquired with the two internal solutions were pooled. The internal solution with 5 mm EGTA was also used for the experiments on mouse cultured neurons (see Fig. 8). The electrode resistance ranged from 3 to 5 MΩ when the pipette was filled with the solution.
Fig. 8.

DSE and WIN55,212-2 (WIN)-induced suppression of EPSCs inCB1+/+ and CB1−/− mice. A, Examples of DSE induced by 5 sec depolarization (Depo) in cultured neurons prepared from CB1+/+ andCB1−/− mice. Two EPSC traces acquired before and 6 sec after depolarization are superimposed. B, Examples of WIN55,212-2-induced suppression of EPSCs in cultured neurons prepared from CB1+/+ and CB1−/− mice. Two EPSC traces acquired before and after application of 100 nm WIN55,212-2 are superimposed. C, The averaged data for DSE before (DSE) and after (DSE/AM281) treatment with 0.3 μm AM281 and suppression of EPSCs by 100 nm WIN55,212-2 or 10 μm baclofen in cultured neurons prepared from CB1+/+ and CB1−/− mice. Each bar represents the averaged value obtained from the indicated number of neuron pairs.
A pair of neurons were whole-cell clamped with two different patch pipettes, and membrane potentials of both cells were held at −80 mV. The presynaptic neuron was stimulated by applying positive-voltage pulses (80 mV, 2 msec) at 0.2–1 Hz, and EPSCs or IPSCs were measured from the postsynaptic neuron with a patch-clamp amplifier (EPC-9/2; Heka Elektronik, Lambrecht/Pfalz, Germany). EPSCs were usually measured in the presence of 5–10 nm TTX to suppress spontaneous firing. IPSCs were measured in the presence of 1 mmkynurenic acid.
Induction of DSE and DSI. To induce DSE and DSI, the postsynaptic neuron was depolarized to 0 mV for the indicated duration (0.5–10 sec) unless otherwise noted. The magnitudes of depolarization-induced suppression were measured as the percentage of the mean amplitude of synaptic currents acquired between 4 and 18 sec after the end of depolarization relative to that acquired for 30 sec before the depolarization. The depression caused by drugs was estimated as the percentage of the mean amplitudes of 5–12 consecutive synaptic currents during drug application relative to that before application. Averaged data from different experiments are presented as mean ± SEM.
CB1 receptor knock-out mouse. CB1 receptor knock-out mice were generated as described previously (Zimmer et al., 1999). Briefly, the coding region of the CB1 gene was replaced between amino acids 32 and 448 with phosphoglycerate kinase-neo in embryonic stem cells. Chimeric mice derived from these cells were bred with C57BL/6J animals. Homozygous mutant (CB1−/−) and wild-type (CB1+/+) mice were produced with heterozygous intermatings. In the present investigation, neonatal [postnatal day 1 (P1)] and juvenile (P9–P12) mice of both sexes were used to prepare cultures and slices, respectively. Animals were housed in groups under standard laboratory conditions (12 hr light/dark cycle) with food and water available ad libitum.
Materials. WIN55,212-2, AM281, SR95531, CNQX, and APV were purchased from Tocris Cookson (Ballwin, MO). Other chemicals were purchased from Sigma (St. Louis, MO). SR141716A was a generous gift from Sanofi Recherche (Montpellier, France). For the perfusion of solutions containing WIN55,212-2, AM281, or SR141716A, different tubes were used to avoid contamination.
RESULTS
Endocannabinoid-mediated DSE in the hippocampus
We first examined whether depolarization of the postsynaptic neuron can induce DSE in hippocampal slices from the rat. When a CA1 pyramidal neuron was depolarized for several seconds under the voltage-clamp mode, the subsequent EPSCs from the depolarized neuron were transiently suppressed. In a CA1 pyramidal neuron shown in Figure1, depolarization from −70 to −30 mV (Fig. 1Aa,b) or 0 mV (Fig. 1Ac,d) for 7 sec was effective at inducing DSE (Fig. 1B). Because the DSE in the cerebellum is shown to be mediated by endocannabinoids (Kreitzer and Regehr, 2001a; Maejima et al., 2001), we subsequently examined whether a CB1 antagonist, SR141716A, could block DSE. In five neurons, a depolarizing voltage pulse (from −70 to 0 mV; 10 sec) was applied before and after addition of 5 μm SR141716A. The depolarization induced a clear suppression of EPSCs in the normal external solution (Fig.2A), whereas the same depolarization caused no significant change in the presence of SR141716A (Fig. 2B). These results clearly indicate that DSE in the hippocampus is also mediated by endocannabinoids that are released from depolarized postsynaptic neurons and suppress the excitatory transmission through activation of cannabinoid receptors, presumably CB1 receptors.
Excitatory transmission is less sensitive to postsynaptic depolarization
We then compared DSE and DSI in the slices from the same animals. As shown in Figure 3, depolarization for 5 sec induced a marked suppression of IPSCs (DSI) (Fig. 3A), whereas the same depolarization induced no significant DSE (Fig.3B). Although subsequent depolarization of a longer duration (10 sec) induced clear DSE (Fig. 3B), it was still less prominent than the DSI induced by the 5 sec depolarization (Fig.3A). These results indicate that excitatory transmission is less sensitive to postsynaptic depolarization than inhibitory transmission.
One possible explanation for these quantitative differences is that the inhibitory presynaptic terminals might be more sensitive to endocannabinoids than the excitatory terminals. In the following experiments, we tested this possibility in cultured hippocampal neurons.
Excitatory presynaptic terminals are less sensitive to cannabinoids
We determined the cannabinoid sensitivities of excitatory and inhibitory synapses using neuron pairs with excitatory or inhibitory synaptic connections. In each pair, the effects of different concentrations of a cannabinoid agonist, WIN55,212-2, on EPSCs or IPSCs were examined. When WIN55,212-2 was applied at 1–1000 nm, the EPSC amplitude was decreased in a dose-dependent manner (Fig. 4A). The suppressing effect was reversed by a cannabinoid receptor antagonist, 0.3 μm AM281 (n = 8) or 0.3 μm SR141716A (n = 3), confirming that WIN55,212-2 acted on cannabinoid receptors. The concentration of WIN55,212-2 that reduced the EPSC amplitude by 50% was between 10 and 100 nm (Fig.4A). Excitatory synapses were relatively homogeneous in terms of sensitivity to WIN55,212-2, and similar results were obtained in another seven pairs. In contrast, inhibitory synapses were heterogeneous and divided into two populations, those sensitive and those insensitive to WIN55,212-2, as reported previously (Ohno-Shosaku et al., 2001). In one population, IPSCs were suppressed by nanomolar concentrations of WIN55,212-2 (Fig. 4B). The concentration of WIN55,212-2 that caused 50% inhibition was between 1 and 10 nm (Fig. 4B). In the other population, IPSCs were totally insensitive to WIN55,212-2, even at concentrations as high as 1000 nm (Fig.4C).
Fig. 4.

Effects of a cannabinoid agonist, WIN55,212-2 (WIN), on EPSCs (A) and IPSCs (B, C). Amplitudes of EPSCs (A) and IPSCs (B, C) are plotted as a function of time. The bath was perfused with a solution containing 1–1000 nm WIN55,212-2 or a cannabinoid antagonist, AM281 (0.3 μm), for the periods indicated by thehorizontal lines. The traces of EPSCs (A) and IPSCs (B, C) acquired before and during application of WIN55,212-2 are superimposed (right).
Figure 5 summarizes data for the sensitivity to WIN55,212-2 of excitatory synapses and the two populations of inhibitory synapses. The WIN55,212-2 concentrations for 50% inhibition were ∼60 nm for EPSCs and 2 nm for the WIN55,212-2-sensitive IPSCs, indicating that EPSCs are ∼30-fold less sensitive than the WIN55,212-2-sensitive IPSCs (Fig. 5A). In the presence of 100 nm WIN55,212-2, the EPSC amplitude was decreased to 23–60% of controls (38.7 ± 5.3%; n = 8) (Fig. 5B, closed circles). In contrast, values for the WIN55,212-2-sensitive IPSCs were all <10% (1.7 ± 0.8%;n = 16) (Fig. 5B, open circles), and those of the WIN55,212-2-insensitive IPSCs were all >90% (99.3 ± 2.8%; n = 8) (Fig. 5B,open triangles). There was no overlap of individual data among these three populations.
Fig. 5.

Summary of the dose-dependent suppressions of EPSCs and IPSCs by WIN55,212-2 (WIN).A, Amplitudes of WIN55,212-2-sensitive IPSCs (open circles), WIN55,212-2-insensitive IPSCs (triangles), and EPSCs (closed circles) plotted against the concentration of WIN55,212-2. In each cell, current amplitudes in the presence of WIN55,212-2 were expressed as the percentages of the control values (dotted line) obtained before application of WIN55,212-2. Eachsymbol represents the average value from the indicatednumber of neuron pairs. B, Individual values of current amplitudes in the presence of 100 nm WIN55,212-2 for WIN55,212-2-sensitive IPSCs (open circles), WIN55,212-2-insensitive IPSCs (triangles), and EPSCs (closed circles).
To test whether WIN55,212-2 causes presynaptic or postsynaptic change in excitatory transmission, we examined paired-pulse plasticity. The paired-pulse ratio of EPSC amplitudes was significantly increased by WIN55,212-2 (Fig. 6A). In the presence of 0.1 and 1 μm WIN55,212-2, the EPSC amplitude decreased by 57 and 77% of controls, and the paired-pulse ratio increased by 34 and 44%, respectively (Fig. 6,closed circles). A decrease in the probability of vesicular transmitter release from presynaptic terminals is generally accompanied by an increase in the paired-pulse ratio (Zucker, 1989). Therefore, the present results indicate that WIN55,212-2 acts on cannabinoid receptors on excitatory presynaptic terminals and causes the suppression of transmitter release, which is in agreement with the previous study (Shen et al., 1996). This mechanism is essentially identical to that for the action of WIN55,212-2 on inhibitory synapses (Katona et al., 1999; Hoffman and Lupica, 2000; Ohno-Shosaku et al., 2001, 2002; Wilson and Nicoll, 2001). We also found that DSE is accompanied by clear increases in the paired-pulse ratio (Fig. 6B,open circles), indicating that DSE is expressed presynaptically as a decrease in excitatory transmitter release.
Fig. 6.

Changes in paired-pulse ratio during suppression of EPSCs by WIN55,212-2 (WIN) (A;B, closed circles) and DSE (B, open circles). A, Examples of EPSCs evoked by paired stimuli with an interpulse interval of 50 msec. Each trace is the average of 10–12 consecutive EPSCs. Traces scaled to the amplitude of the first EPSC are shown at thebottom. The traces acquired before and during bath application of WIN55,212-2 are superimposed. B, The relationships between the reduction in the first EPSC amplitude and the increase in paired-pulse ratio induced by applications of 100 and 1000 nm WIN55,212-2 (closed circles) and the 5 sec and 10 sec depolarizing pulses (DSE). Each symbolrepresents the averaged value from the indicated numberof neuron pairs.
Estimation of local cannabinoid levels during DSE and DSI
Subsequently, we attempted to estimate the local level of endocannabinoids during DSE and DSI as an equivalent concentration of WIN55,212-2. For this purpose, we measured magnitudes of DSI and DSE (Fig. 7) and compared them with those of suppressions induced by WIN55,212-2 (Fig. 5A).
Fig. 7.
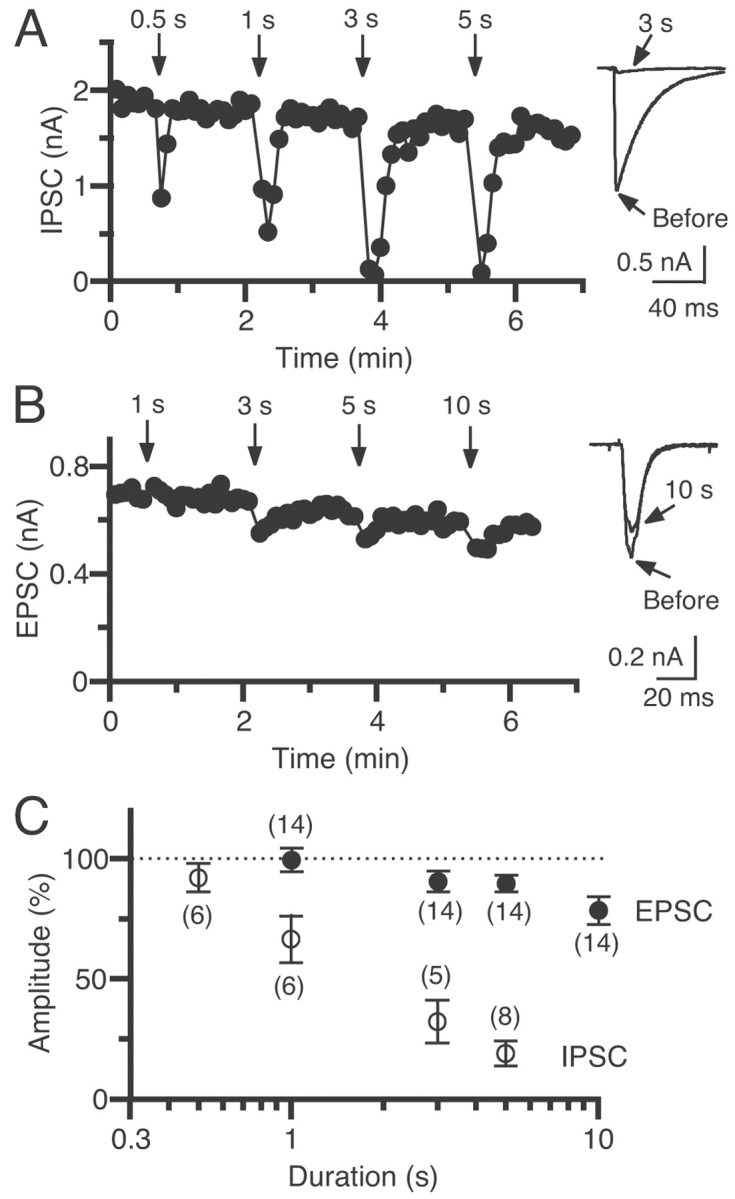
Induction of DSI and DSE depends on the duration of depolarization. A, B, Example of DSI (A) and DSE (B) induced by various depolarizing pulses with durations of 0.5–10 sec. The postsynaptic neuron was depolarized at the times indicated byarrows. Traces of IPSCs (A) or EPSCs (B) acquired before and 7 sec after depolarization with a duration of 3 sec (A) or 10 sec (B) are superimposed on theright. C, The relationships between the depolarizing pulse duration and the relative amplitude of IPSCs (open circles) and EPSCs (closed circles) obtained 6–16 sec after the end of depolarization. The amplitude was normalized to the averaged value (dotted line) before depolarization. Each symbol represents the averaged value obtained from the indicated number of neuron pairs.
DSI was induced by depolarizations with pulse durations of 0.5, 1, 3, and 5 sec at WIN55,212-2-sensitive inhibitory synapses (Fig.7A,C). In a pair shown in Figure 7A, a 0.5 sec depolarization induced significant DSI, and a depolarization of a longer duration induced more prominent DSI. The averaged data show a clear dependence of the DSI magnitude on the depolarizing pulse duration (Fig. 7C, open circles). DSE was induced by depolarizations with pulse durations of 1, 3, 5, and 10 sec in 14 pairs with excitatory connections (Fig. 7B,C). In a pair exemplified in Figure 7B, depolarizing pulses with a duration of >3 sec induced a slight but significant decrease in EPSC amplitudes. The averaged data in Figure 7C show that the 5 sec depolarization of postsynaptic neurons suppressed the IPSC amplitude by 81% (Fig. 7C, open circles) and the EPSC amplitude by 10% (Fig. 7C, closed circles) of control values. These magnitudes of suppression correspond to those for IPSCs and for EPSCs induced by ∼5 nmWIN55,212-2 (Fig. 5A). These results indicate that the local concentration of endocannabinoids around excitatory and inhibitory presynaptic terminals is estimated to reach a level equivalent to several nanomoles of WIN55,212-2 after postsynaptic depolarization for 5 sec. Thus, the quantitative difference between DSE and DSI can be explained by the difference in the cannabinoid sensitivity between excitatory and inhibitory synaptic terminals.
CB1 receptors are responsible for both DSI and DSE in the hippocampus
It was reported recently that both DSI and the WIN55,212-2-induced suppression of IPSCs are absent in hippocampal slices from CB1 knock-out (CB1−/−) mice (Varma et al., 2001; Wilson et al., 2001). We therefore examined whether CB1 receptors mediate the effects of cannabinoids on IPSCs in cultured mouse hippocampal neurons. In neurons prepared from wild-type mice (CB1+/+), DSI (>20% suppression after 5 sec depolarization) was observed in 11 of 25 neuron pairs. In such DSI-positive pairs, WIN55,212-2 (100 nm) was always effective and decreased the IPSC amplitude to 5.2 ± 8.9% of controls (n = 9). In contrast, neither DSI nor WIN55,212-2-induced suppression was observed in cultured neurons from CB1−/− mice. The IPSC amplitude after depolarization was 96.2 ± 3.4% of controls (n = 17), and the amplitude in the presence of 100 nm WIN55,212-2 was 108.4 ± 7.5% (n = 10). These results confirmed that CB1 receptors mediate DSI as well as effects of cannabinoids on inhibitory synapses in cultured hippocampal neurons.
We then examined whether CB1 receptors are also responsible for hippocampal DSE. In cultured neurons from CB1+/+ mice, DSE was clearly induced by a 5 sec depolarization and was completely abolished after treatment with the cannabinoid antagonist AM281 (Fig.8A,C). The EPSC amplitude was strongly suppressed after application of 100 nm WIN55,212-2 (Fig. 8B,C). In neurons from CB1−/− mice, however, neither depolarization nor WIN55,212-2 affected EPSCs (Fig. 8). In some experiments, a longer depolarization (10 sec) and a higher dose of WIN55,212-2 (1 μm) were tested. However, we could not detect any significant effects on EPSCs (n = 5; data not shown). The GABAB agonist baclofen (10 μm) effectively suppressed EPSCs ofCB1−/− neurons (Fig. 8C), indicating that other components required for G-protein-mediated presynaptic inhibition are intact. The involvement of CB1 receptors in DSE was also confirmed in slice preparations. The magnitudes of DSE after a 10 sec depolarization were 6.8 ± 2.3% for CB1−/− (n = 7) and 20.3 ± 5.4% for CB1+/+ (n = 7) mice (p < 0.05). These results indicate that DSE is mediated by CB1 receptors in the hippocampus.
DISCUSSION
In the present study, we report for the first time that DSE can be induced in the hippocampus. Both DSI and DSE were mediated by CB1 receptors, but DSE was less prominent and required longer depolarizations for induction than DSI. This quantitative difference can be explained by the difference in sensitivities to cannabinoids between excitatory and inhibitory synapses. Excitatory transmission was estimated to be ∼30-fold less sensitive to cannabinoids than inhibitory transmission in cultured hippocampal neurons.
Cannabinoid sensitivities of excitatory and inhibitory synapses
Several previous studies show that WIN55,212-2 induces suppression of EPSCs in hippocampal cultures (Shen et al., 1996; Sullivan, 1999) and slices (Misner and Sullivan, 1999). Conversely, other studies show that excitatory transmission in the hippocampus is not suppressed by WIN55,212-2 (Paton et al., 1998) or 2-AG (Stella et al., 1997). One possible explanation for this discrepancy might be the difference in age of the animals used. Most of the studies showing no effect of cannabinoid agonists have been done with older animals. It is possible that cannabinoid modulation of excitatory transmission would be developmentally regulated, as reported recently (Al-Hayani and Davies, 2000). In our preliminary experiments, however, excitatory transmission was still sensitive to WIN55,212-2 in hippocampal slices from 5-week-old rats. Another possibility is that the discrepancy might derive from the difference in recording techniques. The groups reporting the positive effects of WIN55,212-2 applied the patch-clamp technique to cultured neurons (Shen et al., 1996; Sullivan, 1999) or to neurons in slices that were presumably close to the surface (Misner and Sullivan, 1999). Conversely, the groups reporting no effect of cannabinoid agonists measured field potentials (Stella et al., 1997;Paton et al., 1998) that primarily reflected the responses of neurons located in the depth of the slices. Because WIN55,212-2 and 2-AG are lipid in nature, they may not easily diffuse into slices. Thus, it is possible that their local levels around the synapses deep in the slices may have been too low to affect excitatory transmission.
A previous study on hippocampal DSI demonstrated that postsynaptic depolarization affected inhibitory but not excitatory transmission (Wagner and Alger, 1996). The apparent discrepancy between the findings of Wagner and Alger (1996) and the present data may be attributable to the difference in the induction protocol. They used depolarization for 1 sec, which has now turned out to be too short. We found that depolarization with a duration of >7 sec was necessary to induce detectable DSE in slice preparations.
The reasons for the difference in sensitivity to cannabinoids between excitatory and inhibitory transmissions remain elusive. One possibility is that cannabinoid receptor subtypes on presynaptic terminals are different between excitatory and inhibitory synapses. A recent article reports that WIN55,212-2 still suppresses EPSCs in adultCB1−/− mice (Hajos et al., 2001), suggesting an involvement of a novel subtype of cannabinoid receptors. In the present study, however, we could not detect any effects of WIN55,212-2 on either IPSCs or EPSCs in CB1−/− mice. Importantly, neither DSI nor DSE was induced in CB1−/− mice. We therefore conclude that the CB1 receptor mediates both DSI and DSE, at least in the juvenile animals used in the present study. It is possible, however, that the expression of the novel cannabinoid receptor subtype may be developmentally regulated and become functional in older animals. It is also possible that the CB1−/− mouse lines used by Hajos et al. (2001) and in the present study were constructed differently and might be on different backgrounds.
Another possibility for the difference in cannabinoid sensitivity between excitatory and inhibitory synapses is that CB1 receptors are expressed more densely at inhibitory presynaptic terminals than at excitatory ones. Anatomic studies support this possibility. In situ hybridization studies on rats (Matsuda et al., 1993) and mice (Marsicano and Lutz, 1999) suggest that large and moderate amounts of CB1 mRNA are distributed in GABAergic and pyramidal cells, respectively. An immunocytochemical study shows an expression of CB1 receptors in hippocampal pyramidal cells (Pettit et al., 1998). However, other immunocytochemical studies (Katona et al., 1999; Hajos et al., 2000) with a different antibody demonstrate that CB1 receptors are densely localized at subpopulations of GABAergic presynaptic terminals but are absent on glutamatergic neurons. These results suggest that the difference in cannabinoid sensitivity between excitatory and inhibitory transmissions is at least partly attributable to the difference in the amount of CB1 proteins at presynaptic terminals.
Possible physiological significance of DSI and DSE
What could be a functional role of DSI and DSE in the hippocampus? Previous studies show that endocannabinoids are synthesized “on demand” in stimulated neurons and released from them in a Ca2+-dependent manner (Di Marzo et al., 1998; Mechoulam et al., 1998; Piomelli et al., 2000). We have shown that the postsynaptic elevations of [Ca2+]i and DSI had similar time courses in cultured hippocampal neurons (Ohno-Shosaku et al., 2001). Therefore, the amount of released endocannabinoids can directly reflect the activity of postsynaptic neurons and the resultant elevation of [Ca2+]i. If endocannabinoids suppress excitatory and inhibitory inputs to the same extent, they will cause no change in the excitability of postsynaptic neurons. The present study, however, has revealed that excitatory and inhibitory synapses of the hippocampus have different sensitivities to cannabinoids. Whereas excitatory synapses were homogeneous and had moderate sensitivities to WIN55,212-2, inhibitory synapses were dichotomized into two distinct populations, one with a high sensitivity to WIN55,212-2 and the other with no sensitivity. Thus, endocannabinoids can control the balance between excitatory and inhibitory inputs, depending on the local concentration around synapses.
DSI can occur only at CB1-expressing synapses. One of the CB1-expressing neurons is a cholecystokinin (CCK)-containing basket cell (Katona et al., 1999). In contrast, the other type of basket cell, which contains palvalbumin (PV) but not CCK, is devoid of CB1 receptors (Katona et al., 1999). Both types of basket cells form perisomatic synapses on pyramidal neurons. These anatomic data suggest that DSI occurs selectively at CCK-containing but not PV-containing perisomatic synapses on pyramidal cells. In addition to these two types, a third type of inhibitory synapse has been characterized electrophysiologically (Wilson et al., 2001). This type is insensitive to cannabinoids, exhibits no DSI, and presumably originates in distal dendrites because of their slow rise and decay kinetics. This is essentially in agreement with the finding by Martin et al. (2001) that IPSCs can be classified into at least three types: DSI-susceptible IPSC with fast kinetics, DSI-resistant IPSC with fast kinetics, and DSI-resistant IPSC with slow kinetics. To understand the physiological roles of DSI, the heterogeneity of synapses has to be considered carefully in terms of their CB1 expression and locations on pyramidal cells. In addition, it should be noted that the release of cotransmitters might also be modulated by endocannabinoids. The activation of CB1 receptors was found to suppress CCK release in the hippocampus (Beinfeld and Connolly, 2001). If DSI also reduces the release of cotransmitters, DSI can influence neural activity by modulating both fast and slow synaptic events.
We have estimated the local cannabinoid concentration during DSI and DSE in cultured neurons. The magnitudes of DSI and DSE induced by a 5 sec depolarization corresponded to the suppressions induced by ∼5 nm WIN55,212-2 for both IPSCs and EPSCs. These results suggest that at least in our culture system, the difference in the magnitudes of DSI and DSE is determined primarily by presynaptic cannabinoid sensitivities and not by the difference in the local cannabinoid level around presynaptic terminals. However, it should be noted that inputs, synaptic organization, and geometry of neurons may be altered in cultures from the nervous tissue in vivo. Regulation of DSI and DSE may be more complex in the brain. For example, postsynaptic depolarization may induce heterogeneous elevations of [Ca2+]i within single neurons because of heterogeneous distributions of voltage-gated Ca2+ channels and Ca2+ stores along the soma and dendrites. This may cause a heterogeneity of endocannabinoid concentration after depolarization. It is likely that relative contributions of DSE and DSI to neuronal excitability may depend on the geometry of the neuron and distribution of synapses. In addition, recent studies suggest that the activation of postsynaptic metabotropic glutamate receptors (mGluRs) enhances the depolarization-induced release of endocannabinoids (Varma et al., 2001; Ohno-Shosaku et al., 2002). This finding suggests that cannabinoid-mediated modulation may also be influenced by the distribution of mGluRs in neurons.
In addition to the modulation of synaptic transmission, cannabinoid agonists have been reported to exert variable effects on neurons (Di Marzo et al., 1998; Felder and Glass, 1998). These include inhibition of adenylate cyclase (Howlett and Fleming, 1984), inhibition of voltage-gated Ca2+ channels (Mackie and Hille, 1992; Mackie et al., 1995; Twitchell et al., 1997), and activation of inwardly rectifying K+channels (Mackie et al., 1995). These effects can be produced by 1–100 nm WIN55,212-2. It is therefore likely that these effects can work in concert with DSI and DSE to regulate the net excitability of the postsynaptic neuron in vivo.
Footnotes
This work was supported in part by grants-in-aid for scientific research (T.O., M.K.) and special coordination funds for promoting science and technology (M.K.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by grants from the Novartis Foundation (M.K.), the Cell Science Research Foundation (M.K.), and the Sumitomo Foundation (T.O.).
Correspondence should be addressed to Masanobu Kano, Department of Cellular Neurophysiology, Graduate School of Medical Science, Kanazawa University, 13-1 Takara-machi, Kanazawa 920-8640, Japan. E-mail:mkano@med.kanazawa-u.ac.jp.
REFERENCES
- 1.Alger BE, Pitler TA. Retrograde signaling at GABAA-receptor synapses in the mammalian CNS. Trends Neurosci. 1995;18:333–340. doi: 10.1016/0166-2236(95)93923-l. [DOI] [PubMed] [Google Scholar]
- 2.Al-Hayani A, Davies SN. Cannabinoid receptor mediated inhibition of excitatory synaptic transmission in the rat hippocampal slice is developmentally regulated. Br J Pharmacol. 2000;131:663–665. doi: 10.1038/sj.bjp.0703642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beinfeld MC, Connolly K. Activation of CB1 cannabinoid receptors in rat hippocampal slices inhibits potassium-evoked cholecystokinin release, a possible mechanism contributing to the spatial memory defects produced by cannabinoids. Neurosci Lett. 2001;301:69–71. doi: 10.1016/s0304-3940(01)01591-9. [DOI] [PubMed] [Google Scholar]
- 4.Deadwyler SA, Heyser CJ, Michaelis RC, Hampson RE. The effects of delta-9-THC on mechanisms of learning and memory. NIDA Res Monogr. 1990;97:79–93. [PubMed] [Google Scholar]
- 5.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum D, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 6.Diana MA, Levenes C, Mackie K, Marty A. Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci. 2002;22:200–208. doi: 10.1523/JNEUROSCI.22-01-00200.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- 8.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch-clamp recordings from neurons of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 9.Egertova M, Elphick MR. Localisation of cannabinoid receptors in the rat brain using antibodies to the intracellular C-terminal tail of CB1. J Comp Neurol. 2000;422:159–171. doi: 10.1002/(sici)1096-9861(20000626)422:2<159::aid-cne1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Felder CC, Glass M. Cannabinoid receptors and their endogenous agonists. Annu Rev Pharmacol Toxicol. 1998;38:179–200. doi: 10.1146/annurev.pharmtox.38.1.179. [DOI] [PubMed] [Google Scholar]
- 11.Hajos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 12.Hajos N, Ledent C, Freund TF. Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic synaptic transmission in the hippocampus. Neuroscience. 2001;106:1–4. doi: 10.1016/s0306-4522(01)00287-1. [DOI] [PubMed] [Google Scholar]
- 13.Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heyser CJ, Hampson RE, Deadwyler SA. Effects of delta-9-tetrahydrocannabinol on delayed match to sample performance in rats: alterations in short-term memory associated with changes in task specific firing of hippocampal cells. J Pharmacol Exp Ther. 1993;264:294–307. [PubMed] [Google Scholar]
- 15.Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 17.Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase: pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol. 1984;26:532–538. [PubMed] [Google Scholar]
- 18.Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001a;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 20. Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci 21 2001b. RC174:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- 22.Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in ATT20 cells transfected with rat brain cannabinoid receptors. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- 25.Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4224. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- 26.Martin LA, Wei DS, Alger BE. Heterogeneous susceptibility of GABA(A) receptor-mediated IPSCs to depolarization-induced suppression of inhibition in rat hippocampus. J Physiol (Lond) 2001;532:685–700. doi: 10.1111/j.1469-7793.2001.0685e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 28.Matsuda LA, Bonner TI, Lolait SJ. Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol. 1993;327:535–550. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- 29.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminsky NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 30.Mechoulam R, Fride E, Di Marzo V. Endocannabinoids. Eur J Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- 31.Misner DL, Sullivan JM. Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci. 1999;19:6795–6805. doi: 10.1523/JNEUROSCI.19-16-06795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 33.Ohno-Shosaku T, Sawada S, Yamamoto C. Properties of depolarization-induced suppression of inhibitory transmission in cultured rat hippocampal neurons. Pflügers Arch. 1998;435:273–279. doi: 10.1007/s004240050512. [DOI] [PubMed] [Google Scholar]
- 34.Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 35.Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- 36.Paton GS, Pertwee RG, Davies SN. Correlation between cannabinoid mediated effects on paired pulse depression and induction of long term potentiation in the rat hippocampal slice. Neuropharmacology. 1998;37:1123–1130. doi: 10.1016/s0028-3908(98)00096-3. [DOI] [PubMed] [Google Scholar]
- 37.Pettit DA, Harrison MP, Olson JM, Spencer RF, Cabral GA. Immunohistochemical localization of the neural cannabinoid receptor in rat brain. J Neurosci Res. 1998;51:391–402. doi: 10.1002/(SICI)1097-4547(19980201)51:3<391::AID-JNR12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 38.Piomelli D, Giuffrida A, Calignano A, Rodriguez de Fonseca F. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol Sci. 2000;21:218–224. doi: 10.1016/s0165-6147(00)01482-6. [DOI] [PubMed] [Google Scholar]
- 39.Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen M, Piser TM, Seybold VS, Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 42.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 43.Sugiura T, Kodaka T, Nakane S, Miyashita T, Kondo S, Suhara Y, Takayama H, Waku K, Seki C, Baba N, Ishima Y. Evidence that the cannabinoid CB1 receptor is a 2-arachidonoylglycerol receptor. J Biol Chem. 1999;274:2794–2801. doi: 10.1074/jbc.274.5.2794. [DOI] [PubMed] [Google Scholar]
- 44.Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol. 1999;82:1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- 45.Tsou K, Brown S, Sañudo-Reña JC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 46.Tsubokawa H, Ross WN. Muscarinic modulation of spike backpropagation in the apical dendrites of hippocampal CA1 pyramidal neurons. J Neurosci. 1997;17:5782–5791. doi: 10.1523/JNEUROSCI.17-15-05782.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsubokawa H, Offermanns S, Simon MI, Kano M. Calcium-dependent persistent facilitation of spike backpropagation in the CA1 pyramidal neurons. J Neurosci. 2000;20:4878–4884. doi: 10.1523/JNEUROSCI.20-13-04878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 49. Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21 2001. RC188:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagner JJ, Alger BE. Increased neuronal excitability during depolarization-induced suppression of inhibition in rat hippocampus. J Physiol (Lond) 1996;495:107–112. doi: 10.1113/jphysiol.1996.sp021577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 52.Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida T, Hashimoto K, Maejima T, Araishi K, Kano M. The cannabinoid CB1 receptor mediates retrograde signals for depolarization-induced suppression of inhibition in cerebellar Purkinje cells. J Neurosci. 2002;22:1690–1697. doi: 10.1523/JNEUROSCI.22-05-01690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TM. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci USA. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]