

Abstract
Methamphetamine (METH) selectively injures the neurites of dopamine (DA) neurons, generally without inducing cell death. It has been proposed that METH-induced redistribution of DA from the vesicular storage pool to the cytoplasm, where DA can oxidize to produce quinones and additional reactive oxygen species, may account for this selective neurotoxicity. To test this hypothesis, we used mice heterozygous (+/−) or homozygous (−/−) for the brain vesicular monoamine uptake transporter VMAT2, which mediates the accumulation of cytosolic DA into synaptic vesicles. In postnatal ventral midbrain neuronal cultures derived from these mice, METH-induced degeneration of DA neurites and accumulation of oxyradicals, including metabolites of oxidized DA, varied inversely with VMAT2 expression. METH administration also promoted the synthesis of DA via upregulation of tyrosine hydroxylase activity, resulting in an elevation of cytosolic DA even in the absence of vesicular sequestration. Electron microscopy and fluorescent labeling confirmed that METH promoted the formation of autophagic granules, particularly in neuronal varicosities and, ultimately, within cell bodies of dopaminergic neurons. Therefore, we propose that METH neurotoxicity results from the induction of a specific cellular pathway that is activated when DA cannot be effectively sequestered in synaptic vesicles, thereby producing oxyradical stress, autophagy, and neurite degeneration.
Keywords: methamphetamine, VMAT2, oxidative stress, neurodegeneration, dopamine, ventral midbrain, autophagy
Methamphetamine (METH), a widely abused psychostimulant, is neurotoxic for dopamine (DA) neurites (Seiden and Ricaurte, 1987; Cubells et al., 1994). METH promotes a severe loss of dopaminergic axonal arbors and profoundly decreases striatal DA levels. The pathway by which METH induces dopaminergic neurotoxicity appears to parallel that of serotonergic neurotoxicity induced by methylenedioxymethamphetamine (ecstasy) and may share similarities with other neurodegenerative pathways, including that associated with the Parkinson's disease-inducing agentN-methyl-4-phenylpyridinium (MPP+) (Lotharius and O'Malley, 2000). However, METH induces a form of neurodegeneration that is quite distinct from other disorders in that the cell bodies of DA neurons are spared (Ricaurte et al., 1982). Thus, the ability of METH to selectively destroy neurites while leaving neuronal cell bodies intact warrants explanation to understand its neurotoxic mechanism.
The basis for the unusual pattern of METH neurotoxicity may stem from the mechanism by which the amphetamines act to release monoamine neurotransmitters. Whereas cocaine increases the levels of extrasynaptic neurotransmitter by inhibiting DA transporter (DAT)-mediated reuptake of released DA (Sonders et al., 1997), the amphetamines trigger neurotransmitter release from the cytosol to the extracellular space by means of reverse transport through DAT (Sulzer et al., 1995; Jones et al., 1998; Schmitz et al., 2001). Two models have been proposed to explain the role for DA in the actions of METH. In the exchange diffusion model (Fischer and Cho, 1979; Mundorf et al., 1999), amphetamines, as substrates for the DAT (Sonders et al., 1997), act to reverse the transporter conformation so that net DA efflux occurs (Jones et al., 1999). Exchange diffusion predicts that cytosolic DA is decreased by amphetamines, and that METH administration induces extraneuronal DA oxidation (Seiden and Vosmer, 1984; De Vito and Wagner, 1989; Stephans and Yamamoto, 1994). A drawback to this model is that it does not directly explain the specificity of METH degeneration for DA terminals, in that the oxidation of extraneuronal DA would be expected to nonspecifically damage all neighboring neurons, not just dopaminergic ones. In the weak base model (Sulzer et al., 1995), METH acts via both DAT and the vesicular monoamine transporter (VMAT2) to promote the collapse of vesicular proton gradients, redistributing DA from the synaptic vesicle to the cytosol (Mundorf et al., 1999), while preventing vesicular reuptake of cytosolic DA by destroying the driving force for vesicular DA accumulation (Maron et al., 1983). Therefore, this model predicts that METH increases cytosolic levels of DA, leading to DA oxidation within the neuronal cytosol (Cubells et al., 1994; Fumagalli et al., 1999; LaVoie and Hastings, 1999; Lotharius and O'Malley, 2001).
In this study, we used cultured postnatal DA neurons to demonstrate how the particular properties of METH operate together to elicit neurodegeneration, and show that METH induces a specific pathway, autophagy, that is responsible for neurite degeneration.
MATERIALS AND METHODS
Genotyping. Production of VMAT2 mutant mice was performed as described previously (Fon et al., 1997). Tail genomic DNA was used to genotype transgenic VMAT2 mice by PCR. One primer pair (5′-CATCGTGTTCCTCGCGCTGC-3′ and 5′-GGGATGCTGTCACCTGGG-3′) was designed to amplify a 181 bp fragment in the wild-type but not the mutant allele, whereas a second primer pair (5′-CCGCTCCCGATTCGCAGCG-3′ and 5′-GCAGCAGCTTAGCACACTGG-3′) was designed to amplify a 292 bp fragment exclusively in the mutant allele. Results were determined by agarose (1.5%) gel electrophoresis. VMAT2 +/− mice were then mated to generate VMAT2 −/−, +/−, and +/+ mice.
Primary midbrain neuronal cultures. Mouse midbrain cultures derived from postnatal day 1 wild-type or VMAT2 mutant mice were plated onto cortical astrocyte monolayers as described previously (Fon et al., 1997). Approximately 40% of the neurons derived from these midbrain cultures are dopaminergic. Cells were untreated for at least 2 weeks after plating to allow for normal development and elaboration of neurites. Stocks of METH (methamphetamine hydrochloride, 100 mm in 0.1N HCl) were maintained at 4°C until needed. Drugs were applied directly to the culture media and were not removed. All experiments compared cultures derived from matched sets of sister cultures and were repeated at least three times per treatment group.
Immunocytochemistry. Tyrosine hydroxylase (TH) immunostaining on 4% paraformaldehyde-fixed cultures was performed using our standard methods (Mena et al., 1997). Neurons were immunolabeled for GABA as described previously (Mena et al., 1997). Immunostaining was detected by diaminobenzidine/hydrogen peroxide after exposure to biotin-conjugated monoclonal secondary antibodies (anti-mouse and anti-rabbit; 1:200; Vector Laboratories, Burlingame, CA) and a horseradish peroxidase-conjugated avidin/biotin complex (Vector Laboratories). Activated caspase-3 (CM1; Idun Pharmaceuticals, San Diego, CA) immunostaining was performed as described previously (Srinivasan et al., 1998). For GABA immunostaining, the visualization was further enhanced by nickel. To assess the specificity of TH and GABA immunostaining, the primary antibody was omitted, resulting in no detectable signal. In some cases, TH-immunoreactivity was assessed via fluorescence using a monoclonal Texas Red-coupled secondary antibody (Vector Laboratories).
Cell and neurite counts. For cell and neurite counts, neurons were tallied under differential interference contrast (DIC) optics. Effects of METH, nomifensine, and amfonelic acid on the process extension of TH- or GABA-immunopositive neurites were measured using NIH Image (http://rsb.info.nih.gov/nih-image/) by an observer blind to the treatment condition. The number of processes of TH- or GABA-immunopositive neurons were quantified by placing each cell body in the center of a 20× scope field and counting the number of primary neurites that extended to or beyond a 120 μm radius. Frequency distributions for each group were generated by assigning each neuron to a bin corresponding to zero, one, two, three, four, or five neurites per neuron that extended from the cell body beyond 120 μm in length and were analyzed as reported previously (Cubells et al., 1994). From these counts, the average number of neurites per neuron was tallied. Cell count data were subjected to statistical analysis by the ANOVA program, whereas neurite count data, which are nonparametric, were subjected to either the χ2 or Mann–Whitney U tests.
Detection of oxidative stress.2,7-dichlorofluorescein diacetate (DCF; Molecular Probes, Eugene, OR) was prepared in dimethylsulfoxide and stored as 10 mm aliquots at −85°C. Just before use, an aliquot was diluted to 1 μm in minimal essential medium at 32°C. Neuronal cultures were incubated for 15 min, rinsed twice with oxygenated physiological saline, and monitored as described previously using a Zeiss (Thornwood, NY) fluorescein filter set (Cubells et al., 1994). To avoid any artifactual amplification of the fluorescence (Rota et al., 1999), the cells were first brought into focus under DIC optics and fluorescent images were then acquired during a single 200 msec exposure with a 90% neutral density filter. Therefore, there was no previous fluorescent excitation. Fluorescence was measured within 25 × 25 pixel regions of interest and analyzed by NIH Image for pixel intensity.
Analysis of neuronal cell death. For nuclear staining and detection of apoptotic morphology, cultured neurons were rinsed twice in physiological saline, incubated with 30 μmHoechst 33342 (Molecular Probes) for 5 min at 37°C, and then examined using a Zeiss UV filter set. For assessment of cell viability, the LIVE/DEAD Viability/Cytotoxicity kit (Molecular Probes) was used as instructed by the manufacturer.
Detection of autophagic vacuoles. Under light microscopy, autophagic vacuoles were detected with monodansylcadaverine (MDC; Molecular Probes) as described previously (Petersén et al., 2001). Briefly, cultures were incubated with 50 μm MDC for 60 min at 37°C and rinsed twice in physiological saline. For imaging, we used a Zeiss LSM 510 multiphoton laser scanning confocal microscope (excitation, 735 nm; emission, 500–550 nm) equipped with a 40× water immersion objective. In some cases, cultures were fixed after MDC incubation and counterstained for TH using fluorescent Texas Red (rhodamine)-conjugated secondary antibody. Electron microscopy was performed as reported previously (Sulzer et al., 2000).
Measurements of catecholamine levels. HPLC with electrochemical detection (HPLC-EC) was used to measure dopamine and TH activity [vial-dihydroxyphenylalanine (l-DOPA) levels] (Philipp, 1987) as described previously (Pothos et al., 1998, 2000), with some modifications. For dopamine determinations, samples were run on a Supelcosil LC-18 column (Supelco, Bellefonte, PA). The mobile phase consisted of (in mm): 50 sodium acetate, 0.05 EDTA, 0.7 heptanesulfonic acid, and 10% methanol, pH 4.6. For TH activity analyses, cultures were pretreated with 0.1 mml-tyrosine and 10 μmm-hydroxybenzylhydrazine (NSD-1015) for 40 min at 37°C, and samples were analyzed for l-DOPA content on a BioPhase ODS column (BAS, West Lafayette, IN) in a mobile phase composed of (in mm): 50 potassium phosphate, 0.1 EDTA, 0.2 heptanesulfonic acid, and 10% methanol, pH 2.7.
Isolation of cysteinyl-catechols. At 6 d after exposure to vehicle or METH, neuronal cultures were rinsed three times with physiological saline, harvested in 200 μl of 0.3m perchloric acid, binned (n = 3), briefly sonicated, and centrifuged (13,000 rpm, 15 min, 4°C). An aliquot was removed for protein content analysis according to the method of Bradford (1976). The supernatant was removed from the protein pellet and placed in separate tubes. Samples were analyzed for cysteinyl-DA as described previously (Hastings et al., 1996). The detection limit for cysteinyl-DA under these conditions was <0.1 pmol.
RESULTS
METH-induced neurodegeneration diminishes total DA levels but does not block METH-induced DA release
Depletion of striatal DA is often used to assay METH toxicityin vivo. In an effort to validate our in vitromodel of METH neurotoxicity, we compared the total (extracellular plus intracellular) content of DA in control and METH-treated postnatally derived ventral midbrain cultures using HPLC. After prolonged METH exposure, total DA levels were decreased by 75% (Fig.1A), similar to the level of DA depletion in murine models in vivo (for review, see Seiden and Ricaurte, 1987). Short-term amphetamine administration is known to increase extracellular DA levels in dopamine neuronal cultures and slices (Fon et al., 1997; Schmitz et al., 2001). However, METH-induced release of DA has not been reported after a neurotoxic METH regimen in postnatal neuronal cultures. We examined whether short-term METH administration would induce DA release differentially in control cultures and in cultures exposed to long-term (7 d, 100 μm) METH. We exposed control and long-term METH cultures to either physiological saline or short-term (10 μm, 30 min) METH and measured extracellular DA levels. Cultures exposed to long-term METH had twofold higher basal levels of extracellular DA than controls. When the control cultures were treated with a short-term exposure to METH, they released fourfold higher levels of DA compared with basal conditions, whereas long-term METH-treated cultures released 2.5-fold more DA after short-term METH exposure compared with basal conditions (Fig. 1B). Thus, short-term METH engendered the same absolute extracellular DA levels in both control and long-term METH-exposed neurons (Fig.1B), despite the much lower total DA content in long-term METH-exposed cultures. These data indicate that, although long-term METH induces neurodegeneration (Fig.2) and depletes total DA, these neurons retain functional METH-mediated DA release.
Fig. 1.

Effect of METH on ventral midbrain DA levels. Untreated (open bars) and long-term (7 d, 100 μm) METH-treated (filled bars) VMAT2 +/+ midbrain neuronal cultures were exposed to vehicle (basal) or short-term METH stimulus (10 μm, 30 min), and total (A) and extracellular (B) DA levels were measured by HPLC-EC. Long-term exposure to METH diminished total DA levels (A). However, extracellular DA is similar in both untreated and METH-treated cultures aftershort-term METH stimulation (B). Results are shown as the mean ± SEM (n = 15).
Fig. 2.
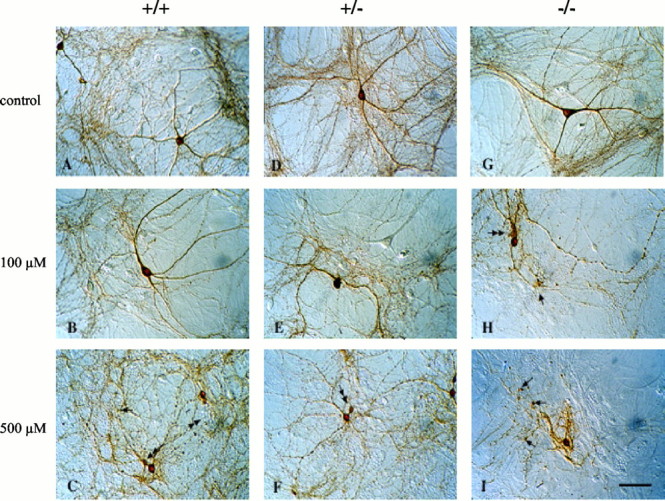
Effect of METH on the morphology of midbrain dopaminergic neurons. TH-immunostained cultures derived from postnatal VMAT2 +/+ (A–C), +/− (D–F), and −/− (G–I) littermate mice are shown. Untreated neurons from all three genotypes display smooth dendrites, an extensive TH neuropil consisting of thin and highly elaborate axons, and small-caliber axonal varicosities. After exposure to 100 μm METH for 7 d (B, E, H), degeneration of neurites occurred in all three genotypes, but particularly in VMAT2 −/− cultures (H). Examples of swollen axonal varicosities are shown (arrows). H shows an example of a pinched-off portion of the cell body, which is a common feature in METH-exposed cultures (double-headed arrow inC, H, F); the remaining area of the cell body is therefore shrunken. After exposure to 500 μm METH for 7 d (C, F, I), neurite degeneration, axonal varicosity swelling, and pinched cell bodies become obvious in all three genotypes, most prominently in the VMAT2 −/− cultures (I). Nondopaminergic neurons were unaffected in all treatments (Fig. 4). Scale bar, 50 μm.
METH neurodegeneration is exacerbated in VMAT2 knock-out DA neurons
Previous primary neuronal culture studies suggest that >98% of DA is normally sequestered in synaptic vesicles (Sulzer et al., 1996), that VMAT2 expression level modulates the amount of DA packaged in midbrain DA synaptic vesicles (Pothos et al., 2000), and that cytosolic DA is required for DA oxidation (Sulzer et al., 2000). To test our hypothesis that reduced sequestration of cytosolic DA in synaptic vesicles would exacerbate METH neurotoxicity, we used VMAT2 −/− mice, which are unable to accumulate DA within synaptic vesicles (Fon et al., 1997). Consistent with our previous study, we found that untreated VMAT2 −/− cultures contained only 3% of the intracellular DA levels of VMAT2 +/+ cultures (Fon et al., 1997). We also found that toxic levels of METH (100 μm for 7 d) elevated intracellular DA by nearly 10-fold in VMAT2 −/− cultures, or to 25% of VMAT2 +/+ culture intracellular DA levels (Table1).
Table 1.
Total cellular levels of DA and cysteinyl-DA (picomoles per milligram of protein) in sister VMAT2 wild-type (+/+) and knock-out (−/−) cultures exposed to vehicle or METH (100 μm, 7 d) as measured by HPLC-EC
| Catecholamine levels (pmol/culture) | +/+ | −/− | ||
|---|---|---|---|---|
| Control | 100 μm METH | Control | 100 μm METH | |
| DA | 24.57 ± 2.75 | 5.09 ± 0.92* | 0.76 ± 0.07 | 6.05 ± 0.59* |
| Cysteinyl-DA | 0.1 ± 0.02 | 0.20 ± 0.03* | <0.1 | 0.47 ± 0.12* |
Data are expressed as mean ± SEM and are derived from at least three independent experiments (*p < 0.05;n = 9).
We have reported previously that 100 μm METH (7 d) selectively promoted degeneration of DA cell neurites, sparing DA cell bodies (Cubells et al., 1994). This concentration is in the range of whole-tissue concentrations of amphetamine, and a similar value in the substantia nigra (73 ± 10 μm) was reported after neurotoxic amphetamine regimens in vivo in rodents (Clausing et al., 1995). We examined whether METH-induced neurite degeneration occurred differentially in dopaminergic ventral midbrain neurons derived from VMAT2 +/+, +/−, and −/− mice. Untreated cultures and cultures treated with METH for 7 d were fixed and immunostained for TH. We found no morphological differences among untreated TH-immunoreactive cultures derived from any of the VMAT2 genotypes (Figs. 2A,D,G, 3). However, neurites of DA neurons were susceptible to METH-induced neurodegeneration in inverse proportion to VMAT2 level (Fig. 3). The affected cells displayed blebbing of the soma, swelling of axonal varicosities, and collapse of neurites. Cultures derived from VMAT2 −/− mice were most sensitive to METH-induced neurodegeneration, displaying primarily neurite remnants (Fig.2H,I).
Fig. 3.

Quantitation of neurite loss from midbrain dopaminergic neurons after METH exposure. Dopaminergic neurons, defined by TH immunoreactivity, were classified by the number of primary neurites extending beyond a 120 μm radius, and results are expressed as the average number of TH-immunopositive neurites per cell (A). Although there was no difference among untreated cultures derived from the genotypes (χ2 = 18.31; df = 12; p = 0.1066), exposure to METH reduced both the number and the extent of TH-positive primary neurites. Both dose-dependent (*p < 0.05) and genotype-dependent (*−p< 0.05) degeneration was evident after 7 d of METH administration, as determined by χ2 goodness-of-fit tests (N = 600–1200 neurons per condition;n > 3). GABAergic neurons, defined by GABA immunoreactivity, were similarly classified (B). METH did not promote neurodegeneration of GABAergic neurons in any of the genotypes (Fig. 4). Open bars, +/+; gray bars, +/−; filled bars, −/−.
Counts of the number of primary DA neurites extending >120 μm from their parent cell bodies confirmed that METH promotes the degeneration of neurites in a concentration-dependent manner (Fig. 3A). Neuronal cultures derived from VMAT2 +/+ mice revealed a dose-dependent reduction in the number of primary neurites. Cultures of VMAT2 +/− DA neurons were more vulnerable than VMAT2 +/+ cultures, because 100 and 500 μm METH resulted in enhanced neurodegeneration of TH-positive neurites, confirming an independent report on the increased vulnerability of VMAT2 +/− mice to METH neurotoxicity (Fumagalli et al., 1999). The most profound neurotoxic effect of METH was evident in neurons derived from VMAT2 −/− mice; exposure to 100 μm METH was as neurotoxic as administration of 500 μm METH to VMAT2 +/+ cultures. The effect of METH on neurodegeneration was specific for DA neurons. GABAergic neurons, which comprise nearly all of the non-DA neurons in these cultures (Mena et al., 1997), were resistant to METH neurotoxicity in all VMAT2 genotypes (Figs. 3B,4).
Fig. 4.
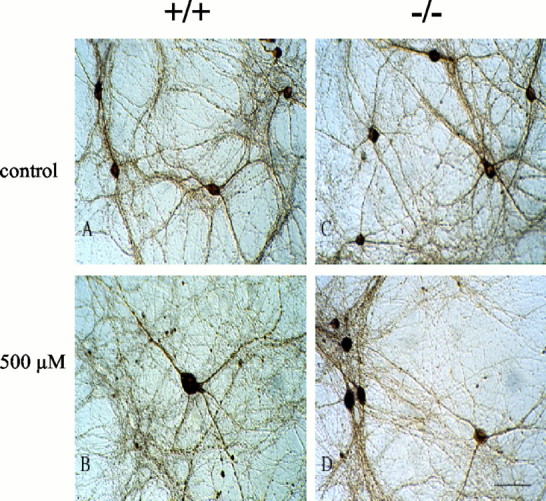
Effect of METH on the morphology of midbrain nondopaminergic neurons. Cultures derived from postnatal VMAT2 +/+, +/− (data not shown), or −/− mice were exposed to 500 μm METH (B, D) or vehicle (A, C) for 7 d and immunostained for GABA. In these cultures, the nondopaminergic neurons are nearly all GABAergic. METH exposure had no significant effect on neurite length or on the number of GABAergic neurons. Scale bar, 50 μm (n = 12).
METH neurotoxicity is dependent on DAT activity
A salient feature of METH neurodegeneration is the requirement for the uptake of METH by DAT, because DAT uptake blockers such as nomifensine and amfonelic acid (Axt et al., 1990; Wan et al., 2000) prevent METH-induced neurotoxicity in vivo, and METH neurotoxicity is absent in DAT −/− mice (Fumagalli et al., 1998). We found that METH-induced neurodegeneration was blocked when the DAT inhibitor nomifensine (20 μm) was administered 30 min before a 7 d, 500 μm METH exposure (Fig. 5). Similar neuroprotection was obtained with amfonelic acid (1 μm) (Fig. 5). These data suggest that our postnatally derived in vitromodel of METH neurodegeneration closely mirrors in vivomodels.
Fig. 5.

METH-induced neurotoxicity is mediated by DAT. Postnatally derived VMAT2 +/+ neuronal cultures were exposed to either vehicle (bar 1), 500 μm METH (bar 2), 20 μm nomifensine (bar 5), or 1 μm amfonelic acid (bar 6), or were pretreated with nomifensine or amfonelic acid 30 min before METH exposure (bars 3 and 4, respectively). After 7 d of exposure, cells were immunostained for TH and assessed as in Figure 3. Nomifensine effectively prevented METH-induced neurodegeneration (Mann–Whitney U test;p = 0.0002; U = 191;n = 18 for METH and n = 19 for METH plus nomifensine); amfonelic acid was also effective (p < 0.0001; U = 273.5;n = 18 for METH and n = 15 for METH plus amfonelic acid). *p < 0.0001 for METH; **DAT inhibitor plus METH.
METH does not promote cell death
To examine whether METH promotes neuronal cell death in cultures derived from VMAT2-deficient mice, we counted living and dead neurons after labeling with calcein AM and ethidium homodimer. Despite the profound degenerative effect on neurites, neuronal death was not observed in any of the VMAT2 genotypes after exposure to 500 μm METH for 7 d (data not shown). Neuronal cultures were immunostained with CM1 for the presence of activated caspase-3, a marker of apoptosis (Srinivasan et al., 1998). There was no increase in CM1 immunostaining with long-term METH exposure (data not shown). These findings were confirmed by nuclear labeling by Hoechst 33342, which revealed the presence of limited, basal levels of apoptosis in all of the genotypes that was not enhanced by METH (data not shown). Thus, by three independent methods, we observed no METH-induced neuronal death.
METH-induced intracellular oxidative stress
Formation of peroxynitrite (Itzhak et al., 1998), peroxide and superoxide (Acikgoz et al., 1998), and hydroxyl radicals (Kita et al., 1999) have all been implicated in METH neurotoxicity. Conversely, overexpression of superoxide dismutase in transgenic mice (Cadet et al., 1994) and pretreatment with antioxidants (Wagner et al., 1985a) reduce METH-induced neurotoxicity. We examined the effect of METH on the production of reactive oxygen species (ROS) in cultured neurons obtained from the VMAT2 genotypes by using DCF, a membrane-permeant fluorogenic compound that, in the presence of peroxynitrite and hydrogen peroxide (Kooy et al., 1997; Possel et al., 1997), becomes fluorescent and trapped in cells after deesterification by cytoplasmic enzymes. When untreated cultures were incubated in DCF, <1% of the neurons from all VMAT2 genotypes were brightly labeled, and only diffuse DCF labeling was observed (Fig.6A). Basal DCF labeling was significantly increased with decreased VMAT2 expression in untreated neurons (Table 2), suggesting that VMAT2 may regulate endogenous oxidative balance in the cytosol. DCF labeling was enhanced in neuronal cultures incubated for 18 hr in 100 μm METH, resulting in diffuse but strong cytoplasmic DCF labeling, with bright spots of fluorescence within varicosities along the axons and in the soma (Fig.6B). VMAT2 +/− cultures exposed to METH revealed a similar pattern of labeling (Fig. 6D). In contrast, VMAT2 −/− neurons exposed to METH displayed intense DCF labeling throughout both the cell bodies and the neurites (Fig.6F), suggesting that VMAT2 −/− neurons generate extremely high intracellular ROS levels after METH exposure (Table 2). These results are in agreement with a recent report in which exposure of primary mesencephalic DA neurons to amphetamine resulted in the induction of DA-dependent, intracellular ROS formation (Lotharius and O'Malley, 2001). As described previously (Cubells et al., 1994), the METH-induced oxidative stress was DA dependent, because cultures derived from the nucleus accumbens, which do not contain DA, were devoid of METH-enhanced DCF labeling (data not shown).
Fig. 6.

Exposure to METH induces intracellular oxidative stress. Living cultures derived from postnatal VMAT2 +/+, +/−, and −/− littermate mice were exposed to 100 μm METH or vehicle alone (control) for 18 hr and viewed under fluorescein optics after DCF labeling. Punctate DCF staining of somata is seen in VMAT2 +/+ and +/− cultures exposed to METH (B, D). Intense staining of varicosities within the neurite is found in the VMAT2 −/− cultures, even in the absence of METH (C). Although METH exposure increased DCF labeling in cultures derived from all three genotypes (Table 2), cultures derived from the VMAT2 −/− mice demonstrated by far the highest levels of oxidative stress (F). The arrow indicates a weakly DCF-labeled neuron. Scale bar, 5 μm.
Table 2.
DCF fluorescence
| +/+ | +/− | −/− | |
|---|---|---|---|
| Control | 113.6 ± 4.6 | 133.8 ± 4.3* | 255.7 ± 26.6* |
| 100 μm METH | 142.1 ± 16.2 | 150.0 ± 7.4 | 1383.6 ± 270.6* |
Mean ± SEM pixel intensities (12 bit resolution), generated from five randomly chosen regions of interest within neurons from four independent experiments in sister cultures exposed to vehicle (control) or METH (100 μm, 18 hr). Basal DCF labeling increases significantly as VMAT2 content decreases in control cultures (Mann–Whitney U test; +/−, U statistic = 305, p = 0.0047; −/−, U statistic = 398, p < 0.0001). After METH exposure, VMAT2 knock-out (−/−) cultures possessed the highest intracellular levels of oxidative stress (U statistic = 75; p = 0.0008). Astrocytes showed negligible staining in all cases (n = 12).
*p < 0.001.
The major oxidative products of DA are DA-quinone and DA-semiquinone. A major stable product of these oxidized DA metabolites is free cysteinyl-DA, which is presumably derived from glutathione conjugated to DA-quinone that is then proteolyzed to free cysteinyl-DA. Cysteinyl-DA may be further metabolized (Zhang and Dryhurst, 1994). DA-quinones and DA-semiquinones can react with proteins and nucleic acids (Hastings and Berman, 1999). Therefore, although free cysteinyl-DA cannot be used to directly compare the amount of DA-quinone produced in the system, its presence confirms that DA-quinone was produced (LaVoie and Hastings, 1999). We found that METH (100 μm for 7 d) increased free cysteinyl-DA in METH-treated cultures by threefold (Table 1), consistent with results from METH-treated rats in vivo (LaVoie and Hastings, 1999). Although cysteinyl-DA levels were undetectable in untreated VMAT2 −/− cultures, METH administration resulted in over twice as much cysteinyl-DA in VMAT2 −/− compared with METH-treated VMAT2 +/+ cultures (Table 1).
METH stimulates TH activity
The DCF results suggested that METH promotes increased intracellular levels of DA-derived oxyradicals, even in the VMAT2 −/− cultures. One explanation for the increase in intracellular DA-derived oxyradicals is via increased DA synthesis, which may occur through enhanced activity of TH. To test whether METH administration increases the synthesis of DA, we monitored the intracellular accumulation of the TH product l-DOPA in the presence of an aromatic acid decarboxylase inhibitor (NSD-1015) that blocks l-DOPA conversion to DA (Ricaurte et al., 1983). Short-term METH administration resulted in a threefold increase in l-DOPA formation compared with controls (Fig.7A). These results are in direct accordance with previous in vivo findings in which the synthesis and release of DA were enhanced in METH-treated degenerating nigrostriatal DA terminals (Ricaurte et al., 1983). To test whether inhibition of vesicular DA uptake affects the METH enhancement of TH activity, we used the VMAT2 blocker reserpine. In reserpine-treated cultures (1 μm, 90 min),l-DOPA accumulation was nearly abolished, whereas METH increased l-DOPA accumulation by fourfold (Fig. 7A). These results suggest that even in the absence of vesicular uptake of DA, METH stimulates the synthesis of DA via increasing TH activity.
Fig. 7.

METH stimulates TH activity in dopaminergic midbrain cultures. A, VMAT2 +/+ ventral midbrain neuronal cultures were exposed to l-tyrosine (100 μm for 40 min) in the presence of the DOPA decarboxylase inhibitor NSD-1015 (10 μm). After incubation and extraction, total l-DOPA levels were measured by HPLC-EC (A). l-DOPA levels were increased by short-term METH stimulation (10 μm, 30 min). METH also promoted the accumulation of l-DOPA, even in the presence of the VMAT2 blocker reserpine (RES). Results are shown as mean ± SEM (n = 15). *p < 0.001. B, Untreated (open bars) and long-term (7 d, 100 μm) METH-treated (filled bars) wild-type midbrain dopaminergic neurons were exposed to vehicle, the TH inhibitor MPT (3 hr, 10 μm), the short-term METH stimulus (10 μm, 30 min), or METH in the presence of ΜPΤ (METH & MPT), and total DA levels were measured by HPLC-EC (B). Results are shown as mean ± SEM (n = 12).
We also used α-methyl-p-tyrosine (MPT; 10 μm for 3 hr), a TH inhibitor, to ascertain the role of DA synthesis in METH-dependent neurotoxicity. In cultures exposed to short-term METH (10 μm, 30 min) in the continuing presence of MPT, the inhibitor significantly reduced total levels of DA by 30% in controls and by 60% in long-term (100 μm, 7 d) METH-treated cultures (Fig.7B). MPT reduced intracellular DA levels by >60% in long-term METH-treated cultures but had no effect on cultures that were not exposed to METH. These data suggest that under basal conditions, neuronal cultures have a very low rate of ongoing TH activity, whereas cultures exposed to long-term METH maintain markedly enhanced DA synthesis. These data also confirm that even damaged DA neurons continue to synthesize DA via upregulation of TH activity. Thus, the DA present in cultures that have undergone METH-induced neurodegeneration appears to be derived primarily from ongoing synthesis.
METH triggers autophagy in DA neurons
Although METH induced high levels of intracellular oxidative stress, we were unable to detect any evidence of cell death or apoptosis in the midbrain cultures. We therefore examined control and METH-treated cultures by electron microscopy. Untreated neurons (Fig.8A,B) had the normal complement of healthy mitochondria (Fig. 8B, white arrow) and endoplasmic reticulum (Fig. 8B, black arrow). Conversely, in cultures exposed to 100 μm METH for 7 d, large electron-dense membranous whorls, multivesicular bodies, and autophagic vacuoles filled the cytoplasm (Fig. 8C, black arrows), but the nuclei appeared healthy and intact (Fig. 8C,D). These features are characteristic of autophagy (for review, see Larsen and Sulzer, 2002).
Fig. 8.
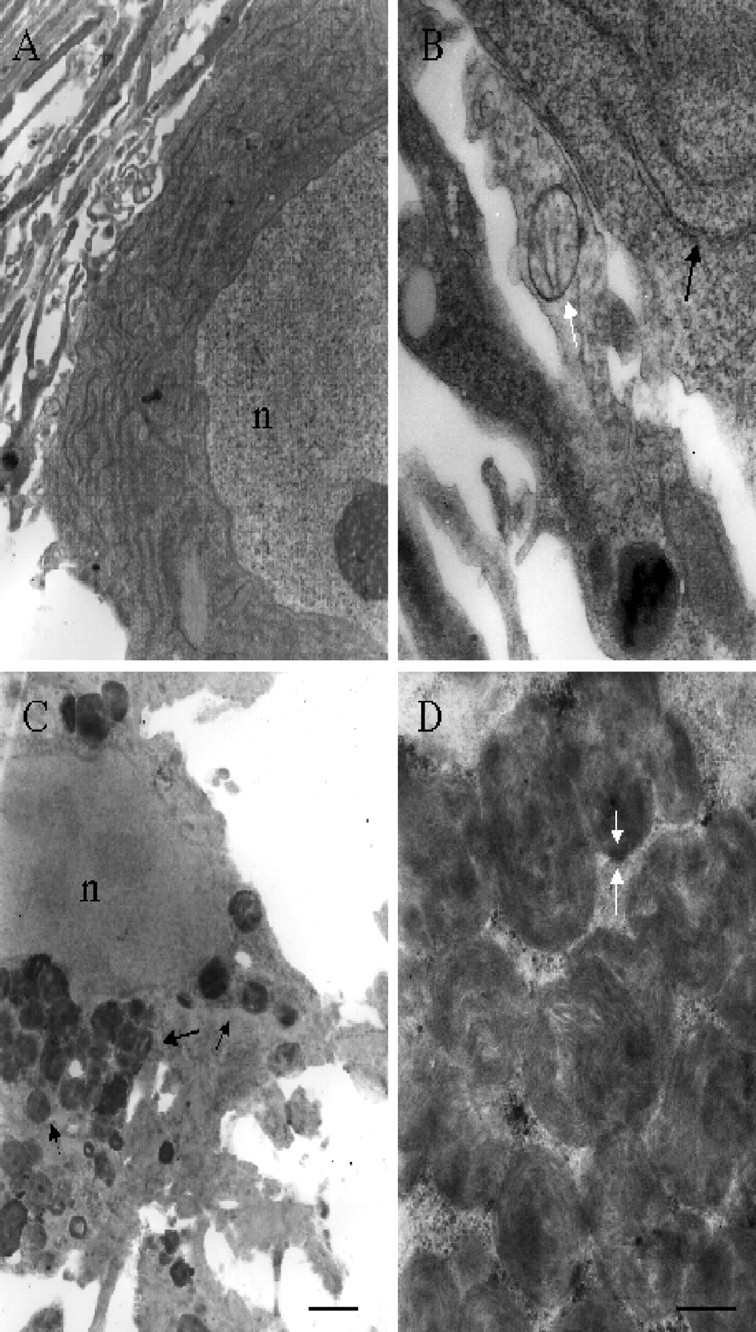
METH induces neuronal autophagy. Electron micrographs of VMAT2 +/+ cultures exposed to either vehicle alone (A, B) or 100 μm METH for 7 d (C, D). Untreated cultures possess healthy nuclei (n) and the usual complement of organelles such as mitochondria and endoplasmic reticulum (B,arrow). Healthy nuclei (n) are also evident in METH-treated cultures (C), but the cytoplasm is nearly devoid of organelles and is instead filled with degradative autophagic vacuoles in the form of membranous whorls (C, arrow, enlarged in D). Scale bars: A, C, 1 μm; B, D, 200 nm.
The autofluorescent dye MDC has been used to exclusively identify autophagic vacuoles in unfixed, living cultures as well as in fixed tissue (Petersén et al., 2001). Therefore, we used this agent to determine whether DA neurons form autophagic vacuoles in the presence of long-term METH. Cells exposed to 100 μm METH for 1 d were loaded with MDC, fixed, and immunostained for TH. A TH-immunoreactive dopaminergic neuron is shown in Figure9. The MDC label (Fig. 9,green) colocalizes with TH (Fig. 9C) and accumulates in the neurites, particularly in distal axonal varicosities (Fig. 9D). We subsequently investigated the time course of METH-induced autophagic vacuole formation. Midbrain cultures were exposed to 100 μm METH for different time intervals and then loaded with MDC as indicated in Figure10. Untreated cultures displayed very faint, basal MDC staining (Fig. 10A). Increasing exposure to METH resulted in numerous autophagic vacuoles, at first throughout the soma and neurites (Fig. 10B,C), but by 7 d, staining was primarily evident in the cell body as the neurites disappeared (Fig. 10D). MDC staining was never present in the nucleus and did not colocalize with GABA-immunoreactive neurons (data not shown).
Fig. 9.
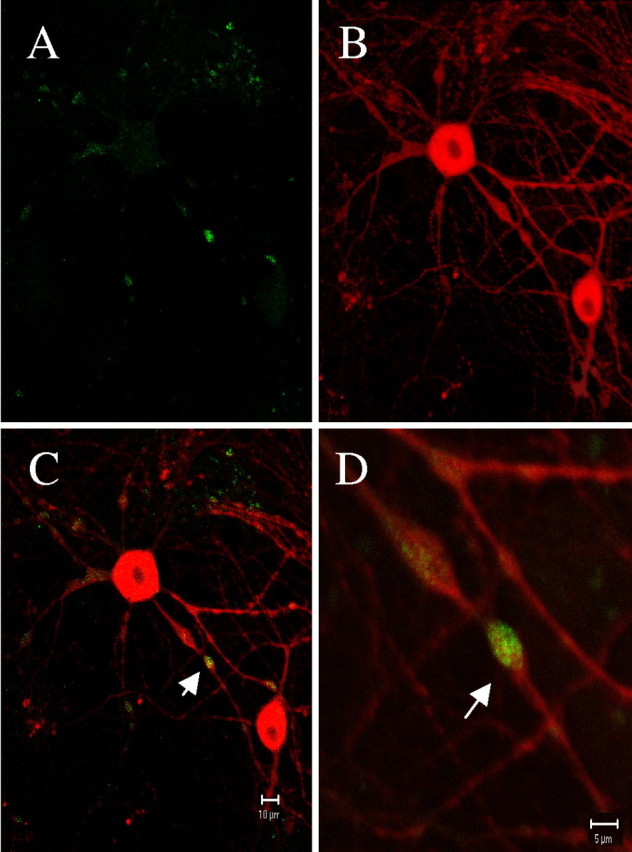
METH-induced autophagic vacuoles are present in DA neurons. Fluorescent two photon micrographs of a VMAT2 +/+ ventral midbrain neuron exposed to 100 μm METH for 1 d. MDC labeling (A) is evident throughout the neuron but is concentrated within neuritic varicosities. The same neuron is identified as dopaminergic with TH immunostaining (B). Arrows (C, D) indicate the localization of MDC-labeled autophagic vacuoles within DA neuronal varicosities. Scale bars: A–C, 10 μm;D, 5 μm.
Fig. 10.

Time-dependent accumulation of autophagic vacuoles. Fluorescent micrographs of a living VMAT2 +/+ ventral midbrain neuron exposed to 100 μm METH for 0 (A), 1 (B), 3 (C), or 7 (D) d and labeled with MDC. MDC label increased over time and eventually accumulated in the neuronal soma. No labeling was ever observed within nuclei. Scale bar, 5 μm.
DISCUSSION
This study proposes a pathway that may underlie the unusual pattern of neurodegeneration elicited by METH (Fig.11). We found that postnatally derived ventral midbrain cultures replicate important attributes of METH neurotoxicity observed in vivo, including (1) loss of DA neurites without neuronal death, (2) reduction in total DA levels, (3) lack of toxicity in nondopaminergic neurons, (4) protection by DAT inhibitors, (5) regulation by VMAT2 expression, and (6) formation of DA-derived oxyradicals. We demonstrated that VMAT2-deficient neurons, which are unable to sequester DA in vesicles, develop high levels of ROS, supporting a role for cytosolic DA in neurodegeneration. We identified a specific pathway, autophagy, which is induced in response to the downstream actions of METH and appears to correlate with the neurite loss. This is in contrast to previous hypotheses that neurite loss occurs nonspecifically, by structural collapse after attenuation of cellular energy stores.
Fig. 11.
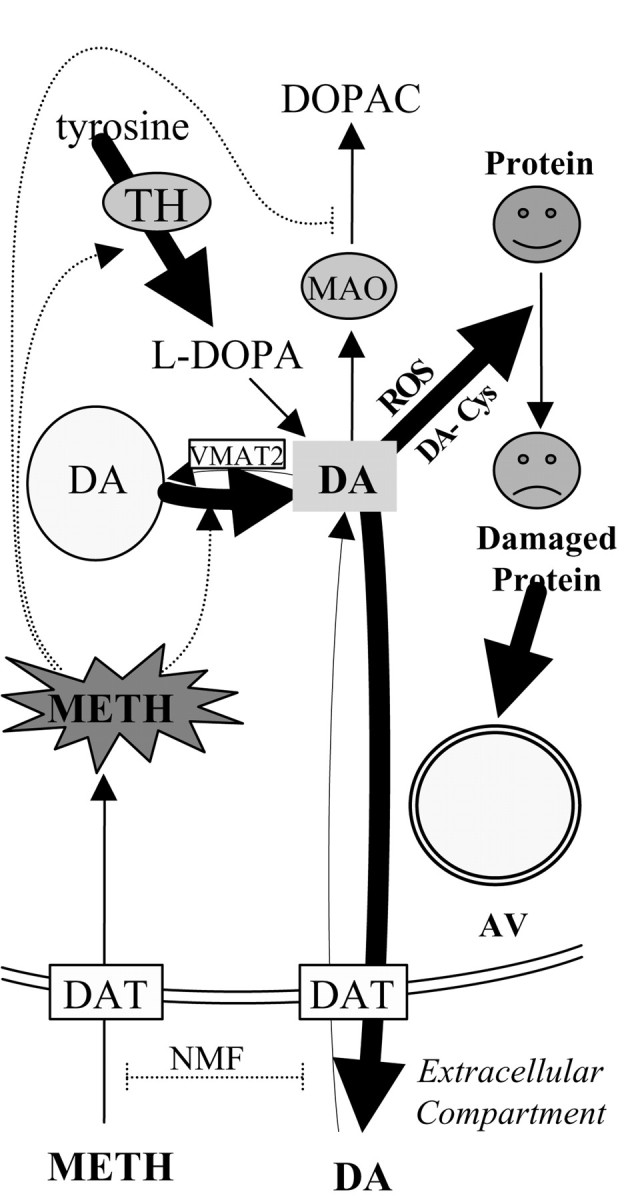
Proposed model for METH-induced neurotoxicity in ventral midbrain DA neurons. METH is taken up by the plasma membrane DAT in a manner inhibited by the uptake blocker nomifensine. Once inside the neuron, METH promotes DA release from synaptic vesicles and stimulates TH activity to synthesize even more DA. Cytosolic DA is metabolized by monoamine oxidase (MAO) to DOPAC and/or released into the extracellular milieu. METH is known to inhibit monoamine oxidase, resulting in additional increases in cytosolic DA. Excess cytosolic DA is oxidized to reactive DA metabolites, such as cysteinyl-DA (DA-Cys), resulting in damaged or dysfunctional proteins and lipids. These damaged constituents are sequestered within autophagic vacuoles (AV) for degradation.
The cause of METH neurotoxicity remains unknown, although roles have been proposed for (1) formation of extracellular oxyradicals by released DA or (2) cytosolic oxyradical stress driven by increased levels of cytosolic DA. We confirmed a role for cytosolic DA oxidation by examining VMAT2-deficient neurons that are unable to accumulate DA in synaptic vesicles. VMAT2 −/− cultures are more susceptible to METH, presumably because neither METH nor DA are safely sequestered in synaptic vesicles while DA synthesis is activated. DA is then metabolized to ROS in the cytosol, as indicated by substantially enhanced intracellular DCF levels and increased levels of cysteinyl-DA. Furthermore, both control and long-term METH-treated neurons displayed similar levels of extracellular DA after short-term METH stimulation, suggesting that enhanced extracellular DA levels with subsequent oxidation (predicted by the exchange diffusion model) are not the predominant means by which METH promotes neurotoxicity, but may be contributing factors. A schematic representation of the mechanisms by which METH promotes increased levels of intraneuronal DA was featured in a recent review (Fleckenstein et al., 2000).
Several investigations have concluded that METH neurotoxicity requires DA, because dopaminergic neurites are selectively vulnerable (Cubells et al., 1994) and TH inhibitors attenuate toxicity (Wagner et al., 1985b; Axt et al., 1990), particularly in VMAT2 +/− animals (Fumagalli et al., 1999). However, a recent in vivo report suggested that endogenous DA is not essential for the expression of METH-induced neurotoxicity (Yuan et al., 2001). The authors reported that the combination of α-MPT and reserpine, a VMAT2 inhibitor, to reduce total DA levels in both synaptic vesicles and cytosol, failed to prevent METH neurotoxicity despite extremely low DA levels. We elected not to use α-MPT on our VMAT2 knock-out or reserpine-treated cultures, because although it is an excellent inhibitor of DA synthesis, it can also be converted to the “false transmitter,” α-methyltyramine (Duffield et al., 1982; Dougan et al., 1983), which is sequestered in synaptic vesicles and released by synaptic vesicle fusion (Dorris, 1976). Thus when MAT2 is blocked or absent, the vesicular sequestration of α-methyltyramine is prevented and could itself lead to cytosolic oxyradical stress, potentially obscuring the importance of DA in METH neurotoxicity. The authors also provided evidence that drugs or manipulations that reduce core temperature protect against METH neurotoxicity, whereas those that increase core temperature promote METH neurotoxicity. Although hyperthermia contributes to METH neurotoxicity in vivo, we do not ascribe temperature effects on neurotoxicity in our in vitro system, because our cultures are maintained at a constant temperature regardless of treatment.
METH neurotoxicity also involves oxidative stress (Di Monte et al., 1996), possibly because of DA autooxidation to neurotoxic DA-quinones (LaVoie and Hastings, 1999). Research on reserpinized animals also revealed elevations in cytoplasmic levels of reactive 5-S-cysteinyl-DA (Fornstedt and Carlsson, 1989). As cysteinyl-DA is derived solely from DA-quinones (Sulzer and Zecca, 2000), our data demonstrate that METH is capable of promoting high levels of cytosolic DA, leading to the induction of intracellular ROS capable of oxidizing DCF. Recent evidence suggests that oxidized DA can react with endogenous cysteinyl residues on DAT and affects DA uptake (Sidell et al., 2001; Whitehead et al., 2001). The requirement for elevated cytosolic DA to produce METH toxicity explains why there was no METH-induced damage of non-DAergic neurons under these conditions, and why METH is selective for DA terminals in the brain.
Our results indicate that there is ongoing DA synthesis in cultures undergoing severe METH-induced neurodegeneration. A parsimonious explanation for increased neurodegeneration in VMAT2-deficient neurons is that because DA cannot be safely sequestered within synaptic vesicles, it remains in the cytosolic and extracellular compartments, where it is subject to oxidation and promotion of oxyradical stress. This would be particularly true for the VMAT2 −/− cultures, which demonstrate high levels of DA synthesis in the presence of METH. An additional contributing factor is that METH itself is sequestered within VMAT2-expressing synaptic vesicles, thereby reducing its downstream effects on cytotoxicity. However, mass spectrophotometric measurements indicate that extracellular METH levels in VMAT +/+ cultures are not reduced after 6 d (Cubells et al., 1994). Therefore, vesicular sequestration does not effectively reduce the levels of METH in culture.
The oxyradical stress controlled by VMAT2 would be expected to occur at terminals, because METH-induced oxyradical stress is expected to be most intense at sites that maintain DA synaptic vesicles. In the neuronal cultures, these small synaptic vesicles are abundant in axonal varicosities (Pothos et al., 1998). In METH-treated cultures, autophagic vacuoles are prominent in these varicosities during the early stages of METH-induced degeneration. It is plausible that the METH-induced overproduction of DA-derived oxyradicals promotes protein damage and/or dysfunction, resulting in the upregulation of autophagic degradation and in the selective degeneration of dopaminergic neurites.
VMAT2 expression has been implicated previously in neuroprotection of dopaminergic neurons (Edwards, 1993; Uhl, 1998), because VMAT2 sequesters the neurotoxin MPP+ away from its primary site of action (Liu et al., 1992). This study indicates that VMAT2 is also protective against METH toxicity. However, because METH itself cannot undergo autooxidation to a quinone or to compounds that might react with DCF, the neuroprotection appears primarily to be caused by VMAT2-mediated vesicular sequestration of DA itself. METH interaction with DAT would be required by both the exchange diffusion and weak base models for amphetamine action (Pifl et al., 1995) and is consistent with previous findings indicating a role for DAT in METH toxicity (Fumagalli et al., 1998; Wan et al., 2000) as well as with our findings that the DAT inhibitors nomifensine and amfonelic acid provide neuroprotection. It appears that a careful balance between these transport systems, as well as metabolic systems including monoamine oxidase, glutathione conjugation, and autophagy, are important for protecting against severe consequences of elevated cytosolic DA (Edwards, 1993; Uhl, 1998; Hastings and Berman, 1999; Sulzer and Zecca, 2000).
Footnotes
We are grateful for support from the National Parkinson's Foundation (K.E.L.), the Parkinson's Disease Foundation (D.S.), the National Institute on Drug Abuse (D.S., R.H.E.), and the National Institute of Neurological Disorders and Stroke (Udall Parkinson's Center of Excellence) (D.S.). We thank Drs. Eugene Mosharov and Theresa Swayne for assistance with the two photon microscope; Drs. Serge Przedborski, Roland Staal, Yvonne Schmitz, and Dmitriy Markov for helpful discussion; Mary Schoenebeck for performing electron microscopy; and Chao Annie Yuan and Gerald Behr for excellent technical assistance.
Correspondence should be addressed to Dr. David Sulzer, Black Building, Room 305, 650 West 168th Street, New York, NY 10032. E-mail:ds43@columbia.edu.
REFERENCES
- 1.Acikgoz O, Gonenc S, Kayatekin BM, Uysal N, Pekcetin C, Semin I, Gure A. Methamphetamine causes lipid peroxidation and an increase in superoxide dismutase activity in the rat striatum. Brain Res. 1998;813:200–202. doi: 10.1016/s0006-8993(98)01020-8. [DOI] [PubMed] [Google Scholar]
- 2.Axt KJ, Commins DL, Vosmer G, Seiden LS. α-Methyl-p-tyrosine pretreatment partially prevents methamphetamine-induced endogenous neurotoxin formation. Brain Res. 1990;515:269–276. doi: 10.1016/0006-8993(90)90606-c. [DOI] [PubMed] [Google Scholar]
- 3.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 4.Cadet JL, Sheng P, Ali S, Rothman R, Carlson E, Epstein C. Attenuation of methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- 5.Clausing P, Gough B, Holson RR, Slikker W, Bowyer JF. Amphetamine levels in brain microdialysate, caudate/putamen, substantia nigra, and plasma after dosage that produces either behavioral or neurotoxic effects. J Pharmacol Exp Ther. 1995;274:614–621. [PubMed] [Google Scholar]
- 6.Cubells J, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- 8.Di Monte DA, Royland JE, Jakowec MW, Langston JW. Role of nitric oxide in methamphetamine neurotoxicity: protection by 7-nitroindazole, an inhibitor of neuronal nitric oxide synthase. J Neurochem. 1996;67:2443–2450. doi: 10.1046/j.1471-4159.1996.67062443.x. [DOI] [PubMed] [Google Scholar]
- 9.Dorris R. Release of 3H-α-methyl-m-tyramine from rat striatum in vitro. Eur J Pharmacol. 1976;35:225–228. doi: 10.1016/0014-2999(76)90321-6. [DOI] [PubMed] [Google Scholar]
- 10.Dougan D, Duffield A, Duffield P, Wade D. The effects of (+)-amphetamine, α-methyltyrosine, and α-methylphenylalanine on the concentrations of m-tyramine in rat striatum. Br J Pharmacol. 1983;80:309–314. doi: 10.1111/j.1476-5381.1983.tb10035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duffield P, Dougan D, Wade D, Duffield A. Absence of α-methyldopamine in rat striatum after chronic administration of d-amphetamine. Life Sci. 1982;30:1701–1705. doi: 10.1016/0024-3205(82)90302-2. [DOI] [PubMed] [Google Scholar]
- 12.Edwards RH. Neuronal degeneration and the transport of neurotransmitters. Annu Neurol. 1993;34:638–645. doi: 10.1002/ana.410340504. [DOI] [PubMed] [Google Scholar]
- 13.Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J Pharmacol Exp Ther. 1979;208:203–209. [PubMed] [Google Scholar]
- 14.Fleckenstein AE, Gibb JW, Hanson GR. Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity. Eur J Pharmacol. 2000;406:1–13. doi: 10.1016/s0014-2999(00)00639-7. [DOI] [PubMed] [Google Scholar]
- 15.Fon EA, Pothos EN, Sun B-C, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 16.Fornstedt B, Carlsson A. A marked rise in 5-S-cysteinyl-dopamine levels in guinea pig striatum following reserpine treatment. J Neural Transm. 1989;76:155–161. doi: 10.1007/BF01578755. [DOI] [PubMed] [Google Scholar]
- 17.Fumagalli F, Gainetdinov RR, Valenzano KJ, Caron MG. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci. 1998;18:4861–4869. doi: 10.1523/JNEUROSCI.18-13-04861.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knock-out mice. J Neurosci. 1999;19:2424–2431. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hastings TG, Berman SB. Dopamine-induced toxicity and quinone modification of proteins: implication for Parkinson's disease. In: Creveling CR, editor. Role of catechol quinone species in cellular toxicity. F. P. Graham Publishing; Johnson City, TN: 1999. pp. 69–89. [Google Scholar]
- 20.Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itzhak Y, Gandia C, Huang PL, Ali SF. Resistance of neuronal nitric oxide synthase-deficient mice to methamphetamine-induced dopamine neurotoxicity. J Pharmacol Exp Ther. 1998;284:1040–1047. [PubMed] [Google Scholar]
- 22.Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones SR, Joseph JD, Barak LS, Caron MG, Wightman RM. Dopamine neuronal transport kinetics and effects of amphetamine. J Neurochem. 1999;73:2406–2414. doi: 10.1046/j.1471-4159.1999.0732406.x. [DOI] [PubMed] [Google Scholar]
- 24.Kita T, Takahashi M, Kubo K, Wagner GC, Nakashima T. Hydroxyl radical formation following methamphetamine administration to rats. Pharmacol Toxicol. 1999;85:133–137. doi: 10.1111/j.1600-0773.1999.tb00080.x. [DOI] [PubMed] [Google Scholar]
- 25.Kooy NW, Royall J, Ischiropoulos H. Oxidation of 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1997;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- 26.Larsen K, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002;17:897–908. doi: 10.14670/HH-17.897. [DOI] [PubMed] [Google Scholar]
- 27.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenbergy D, Brecha N, Edwards RH. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70:539–551. doi: 10.1016/0092-8674(92)90425-c. [DOI] [PubMed] [Google Scholar]
- 29.Lotharius J, O'Malley KL. The parkinsonism-inducing drug l-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation. A novel mechanism of toxicity. J Biol Chem. 2000;275:38581–38588. doi: 10.1074/jbc.M005385200. [DOI] [PubMed] [Google Scholar]
- 30.Lotharius J, O'Malley KL. Role of mitochondrial dysfunction and dopamine-dependent oxidative stress in amphetamine-induced toxicity. Ann Neurol. 2001;49:79–89. doi: 10.1002/1531-8249(200101)49:1<79::aid-ana11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 31.Maron RY, Stern BI, Kanner BI, Schuldiner S. Functional asymmetry of the amine transporter from chromaffin granules. J Biol Chem. 1983;258:11476–11481. [PubMed] [Google Scholar]
- 32.Mena MA, Davila V, Sulzer D. Neurotrophic effects of l-DOPA in postnatal midbrain dopamine neuron/cortical astrocyte cocultures. J Neurochem. 1997;69:1398–1408. doi: 10.1046/j.1471-4159.1997.69041398.x. [DOI] [PubMed] [Google Scholar]
- 33.Mundorf ML, Hochstetler SE, Wightman RM. Amine weak bases disrupt vesicular storage and promote exocytosis in chromaffin cells. J Neurochem. 1999;73:2397–2405. doi: 10.1046/j.1471-4159.1999.0732397.x. [DOI] [PubMed] [Google Scholar]
- 34.Petersén Å, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, Sulzer D. Expanded CAG repeats in exon 1 of the Huntington's disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet. 2001;10:1243–1254. doi: 10.1093/hmg/10.12.1243. [DOI] [PubMed] [Google Scholar]
- 35.Philipp E. Assay for tyrosine hydroxylase in hypothalamic homogenates using high-performance liquid chromatography with electrochemical detection. J Chromatogr. 1987;419:27–36. doi: 10.1016/0378-4347(87)80263-3. [DOI] [PubMed] [Google Scholar]
- 36.Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA. Mechanism of the dopamine-releasing actions of amphetamine and cocaine: plasmalemmal dopamine transporter versus vesicular monoamine transporter. Mol Pharmacol. 1995;47:368–373. [PubMed] [Google Scholar]
- 37.Possel H, Noack H, Augustin W, Keilhoff G, Wolf G. 2,7-dihydrochlorofluorescein diacetate as a fluorescent marker for peroxynitrate formation. FEBS Lett. 1997;416:175–178. doi: 10.1016/s0014-5793(97)01197-6. [DOI] [PubMed] [Google Scholar]
- 38.Pothos E, Davila V, Sulzer D. Presynaptic recording of quanta from midbrain dopamine neurons and modulation of the quantal size. J Neurosci. 1998;18:4106–4118. doi: 10.1523/JNEUROSCI.18-11-04106.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pothos E, Larsen K, Krantz D, Liu Y-J, Edwards R, Sulzer D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci. 2000;20:7297–7306. doi: 10.1523/JNEUROSCI.20-19-07297.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- 41.Ricaurte GA, Seiden LS, Schuster CR. Increased dopamine metabolism in the rat neostriatum after toxic doses of d-methylamphetamine. Neuropharmacology. 1983;22:1383–1388. doi: 10.1016/0028-3908(83)90228-9. [DOI] [PubMed] [Google Scholar]
- 42.Rota C, Chignell CF, Mason RP. Evidence for free radical formation during the oxidation of 2′,7′-dichlorofluorescin to the fluorescent dye 2′,7′-dichlorofluorescein by horseradish peroxidase: possible implications for oxidative stress measurements. Free Radic Biol Med. 1999;27:873–881. doi: 10.1016/s0891-5849(99)00137-9. [DOI] [PubMed] [Google Scholar]
- 43.Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seiden L, Ricaurte G. Neurotoxicity of methamphetamine and related drugs. In: Meltzer H, editor. Pyschopharmacology: the third generation of progress. Raven; New York: 1987. pp. 359–366. [Google Scholar]
- 45.Seiden LS, Vosmer G. Formation of 6-hydroxydopamine in caudate nucleus of the rat brain after a single large dose of methylamphetamine. Pharmacol Biochem Behav. 1984;21:29–31. doi: 10.1016/0091-3057(84)90125-4. [DOI] [PubMed] [Google Scholar]
- 46.Sidell K, Olson S, Ou J, Zhang Y, Amarnath V, Montine T. Cysteine and mercapturate conjugates of oxidized dopamine are in human striatum but only the cysteine conjugate impedes dopamine trafficking in vitro and in vivo. J Neurochem. 2001;79:510–521. doi: 10.1046/j.1471-4159.2001.00586.x. [DOI] [PubMed] [Google Scholar]
- 47.Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J Neurosci. 1997;17:960–974. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srinivasan A, Roth K, Sayers R, Schindler K, Wong A, Fritz L, Tomaselli K. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- 49.Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- 50.Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotoxicity Res. 2000;1:181–195. doi: 10.1007/BF03033289. [DOI] [PubMed] [Google Scholar]
- 51.Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sulzer D, St. Remy C, Rayport S. Reserpine inhibits amphetamine action in ventral midbrain culture. Mol Pharmacol. 1996;49:338–342. [PubMed] [Google Scholar]
- 53.Sulzer D, Bogulavsky J, Larsen K, Karatekin E, Kleinman M, Turro N, Krantz D, Edwards R, Greene L, Zecca L. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci USA. 2000;97:11869–11874. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uhl GR. Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson's disease. Ann Neurol. 1998;43:555–560. doi: 10.1002/ana.410430503. [DOI] [PubMed] [Google Scholar]
- 55.Wagner GC, Carelli RM, Jarvis MF. Pretreatment with ascorbic acid attenuates the neurotoxic effects of methamphetamine in rats. Res Commun Chem Pathol Pharmacol. 1985a;47:221–228. [PubMed] [Google Scholar]
- 56.Wagner GC, Lucot JB, Schuster CR, Seiden LS. α-Methyltyrosine attenuates and reserpine increases methamphetamine-induced neuronal changes. Brain Res. 1985b;270:285–288. doi: 10.1016/0006-8993(83)90602-9. [DOI] [PubMed] [Google Scholar]
- 57.Wan FJ, Shiah IS, Lin HC, Huang SY, Tung CS. Nomifensine attenuates d-amphetamine-induced dopamine terminal neurotoxicity in the striatum of rats. Chin J Physiol. 2000;43:69–74. [PubMed] [Google Scholar]
- 58.Whitehead R, Ferrer J, Javitch J, Justice J. Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J Neurochem. 2001;76:1242–1251. doi: 10.1046/j.1471-4159.2001.00125.x. [DOI] [PubMed] [Google Scholar]
- 59.Yuan J, Callahan BT, McCann UD, Ricaurte GA. Evidence against an essential role of endogenous brain dopamine in methamphetamine-induced dopaminergic neurotoxicity. J Neurochem. 2001;77:1338–1347. doi: 10.1046/j.1471-4159.2001.00339.x. [DOI] [PubMed] [Google Scholar]
- 60.Zhang F, Dryhurst G. Effects of l-cysteine on the oxidation chemistry of dopamine: new reaction pathways of potential relevance to idiopathic Parkinson's disease. J Med Chem. 1994;37:1084–1098. doi: 10.1021/jm00034a006. [DOI] [PubMed] [Google Scholar]