

Abstract
It has been shown recently that extinction of conditional fear does not depend acutely on NMDA-type glutamate receptors, although other evidence has led to the hypothesis that L-type voltage-gated calcium channels (LVGCCs) play a role in conditional fear. We therefore tested the role of LVGCCs in the acquisition, expression, and extinction of conditional fear of cue and context in mice. Using systemic injections of two LVGCC inhibitors, nifedipine and nimodipine, which both effectively cross the blood–brain barrier, we show that LVGCCs are essential for the extinction, but not for the acquisition or expression, of conditional fear in mice.
Keywords: fear, acquisition, extinction, expression, freezing, L-type, calcium channel, nifedipine, nimodipine, state dependence
Fear conditioning is an important form of behavioral plasticity that has been correlated to changes in synaptic strength in the amygdala (Rogan and LeDoux, 1995; Rogan et al., 1997). Both the acquisition and extinction of conditional fear are forms of active learning. The acquisition of conditional fear requires the establishment of a novel association by pairing an initially neutral conditional stimulus (CS), such as a tone, with an intrinsically aversive unconditional stimulus (US), typically a mild foot shock. Although extinction, the reduction of conditional responding after repeated exposures to the CS alone, might initially appear to be a passive decay or erasure of this association, many observations indicate that extinction is new inhibitory learning, which leaves the original memory intact. Conditional fear responding shows the following: spontaneous recovery with time (Baum, 1988), reinstatement after unpaired US presentations (Rescorla and Heth, 1975), and renewal with context change (Bouton and King, 1983). These observations indicate that the original association is not lost during extinction but rather is suppressed by new, context-dependent learning, which is likely attributable to plasticity at separate synapses from those mediating acquisition.
Many recent studies have investigated the molecular basis of fear conditioning and its extinction. Accumulating evidence indicates that fear acquisition and expression require NMDA-type glutamate receptor (NMDAR) activity in the amygdala (Miserendino et al., 1990; Kim et al., 1991, 1992; Falls et al., 1992; Tang et al., 1999; Rodrigues et al., 2001). There is also evidence that extinction of fear depends on NMDARs (Falls et al., 1992; Baker and Azorlosa, 1996; Tang et al., 1999). However, the NMDAR inhibitors used in these experiments may also have altered basal synaptic transmission in the amygdala, and thus, fear expression, which may be required for extinction (Li et al., 1995;Maren et al., 1996; Lee and Kim, 1998). A recent experiment demonstrated that NMDAR activity is necessary for the long-term retention of extinction memories but is not required for the generation of extinction acutely (Santini et al., 2001). Thus, although it seems clear that NMDAR activity is required for the formation of fear acquisition memories, considerably less is known about the molecules that mediate the induction of fear extinction.
Recently, a form of NMDAR-independent long-term potentiation (LTP) has been described in synapses between thalamic afferents and neurons in the lateral amygdala (Weisskopf et al., 1999). This LTP depends, instead, on L-type voltage-gated calcium channels (LVGCCs). Because these synapses have also been implicated in auditory fear conditioning (Rogan and LeDoux, 1995), amygdaloid LVGCC-LTP is an important candidate for a mechanism that may underlie some aspect of conditional fear (Blair et al., 2001; Bauer et al., 2002). In this paper, we tested the specific hypotheses that LVGCC activity is required for the (1) acquisition, (2) expression, and (3) extinction of conditional fear in mice. To do so, we used systemic injections of two dihydropyridine LVGCC antagonists with good penetration through the blood–brain barrier, nifedipine and nimodipine. Our results indicate that LVGCCs are necessary for the extinction of conditional fear but are not required for its acquisition or expression.
MATERIALS AND METHODS
Subjects
Naïve 12- to 20-week-old C57BL/6 male mice (Taconic Farms, Germantown, NY) were housed four per cage, were maintained on a 12 hr light/dark schedule, and were allowed access to food and waterad libitum. All testing was conducted during the light phase in illuminated testing rooms following protocols approved by the Institutional Animal Care and Use Committee of the University of California, Los Angeles.
Drugs
The LVGCC antagonists nifedipine (1.25–80 mg/kg) and nimodipine (4–16 mg/kg; Sigma, St. Louis) were sonicated into 100% Cremophor EL (BASF, Mt. Olive, NJ). PBS was added to make the final vehicle 10% Cremophor–90% PBS. The highest nifedipine doses were partly suspensions, and care was taken to thoroughly mix the drugs before injecting them. Mice were injected subcutaneously 20 or 50 min before behavioral testing (10 ml/kg). Drug pretreatment times were chosen based on pilot studies and previous reports of systemic administration in rodents (Janicki et al., 1988; Larkin et al., 1992).
Conditioning apparatus
Two contexts (A and B), in separate rooms, were used for all behavioral fear testing. Shuttle box compartments (Med Associates, St. Albans, VT) measuring 20.3 × 15.9 × 21.3 cm served as context A, and conditioning boxes (Med Associates) measuring 30.5 × 24.1 × 21 cm served as context B. Both contexts had two transparent walls and stainless steel grid floors (3.2 mm in diameter, 8 mm centers); however, the grid floors in context B were covered with flat white acrylic inserts to minimize context generalization. Context A was wiped down before testing with 10% ethanol, and context B was wiped down with 10% methanol. Individual video cameras were mounted in the ceiling of each chamber and connected via a quad processor to a standard video cassette recorder and monitor for videotaping and scoring of freezing. Grid floors were connected to a scrambled shock source (Med Associates). Auditory stimuli (Med Associates) were delivered via a speaker in the chamber wall. Delivery of stimuli was controlled with a personal computer and Med-PC software through a SmartCTL Interface System (DIG-716; Med Associates). Background white noise was maintained at 62 dB throughout behavioral testing.
Open field
Spontaneous locomotor activity was monitored by placing mice in one of four chambers (40 × 40 × 40 cm) and allowing them to freely explore for 60 min. Chambers had white floors and two white walls; the remaining two walls were transparent. A video camera was mounted above the chambers, and total distance traveled was tracked with a personal computer and software (Poly-Track Video Tracking System, Chromotrack version 4.02; San Diego Instruments, San Diego, CA) and expressed in arbitrary units.
Conditional fear testing
Cue fear acquisition. Experiments investigating the effects of LVGCC antagonists on the acquisition of cue fear consisted of two phases: fear acquisition (context A) and testing (context B). After injections, conditional fear was induced by presenting audible cues (CS: white noise, 2 min, 80 dB) that coterminated with mild foot shocks (US: shock, 2 sec, 0.7 mA). Two minute stimulus-free periods preceded, separated, and followed the pairings. Most experiments used five CS–US pairings; however, experiments using one (Fig.1F) or two (Figs.2E–G,3B,C) pairings were also conducted to ensure that LVGCC blockers did not impair fear acquisition with weaker training protocols. After allowing 1 d for memory consolidation, cue fear was tested by presenting one, continuous, 2 min CS after a 2 min acclimation. In two cases in which reductions in fear acquisition were observed, a second test was conducted 3 hr later to determine whether the reductions reflected state-dependent memory retrieval (Fig.2D,G). For these state-dependence tests, mice were reinjected and subjected to an identical test of cue fear. In a separate test of state dependence, using a 2 × 2 intersubject design, mice were injected with vehicle or nimodipine (16 mg/kg) (Fig. 3) 20 min before both training and testing for fear expression 24 hr later.
Fig. 1.
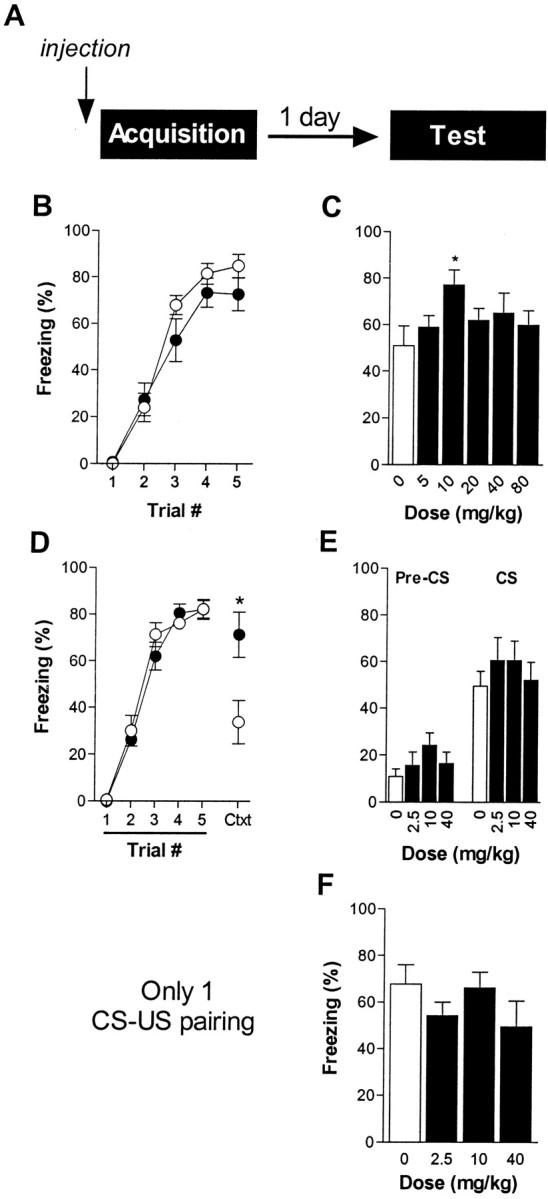
Nifedipine does not affect cue fear acquisition.A, Experimental design (7–8 mice per group).Open circles, Vehicle; filled circles, nifedipine. B, Freezing during the five cues that preceded each shock for mice injected with nifedipine (40 mg/kg dose shown, 20 min pretreatment) or vehicle. C, Freezing 24 hr later during a 2 min, drug-free CS test. D, Freezing during the five cues that preceded each shock for mice injected with nifedipine (40 mg/kg dose shown, 50 min pretreatment) or vehicle. Also included is freezing during the 2 min stimulus-free period after the last CS–US pairing (Ctxt). E, Freezing 24 hr later during a 2 min acclimation period (Pre-CS), followed by a 2 min, drug-free CS test. F, Freezing during a 2 min, drug-free CS test, 24 hr after injections (50 min pretreatment) and a single cue–shock pairing. *p< 0.05 versus vehicle.
Fig. 2.

Nimodipine does not affect cue fear acquisition, although recall appears to be state dependent. A, Experimental design (8 mice per group). B, Freezing during the five cues that preceded each shock for mice injected with nimodipine (15 mg/kg, 20 min pretreatment) or vehicle.C, Freezing 24 hr later during a 2 min, drug-free CS test. D, Freezing during a second state-dependence test (3 hr after test 1) after reinjections of drug or vehicle (20 min pretreatment). E–G, Repeat of the above experiment (A–C) using a weaker training protocol (2 cue–shock pairings). *p < 0.05 versus vehicle.
Fig. 3.

Recall of fear is partially state dependent with nimodipine. A, Experimental design (8 mice per group; between-subjects design). B, Freezing during the two cues that preceded each shock for mice injected with nimodipine (16 mg/kg, 20 min pretreatment; n = 16) or vehicle (n = 16). Freezing 24 hr later during a 2 min CS test, 20 min after drug or vehicle injections. One-half of the mice from each of the treatment groups of the previous day received nimodipine (16 mg/kg) or vehicle. *p < 0.05 versus 0–0 group; +p < 0.05 versus 16–0 group.
Cue fear extinction. Experiments investigating the effects of LVGCC antagonists on cue fear extinction consisted of three phases: fear acquisition (context A), fear extinction (context B), and testing (context B), each separated by 1 d to allow for memory consolidation. In all experiments, cue fear was induced in nondrugged, naïve mice with the five-pairing protocol described above. Mice were matched into equivalent treatment groups based on freezing during the third training CS. One day later, after injections, mice were placed in context B and allowed to acclimate for 2 min. Extinction was induced with 60 2 min CS presentations [5 sec intertrial interval (ITI)]. Additionally, nonextinguished (retention control) mice were injected with vehicle and placed in context B for an equivalent period of time but not exposed to any CS presentations. One day after extinction, all mice were returned to context B in the drug-free state. After a 2 min acclimation, freezing was assessed during a 2 min, continuous CS presentation. In a subsequent experiment (see Fig. 7) examining the potential for state-dependent recall of extinction memories, fear acquisition, extinction, and testing were conducted as described above; however, mice were reinjected 20 min before the final test with drug or vehicle.
Fig. 7.

Extinction with LVGCC blockers is not state dependent. A, Experimental design (7–8 mice per group). The 3 d experiment was identical to previous experiments (Figs. 5, 6), with the exception that drugs and vehicle were injected 20 min before both day 2 extinction and the day 3 test.B, Freezing during a 2 min CS test, 24 hr after extinction. *p < 0.05 versus retention control (RC); +p < 0.05 versus vehicle–extinction.
Context fear acquisition. Experiments investigating the effects of LVGCC antagonists on context fear acquisition consisted of two phases, both conducted in context B with the white inserts removed. After injections, mice were placed in the chambers in which they received five 0.7 mA, 2 sec unsignaled foot shocks. Two minute stimulus-free periods preceded, separated, and followed the foot shocks. Twenty-four hours later, mice were returned to the same chambers for a 5 min test of context fear.
Context fear extinction. The experiment investigating the effects of LVGCC antagonists on context fear extinction consisted of three phases, all conducted in context B with the white inserts removed. Context fear was induced in naïve, untreated mice with the five-shock protocol described above. Mice were then matched into equivalent treatment groups based on freezing during the 2 min period after the fifth shock. One day later, mice were injected and returned the conditioning chambers for a 120 min shock-free session. Nonextinguished retention control mice were injected with vehicle and placed in different chambers (see Materials and Methods, Open field) for 120 min. One day after extinction, all mice were returned to the context B chambers for a 5 min drug-free test session.
Statistical analyses
Behavioral freezing, the absence of all nonrespiratory movements, was rated during all phases by a blinded, experienced investigator using a 5 sec instantaneous time sampling technique. Percentage of freezing scores were calculated for each mouse, and data represent mean ± SEM freezing percentages for groups of mice during specified time bins. Total session means and individual CS exposures were analyzed with one-way ANOVA and planned post hoc Dunnett's test comparisons. Student's t tests were used to analyze experiments with only two treatment groups. Multiple trial data were analyzed with matched two-way ANOVA and Bonferroni's post hoc tests to compare individual time points. Differences were considered significant if p < 0.05.
RESULTS
LVGCC inhibitors do not prevent the acquisition or retention of conditional cue fear
To test whether LVGCC activity is required for cue fear acquisition, we injected nifedipine 20 min before a moderate training protocol (five CS–US pairings). We generated a dose–response curve for nifedipine (5–80 mg/kg) and assessed freezing during acquisition and, 24 hr later, during a drug-free retention test. Acute acquisition of freezing was unaffected by nifedipine administration; mice injected with vehicle or nifedipine (40 mg/kg) froze identically during the CS preceding each shock (for drug,F(4,65) = 3.22, p = 0.08; for drug × trial interaction,F(1,65) = 1.05, p = 0.39) (Fig. 1B). After allowing 24 hr for consolidation of learning, the mice were presented with the CS in a novel context. None of the drug doses reduced freezing during this test (F(5,42) = 1.64,p = 0.17) (Fig. 1C). In fact, mice injected previously with 10 mg/kg nifedipine froze slightly, but significantly, more than mice injected previously with vehicle (p < 0.05).
Because the rate of absorption and brain penetration of systemically administered nifedipine varies depending on its vehicle (Larkin et al., 1992), we also generated an abbreviated dose–response curve with a 50 min pretreatment (2.5–40 mg/kg). Again, we saw no impairment of fear acquisition or retention with nifedipine administration [for 40 mg/kg nifedipine during acquisition, F(1,35)= 0.88, p = 0.36; for the drug × trial interaction, F(4,35) = 0.57,p = 0.69 (Fig. 1D); for the retention test, F(3,28) = 0.48,p = 0.70 (Fig. 1E)]. To confirm that confounding effects of context conditioning or generalization were not somehow obscuring a blockade of acquisition, we also scored context freezing in this experiment, both in the last 2 min stimulus-free period of training and during the 2 min pre-CS period of testing on day 2 in context B. There was no evidence that nifedipine impaired conditioning to the training context during CS–US pairings. In fact, nifedipine-treated mice froze nearly twice as much as control mice during the final 2 min stimulus-free period of training (p < 0.05 vs vehicle) (Fig.1D, Ctxt). Because nifedipine does not induce freezing or potentiate context conditioning when the shocks are unsignaled (Fig. 4), these data suggest that nifedipine may retard negative contingency learning (i.e., that the CS, not the context, predicts US delivery). During the retention test, all groups showed a small amount of context generalization. However, freezing before the CS delivery was low and statistically undistinguishable for all groups (F(3,28) = 1.30,p = 0.29) (Fig. 1E,Pre-CS) and is unlikely to have obscured a blockade of long-term cue fear acquisition. Last, to eliminate the possibility that our lack of effect with nifedipine was attributable to an overly strong training protocol, we injected the drug (2.5–40 mg/kg) 50 min before a single CS–US pairing. This protocol does not allow the measurement of short-term acquisition, but again we detected no significant reduction in retained conditional fear 24 hr later (F(3, 28) = 1.19, p = 0.33) (Fig.1F).
Fig. 4.

LVGCC inhibitors do not prevent acquisition of context fear. A, Experimental design (7–8 mice per group). B, Freezing shown in 2 min blocks during the entire 12 min session for mice injected with nifedipine (40 mg/kg, 50 min pretreatment) or vehicle. Unsignaled foot shocks occurred at the 2nd, 4th, 6th, 8th, and 10th minutes. C, Freezing 24 hr later during a 5 min drug-free context exposure. D,E, Identical experiment conducted with nimodipine (16 mg/kg, 20 min pretreatment).
To ensure that our failure to block acquisition was not specific to nifedipine, we tested nimodipine (15 mg/kg, 20 min pretreatment), another LVGCC antagonist. Nimodipine had no effect on the acute acquisition of fear measured by freezing during the CS that preceded each of the five shocks (for drug,F(1,35) = 0.07, p = 0.79; for drug × trial interaction,F(4,35) = 0.65, p = 0.63) (Fig. 2B). When tested for retention of conditional fear on the next day in a drug-free state, animals treated with nimodipine froze slightly less than vehicle-treated controls; however, this effect was not significant (t(14) = 1.35, p = 0.20) (Fig. 2C). State-dependent effects on learning have been reported for a number of pharmacological agents (Connelly et al., 1975; Jackson et al., 1992; Blokland et al., 1998), suggesting that such drugs provide a salient internal context that contributes to the learned cue association. We therefore assayed the same mice for freezing again, 3 hr after the first test, 20 min after reinjections. Nimodipine-treated animals now showed the same levels of freezing as vehicle-treated animals (t(14) = 0.58,p = 0.57) (Fig. 2D). Thus, nimodipine did not interfere with the process of fear conditioning, although our data indicates that it made the recall of fear partially state dependent. Again, to rule out the possibility that the five-shock training protocol represented overtraining that might obscure a subtle role for LVGCC in the acquisition of conditional fear, we also tested the role of nimodipine on a weaker training protocol, using only two CS–US pairings. There were no differences in freezing during the CS that preceded each shock (for drug,F(1,14) = 0.11, p = 0.74; for drug × trial interaction,F(1,14) = 0.84, p = 0.38) (Fig. 2E), and nimodipine again decreased freezing slightly, although this time significantly, when assayed 1 d after training in the drug-free state (t(14) = 2.32, p < 0.05) (Fig. 2F). Again, the difference disappeared when the animals were reinjected before a second fear assay (t(14) = 1.69, p = 0.11) (Fig. 2G).
We had not anticipated state-dependent recall of cue fear with nimodipine, and the initial experiments were not designed to test this. Therefore, we next assessed state dependence directly in a between-subjects design, using two CS–US pairings for training followed after 1 d by a test CS presentation in a novel context. Eight mice per group received an injection before both training and testing, in a 2 × 2 design: either nimodipine (16 mg/kg) before training and then nimodipine before testing, or vehicle both days, or vehicle first followed by nimodipine, or vice versa. As seen before, nimodipine had no effect on freezing during the tones that preceded each shock during acquisition (for drug,F(1,30) = 0.03, p = 0.86; for drug × trial interaction,F(1,30) = 0.01, p = 0.93) (Fig. 3B). Again, nimodipine treatment before acquisition reduced freezing 24 hr later compared with vehicle-treated mice (0–0 vs 16–0, p < 0.01) (Fig. 3C). However, freezing in mice that received nimodipine before both the acquisition and test sessions was indistinguishable from vehicle treated mice (0–0 vs 16–16, p > 0.05), supporting the hypothesis that nimodipine makes cue fear recall partially state dependent. Importantly, nimodipine injections before testing did not increase freezing in mice that were trained with vehicle (0–0 vs 0–16, p > 0.05). Thus, although recall of fear is state dependent with nimodipine, expression of fear is unaltered.
LVGCC inhibitors do not prevent the acquisition or retention of conditional context fear
We also examined the effects of nifedipine (40 mg/kg) and nimodipine (16 mg/kg) on acquisition of context fear in separate experiments (Fig. 4). Acutely, mice injected with either nifedipine or vehicle acquired context fear (for time,F(5,78) = 10.12, p < 0.01), although nifedipine-treated mice appeared to learn at a slightly slower rate, and the effect of drug treatment was statistically significant (F(1,78) = 8.33,p < 0.01) (Fig. 4B). However, the effect was small because freezing by nifedipine-treated mice was never statistically different at any single time point (pvalues >0.05), and the group × time interaction was statistically insignificant (F(5,78) = 0.87, p = 0.51). Nimodipine-treated mice were indistinguishable from vehicle-treated mice in the acquisition of context fear (for time, F(5,84) = 17.27, p < 0.01; for drug,F(1,84) = 2.46, p = 0.12; for the drug × time interaction,F(5,84) = 0.83, p = 0.54) (Fig. 4D). When tested drug-free 24 hr later, both nifedipine- and nimodipine-treated mice froze the same as vehicle-treated mice, indicating that retention of context fear was unimpaired by LVGCC blockade (t(13) = 0.74, p = 0.47 andt(14) = 0.29 ,p = 0.78, respectively) (Fig.4C,E).
LVGCC inhibitors block extinction but not expression of conditional cue fear
Using five CS–US pairings for training groups of naïve mice, we next tested the effect of LVGCC inhibitors on the expression and extinction of cue fear. One day after training, mice were injected with drug or vehicle, placed in a novel context, and exposed to 60 2 min CS (5 sec ITI). Retention control mice were injected with vehicle and placed in the extinction chambers for an equivalent period of time but were not exposed to any CS. Expression of conditional fear was assessed by measuring freezing during the first CS exposure of the day 2 session, before any extinction could occur. Acute extinction was assessed during the first 15 CS exposures, and retained extinction was assessed during a single 2 min CS exposure in the same context 1 d later. The 60 CS protocol generated substantial persistent extinction in vehicle-treated mice compared with retention controls (p values <0.05) (Figs.5C,E,6C,E). Neither inhibitor affected the expression of conditional fear during the first 2 min exposure to the cue (p values >0.05 compared with vehicle) (Figs.5B,D,6B,D). Furthermore, nifedipine did not affect pre-CS freezing to the novel context before cue exposure began (t14 = 1.1, p = 0.28) (Fig. 5D, Pre) or pre-CS freezing on the test of extinction (F(2,21) = 1.64,p = 0.22) (Fig. 5E, Pre-CS). However, whereas vehicle-treated mice showed a progressive decline in freezing with repeated CS exposures, freezing by mice treated with LVGCC blockers remained elevated in all four experiments. Although mice treated with 40 mg/kg nifedipine (20 min pretreatment) appear to begin to extinguish, they rapidly return to initial freezing levels, whereas controls show continuing declines (for drug,F(1,105) = 39, p < 0.01; for drug × trial interaction,F(14,105) = 2.27, p < 0.01) (Fig. 5B). Furthermore, freezing during each of the CS was never significantly less than freezing during the first CS (F(14,105) = 1.61, p = 0.09). In contrast, vehicle controls show a significant reduction in freezing by the seventh CS presentation (F(14,105) = 4.71, p< 0.01). Nevertheless, a trend toward some early extinction suggested that nifedipine may be more efficient with a longer pretreatment, and we repeated both our extinction experiment and our acquisition experiments (see above) with longer pretreatments. When injected with nifedipine (40 mg/kg) 50 min before extinction began, there was no hint of acute extinction in the nifedipine-treated group (for drug,F(1,105) = 228, p < 0.01; for drug × trial,F(14,105) = 2.05, p < 0.05) (Fig. 5D). To verify that the blockade of extinction was not particular to nifedipine, we also tested nimodipine (15 mg/kg), and it, too, blocked acute extinction entirely (for drug,F(1,165) = 220, p < 0.01; for drug × trial,F(14,165) = 5.05, p < 0.01) (Fig. 6B). A dose–response curve was then generated for nimodipine (4–16 mg/kg), and an identical result was also obtained for the 16 mg/kg dose (for drug,F(1,165) = 179, p < 0.01; for drug × trial,F(14,165) = 1.72, p = 0.06) (Fig. 6D); acute extinction was blocked entirely.
Fig. 5.

Nifedipine blocks extinction, but not expression, of cued fear. A, Experimental design. Extinction sessions began 24 hr after cue fear acquisition (5 cue–shock pairings). B, Freezing during the first 15 CS presentations, after injections of nifedipine (40 mg/kg dose shown, 20 min pretreatment) or vehicle. C, Freezing 24 hr later during a 2 min, drug-free CS test (n values = 8 for extinction groups). Retention control mice (RC;n = 16) were injected with vehicle and placed in the extinction chamber on day 2 but were not exposed to any CS.D, E, Identical experiment with a longer pretreatment (50 min) of a single nifedipine dose (40 mg/kg) or vehicle (n values = 8). Freezing was also scored during a 2 min acclimation period (Pre-CS). *p < 0.05 versus retention control; +p < 0.05 versus vehicle–extinction.
Fig. 6.

Nimodipine blocks extinction, but not expression, of cued fear. A, Experimental design (12 mice per group). Extinction sessions began 24 hr after cue fear acquisition (5 cue–shock pairings). B, Freezing during the first 15 CS presentations, after injections of nimodipine (15 mg/kg, 20 min pretreatment) or vehicle. C, Freezing 24 hr later during a 2 min, drug-free CS test. Retention control mice (RC) were injected with vehicle and placed in the extinction chamber on day 2 but were not exposed to any CS. D, E, Identical experiment with several doses of nimodipine. Only the 16 mg/kg dose is shown for acute extinction. *p < 0.05 versus retention control; +p < 0.05 versus vehicle–extinction.
Consistent with the acute results, when the same mice were tested drug free after 1 d for consolidation, both LVGCC antagonists had completely blocked the extinction seen in vehicle-treated mice (p < 0.01 for 40 mg/kg nifedipine, 20 min pretreatment; p < 0.01 for 40 mg/kg nifedipine, 50 min pretreatment; p values <0.05 for 15 and 16 mg/kg nimodipine, 20 min pretreatment). Additionally, freezing in these groups was statistically equivalent to nonextinguished retention controls (p > 0.05 for 20, 40, and 80 mg/kg nifedipine, 20 min pretreatment; p > 0.05 for 40 mg/kg nifedipine, 50 min pretreatment; and p values >0.05 for nimodipine 4, 8, 15, and 16 mg/kg) (Figs.5C,E,6C,E).
It has been shown previously that extinction generated in the presence of benzodiazepines can be state dependent (Bouton et al., 1990). Thus, although no persistent extinction is seen when rats extinguished in the presence of benzodiazepines are tested in the absence of drugs, extinction can be uncovered by administering benzodiazepines again before the extinction test. We therefore tested whether such state-dependent extinction might occur with LVGCC antagonist treatment, by both extinguishing and testing after drug injections. There was no evidence of state-dependent extinction; mice given extinction training in the presence of nifedipine or nimodipine and then reinjected with drug before testing showed freezing no lower than retention controls (p values >0.05) and froze significantly more than mice extinguished and tested in the presence of vehicle (p values <0.05) (Fig.7). Nifedipine-treated animals showed a trend to more freezing than retention controls in the final drugged test; however, this effect was not statistically significant (p > 0.05).
LVGCC inhibitors block extinction but not expression of conditional context fear
To further explore the effects of LVGCC blockade, we also tested the effects of nifedipine (40 mg/kg) and nimodipine (16 mg/kg) directly on the expression and extinction of context fear (Fig.8). Neither inhibitor altered the expression of freezing as measured during the first 5 min block of the extinction session (p values >0.05 vs vehicle) (Fig.8B). Freezing was assessed during the first 30 min of the 120 min extinction session. As with cue fear, both LVGCC inhibitors blocked the acute extinction of context fear evident in vehicle-treated mice (nifedipine: for group, F(1,90) = 45.7, p < 0.01; for time,F(5,90) = 10.9, p < 0.01; for the group × time interaction,F(5,90) = 8.3, p < 0.01; nimodipine: for group, F(1,90) = 54.2, p < 0.01; for time,F(5,90) = 19.6, p < 0.01; for the group × time interaction,F(5,90) = 5.7, p < 0.01) (Fig. 8B). Likewise, vehicle-treated mice showed significant long-term extinction compared with retention control mice when tested 24 hr later (for group,F(1,150) = 20.7, p < 0.01) (Fig. 8C). Treatment during context exposure with nifedipine or nimodipine completely blocked long-term extinction of context fear; freezing in these groups was indistinguishable from nonextinguished retention control mice (nifedipine vs vehicle, F(1,150) = 33.1, p < 0.01; nimodipine vs vehicle,F(1,145) = 22.1, p < 0.01; nifedipine vs retention control,F(1,150) = 0.24, p = 0.62; nimodipine vs retention control,F(1,145) = 0.27, p = 0.60). All mice showed extinction during this final 5 min drug-free test (main effect for time,F(4,295) = 15.9, p < 0.01); however, there was no indication that the groups extinguished at different rates during this test (main effect for group × time interaction, F(12,295) = 0.31,p = 0.99).
Fig. 8.

LVGCC inhibitors block extinction, but not expression, of context fear. A, Experimental design (16 mice per group). Extinction was conducted 24 hr after context fear acquisition (5 unsignaled foot shocks, 2 min ITI). B, Freezing in 5 min blocks for the first 30 min of context exposure (120 total). Mice were injected with nifedipine (40 mg/kg, 50 min pretreatment), nimodipine (16 mg/kg, 20 min pretreatment), or vehicle. Retention control mice were injected with vehicle and placed in dissimilar chambers for 120 min. C, Freezing 24 hr later during a 5 min drug-free context fear test (data were lost for 1 mouse in the nimodipine group attributable to a camera failure).
Spontaneous locomotor activity and LVGCC blockers
To rule out the possibility that our behavioral effects with the LVGCC inhibitors were attributable to gross reductions in movement, we next assessed the effects of nifedipine and nimodipine on spontaneous locomotor activity in a novel open field. Mice were injected with vehicle, nifedipine (40 mg/kg), or nimodipine (16 mg/kg) 20 min before a 60 min session. One day later, mice were returned to the same chambers and allowed to freely explore again for 60 min, although now in the drug-free state. Total distance traveled (arbitrary units) was recorded in each session. The distance traveled by nifedipine-treated mice was no different from vehicle-treated mice in either session (p values >0.05) (Fig.9). Nimodipine-treated mice showed suppressed locomotion acutely (test 1, p < 0.01) but normal locomotion the next day (test 2, p > 0.05).
Fig. 9.

Effects of nifedipine and nimodipine on spontaneous locomotor activity in an open field. A, Experimental design (6 mice per group). B,Left, Total distance traveled during a 60 min session in a novel chamber (arbitrary units). Mice were injected 20 min before the session with nifedipine (40 mg/kg), nimodipine (16 mg/kg), or vehicle.Right, An identical test of the same mice 24 hr later in the drug-free state. p < 0.05 versus vehicle.
DISCUSSION
We presented data that point to a distinction in the molecular mechanisms underlying acquisition and extinction of conditional fear in mice. Blockade of LVGCCs effectively prevents extinction in a dose-related manner (Figs. 5-8), both acutely (during extinction exposures) and persistently (1 d after exposures). These effects are not state dependent; no hidden extinction is uncovered by treating mice with the same drugs at testing as during extinction exposures (Fig. 7). LVGCC activity thus appears to be essential to induce conditional fear extinction. To our knowledge, this is the first intervention reported to completely block the induction of extinction. Because it has been shown recently that NMDAR activity is not required for extinction induction (Santini et al., 2001), this work provides a candidate induction mechanism: calcium entry through LVGCCs may initiate plasticity underlying extinction memories.
On the other hand, doses of LVGCC inhibitors that completely block extinction fail to prevent conditional fear acquisition (Figs. 1-4). For cue fear, drug-injected animals showed a pattern of increasing freezing with training trials identical to that of vehicle-treated controls, indicating that acute fear learning is independent of LVGCC activity. Across a wide range of nifedipine doses and two pretreatment durations, conditional fear 1 d later was at least as great as that of controls. These data indicate that long-term retention of fear learning does not require LVGCC activity and that the failure to block acquisition was not attributable to delayed absorption or early metabolism of the drug. Whereas drug-free freezing was sometimes significantly lower in the long-term retention test 1 d later in animals treated with nimodipine, this was clearly a state-dependent effect on recall (Figs. 2, 3). When nimodipine was reinjected (Fig. 2) or injected before both training and testing (Fig. 3), mice showed conditional freezing identical to that expressed by animals doubly injected with vehicle. Normal fear acquisition occurred with both moderate and weak training protocols, indicating that it was not overtraining that prevented detection of a role for LVGCCs in fear acquisition.
LVGCC inhibitors also failed to block context fear acquisition (Figs.1, 4). The rate of acute context fear acquisition in nimodipine-treated mice was indistinguishable from vehicle-treated mice. Nifedipine-treated mice appear to acquire context fear more slowly in the absence of auditory CS, but the difference in acquisition rate was not significant, and animals achieved the same final freezing levels. The 24 hr retention test was unambiguous; neither LVGCC inhibitor prevented the long-term acquisition of context fear (Fig. 4), and nifedipine caused no significant increase in context generalization of fear (Fig. 1). This last finding may be especially relevant to the present studies, because extinction expression is context dependent (Bouton and Bolles, 1979). Our context experiments indicate that LVGCC inhibitors do not impair context-dependent learning and that their blockade of extinction is probably not by preventing associations with the context of extinction.
Several of our findings make it clear that LVGCCs are also not required for the expression of conditional fear. Vehicle- and drug-treated mice acquire freezing to the tone at the same rate (Figs. 1-4). Furthermore, initial freezing levels during the extinction sessions were equivalent for drug-injected and vehicle-injected mice (Figs. 5,6, 8). Finally, in the state-dependence test of acquisition, animals injected first with vehicle and then with nimodipine show no difference in freezing from those injected with vehicle twice (Fig. 3). These findings argue strongly that the drugs interfere neither with the detection of the CS or US nor with expression of conditional fear (freezing).
We also tested whether nonspecific effects of the drugs on locomotion could account for our results. In an open field test, nimodipine, but not nifedipine, acutely decreased total distance traveled, but neither affected locomotion the next day (Fig. 9). Thus, affects on locomotion cannot account for the blockade of extinction in our drug-free tests. It is also unlikely that our blockade of acute extinction is a result of reduced locomotion because nifedipine blocks extinction acutely but does not reduce locomotion. In addition, nimodipine-injected animals were never scored with increased freezing acutely compared with vehicle-injected animals (Figs. 2-4, 6, 8). These results confirm that freezing is behaviorally distinct from reduced locomotion and that the acute effects of nimodipine on locomotion neither confounded our freezing scores nor accounted for the persistent blockade of extinction.
Forms of LVGCC-dependent synaptic modification have been described in a number of synapses in the brain (Johnston et al., 1992; Huang and Malenka, 1993; Christie and Abraham, 1994; Huber et al., 1995; Zhuo and Hawkins, 1995; Kurotani et al., 1996; Izumi and Zorumski, 1998; Kapur et al., 1998; Morgan and Teyler, 2001; Zakharenko et al., 2001). Usually, these synapses also show NMDAR-dependent LTP. However, LTP at synapses between thalamic afferents and neurons in the lateral amygdala is NMDAR independent (Weisskopf et al., 1999). This LTP depends, instead, on LVGCCs, because it can be blocked by nifedipine. Consistent with this, few previous reports indicate a clear dependence of learning on LVGCCs (Lee and Lin, 1991; Deyo et al., 1992; Borroni et al., 2000). To the contrary, many studies indicate that LVGCC blockade promotes learning rather than blocking it (Disterhoft et al., 1993; Vetulani et al., 1993; Fulga and Stroescu, 1997; Quevedo et al., 1998; Quartermain et al., 2001). We also show a significantly increased acquisition of fear with 10 mg/kg nifedipine (Fig. 1C) and trends toward better learning at other doses in acquisition and in extinction (Figs.1C, 4C). Previous investigators hypothesize various explanations for the paradoxical enhancement of learning by LVGCC blockade, including compensatory cellular changes (Quevedo et al., 1998), low concentrations of antagonists acting to hold channels open rather than closing them (Fulga and Stroescu, 1997), or nonspecific vasodilatory effects (Vetulani et al., 1993). Importantly, this learning enhancement has been observed repeatedly to disappear as the dose of LVGCC inhibitors increases, suggesting that it results from modulation rather than complete blockade of the channels.
However, in the present studies, we did not pursue the nootropic effects of low doses of LVGCC inhibitors, because we wanted to determine when LVGCC-dependent plasticity was required in fear learning. We, therefore, chose high drug doses to maximally inhibit LVGCCs. The results demonstrate a robust blockade of one type of inhibitory learning (extinction) with no effect on a type of excitatory learning (acquisition). The fact that LVGCCs are implicated in extinction but not in acquisition of conditional fear, whereas NMDARs are implicated in both (Falls et al., 1992; Baker and Azorlosa, 1996;Tang et al., 1999), raises questions about the need for this extra molecule in extinction learning. We hypothesize that LVGCCs are needed in extinction but not in acquisition, because no CS–US pairing occurs during extinction. LVGCCs may allow plasticity to occur after presentation of CS alone, a hypothesis we hope to test using other forms of CS-alone learning, such as latent inhibition (Lubow, 1973).
Although systemic injections cannot support any anatomical hypothesis about the sites at which these inhibitors have their effect on extinction, other evidence suggests that the amygdala may be the relevant location. First, whereas long-lasting extinction may depend on areas of prefrontal cortex (Morgan et al., 1993), the induction of extinction proceeds normally in animals with frontal lesions (Quirk et al., 2000). Similarly, extinction induction occurs normally in animals with hippocampal lesions (Frohardt et al., 2000). The amygdala, on the other hand, clearly plays a role in extinction, because intraparenchymal infusions of NMDAR or mitogen-activated protein kinase inhibitors there block extinction (Falls et al., 1992; Lu et al., 2001). Furthermore, the identification of LVGCC-dependent, but NMDAR-independent, LTP in the thalamo-amygdala pathway (Weisskopf et al., 1999) has led to the hypothesis that this LTP is crucial for fear conditioning (Blair et al., 2001), and a very recent paper from this group shows an attenuation of cue fear acquisition with the LVGCC blocker verapamil (Bauer et al., 2002). We cannot account entirely for the inconsistency of these results with our own. Our data argue strongly against the importance of LVGCC-dependent LTP, whether in amygdala or elsewhere in the brain, in the acquisition of conditional fear. Two potential confounds may account for the inconsistency. First, it is difficult to compare intraparenchymal infusions of verapamil with our systemic administrations, and, second, no test of state-dependent recall was performed by Bauer et al. Because our data clearly indicate that LVGCCs participate in the extinction of conditional fear at doses that do not affect acquisition, we expect that intra-amygdala infusions of LVGCC inhibitors will also block extinction of conditional fear at doses that fail to block acquisition. We are currently testing this hypothesis with intraparenchymal administrations and performing state-dependence controls to resolve the apparent inconsistency. However, it should be noted that LVGCCs are ubiquitous in the brain, and LVGCC-dependent plasticity outside the amygdala may well make the relevant contribution to extinction memory formation.
The demonstration that LVGCCs are required for extinction but not acquisition of conditional fear suggests that it may be possible to identify cells, synapses, or molecular pathways specific to extinction. Because extinction is the explicit model for behavior therapy (Wolpe, 1969), the most efficacious treatment for human anxiety disorders, this discovery also holds out hope for the development of new drugs that can make such therapy easier and more effective by selectively facilitating the extinction of fear.
Footnotes
This work was supported in part by a National Alliance for Research on Schizophrenia and Depression Young Investigator Award and by the Forest Award of the West Coast College of Biological Psychiatry (M.B.). We thank Aaron Blaisdell, Gene Gurkoff, Kelsey Martin, Tom O'Dell, and Alcino Silva for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Mark Barad, Department of Psychiatry and Biobehavioral Sciences, University of California, Los Angeles, 3506 Gonda, 695 Charles Young Drive South, Los Angeles, CA 90095-1761. E-mail: mbarad@mednet.ucla.edu.
REFERENCES
- 1.Baker JD, Azorlosa JL. The NMDA antagonist MK-801 blocks the extinction of Pavlovian fear conditioning. Behav Neurosci. 1996;110:618–620. doi: 10.1037//0735-7044.110.3.618. [DOI] [PubMed] [Google Scholar]
- 2.Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baum M. Spontaneous recovery from the effects of flooding (exposure) in animals. Behav Res Ther. 1988;26:185–186. doi: 10.1016/0005-7967(88)90118-0. [DOI] [PubMed] [Google Scholar]
- 4.Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem. 2001;8:229–242. doi: 10.1101/lm.30901. [DOI] [PubMed] [Google Scholar]
- 5.Blokland A, Prickaerts J, Honig W, de Vente J. State-dependent impairment in object recognition after hippocampal NOS inhibition. NeuroReport. 1998;9:4205–4208. doi: 10.1097/00001756-199812210-00037. [DOI] [PubMed] [Google Scholar]
- 6.Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ. Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J Neurosci. 2000;20:9272–9276. doi: 10.1523/JNEUROSCI.20-24-09272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouton ME, Bolles RC. Contextual control of the extinction of conditioned fear. Learn Motiv. 1979;10:445–466. [Google Scholar]
- 8.Bouton ME, King DA. Contextual control of the extinction of conditioned fear: tests for the associative value of the context. J Exp Psychol Anim Behav Process. 1983;9:248–265. [PubMed] [Google Scholar]
- 9.Bouton ME, Kenney FA, Rosengard C. State-dependent fear extinction with two benzodiazepine tranquilizers. Behav Neurosci. 1990;104:44–55. doi: 10.1037//0735-7044.104.1.44. [DOI] [PubMed] [Google Scholar]
- 10.Christie BR, Abraham WC. L-type voltage-sensitive calcium channel antagonists block heterosynaptic long-term depression in the dentate gyrus of anaesthetized rats. Neurosci Lett. 1994;167:41–45. doi: 10.1016/0304-3940(94)91023-5. [DOI] [PubMed] [Google Scholar]
- 11.Connelly JF, Connelly JM, Phifer R. Disruption of state-dependent learning (memory retrieval) by emotionally-important stimuli. Psychopharmacologia. 1975;41:139–143. doi: 10.1007/BF00421071. [DOI] [PubMed] [Google Scholar]
- 12.Deyo RA, Nix DA, Parker TW. Nifedipine blocks retention of a visual discrimination task in chicks. Behav Neural Biol. 1992;57:260–262. doi: 10.1016/0163-1047(92)90262-3. [DOI] [PubMed] [Google Scholar]
- 13.Disterhoft JF, Moyer JR, Jr, Thompson LT, Kowalska M. Functional aspects of calcium-channel modulation. Clin Neuropharmacol. 1993;16:S12–S24. doi: 10.1097/00002826-199316001-00003. [DOI] [PubMed] [Google Scholar]
- 14.Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frohardt RJ, Guarraci FA, Bouton ME. The effects of neurotoxic hippocampal lesions on two effects of context after fear extinction. Behav Neurosci. 2000;114:227–240. doi: 10.1037//0735-7044.114.2.227. [DOI] [PubMed] [Google Scholar]
- 16.Fulga IG, Stroescu V. Experimental research on the effect of calcium channel blockers nifedipine and verapamil on the anxiety in mice. Rom J Physiol. 1997;34:127–136. [PubMed] [Google Scholar]
- 17.Huang YY, Malenka RC. Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the induction of LTP. J Neurosci. 1993;13:568–576. doi: 10.1523/JNEUROSCI.13-02-00568.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huber KM, Mauk MD, Kelly PT. Distinct LTP induction mechanisms: contribution of NMDA receptors and voltage-dependent calcium channels. J Neurophysiol. 1995;73:270–279. doi: 10.1152/jn.1995.73.1.270. [DOI] [PubMed] [Google Scholar]
- 19.Izumi Y, Zorumski CF. LTP in CA1 of the adult rat hippocampus and voltage-activated calcium channels. NeuroReport. 1998;9:3689–3691. doi: 10.1097/00001756-199811160-00022. [DOI] [PubMed] [Google Scholar]
- 20.Jackson A, Koek W, Colpaert FC. NMDA antagonists make learning and recall state-dependent. Behav Pharmacol. 1992;3:415–421. doi: 10.1097/00008877-199208000-00018. [DOI] [PubMed] [Google Scholar]
- 21.Janicki PK, Siembab D, Paulo EA, Krzascik P. Single-dose kinetics of nifedipine in rat plasma and brain. Pharmacology. 1988;36:183–187. doi: 10.1159/000138382. [DOI] [PubMed] [Google Scholar]
- 22.Johnston D, Williams S, Jaffe D, Gray R. NMDA-receptor-independent long-term potentiation. Annu Rev Physiol. 1992;54:489–505. doi: 10.1146/annurev.ph.54.030192.002421. [DOI] [PubMed] [Google Scholar]
- 23.Kapur A, Yeckel MF, Gray R, Johnston D. L-Type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JJ, DeCola JP, Landeira-Fernandez J, Fanselow MS. N-methyl-d-aspartate receptor antagonist APV blocks acquisition but not expression of fear conditioning. Behav Neurosci. 1991;105:126–133. doi: 10.1037//0735-7044.105.1.126. [DOI] [PubMed] [Google Scholar]
- 25.Kim JJ, Fanselow MS, DeCola JP, Landeira-Fernandez J. Selective impairment of long-term but not short-term conditional fear by the N-methyl-d-aspartate antagonist APV. Behav Neurosci. 1992;106:591–596. doi: 10.1037//0735-7044.106.4.591. [DOI] [PubMed] [Google Scholar]
- 26.Kurotani T, Higashi S, Inokawa H, Toyama K. Protein and RNA synthesis-dependent and -independent LTPs in developing rat visual cortex. NeuroReport. 1996;8:35–39. doi: 10.1097/00001756-199612200-00008. [DOI] [PubMed] [Google Scholar]
- 27.Larkin JG, Thompson GG, Scobie G, Forrest G, Drennan JE, Brodie MJ. Dihydropyridine calcium antagonists in mice: blood and brain pharmacokinetics and efficacy against pentylenetetrazol seizures. Epilepsia. 1992;33:760–769. doi: 10.1111/j.1528-1157.1992.tb02358.x. [DOI] [PubMed] [Google Scholar]
- 28.Lee EH, Lin WR. Nifedipine and verapamil block the memory-facilitating effect of corticotropin-releasing factor in rats. Life Sci. 1991;48:1333–1340. doi: 10.1016/0024-3205(91)90530-o. [DOI] [PubMed] [Google Scholar]
- 29.Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci. 1998;18:8444–8454. doi: 10.1523/JNEUROSCI.18-20-08444.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li XF, Phillips R, LeDoux JE. NMDA and non-NMDA receptors contribute to synaptic transmission between the medial geniculate body and the lateral nucleus of the amygdala. Exp Brain Res. 1995;105:87–100. doi: 10.1007/BF00242185. [DOI] [PubMed] [Google Scholar]
- 31. Lu KT, Walker DL, Davis M. Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J Neurosci 21 2001. RC162(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lubow RE. Latent inhibition. Psychol Bull. 1973;79:398–407. doi: 10.1037/h0034425. [DOI] [PubMed] [Google Scholar]
- 33.Maren S, Aharonov G, Stote DL, Fanselow MS. N-methyl-d-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 34.Miserendino MJ, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 35.Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- 36.Morgan SL, Teyler TJ. Electrical stimuli patterned after the theta-rhythm induce multiple forms of ltp. J Neurophysiol. 2001;86:1289–1296. doi: 10.1152/jn.2001.86.3.1289. [DOI] [PubMed] [Google Scholar]
- 37.Quartermain D, deSoria VG, Kwan A. Calcium channel antagonists enhance retention of passive avoidance and maze learning in mice. Neurobiol Learn Mem. 2001;75:77–90. doi: 10.1006/nlme.1999.3958. [DOI] [PubMed] [Google Scholar]
- 38.Quevedo J, Vianna M, Daroit D, Born AG, Kuyven CR, Roesler R, Quillfeldt JA. L-type voltage-dependent calcium channel blocker nifedipine enhances memory retention when infused into the hippocampus. Neurobiol Learn Mem. 1998;69:320–325. doi: 10.1006/nlme.1998.3822. [DOI] [PubMed] [Google Scholar]
- 39.Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rescorla RA, Heth CD. Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process. 1975;1:88–96. [PubMed] [Google Scholar]
- 41.Rodrigues SM, Schafe GE, LeDoux JE. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci. 2001;21:6889–6896. doi: 10.1523/JNEUROSCI.21-17-06889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15:127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 43. Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390 1997. 604 607 [Erratum (1998) 391:818] [DOI] [PubMed] [Google Scholar]
- 44.Santini E, Muller RU, Quirk GJ. Consolidation of extinction learning involves transfer from NMDA-independent to NMDA-dependent memory. J Neurosci. 2001;21:9009–9017. doi: 10.1523/JNEUROSCI.21-22-09009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 46.Vetulani J, Battaglia M, Castellano C, Sansone M. Facilitation of shuttle-box avoidance behaviour in mice treated with nifedipine in combination with amphetamine. Psychopharmacology. 1993;113:217–221. doi: 10.1007/BF02245700. [DOI] [PubMed] [Google Scholar]
- 47.Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci. 1999;19:10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolpe J. The practice of behavior therapy, Ed 1. Pergamon; New York: 1969. [Google Scholar]
- 49.Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- 50.Zhuo M, Hawkins RD. Long-term depression: a learning-related type of synaptic plasticity in the mammalian central nervous system. Rev Neurosci. 1995;6:259–277. doi: 10.1515/revneuro.1995.6.3.259. [DOI] [PubMed] [Google Scholar]