

Abstract
Hypoxia-inducible factor-1 (HIF-1) is a transcription factor that regulates the adaptive response to hypoxia in mammalian cells. It consists of a regulatory subunit HIF-1α, which accumulates under hypoxic conditions, and a constitutively expressed subunit HIF-1β.
In this study we analyzed HIF-1α expression in the rat cerebral cortex after transient global ischemia induced by cardiac arrest and resuscitation. Our results showed that HIF-1α accumulates as early as 1 hr of recovery and persists for at least 7 d.
In addition, the expression of HIF-1 target genes, erythropoietin and Glut-1, were induced at 12 hr to 7d of recovery. A logical explanation for HIF-1α accumulation might be that the brain remained hypoxic for prolonged periods after resuscitation. By using the hypoxic marker 2-(2-nitroimidazole-1[H]-y1)-N-(2,2,3,3,3-pentafluoropropyl)-acetamide (EF5), we showed that the brain is hypoxic during the first hours of recovery from cardiac arrest, but the tissue is no longer hypoxic at 2 d. Thus, the initial ischemic episode must have activated other nonhypoxic mechanisms that maintain prolonged HIF-1α accumulation. One such mechanism might be initiated by insulin-like growth factor-1 (IGF-1). Our results showed that IGF-1 expression was upregulated after cardiac arrest and resuscitation. In addition, we showed that IGF-1 was able to induce HIF-1α in pheochromocytoma cells and cultured neurons as well as in the brain of rats that received intracerebroventricular and systemic IGF-1 infusion. Moreover, infusion of a selective IGF-1 receptor antagonist abrogates HIF-1α accumulation after cardiac arrest and resuscitation. Our study suggest that activation of HIF-1 might be part of the mechanism by which IGF-1 promotes cell survival after cerebral ischemia.
Keywords: global cerebral ischemia, hypoxia-inducible factor-1α, insulin-like growth factor-1, hypoxia-inducible genes, cardiac arrest, brain hypoxia
Mammalian cells are able to sense a decrease in oxygen tension and respond by activating adaptive mechanisms, including activation of several hypoxia-inducible genes such as erythropoietin (Epo) and vascular endothelial growth factor (VEGF). Many of these genes are regulated by the hypoxia-inducible factor-1 (HIF-1), a heterodimeric transcription factor consisting of two subunits, HIF-1α and HIF-1β (Wang and Semenza, 1995). Both subunits belong to a family of basic helix-loop-helix transcription factors and are required for DNA binding and transactivation of target genes (Wang et al., 1995). HIF-1β is a common binding partner for other members of the family, and it is constitutively expressed. HIF-1α is unique to HIF-1 and its expression is primarily regulated by oxygen tension (Semenza, 1999). During normoxia, the HIF-1α gene is expressed continuously; however, the HIF-1α protein is rapidly degraded by the ubiquitin–proteosome system (Salceda and Caro, 1997;Huang et al., 1998). During hypoxia or in the presence of iron chelators, degradation of HIF-1α is suppressed, and thereby it rapidly accumulates in the nucleus (Jiang et al., 1996; Jewell et al., 2001).
In addition to hypoxia, many reports have shown that some growth factors such as insulin, insulin-like growth factor (IGF)-1 and IGF-2 can induce HIF-1α accumulation and HIF-1 DNA binding activity in different cell types (Zelzer et al., 1998; Feldser et al., 1999; Zundel et al., 2000).
In the adult CNS, IGF-1 is involved in the response of neuronal tissue to injury and during various neurodegenerative conditions (Torres-Aleman, 2000). The expression of IGF-1 and its binding proteins are altered in response to brain ischemia (Lee et al., 1996; Schwab et al., 1997; Beilharz et al., 1998), and exogenous administration of IGF-1 significantly reduces neuronal loss in different models of cerebral ischemia (Johnston et al., 1996; Tagami et al., 1997; Guan et al., 2000; Wang et al., 2000). Furthermore, it has been shown that IGF-1 protects cultured neurons against diverse forms of injury, including hypoxia and oxidative stress (Heck et al., 1999; Yamaguchi et al., 2001).
In this study we analyzed the expression of HIF-1α in the rat cerebral cortex after transient global ischemia induced by cardiac arrest and resuscitation. Our results showed that HIF-1α accumulates as early as 1 hr of recovery and unexpectedly persists for at least 7 d. We used the in vivo hypoxia marker EF5 to determine whether brain hypoxia could explain persistent HIF-1α accumulation. In addition, we analyzed the expression of Von Hippel Lindau protein (pVHL) and the activity of the 26S proteosome to assess changes in the HIF-1α degradation pathway. Our data revealed that neither hypoxia nor impairment in the degradation machinery could explain a sustained expression of HIF-1α. Thus, the initial ischemic episode must have activated other mechanisms that produced prolonged HIF-1α accumulation. One such mechanism might be initiated by IGF-1 that was found to be upregulated in the brain after cardiac arrest and resuscitation. To support this hypothesis, we studied the ability of IGF-1 to induce HIF-1α in vitro and in vivo. In addition, we tested whether selective inhibition of the IGF-1 receptor (IGF-1R) abrogates sustained HIF-1α accumulation after transient global ischemia.
MATERIALS AND METHODS
Induction of transient global ischemia by cardiac arrest and resuscitation. Transient global cerebral ischemia was produced by a modification of the cardiac arrest model described by Crumrine and LaManna (1991). Male Wistar rats (300–350 gm) were anesthetized with 2.5% halothane/70% nitrous oxide/30% oxygen. The ventral tail artery was cannulated to monitor systemic arterial pressure and to obtain samples of blood gases and pH measurements. In addition, a catheter was inserted through the external jugular vein into the right atrium, and body temperature was maintained at 37°C by a feedback-controlled infrared heat lamp. Animals were allowed to recover completely from anesthesia for at least 1 hr. Cardiac arrest was induced by a sequential intra-atrial injection of d-tubocurare (0.3 mg) and ice-cold KCl solution (0.5 m, 0.12 ml/100 gm of body weight). Resuscitation efforts began after 7 min of arrest. For this purpose, rats were orotracheally intubated for mechanical ventilation accompanied by chest compression. Once spontaneous heartbeat returned, a small dose of epinephrine (2 μg) was administered to achieve a mean arterial blood pressure of at least 80 mmHg. The duration of ischemia was between 11 and 13 min and was defined as the period between the decrease of blood pressure to zero and its return to 80% of pre-arrest value. Ventilation was adjusted to achieve normoxia and normocapnia until rats regained spontaneous respiration. In the sham-operated animals, the same surgical procedure was performed without cardiac arrest and resuscitation. Animals were killed by decapitation at various durations of recovery.
Immunodetection of the hypoxic marker EF5. To map brain hypoxic regions after transient global cerebral ischemia, EF5, a marker for hypoxia, was administered intravenously (10 mg) using an infusion pump (Harvard Apparatus) at a rate of 150 μl/min for 20 min.
Rat subjected to transient ischemia were infused with EF5 at 15 min or 2 d after resuscitation from cardiac arrest and were killed 45 min after infusion. Sham-operated rats received a similar treatment. In addition, a group of rats were infused with EF5 while being exposed to different oxygen concentrations (21, 16, 14, 12, 10, and 8% O2) for 4 hr. Once animals were killed, the brains were quickly removed and frozen at −70°C. To detect EF5 adducts, 10 μm coronal sections were incubated with an ELK3–51 monoclonal Cy3-conjugated antibody (provided by S. Evans, University of Pennsylvania, Philadelphia, PA) according to the procedure described byEvans et al. (1995).
Proteasome chymotrypsin-like activity. Chymotrypsin-like activity of the proteasome was assayed using the fluorogenic peptide succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (LLVY-MCA) (Sigma, St. Louis, MO). After rapid dissection, brain cortex was homogenized in lysis buffer (10 mm Tris-HCl, pH 7.8, 0.5 mm DTT, 5 mm ATP, 0.035% SDS, and 5 mm MgCl2). Assays were performed with 100 μg/100 μl aliquots from the resulting lysate supplemented with 40 μm of LLVY-MCA. After incubation at 37°C for 30 min, the substrate was quenched by addition of 300 μl of ethanol followed by 2.0 ml of H2O. Proteolytic activity (release of MCA) was measured using a Perkin-Elmer LS-5B spectrofluorimeter at 380 nm excitation and 440 nm emission using free MCA as the standard for quantification. Background fluorescence was determined by incubating lysates with the proteasome inhibitor lactacystin (50 μm) for 30 min before adding the substrate.
In vivo administration of IGF-1 and IGF-1R antagonist.Male Wistar rats received systemic (n = 4) or intracerebroventricular (n = 3) infusion of human IGF-1 (generously provided by A. F. Parlow, National Hormone Peptide Program) dissolved in PBS. Systemic infusion was performed by using a subcutaneous mini-osmotic pump (Alzet 2001; at a dose of 50 μg · kg−1 · d−1, rate of infusion 1 μl/hr) for 7 d according to the protocol described by Carro et al. (2000). At the end of the infusion, blood samples (1 ml) were collected for ELISA analysis of IGF-1. Animals were killed, and the brains were removed and frozen at −80°C. Intracerebroventricular IGF-1 was administered by using a stainless steel cannula (Alzet) implanted into the left lateral ventricle. Stereotaxic coordinates used were 0.4 mm posterior to bregma, 1.5 mm lateral to the midline, and 4 mm ventral to the pial surface, as established by Paxinos and Watson (1997). The cannula was connected to a subcutaneous osmotic pump (Alzet 2001) filled with IGF-1 (0.25 μg/μl). After 7 d of infusion, animals were killed and perfused-fixed, and the brains were processed for HIF-1α immunohistochemistry.
In addition, a group of rats were subjected to cardiac arrest and resuscitation and subsequently received a continuous intracerebroventricular infusion of saline or 25 μg of JB-1, a selective antagonist for IGF-1R (Bachem, San Carlos, CA). For this experiment, a cannula was implanted as described above and connected to an osmotic pump (Alzet 1003D; delivery rate 1 μl/hr). After 4 d of infusion, animals were killed, and brains were processed for HIF-1 immunohistochemistry.
IGF-1 sandwich ELISA. Total circulating IGF-1 was measured using ELISA with a mouse monoclonal anti-IGF-1 antibody (Upstate Biotechnology, Waltham, MA). Microtiter plates were coated overnight with these captured antibodies at 4°C. After the plates were washed with TBS, 25 μl of IGF-1 standards or acid-extracted serum samples were added to each well and incubated at room temperature for 2 hr. After several washes, 1 μl of biotinylated anti-IGF-1 antibody (R&D Systems, Minneapolis, MN) with 0.5% normal goat serum was added and incubated for 2 hr. Immunodetection was accomplished using streptavidin-HRP (R&D Systems) and the chromogen orthophenylene diamine (Sigma). Plates were read at 490 nm, and results were expressed as nanograms of IGF-1 per milliliters of plasma.
Cell cultures and experimental treatments. Primary neuronal cultures were prepared as described previously (Brewer, 1995) from embryonic day 18 Wistar rats. Dissociated cortical neurons were plated in six-well plates (coated with poly-l-lysine) under serum-free conditions in neurobasal medium supplemented with B27 (Invitrogen, Carlsbad, CA). On the fourth day of plating, one-half of the medium was changed to B27/neurobasal without glutamate. Experiments were performed in cells that had been in culture for 6–8 d. Rat pheochromocytoma (PC12) cells were grown in Roswell Park Memorial Institute 1640 culture medium (containingl-glutamine) supplemented with 5% horse serum, 10% fetal bovine serum, 100 U/ml penicillin, 100 mg/ml streptomycin on collagen-coated dishes. Before IGF-1 treatment, PC12 cells were detached and seeded in poly-l-lysine (10 μg/ml)-coated plates in a 2% serum medium for 48 hr. PC12 and primary neurons were exposed to hypoxia (1% O2, 5% CO2, 94% N2) for 4 hr in Plexiglas modular chambers. Cells incubated under standard normoxic conditions (5% CO2/95% room air) were used as controls. To study the effect of IGF-1 on HIF-1α expression, PC12 and neurons were treated with 100 nm recombinant human IGF-1 (Calbiochem, San Diego, CA) or vehicle for 24 hr.
RNase protection assay and Northern blot analysis. Total RNA was isolated from brain cortex using the RNAgents Isolation System (Promega, Madison WI) according to the manufacturer's recommendations. A PCR fragment (468 bp) containing IGF-1 exon 2 and a portion of exons 1 and 3 was generated by RT-PCR using forward primer 5′-AAGCCTACAAAGTCAGCTCG-3′ and reverse primer 5′-GGTCTTGTTTCCTGCACTTC-3′. The resulting cDNA was subcloned into pGEM-4Z (Promega) and sequenced. To synthesize radiolabeled IGF antisense riboprobe, the plasmid was linearized with EcoR1 and transcribed with T7 RNA polymerase in the presence of [α32P]UTP using the MAXIscriptin vitro transcription kit (Ambion, Austin, TX). Then, the probe was treated with Dnase I and purified through PAGE. Rnase protection assay (RPA) was performed according to the instructions provided with the RPAIII kit (Ambion), using 20 μg of total RNA. Protected RNA fragments were resolved by electrophoresis on denaturing urea-polyacrylamide gels (5%) and visualized by autoradiography. As an internal control, a riboprobe for β-actin was used. Northern blot analysis was performed as previously described (Chavez et al., 2000) using 10 μg of total RNA. Specific cDNA probes for rat VEGF and Glut-1 were purchased from Research Genetics (Huntsville, AL). Epo cDNA was generated by RT-PCR using the following primers: sense 5′-CTCT GGGCCTCCCAGTC-3′ and antisense 5′-TGTTCGGAGTGGAGCAG-3′.
Western blot analysis and immunoprecipitation. Brain cortical samples were homogenized in lysis buffer (20 mm HEPES, pH 7.5, 1.5 mmMgCl2, 0.2 mm EDTA, 0.1m NaCl) supplemented with 0.2 mm dithiothreitol, and protease inhibitors (1 μg/ml leupeptin, 0.5 μg/ml aprotinin, 1.5 μg/ml pepstatin, 0.5 mm PMSF, 1 mmNa3VO4). After adding NaCl to a final concentration of 0.45 m, homogenates were centrifuged at 20,000 × g for 15 min at 4°C. Supernatants were collected and mixed with an equal volume of homogenization buffer containing 40% (v/v) glycerol. Cultured cells were washed with PBS, harvested, and centrifuged at 1000 ×g for 5 min. The cell pellet was resuspended in lysis buffer and processed as described above. Brain cortical lysates (200 μg protein) or cell lysates (50 μg protein) were electrophoresed on 7.5% (HIF-1α and HIF-1β) or 15% SDS-polyacrylamide gels (IGF-1, VHL) and transferred to nitrocellulose membrane by standard procedures. After blocking with non-fat dry milk, membranes were incubated with the following primary antibodies: HIF-1α mouse monoclonal antibody (Novus Biologicals, Littleton, CO), IGF-1 mouse monoclonal antibody (Upstate Biotechnology), goat polyclonal antibody against β-chain of the IGF-1 receptor (Santa Cruz Biotechnology, Santa Cruz, CA), VHL monoclonal antibody (Pharmigen, Carlsbad, CA), and actin goat polyclonal antibody (Santa Cruz Biotechnology). Crude nuclear extracts of Hep3B cells (American Type Culture Collection, CRL-1830) exposed to 1 or 20% oxygen were used as positive controls (30 μg of protein) in HIF-1α Western blot analysis.
For immunoprecipitation of the IGF-1Rβ subunit, cell lysates (50 μg) were incubated at 4°C with anti-IGF-1Rβ (2 μg; Santa Cruz Biotechnology). Protein G-agarose was added, and samples were incubated for 2 hr at 4°C. The pellet was collected by centrifugation (5000 rpm, 5 min), washed with PBS, and boiled in Laemmli sample buffer. Samples were resolved in 7.5% SDS-PAGE, and Western blots were performed using an anti-phosphotyrosine-specific antibody (1:500; Santa Cruz). These blots were stripped and reprobed with anti-IGF-1R antibody (1:500, Santa Cruz Biotechnology). Signals were visualized by enhanced chemiluminescence (Amersham), and autoradiographic results were quantified by densitometry. Protein concentrations were determined by Bradford protein assay with bovine serum albumin as standard (Bio-Rad, Hercules, CA).
HIF-1α and IGF-1 immunohistochemistry.Anesthetized rats were perfused transcardially with ice-cold PBS followed by 4% paraformaldehyde. Brains were removed, postfixed in 2% paraformaldehyde for 24 hr, and embedded in paraffin. Coronal sections (6 μm) were deparaffinized, hydrated, and subjected to antigen retrieval at 90°C for 20 min using Target Retrieval Solution (Dako, Carpinteria, CA). Sections were incubated with 10% normal serum for 2 hr and subsequently were incubated overnight at 4°C with a mouse monoclonal antibody against HIF-1α (1:200; Novus Biologicals). HIF-1α-positive cells were visualized with biotinylated secondary anti-mouse antibody (Vector, Burlingame, CA) and streptavidin conjugated with Oregon Green (Molecular Probes, Eugene, OR).
To identify some of the cells expressing HIF-1α, double immunolabeling using two cellular markers, neuronal-nuclei specific (NeuN) and glial fibrillary acidic protein (GFAP) were performed. The polyclonal anti-GFAP (1:500; Santa Cruz Biotechnology) and anti-NeuN (1:500; Chemicon, Temecula, CA) were detected using Texas Red-conjugated secondary antibodies (Vector). For IGF-1 and HIF-1α double labeling, sections were first processed for HIF-1α staining as described above, and then for IGF-1 with a monoclonal anti-IGF-1 antibody (1:200; Upstate Biotechnology). Immunodetection was accomplished using a biotin-conjugated anti-mouse antibody and streptavidin conjugated with Cy3 (Jackson ImmunoResearch, West Grove, PA).
Statistical analysis. Data are presented as mean ± SD. Statistical comparisons among groups were made using a one-way ANOVA test with Tukey correction. A p < 0.05 was considered statistically significant.
RESULTS
Cardiac arrest and resuscitation induce HIF-1α accumulation in the brain cortex and upregulation of HIF-1 target genes
HIF-1α migrates with a characteristic diffuse pattern (∼120 kDa) probably corresponding to post-translational modifications of this protein (Fig. 1A). Under normoxic conditions, HIF-1α is continually synthesized but rapidly degraded by the ubiquitin–proteosome system. Accordingly, HIF-1α was barely detected in brain cortical samples of sham-operated animals. Transient global cerebral ischemia induced by cardiac arrest and resuscitation led to a rapid accumulation of HIF-1α in the brain cortex. This accumulation was observed at 1 hr after resuscitation, persisted for at least 7 d, and subsided by 14 d of recovery. On the other hand, the levels of the constitutively expressed HIF-1β subunit were not affected by cardiac arrest and resuscitation.
Fig. 1.

HIF-1α accumulation and induction of HIF-1 target genes in the cerebral cortex of rats subjected to transient global cerebral ischemia. A, Western blots showing HIF-1α and HIF-1β protein levels in cortical samples from sham-operated (C) and rats subjected to ∼12 min of cardiac arrest and resuscitation and allowed to recover for 1 hr or up to 30 d (1h–30d). Crude nuclear extracts from Hep3B cells exposed to 21% oxygen (−) or 1% O2 (+) were used as a negative and positive control, respectively. β-actin immunoblot was used to document equal protein loading. B, Northern blots showing transient induction of Epo and Glut-1 mRNA levels in the brain cortex of sham-operated rats (C) and cardiac arrest/resuscitated rats (1h–30d). Northern blot of β-actin served as the sample-loading control.
In addition, we analyzed the expression of two HIF-1 target genes, Epo and Glut-1, by Northern blot analysis. Our results showed that both targets were induced at 12 hr and remained elevated up to 7 d of recovery. At 14 d of recovery, when HIF-1α was no longer present, mRNA levels of both targets returned to control levels (Fig.1B).
Immunolocalization of HIF-1α
The results shown in Figure 2demonstrate that HIF-1α immunoreactivity was nondetectable in control brain sections (Fig. 2A). Intense nuclear immunostaining was observed at 1 d after cardiac arrest, mainly in neurons (Fig. 2B), as indicated by colocalization with the neuronal-specific marker NeuN (Fig.2C,D). In addition, immunopositive flat nuclei probably corresponding to endothelial cells were also observed in some blood vessels (Fig. 2C). A similar pattern of neuronal staining was observed after 1 hr and 2 and 4 d of recovery from cardiac arrest (data not shown). Although we did not observe obvious colocalization of HIF-1α-positive cells with GFAP, we could not rule out the possibility that glial cells are also expressing HIF-1α after cardiac arrest (data not shown).
Fig. 2.

HIF-1α immunostaining in the cerebral cortex at 1 d of recovery from cardiac arrest. A, HIF-1α was not detected in sham-operated rat brain. B, Positive HIF-1α immunostaining (green) in the brain cortex at 1 d of recovery in cells with rounded nuclei characteristic of neurons. C, D, Double staining for HIF-1α (green) and the neuronal-specific marker NeuN (red) showed partial colocalization. In addition, flat nuclei associated with small blood vessels stained for HIF-1α, probably corresponding to endothelial cells (C, arrow). Scale bar, 100 μm.
In vivo analysis of brain hypoxia after cardiac arrest and resuscitation
Because HIF-1α accumulation is triggered primarily by hypoxia, it was necessary to determine whether the brain remained hypoxic for a prolonged period after transient global ischemia. For this purpose we used the nitroimidazole EF5 as an in vivo hypoxia marker. In our model of transient global cerebral ischemia, we found a strong EF5 adduct binding at 1 hr of recovery from cardiac arrest, indicating the presence of tissue hypoxia at that time. EF5 binding was especially prominent in cortex, hippocampus, and white matter. In contrast, no hypoxic regions were found in the brain at 2 d after cardiac arrest (Fig. 3). This result showed that the tissue remained hypoxic by the first hour of recovery, probably because of a decreased cerebral blood flow reported previously. However, at 2 d of recovery, the brain was no longer hypoxic, suggesting that the HIF-1α accumulation that is found at that time cannot be caused by continued tissue hypoxia.
Fig. 3.
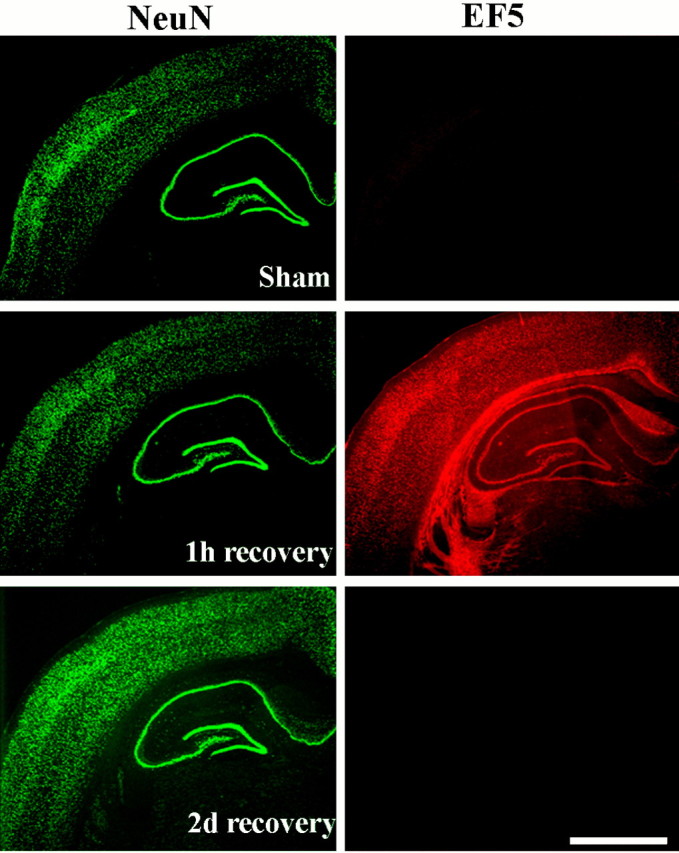
Immunohistochemistry for the in vivo hypoxia marker EF5 showing weak signal (red) in the sham-operated rat brain but strong signal at 1 hr of recovery from cardiac arrest and resuscitation. At 48 hr of recovery, EF5 adduct binding was comparable with the sham-control. The same sections were immunostained for the neuronal marker NeuN (green). The nitroimidazole compound EF5 was injected intravenously as described in Materials and Methods. Scale bar, 1 mm.
To test the sensitivity of EF5 as a hypoxic marker, we compared HIF-1α levels and EF5 binding in the brain of rats exposed to different oxygen concentrations. Our results showed that EF5 binding appears to be graded with hypoxic severity. EF5 signal was detected only at 12% oxygen exposure or below, whereas no signal was detected at 14% (data not shown) or 21% O2 (Fig.4A). In the same samples, significant brain HIF-1α accumulation was detected only after exposure to 12% O2 or below (Fig.4B). Thus, our result demonstrates that EF5 sensitivity correlates with HIF-1α expression induced by hypoxia. Taken together, these experiments show that after a transient ischemic episode the rat brain does not remain hypoxic for a prolonged period, at least not to an extent that could explain sustained HIF-1α accumulation. Although hypoxia can be considered the primary activator of HIF-1α during the early phase of recovery, it is likely that a nonhypoxic mechanism is responsible for the sustained HIF-1α accumulation after 2 d of recovery from cardiac arrest.
Fig. 4.

Detection of the hypoxia marker EF5 and HIF-1α accumulation in the brain of animals exposed to varying oxygen concentrations. A, Increasing brain EF5 adduct binding (red) with increasing severity of hypoxia (4 hr exposure at 21, 12, or 8% oxygen). The same sections were immunostained for NeuN (green). Scale bar, 1 mm. B, Immunoblots showing increasing HIF-1α accumulation but unchanged HIF-1β expression in the brain cortex with decreasing oxygen concentrations. β-actin immunoblot shows equal protein loading.
Von Hippel Lindau protein expression and 26S proteosome activity
To determine whether the metabolic pathway that degrades HIF-1α is affected by transient cerebral ischemia, we studied pVHL expression and the 26S proteasome activity. During normoxia, pVHL in association with elongin B and elongin C, binds to HIF-1α subunits, and targets them for ubiquitination and subsequent degradation by the proteosome (Maxwell et al., 1999; Cockman et al., 2000). Hence, changes in pVHL expression or impairment of the proteasome can potentially affect the rate of HIF-1α degradation. Immunoblot analysis showed that pVHL expression was not affected by cardiac arrest and reperfusion in the brain cortex (Fig. 5A). Chymotrypsin proteasome activity was transiently and moderately decreased by ∼25% at 1 hr, 12 hr, and 1 d of recovery from cardiac arrest compared with control cortical samples. However, proteasome activity was no longer impaired at 2–7 d of recovery (Fig.5B). Despite normal levels of pVHL protein expression, these results suggest that the transient inhibition of proteosome activity may contribute to HIF-1α accumulation only in the early phase of recovery from cardiac arrest.
Fig. 5.

Cardiac arrest and resuscitation did not affect pVHL expression but induced transient reduction in proteosome activity.A, Western blot showing pVHL protein levels in the cerebral cortex of control (C) and resuscitated animals (1h–30d recovery after cardiac arrest). B, Graph representing changes in the chymotrypsin-like activity of the proteosome after transient cerebral ischemia (1h–7d recovery). Results are shown as mean ± SD of three different samples per time point (*p < 0.05 compared with control).
Induction of brain IGF-1 expression but not IGF-1R after cardiac arrest and resuscitation
Although hypoxia is considered to be the main stimulus for HIF-1α accumulation, other nonhypoxic-related factors such as insulin, IGF-1, IGF-2, and EGF have been shown to increase HIF-1α protein levels in certain cell types (Zelzer et al., 1998; Feldser et al., 1999; Zhong et al., 2000; Zundel et al., 2000).
We analyzed the expression of IGF-1 that was previously reported to be responsive to ischemia/reperfusion brain injury. Ribonuclease protection assay was used to quantify the levels of IGF-I mRNA. The antisense riboprobe used for this experiment protects a region common to IGF-1A and IGF-1B isoforms, generating a single protected fragment. Low basal levels of IGF-1 mRNA and protein were detected in control cortical samples. IGF-1 mRNA levels were not affected during the first 12 hr of recovery from cardiac arrest but were markedly induced at 1 and 2 d of recovery and started to decrease at 7 d (Fig.6A). IGF-1 protein levels were also induced, and as expected, this induction was delayed compared with the mRNA response. A nearly fourfold increase was observed at 2–7 d of recovery, but this increase subsided at 14 d of recovery (Fig. 6B). In addition, our results showed no changes in the expression of the IGF-1 receptor β subunit during the recovery period (Fig. 6B).
Fig. 6.

Upregulation of IGF-1 mRNA and protein levels in the cerebral cortex after cardiac arrest and resuscitation.A, Ribonuclease protection assay for IGF-1 and β-actin (loading control) showing a significant induction of IGF-1 mRNA in the cerebral cortex at 1–7 d of recovery compared with control (C). B, Western blot showing increased IGF-1 protein levels in the cortex at 2–7 d of recovery from cardiac arrest. β-actin was used as a control for equal protein loading. Comparable IGF-1R β-subunit protein levels in the cerebral cortex of control and resuscitated animals (1h–7d of recovery). C, Graph representing the levels of endogenous circulating rat IGF-1 measured by ELISA in the plasma of rats exposed to cardiac arrest and resuscitation. The apparent increase in IGF-1 levels in the experimental samples (1h–4d of recovery) did not reach statistical significance when compared with the control samples. Results are shown as the mean ± SD of duplicate samples from three different animals.
To determine whether an increased uptake of IGF-1 from the plasma could contribute to our findings showing an increase in brain IGF-1, we measured endogenous circulating IGF-1 levels using an ELISA assay in rats subjected to cardiac arrest and allowed to recover for up to 4 d. Our results showed that the IGF-1 plasma levels did not change significantly after cardiac arrest compared with controls (Fig.6C). Although we cannot rule out a contribution from the IGF-1 circulating pool, the fact that its mRNA levels are significantly elevated suggests that the greater part of the increase in brain IGF-1 is likely caused by an upregulation of its expression in the tissue itself. Taken together, our results showed that the delayed brain IGF-1 induction after transient global ischemia temporally correlated with HIF-1α accumulation.
The mechanism underlying the delayed upregulation of IGF-1 is not known; however, we can speculate that hypoxia is not involved because by 2 d of recovery, EF5 binding was not detected. In fact, we found that the expression of IGF-1 (mRNA and protein) does not change in the brain of rats exposed to hypobaric hypoxia [0.5 atmosphere (ATM), equivalent to 10% O2] for 1 hr to 7 d (Fig.7A,B).
Fig. 7.

Expression of IGF-1 in the rat cerebral cortex after prolonged hypoxia. A, Ribonuclease protection assay showing that hypobaric hypoxia (0.5 ATM, equivalent to 10% O2 for 1 hr–7 d) did not affect IGF-1 mRNA levels in the brain cortex compared with normoxic control (C).B, Immunoblot showing no changes in IGF-1 protein levels in the brain cortex during hypobaric hypoxia (1h–7d).
IGF-1 colocalized with HIF-1α
Control sections showed weak IGF-1 immunoreactivity in the cortex mainly associated with neurons (data not shown). This immunostaining was enhanced after 4 d of recovery from cardiac arrest and resuscitation. No apparent staining of glial cells was observed (Fig.8A). Double immunostaining for IGF-1 and HIF-1α indicates that most neurons expressing IGF-1 also express HIF-1α (Fig.8A,B). However, many HIF-1α-positive neurons did not stain for IGF-1. No obvious colocalization of IGF-1 and HIF-1α immunostaining was observed in small blood vessels.
Fig. 8.

IGF-1 immunostaining of the rat cerebral cortex after transient global cerebral ischemia. A, IGF-1 expression (red) was apparently restricted mainly to neurons at 4 d of recovery from cardiac arrest. B, Double staining for HIF-1α (green) showing colocalization with some IGF-1-positive neurons (arrows). Scale bar, 100 μm.
IGF-1 induces HIF-1α accumulation and HIF-1 target genes in PC12 cells and primary cortical neurons
We examined the ability of IGF-1 to induce HIF-1 activation in PC12 cells and primary cortical neurons. Treatment of these cells with IGF-1 (100 nm, 24 hr) under normoxic conditions induced a significant accumulation of HIF-1α (n = 3). However, the effect of IGF-1 on HIF-1α accumulation was less compared with hypoxia (1% O2). In contrast, neither hypoxia nor IGF-1 affected HIF-1β expression in either cell type (Fig.9A). In addition, we analyzed the expression of two HIF-1 target genes, Glut-1 and VEGF, by Northern blot analysis. Our results showed that both targets were significantly induced by IGF-1 treatment. This induction was less pronounced compared with hypoxia (Fig. 9B). To show that IGF-1 was biologically active, we measured ligand-induced tyrosine phosphorylation of IGF-1R in PC12 and primary neurons by immunoprecipitation with IGF-1Rβ subunit antibody followed by immunoblotting with either anti-phosphotyrosine or anti-IGF-1Rβ antibodies (n = 3). We found an increase in tyrosine-phosphorylated IGF-1Rβ subunit in PC12 and primary neurons treated with IGF-1, demonstrating the activation of the receptor. No changes in IGF-1Rβ levels were observed under these conditions (Fig. 9C).
Fig. 9.

IGF-1 induces HIF-1α accumulation and expression of Glut-1 and VEGF in PC12 cells and primary cultures of cortical neurons. A, Western blot showing that hypoxia (1% O2 for 24 hr) and IGF-1 (100 nm, 24 hr) treatment were able to induce HIF-1α accumulation in PC12 and primary cortical neurons. HIF-1β and β-actin protein levels were not affected by either treatment. B, Northern blot analysis showing induction of Glut-1 and VEGF mRNA levels by hypoxia (1% O2 for 24 hr) or IGF-1 treatment (100 nm, 24 hr) in PC12 and cortical neurons. C, To determine whether IGF-1 treatment (100 nm, 24 hr) results in the activation of the IGF-1R, samples were immunoprecipitated with anti-IGF-1Rβ antibody and Western blot was performed with a phosphotyrosine-specific antibody (pTyr). The same membrane was used for IGF-1Rβ Western blot. D, Western blot analysis of HIF-1α in PC12 cells exposed to normoxia (N; 21% O2), hypoxia (H; 1%O2 for 4 hr), or IGF-1 (100 nm for 24 hr) with or without anti-IGF-1 antibody (Ab; 0.25 μg/ml for 24 hr).
To demonstrate that IGF-1 is not involved in the hypoxia-dependent induction of HIF-1α, we exposed PC12 cells to hypoxia (1% O2, 24 hr) or IGF-1 (100 nm, 24 hr) in the absence or presence of an anti-IGF-1 antibody (0.25 μg/ml). This neutralizing antibody did not affect the accumulation of HIF-1α during hypoxia, but it reduced significantly the expression of HIF-1α in response to IGF-1 (Fig. 9D).
Peripheral infusion of IGF-1 induces HIF-1α accumulation and expression of HIF-1α target genes in the rat brain
Considering that peripheral administration of recombinant IGF-1 exerts potent therapeutic effects in several models of brain damage, including brain ischemia, we studied the effects of systemic administration of IGF-1 on HIF-1α accumulation in the brain of normal rats. As shown in Figure10A, subcutaneous administration of recombinant IGF-1 with an osmotic minipump for 7 d resulted in a significant accumulation of HIF-1α in the cerebral cortex. Conversely, the animals infused only with the vehicle did not show HIF-1α accumulation. The induction of HIF-1α and HIF-1 target genes by IGF-1 was comparable to the effect of transient global ischemia.
Fig. 10.

Effect of peripheral infusion of human recombinant IGF-1 on HIF-1α accumulation and the expression of target genes in the rat cerebral cortex. A, Western blots showing HIF-1α protein levels in the cerebral cortex after hypoxia (H; 4 hr of 8%O2), 4 d of recovery after cardiac arrest (CA), and 7 d of continuous IGF-1 infusion (IGF). HIF-1β protein levels did not change in response to either treatment. β-Actin was used as a control for equal protein loading. C, Control samples for each experimental treatment. B, Northern blot analysis of Epo and Glut-1 mRNA levels in the brain cortex after hypoxia (H), transient global ischemia (CA), and IGF-1 infusion at conditions similar to those described in A.
To demonstrate that HIF-1 is transcriptionally active after IGF-1 treatment, we analyzed the expression of the HIF-1 target genes, Epo and Glut-1, by Northern blot analysis. Our results showed that both targets were significantly induced in the brain of rats treated with IGF-1 (Fig. 10B). The dose used in this study resulted in a twofold increase in total circulating IGF-1 from 174.17 ± 25.02 to 366.65 ± 62.11 ng/ml of plasma after 7 d of infusion (p < 0.05). This amount was comparable to previous studies using similar doses, and those levels of IGF-1 were enough to induce other effects in the rat brain (Fernandez et al., 1998).
Infusion of IGF-1 in the lateral ventricle induced HIF-1α accumulation in the rat brain
Although peripherally infused IGF-1 has been shown to cross the blood–brain barrier (Reinhardt and Bondy, 1994), we could not rule out a systemic effect of IGF-1 that can secondarily affect HIF-1α expression in the brain. To demonstrate a direct effect of IGF-1 on HIF-1α expression in the rat brain, we infused IGF-1 in the lateral ventricle of normal rats and analyzed HIF-1α expression in the periventricular area by immunohistochemistry. Our analysis revealed that HIF-1α-positive cells were present in the parenchymal tissue surrounding the lateral ventricular on the infused side (Fig.11B) but not on the contralateral site, indicating that IGF-1 did not diffuse contralaterally. The number of HIF-1α positive-cells decreased as a function of distance from the lateral ventricle. Double labeling with NeuN showed that most of the cells positive for HIF-1α were neurons (Fig. 11C,D).
Fig. 11.

Effect of intraventricular infusion of IGF-1 on HIF-1α accumulation. A, Schematic coronal section showing site of IGF-1 infusion into the lateral ventricle.B, Immunofluorescence staining of HIF-1α showing positive cells (green) in the striatum parenchyma adjacent to the infused lateral ventricle. C, Double staining for the neuronal marker NeuN (red) of the section shown in B. D, Merged images fromB and C showing that most HIF-1α-positive cells were neurons (yellow).LV, Lateral ventricle. Scale bar, 100 μm.
Selective inhibition of IGF-1R after cardiac arrest and resuscitation abrogates HIF-1α accumulation
To confirm the role of IGF-1 in the delayed and persistent activation of HIF-1α in the brain after transient global cerebral ischemia, the biological activity of the IGF-1R was neutralized by the intracerebroventricular administration of the selective antagonist JB-1. Figure 12 depicts representative examples of HIF-1α immunoreactivity in the brain of rats that were subjected to cardiac arrest and resuscitation and subsequently received continuous intracerebroventricular infusion of saline or JB-1 for 4 d during the recovery phase. The brain of the saline-treated rats exhibited widespread neuronal HIF-1α staining in the cerebral cortex, the caudoputamen, hippocampal and septal formation, and other parenchymal regions in both hemispheres (Fig.12A). Injection of the selective IGF-1R antagonist into the lateral ventricle caused a significant reduction of HIF-1α immunoreactivity in regions ipsilateral to the site of the injection. This inhibitory effect of JB-1 was highest in the right caudoputamen, in the cerebral cortex, and in regions neighboring the ventricle walls (Fig. 12B). The effect of JB-1 decreased in distant structures from the site of injection, which may be attributed to the slow diffusion or to a decreased half-life of this peptide in the CSF.
Fig. 12.

Effect of intracerebroventricular infusion of JB-1 on HIF-1α accumulation in the rat brain after cardiac arrest and resuscitation. A, HIF-1α immunostaining in the parenchyma adjacent to the lateral ventricle after intracerebroventricular infusion of saline after cardiac arrest and resuscitation. B, HIF-1α immunostaining in a similar region as in A showing absence of HIF-1α-positive cells after selective inhibition of the IGF-1R by intracerebroventricular infusion of JB-1 for 4 d after cardiac arrest and resuscitation. C, D, Double staining for NeuN of the same sections shown in A andB, respectively. LV, Lateral ventricle. Scale bar, 100 μm.
DISCUSSION
In our model of cardiac arrest, rats regained spontaneous respiration within 3 hr after resuscitation and regained consciousness before 12 hr. Resuscitated rats were usually able to move and feed themselves by 24 hr. The major pathophysiological effects of ∼12 min of global ischemia induced by cardiac arrest and resuscitation in the rat have been reported previously (Crumrine and LaManna, 1991). These include an initial arterial acidosis, hypertension, and hemoconcentration that returned to pre-ischemic levels between 30 and 180 min after reperfusion. In addition, changes in brain blood flow include an initial hyperemia followed by a secondary hypoperfusion (Crumrine and LaManna, 1991; Lauro et al., 1999).
Transient global cerebral ischemia induced by cardiac arrest and resuscitation leads to delayed neuronal death as indicated by a loss of CA1 hippocampal neurons (Crumrine and LaManna, 1991). In response to this metabolic stress, the expression of VEGF is upregulated as early as 1 hr of recovery and lasts for up to 4 d (Pichiule et al., 1999; Jin et al., 2000). This gene has a hypoxic response element in its promoter and is regulated by the transcription factor HIF-1 (Forsythe et al., 1996). To determine whether activation of HIF-1 was responsible for an increased expression of VEGF, we studied the expression of the regulatory subunit HIF-1α. Our results showed that HIF-1α accumulates within 1 hr after resuscitation from cardiac arrest, and unexpectedly, it remains elevated for >1 week. In addition to VEGF induction, we showed that other HIF-1 target genes such as Epo and Glut-1 are also transiently induced. Thus, reversible global ischemia leads to the activation of HIF-1 and the expression of its target genes.
Next, we attempted to determine the mechanism responsible for prolonged HIF-1α accumulation. The most logical explanation would be that HIF-1 is responding to a tissue hypoxic signal. In fact, cerebral blood flow was found severely reduced in the first hour (16% of control) and remained approximately half of the control value at 6 hr of recovery (Crumrine and LaManna, 1991). This hypoperfusion is likely to decrease oxygen delivery that might induce HIF-1 activation in the early period of recovery. Nevertheless, it is unlikely that cerebral blood flow remained low during longer recovery periods (2 d or more). To determine whether the brain remained hypoxic for a prolonged period after transient ischemia, we used the hypoxic marker EF5. This 2-nitroimidazole drug has been used extensively to detect tissue hypoxia because the rate of its bioreductive metabolism is inversely dependent on oxygen partial pressure. Intracellular metabolism of EF5 leads to its covalent binding with several molecules. These bound adducts can be detected with specific antibodies (Evans et al., 1995). Our results showed EF5 binding in the brain at 1 hr but not at 2 d of recovery from cardiac arrest.
To test the sensitivity of EF5 as a hypoxic marker in the brain, we exposed rats to different oxygen concentrations and compared the brain EF5 signal with HIF-1α accumulation. We found that EF5 binding correlates with HIF-1α accumulation at 12% or lower oxygen concentrations. However, both EF5 binding and HIF-1α were barely detected at 14–21% oxygen. Thus, the lack of EF5 binding at 2 d of recovery indicates that the brain was no longer hypoxic. Other nonhypoxic mechanisms must be responsible for the unexpected prolonged HIF-1α induction.
Considering that impairment of the HIF-1α degradation machinery can lead to HIF-1α accumulation, we analyzed the activity of the 26S proteosome complex as well as the expression of the tumor suppressor pVHL. We found a slight transient reduction of the proteosome activity in the first hours of recovery from cardiac arrest as indicated by a 25% decrease in chymotrypsin-like activity, whereas pVHL protein levels in the cerebral cortex were unchanged after cardiac arrest. These observations suggest that HIF-1α accumulation in the first hours of recovery might be caused by hypoxic stress caused by a reduced brain blood flow in addition to a transient inhibition of the 26S proteosome. However, none of these conditions persisted for >2 d after cardiac arrest.
To attempt to explain the accumulation of HIF-1α after 2 d of recovery, we focused our attention on IGF-1. It was reported previously that this growth factor activated HIF-1α in certain cell types under normoxic conditions (Zelzer et al., 1998; Feldser et al., 1999; Zundel et al., 2000). In the CNS, IGF-1 is of particular interest because it is locally synthesized, and systemic IGF-1 can cross the blood–brain barrier (Reinhardt and Bondy, 1994; Pan and Kastin, 2000). IGF-1 and its receptor are known to be present and active in the mature nervous system. However, brain IGF-1 levels are low compared with peripheral tissues such as the liver (Daughaday and Rotwein, 1989). Previous reports have shown that IGF-1, its receptors, and binding proteins are all upregulated in response to ischemia and other neurodegenerative conditions (Schwab et al., 1997; Beilharz et al., 1998; Fernandez et al., 1998). This upregulation appears to provide neuroprotection, because exogenous IGF-1 both in vivo and in vitroprotects neurons from different types of injury (Tagami et al., 1997;Heck et al., 1999; Guan et al., 2000; Wang et al., 2000; Yamaguchi et al., 2000).
In this study, we have shown that IGF-1 gene expression was transiently upregulated in the cerebral cortex after cardiac arrest and resuscitation. Induction of IGF-1 expression occurred between 2 and 7 d of recovery and temporally correlated with the persistent accumulation of HIF-1α during the same period. In contrast, the circulating levels of IGF-1 did not change significantly.
To test the ability of IGF-1 to induce HIF-1 activation in neural cells, we studied the effect of IGF-1 on the accumulation of HIF-1α and the expression of HIF-1 target genes in vitro andin vivo. Our results showed that IGF-1 induced HIF-1α accumulation in cultured neurons and PC12 cells. In addition, we showed that peripheral and intracerebroventricular infusion of IGF-1 in rats resulted in accumulation of HIF-1α in the brain. Furthermore, we showed that the HIF-1 target genes, VEGF, Epo, and Glut-1, are all induced after IGF-1 treatment, indicating that HIF-1 is transcriptionally active. These lines of evidence demonstrate that IGF-1 can induce activation of HIF-1 in the CNS. Accordingly, IGF-1 might be responsible for the sustained HIF-1α accumulation after cardiac arrest and resuscitation. To prove this, we used an IGF-1 analog that selectively inhibits the autophosphorylation of the IGF-1R. This antagonist, JB-1, has been used previously to inhibit the IGF-1 pathway in the rat brain and in cell culture systems (Pietrzkowski et al., 1992; Quesada and Etgen, 2002). Our results showed that intracerebroventricular infusion of JB-1 during the recovery phase from cardiac arrest and resuscitation inhibited HIF-1α accumulation. Thus, the delayed induction of IGF-1 expression and the subsequent activation of the IGF-1R receptor after cerebral ischemia seem to be required for the long-lasting HIF-1 activation.
Our study does not provide evidence for the mechanism underlying the induction of IGF-1 after brain ischemia; however, this induction seems not to be oxygen dependent because by 2 d of recovery the brain is no longer hypoxic, as indicated by EF5 binding analysis. Consistent with this notion, even prolonged exposure to hypoxia (up to 7 d, ∼10% oxygen), previously found to induce HIF-1 activation (Chavez et al., 2000), does not induce IGF-1 in the rat brain. Interestingly, in cultured PC12 cells, a neutralizing anti-IGF-1 antibody did not affect the hypoxia-induced HIF-1α accumulation, whereas it blocked the IGF-1-mediated HIF-1α accumulation, as expected. Thus, IGF-1 and hypoxia activate two distinct and independent mechanisms of HIF-1 activation.
Whether activation of HIF-1 has a role in neuronal survival or contributes to cell death during ischemia and other CNS insults is still a matter of discussion. The picture emerging from studies with different models of CNS injury is that activation of HIF-1 is part of an adaptive mechanism that might allow cell survival during hypoxia and after an ischemic insult. HIF-1 target genes that may mediate neuronal survival after ischemia include glycolytic enzymes, erythropoietin, Glut-1, and VEGF (Bergeron et al., 1999; Zaman et al., 1999;Marti et al., 2000; Digicaylioglu and Lipton, 2001). Additional evidence supporting the neuroprotective role of HIF-1 comes from studies using iron chelators and cobalt chloride. These compounds seem to exert neuroprotective effects against oxidative stress in neural cells in part by activating HIF-1 and its target genes (Zaman et al., 1999; Bergeron et al., 2000). In contrast, some studies have documented HIF-1α pro-apototic effects in embryonic stem cells or cortical neurons under hypoxic conditions. It appears that HIF-1 mediates upregulation as well as stabilization of p53 and downregulation of Bcl-2 leading to cell death (An et al., 1998;Carmeliet et al., 1998; Halterman et al., 1999). These apparent contradictory observations suggest that the effect of HIF-1 induction may depend on the cell type, the developmental stage of the cell, or the death stimulus.
In conclusion, after transient global cerebral ischemia, the sustained HIF-1 activation seems to be regulated through hypoxia-dependent and -independent mechanisms. The subsequent induction of HIF-1 target genes may be part of the intrinsic neuroprotective mechanism aimed at attenuating damage as a result of oxidative stress and disruption of energy metabolism in the brain. Moreover, our results suggest that IGF-1 might exert its neuroprotective effects in different types of CNS injury at least in part by activating HIF-1 and its target genes.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS-38632 and NS-37111. We thank Dr. Cameron Koch (University of Pennsylvania) for his advice and assistance with the EF5 staining. We also thank Dr. W. David Lust (Department of Neurological Surgery, Case Western Reserve University) for suggestions and critical reviews of this manuscript and Sandy Hufeisen for technical assistance with the cell culture work. The human recombinant IGF-1 was generously provided by A. F. Parlow from the National Institute of Diabetes and Digestive and Kidney Diseases National Hormone and Peptide Program (Harbor-University of California Los Angeles Medical Center). EF5 was a gift from S. Evans (University of Pennsylvania).
Correspondence should be addressed to Dr. Joseph C. LaManna, Department of Neurology (BRB525), Case Western Reserve University, School of Medicine, 10900 Euclid Avenue, Cleveland OH 44106-4938. E-mail:JCL4@po.cwru.edu.
REFERENCES
- 1.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392:405–408. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 2.Beilharz EJ, Russo VC, Butler G, Baker NL, Connor B, Sirimanne ES, Dragunow M, Werther GA, Gluckman PD, Williams CE, Scheepens A. Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic-ischemic injury. Brain Res Mol Brain Res. 1998;9:119–134. doi: 10.1016/s0169-328x(98)00122-3. [DOI] [PubMed] [Google Scholar]
- 3.Bergeron M, Yu AY, Solway KE, Semenza GL, Sharp FR. Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur J Neurosci. 1999;11:4159–4170. doi: 10.1046/j.1460-9568.1999.00845.x. [DOI] [PubMed] [Google Scholar]
- 4.Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–296. [PubMed] [Google Scholar]
- 5.Brewer GJ. Serum-free B27/Neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 7.Carro E, Nunez A, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J Neurosci. 2000;20:2926–2933. doi: 10.1523/JNEUROSCI.20-08-02926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chavez JC, Agani F, Pichiule P, LaManna JC. Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J Appl Physiol. 2000;89:1937–1942. doi: 10.1152/jappl.2000.89.5.1937. [DOI] [PubMed] [Google Scholar]
- 9.Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 10.Crumrine RC, LaManna JC. Regional cerebral metabolites, blood flow, plasma volume, and mean transit time in total cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1991;11:272–282. doi: 10.1038/jcbfm.1991.59. [DOI] [PubMed] [Google Scholar]
- 11.Daughaday WH, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989;10:68–91. doi: 10.1210/edrv-10-1-68. [DOI] [PubMed] [Google Scholar]
- 12.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between JAK2 and NF-kappa B signaling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 13.Evans SM, Joiner B, Jenkins WT, Laughlin KM, Lord EM, Koch CJ. Identification of hypoxia in cells and tissues of epigastric 9L rat glioma using EF5 [2-(2-nitro-1H-imidazol-1-yl)-N-(2,2,3,3,3-pentafluoropropyl) acetamide. Br J Cancer. 1995;72:875–882. doi: 10.1038/bjc.1995.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999;59:3915–3918. [PubMed] [Google Scholar]
- 15.Fernandez AM, de la Vega AG, Torres-Aleman I. Insulin-like growth factor I restores motor coordination in a rat model of cerebellar ataxia. Proc Natl Acad Sci USA. 1998;95:1253–1258. doi: 10.1073/pnas.95.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan J, Bennet TL, George S, Waldvogel HJ, Faull RL, Gluckman PD, Keunen H, Gunn AJ. Selective neuroprotective effects with insulin-like growth factor-1 in phenotypic striatal neurons following ischemic brain injury in fetal sheep. Neuroscience. 2000;95:831–839. doi: 10.1016/s0306-4522(99)00456-x. [DOI] [PubMed] [Google Scholar]
- 18.Halterman MW, Miller CC, Federoff HJ. Hypoxia-inducible factor-1α mediates hypoxia-induced delayed neuronal death that involves p53. J Neurosci. 1999;19:6818–6824. doi: 10.1523/JNEUROSCI.19-16-06818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heck S, Lezoualc'h F, Engert S, Behl C. Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor kappaB. J Biol Chem. 1999;274:9828–9835. doi: 10.1074/jbc.274.14.9828. [DOI] [PubMed] [Google Scholar]
- 20.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jewell UR, Kvietikova I, Scheid A, Bauer C, Wenger RH, Gassmann M. Induction of HIF-1alpha in response to hypoxia is instantaneous. FASEB J. 2001;15:1312–1314. [PubMed] [Google Scholar]
- 22.Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. 1996;271:C1172–1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- 23.Jin KL, Mao XO, Nagayama T, Goldsmith PC, Greenberg DA. Induction of vascular endothelial growth factor and hypoxia-inducible factor-1alpha by global ischemia in rat brain. Neuroscience. 2000;99:577–585. doi: 10.1016/s0306-4522(00)00207-4. [DOI] [PubMed] [Google Scholar]
- 24.Johnston BM, Mallard EC, Williams CE, Gluckman PD. Insulin-like growth factor-1 is a potent neuronal rescue agent after hypoxic-ischemic injury in fetal lambs. J Clin Invest. 1996;97:300–308. doi: 10.1172/JCI118416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauro KL, Kabert H, LaManna JC. Methyl isobutyl amiloride alters regional brain reperfusion after resuscitation from cardiac arrest in rats. Brain Res. 1999;831:64–71. doi: 10.1016/s0006-8993(99)01394-3. [DOI] [PubMed] [Google Scholar]
- 26.Lee WH, Wang GM, Seaman LB, Vannucci SJ. Coordinate IGF-I and IGFBP5 gene expression in perinatal rat brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 1996;16:227–236. doi: 10.1097/00004647-199603000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Marti HH, Bernaudin M, Petit E, Bauer C. Neuroprotection and angiogenesis: dual role of erythropoietin in brain ischemia. News Physiol Sci. 2000;15:225–229. doi: 10.1152/physiologyonline.2000.15.5.225. [DOI] [PubMed] [Google Scholar]
- 28.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumor suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 29.Pan W, Kastin AJ. Interactions of IGF-1 with the blood-brain barrier in vivo and in situ. Neuroendocrinology. 2000;72:171–178. doi: 10.1159/000054584. [DOI] [PubMed] [Google Scholar]
- 30.Paxinos G, Watson C. The rat brain in stereotaxis coordinates, Ed 2. Academic; New York: 1986. [Google Scholar]
- 31.Pichiule P, Chavez JC, Xu K, LaManna JC. Vascular endothelial growth factor upregulation in transient global ischemia induced by cardiac arrest and resuscitation in rat brain. Brain Res Mol Brain Res. 1999;74:83–90. doi: 10.1016/s0169-328x(99)00261-2. [DOI] [PubMed] [Google Scholar]
- 32.Pietrzkowski Z, Wernicke D, Porcu P, Jameson BA, Baserga R. Inhibition of cellular proliferation by peptide analogues of insulin-like growth factor 1. Cancer Res. 1992;52:6447–6451. [PubMed] [Google Scholar]
- 33.Quesada A, Etgen AM. Functional interactions between estrogen and insulin-like growth factor-I in the regulation of α 1B-adrenoceptors and female reproductive function. J Neurosci. 2002;22:2401–2408. doi: 10.1523/JNEUROSCI.22-06-02401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reinhardt RR, Bondy CA. Insulin-like growth factors can cross the blood-brain barrier. Endocrinology. 1994;135:1753–1761. doi: 10.1210/endo.135.5.7525251. [DOI] [PubMed] [Google Scholar]
- 35.Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 36.Schwab S, Spranger M, Krempien S, Hacke W, Bettendorf M. Plasma insulin-like growth factor I and IGF binding protein 3 levels in patients with acute cerebral ischemic injury. Stroke. 1997;28:1744–1748. doi: 10.1161/01.str.28.9.1744. [DOI] [PubMed] [Google Scholar]
- 37.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 38.Tagami M, Ikeda K, Nara Y, Fujino H, Kubota A, Numano F, Yamori Y. Insulin-like growth factor-1 attenuates apoptosis in hippocampal neurons caused by cerebral ischemia and reperfusion in stroke-prone spontaneously hypertensive rats. Lab Invest. 1997;76:613–617. [PubMed] [Google Scholar]
- 39.Torres-Aleman I. Serum growth factors and neuroprotective surveillance. Mol Neurobiol. 2000;21:153–160. doi: 10.1385/mn:21:3:153. [DOI] [PubMed] [Google Scholar]
- 40.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 41.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang JM, Hayashi T, Zhang WR, Sakai K, Shiro Y, Abe K. Reduction of ischemic brain injury by topical application of insulin-like growth factor-I after transient middle cerebral artery occlusion in rats. Brain Res. 2000;859:381–385. doi: 10.1016/s0006-8993(00)02008-4. [DOI] [PubMed] [Google Scholar]
- 43.Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- 44.Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21 (waf1/cip1), and erythropoietin. J Neurosci. 1999;19:9821–9830. doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J. 1998;17:5085–5094. doi: 10.1093/emboj/17.17.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 47.Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391–396. [PMC free article] [PubMed] [Google Scholar]