

Abstract
Huntington's disease is a genetic neurodegenerative disorder characterized clinically by both motor and cognitive impairments and striatal lesions. At present, there are no pharmacological treatments able to prevent or slow its development. In the present study, we report the neuroprotective effect of adenosine amine congener (ADAC), a specific A1 receptor agonist known to be devoid of any of the side effects that usually impair the clinical use of such compounds. Remarkably, in a rat model of Huntington's disease generated by subcutaneous infusion of the mitochondrial inhibitor 3-nitropropionic acid (3NP), we have observed that an acute treatment with ADAC (100 μg · kg−1 · d−1) not only strongly reduces the size of the striatal lesion (−40%) and the remaining ongoing striatal degeneration (−30%), but also prevents the development of severe dystonia of hindlimbs. Electrophysiological recording on corticostriatal brain slices demonstrated that ADAC strongly decreases the field EPSP amplitude by 70%, whereas it has no protective effect up to 1 μm against the 3NP-induced neuronal death in primary striatal cultures. This suggests that ADAC protective effects may be mediated presynaptically by the modulation of the energetic impairment-induced striatal excitotoxicity. Altogether, our results indicate that A1 receptor agonists deserve further experimental evaluation in animal models of Huntington's disease.
Keywords: Huntington's disease, 3-nitropropionic acid, adenosine, A1 receptor, neuroprotection, striatum, cell death
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder characterized by both motor and cognitive symptoms (Brouillet et al., 1999). HD is caused by a mutation located within the IT15 gene, encoding for the Huntingtin protein, leading to an abnormal (CAG)n repeat in the 5′ coding sequence (The Huntington's Disease Collaborative Research Group, 1993). Histologically, this produces the formation of neuronal intranuclear inclusions (DiFiglia et al., 1997) and, for unclear reasons, the preferential loss of the striatal GABAergic projecting medium-sized spiny neurons (Vonsattel et al., 1985; Sieradzan and Mann, 2001). Despite recent promising advances in the fields of cellular and gene therapies (Hantraye et al., 1992; Emerich et al., 1997; Palfi et al., 1998; Bachoud-Levi et al., 2000; Mittoux et al., 2000; De Almeida et al., 2001), there are no available therapeutic tools to delay the onset of the symptoms and the associated neurodegeneration.
The striatal bioenergetic impairment suggested to occur in HD is thought to cause deleterious secondary excitotoxicity (Brouillet et al., 1999). Consistently, neurotoxicity of systemic administration of 3-nitropropionic acid (3NP), an irreversible inhibitor of succinate dehydrogenase known to produce striatal lesions in rats and nonhuman primates (Beal et al., 1993; Brouillet et al., 1995, 1998, 1999; Palfi et al., 1996; Dautry et al., 1999), has been thought to involve glutamate neurotoxicity (Beal et al., 1993; Schulz et al., 1996; Guyot et al., 1997a). Interestingly, in addition to the striatal lesion, 3NP also produces other features of HD such as movement (chorea, dystonia) and cognitive (perseveration) disorders, specific loss of spiny projection neurons and sparing of NADPH-diaphorase-expressing cells (Beal et al., 1993; Guyot et al., 1997a; Vis et al., 1999; El Massioui et al., 2001).
Adenosine is a purinergic messenger known to reduce neuronal activity through activation of high-affinity receptors (Sebastiao and Ribeiro, 2000; Dunwiddie and Masino, 2001). Many studies previously reported the neuroprotective properties of adenosine, and more especially of A1 receptor agonists, in ischemic/hypoxic or epileptic conditions (Rudolphi et al., 1992; De Mendonca et al., 2000;Dunwiddie and Masino, 2001). These neuroprotective effects are thought to be related to the inhibitory function that presynaptic A1 receptors exert on the release of excitatory amino acids, restraining, in particular, the activation of NMDA receptors (Fredholm and Dunwiddie, 1988; Palmer and Stiles, 1995). Unfortunately, the potential therapeutic use of A1 adenosine agonists is particularly impaired by cardiovascular side effects (Williams, 1993; White et al., 1996), and the opposite outcome is observed after acute and chronic regimen (Von Lubitz et al., 1994a,b; De Sarro et al., 1996; Jacobson et al., 1996); however, some A1 receptor agonists, devoid of any deleterious cardiovascular effects, have been described recently (Bischofberger et al., 1997). Interestingly, among them it was shown that low concentrations of adenosine amine congener (ADAC) (Von Lubitz et al., 1996a,b) can efficiently protect hippocampal neurons against cerebral ischemia after both acute and chronic treatments (Von Lubitz et al., 1999). This attractive compound thus deserves evaluation, especially in HD models for which A1 receptor agonists have never been evaluated.
Recently, it was reported that Lewis rats, unlike the Sprague Dawley strain, respond homogeneously to 3NP and develop topologically reproducible striatal lesions, providing a suitable HD model for neuroprotective studies (Ouary et al., 2000; Blum et al., 2001, 2002;Mittoux et al., 2002). In the present work, we thus aimed to determine the potential neuroprotective effect of either chronic or acute treatment with the A1 receptor agonist ADAC against the striatal lesions induced by 3NP in Lewis rats.
MATERIALS AND METHODS
Animals
Adult male Lewis rats (IFFA Credo, Reims, France), 12 weeks of age, weighing 320–380 gm were used in this study. Animals were housed three per cage and maintained in a temperature- and humidity-controlled room on a 12 hr light/dark cycle with food and water ad libitum. The number of animals was kept to the minimum, and all efforts to avoid animal suffering were made in accordance with the standards of the Institutional Ethical Committee of the School of Medicine of Université Libre de Bruxelles.
Surgery and 3NP treatment
Rats were anesthetized with a mixture containing xylazine hydrochloride (Rompun, Bayer; 4.5 mg/kg) and ketamine hydrochloride (Imalgene, Merial; 90 mg/kg). In the 3NP-treated animals, an incision was made below the base of the neck and a 2mL1 Alzet osmotic minipump (delivering 10 μl/hr for 7 d; IFFA Credo) containing 3-nitropropionic acid (Fluka) was positioned under the skin. 3NP was dissolved in 0.1 m PBS, pH 7.4, adjusted to pH 7.3–7.4 with 5N NaOH. The final concentration of 3NP in the pump was adjusted to the weight of the rats on the day of implantation to exactly deliver 56 mg · kg−1 · d−1. Sham rats and animals treated with ADAC alone underwent all of the surgical procedures (without minipump implantation).
All rats were killed after 5 d of 3NP subcutaneous infusion. It is noteworthy that the time they were killed was chosen according to the known kinetics of striatal lesion occurrence in this particular model, because it was determined previously that macroscopic lesions induced by 3NP are detected 5 d after the beginning of the toxic treatment (Ouary et al., 2000; Blum et al., 2001).
ADAC treatment
Two separate experiments were performed (Fig.1). In the first one, 28 rats (sham,n = 7; vehicle/3NP, n = 7; ADAC−8/5, n = 7; ADAC−8/5/3NP, n = 7) were used to study the potential protective effect of chronic administration of ADAC against the neurotoxic effects of 3NP. In this protocol, ADAC injections began 8 d before the onset of 3NP intoxication and were continued until the animals were killed (13 d in all; last injection 6 hr before animals were killed). ADAC solution was prepared as follows: 6 mg of ADAC (Sigma) were first dissolved in 200 μl of 1N HCl and then added to 120 ml of 0.1 m PBS, pH 7.4, to reach a final concentration of 50 μg/ml. Before each injection, the solution was heated to 37°C for 15–30 min to ensure complete dissolution. Vehicle solution (200 μl of 1N HCl in 120 ml of 0.1m PBS) was processed similarly. The final pH of all solutions was 7.3–7.4. The volume of the ADAC solution injected intraperitoneally was adjusted to the weight of rats to deliver exactly 100 μg · kg−1 · d−1(200 μl/100 gm of weight), a supramaximal dose that has been shown previously to be protective against cerebral ischemia after both chronic and acute administration (Von Lubitz et al., 1999) without any cardiovascular side effects (Von Lubitz et al., 1996b).
Fig. 1.

Schematic drawing representing the experimental protocols used for chronic and acute administrations of ADAC in the control and 3NP-treated rats.
In the second experiment, 50 rats were used to study the potential neuroprotective effects of acute treatments with ADAC (sham,n = 7; 3NP/vehicle, n = 9; ADAC alone injected at days 3, 4, and 5 = ADAC3/5group, n = 7; 3NP-treated rats injected with ADAC at days 3, 4 and 5 = 3NP/ADAC3/5,n = 10; ADAC alone injected at days 4 and 5 = ADAC4/5 group, n = 7; 3NP-treated rats injected with ADAC at days 4 and 5 = 3NP/ADAC4/5, n = 10). ADAC injections were performed as described above (last injection 6 hr before animals were killed).
Tissue post-processing
All rats were killed by decapitation, and their brains were quickly removed. The two cerebral hemispheres were separated. One was frozen in 2-methylbutane cooled by dry ice (−40°C), and the other was embedded in paraffin. The frozen tissue was cut at 16 μm thickness on a cryostat (Leitz), and the serial coronal sections were mounted onto poly-l-lysine or gelatin-coated slides and stored at −20°C until use. Paraffin-embedded brains were cut at 10 μm using a microtome (Historange), and the coronal sections were mounted on gelatin-coated slides in glutamine albumin (BDH Chemicals, Poole, UK) for immunohistochemistry.
Behavioral analysis
Controls and 3NP-treated animals were evaluated every day for both weight loss and motor impairment. For the latter, we used a quantitative neurological scale as described previously (Guyot et al., 1997a; Ouary et al., 2000; Blum et al., 2001, 2002; Mittoux et al., 2002). Briefly, behavioral abnormalities were determined according to the presence and severity of motor symptoms consisting of dystonia (intermittent dystonia of one hindlimb, score = 1; intermittent dystonia of two hindlimbs, score = 2; permanent dystonia of hindlimbs, score = 3), gait abnormalities consisting mainly of an uncoordinated and wobbling gait (score = 1), and recumbency (animals lying on one side but showing uncoordinated movements when stimulated, score = 1; near-death recumbency characterized by almost complete paralysis with rapid breathing, score = 2). Additionally, the capabilities of animals to grasp a cage grid with forepaws (unable = 1) or to remain on a small platform (9 × 5 cm) for >10 sec (unable = 1) were determined. The final neurological score was assessed as the sum of the above individual scores (minimum = 0, normal animal; score = 8, animal showing near-death recumbency).
NeuN immunohistochemistry
Paraffin sections were successively dipped in toluol and 100% alcohol. After quenching of endogenous peroxidases (0.3% hydrogen peroxide in methanol for 30 min), sections were rehydrated by 90 and 70% ethanol and then water. After a 10 min microwave treatment in citrate buffer (0.01 m, pH 6), slides were rinsed in PBS and incubated for 10 min in 10% normal horse serum (Invitrogen). Sections were then incubated overnight at 4°C with mouse monoclonal anti-NeuN antibody [1:300 in 1% normal horse serum (Chemicon, Temecula, CA) MAB377]. After two washes, they were incubated further for 15 min with 10% normal horse serum and then for 30 min with biotinylated donkey anti-mouse secondary antibody (1:200 in 1% normal horse serum; Jackson ImmunoResearch, West Grove, PA). After two washes, the signal was revealed by the ABC method (Vector Laboratories) and diaminobenzidine (Dako).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining
Detection of nuclei presenting DNA-strand breaks was obtained by the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) method. Frozen sections mounted on poly-l-lysine-coated slides were postfixed for 30 min in 4% paraformaldehyde, rinsed three times in PBS, and successively treated at room temperature by a 0.1% citrate and 1% Triton X-100 buffer for 5 min, rinsed in PBS, and incubated for an additional 5 min with proteinase K (10 μg/ml in PBS, pH 7.4). TUNEL reaction (30 min at 37°C) was performed using a commercial kit according to the manufacturer's instructions (Roche). Positive-labeled cells were observed under epifluorescence using a Zeiss microscope connected to an acquisition system. Quantification of TUNEL-positive cells was performed at 40× magnification on four digitalized fields located in the dorsolateral part of the striatum at the level of bregma approximately +0.48 mm according to the atlas of Paxinos and Watson (1990) (Fig. 2) [region of interest 2 (ROI 2)].
Fig. 2.

Schematic drawing showing the ROIs (1-6) delimited for quantification: 1, whole striatum; 2, dorsolateral striatum; 3, dorsomedial striatum;4, ventrolateral striatum; 5, outer layers of the cortex; 6, overlying cortex.
Semiquantitative measurement of succinate dehydrogenase activity
Measurement of succinate dehydrogenase (SDH) activity in control and 3NP-treated rats was performed as described previously (Brouillet et al., 1998). Frozen sections mounted on poly-l-lysine-coated slides were air dried and then incubated for 15 min in 0.1 m PBS (pH 7.4, 0.9% NaCl) at 37°C followed by incubation in 0.3 mm nitroblue tetrazolium (Sigma), 0.05 m sodium succinate (Sigma), and 0.05 m phosphate buffer, pH 7.6, for 30 min at 37°C. Finally, sections were rinsed successively for 5 min in cold PBS and deionized water and dried at room temperature. The image of each section was acquired, and the quantification was performed as described previously (Brouillet et al., 1998; Blum et al., 2001) using NIH image software according to the ROIs (1–3 and 5) presented in Figure 2. Results were expressed as the percentage mean ± SEM of mean sham value.
Enkephalin mRNA in situ hybridization
The hybridization technique was adapted from previous reports from our lab (Schiffmann and Vanderhaeghen, 1993; Dassesse et al., 1999). The sections mounted on RNase free poly-l-lysine-coated slides were fixed in 4% freshly prepared paraformaldehyde for 30 min and rinsed in 0.1 mM PBS. All sections were dehydrated and dipped for 3 min in chloroform. After air drying, the sections were incubated overnight at 42°C with 0.35 × 106 cpm per section of35S-labeled probes diluted in hybridization buffer, which consisted of 50% formamide, 4× SSC (1× SSC: 0.15 m NaCl, 0.015 m sodium citrate, pH 7.4), 1× Denhardt's solution (0.02% each of polyvinylpyrrolidone, bovine serum albumin, Ficoll), 1% sarcosyl, 0.02 m sodium phosphate at pH 7.4, 10% dextran sulfate, yeast tRNA at 500 μg/ml, salmon sperm DNA at 100 μg/ml, and 60 mm dithiothreitol. Compounds were provided by Sigma. After hybridization, the sections were rinsed for 4 × 15 min in 1× SSC at 55°C, dehydrated, and covered with Hyperfilm-βmax film (Amersham) for 2 or 3 weeks. The oligonucleotide enkephalin probe (5′-GTGTGCATGCCAGGAAGTTGATGTCGCCGGGACGTACCAGGCGG-3′) was synthesized on an Applied Biosystems 381A DNA synthesizer. It was labeled with α-35S dATP (DuPont NEN) at its 3′ end by terminal DNA deoxynucleotidylexotransferase (Invitrogen) and purified with a G50 column (Pharmacia Biosciences) according to the manufacturer's instructions.
In vitro receptor autoradiography
A1 receptor binding autoradiography was performed as described previously (Dassesse et al., 2001) using a single concentration of radioligand. The gelatin-coated slides, stored at −20°C until use, were brought to room temperature 30 min before the autoradiographic experiments.
The sections were preincubated for 30 min at 37°C in preincubation buffer [170 mm Tris-HCl, pH 7.4, 1 mm EDTA, 2 UI/ml adenosine deaminase (ADA)] and incubated for 2 hr at room temperature in buffer containing 170 mm Tris-HCl, pH 7.4, 1 mm MgCl2, 2 UI/ml ADA, and 0.5 nm of the A1 antagonist [3H]-8-cyclopentyl-1,3-dipropylxanthine (DPCPX) (120.0 Ci/mmol; DuPont NEN). Nonspecific binding of the3H-labeled ligand was assessed by the addition of 20 μmR(−)N6-2-phenylisopropyladenosine (R-PIA). Slides were washed three times for 15 min in ice-cold 170 mm Tris-HCl, pH 7.4 buffer, dipped in ice-cold distilled water, dried under a stream of cold air, and exposed to3H-Hyperfilms (Amersham) for 6 weeks.
Image analysis
Digitalized images with 256 gray levels were generated from the autoradiograms with the public domain NIH image 1.61 program (National Institutes of Health, Bethesda, MD), a Power Macintosh G3, and a CCD video camera (Dage-MTI) with fixed gain and black level. For the quantification of in situ hybridization and binding, depending on the marker studied, the average optical densities were measured on x-ray film autoradiograms in three different striatal regions (ROIs 1, 2, and 3) (Fig. 2) and in the overlying cortex (ROI 6) (Fig. 2). For in situ hybridization analysis, on each section an averaged optical density of the background level was subtracted from that of the measured areas to obtain corrected values. For quantification of binding autoradiography, specific binding was determined by subtracting the nonspecific from the total binding. Analyses were performed at the transverse level ∼0.48 mm rostral to bregma, according to the rat brain atlas of Paxinos and Watson (1990). Results were expressed as the percentage mean ± SEM of mean sham value.
Electrophysiology
Electrophysiological experiments were performed as described previously (D'Alcantara et al., 2001). Wistar rats (15–30 d old) were used (n = 5). Neostriatal slices were prepared as follows. Animals were anesthetized with ether and decapitated. The brain was immediately removed, transferred to ice-cold modified Krebs' solution, and cut in coronal blocks that were then glued to the stage of a Vibratome (Leica) with cyanoacrylate glue. The composition of the solution was (in mm): 124 NaCl, 3 KCl, 1.2 NaH2PO4, 1 MgCl2, 2 CaCl2, 10 glucose, and 26 NaHCO3, and it was gassed continuously with a 95% O2, 5% CO2mixture. Coronal slices (300 μm) taken ∼0.48 mm rostral to the bregma according to the brain atlas of Paxinos and Watson (1990) were cut from these tissue blocks. Slices were allowed to recover for ≥1 hr in the same oxygenated solution at 32–34°C before recording. A single slice was transferred to a recording chamber (3–4 ml volume) and submerged in a continuously flowing extracellular solution (21–24°C, 2–3 ml/min) gassed with a 95% O2, 5% CO2 mixture. For recording, this modified Krebs' solution also contained 25 μmpicrotoxin, a GABA receptor antagonist, to isolate excitatory potentials. The slices were allowed to further recover for 30–60 min at room temperature in the picrotoxin-containing solution before the recording procedure was started. Standard field potential recording techniques were used. Electrodes (2–8 MΩ) pulled out from borosilicate capillaries were filled with the same solution without picrotoxin. Extracellular potentials were amplified using a World Precision Instrument DAM 80 amplifier, displayed on an oscilloscope, and digitized using the Bio-logic LM-200 interface. Slices were stimulated by a 50 μsec current pulse delivered through bipolar tungsten electrodes and controlled by a Master 8 pulse generator (AMPI). The stimulation electrode was located in the dorsolateral striatum close to the recording electrode (0.2–1.5 mm). For all experiments, data were filtered at 1 kHz, digitized at 6–7 kHz, and collected using the Bio-logic Acquis1 acquisition program, which provided an on-line analysis of the amplitude of the rising phase of the field EPSP (fEPSP). Stimuli were given at 0.1 Hz, and points on the illustrated figures represent the means of data collected from all traces in bins of 1 min. In electophysiological protocols, the input stimulation was calibrated to obtain half-maximal fEPSP. Numerical data were expressed as mean ± SEM. Depending on experimental protocols, the extracellular solution was modified by addition of ADAC (diluted at the same concentration as for in vivo experiments, i.e., 50 μg/ml, ∼86 μm).
Cell culture, treatments, and viability assay
Primary cultures of striatal neurons were obtained from 17- to 18-d-old Wistar rat embryos and prepared as described (Schiffmann et al., 1998). Cells were cultured in Neurobasal medium supplemented with B27 containing 200 mm glutamine. Cells were treated with 3NP at 7 DIV. ADAC treatment (10−2m stock solution in DMSO, diluted in culture medium) was performed 60 min before the addition of 3NP. Cell viability was assessed by MTT assay, 3 d after 3NP treatment as follows. Cells were cultured for 4 hr in the presence of MTT (5 mg/ml; Sigma). The reaction was stopped by adjunction of DMSO. The optical density was measured at a wavelength of 540 nm on a Titertek Multiskan MCC/340 (ICN Biomedicals, Costa Mesa, CA).
Determination of protein kinase A activity
In vivo treatments. Lewis rats (12 weeks old) were treated either by vehicle (n = 3) or by one daily injection of ADAC for 3 d (n = 3). As a control for desensitization, one rat was chronically treated with ADAC for 13 d. At the end of treatments, animals were killed by decapitation. Their striata were dissected out and homogenized with a glass tissue blender in 10 vol of ice-cold extraction buffer (25 mm Tris-HCl, pH 7.4, 1 mmEDTA, 1 mm DTT) containing a protease inhibitor mixture (Complete, Roche Molecular Biochemicals, Mannheim, Germany). Forskolin (10−4m) was incubated with striatal homogenates for 10 min at 30°C. ADAC (10−5m) treatment of the homogenates was started 10 min before incubation with forskolin.
In vitro treatments. Striatal neurons were treated with forskolin (10−4m) for 1 hr at 37°C in a 5% CO2atmosphere. ADAC (10−6m) treatment was started 30 min before this incubation. Cells were then washed with warm PBS and homogenized in lysis buffer (M-PER, Pierce, Rockford, IL; 6 × 106 cells in 400 μl) containing a protease inhibitor mixture (Complete, Roche Molecular Biochemicals).
Protein kinase A assay. Soluble extracts from rat striata or striatal neurons were separated by centrifugation (15,000 ×g, 10 min, 4°C), and the protein concentration was determined using MicroBCA Protein Assay (Pierce). PepTag assay for nonradioactive detection of cAMP-dependent protein kinase was performed consistently following the manufacturer's instructions (Promega, Madison, WI). Briefly, all reaction components were combined on ice, and protein kinase A (PKA) activity was assayed at 30°C for 30 min, in a final volume of 25 μl of the following mixture: 5 μl of reaction buffer, 5 μl (0.4 μg/μl) f-kemptide, 1 μl of anti-protease solution, and sample striatal extract (2 μg protein) or sample neuron lysate (10 μg protein). For each condition, reactions were performed in triplicate. Reactions were stopped after 30 min by placing the tubes in a water bath at 95°C for 10 min and then either immediately loaded for electrophoresis or frozen at −20°C until use. Samples were loaded on a 0.8% agarose gel prepared in 50 mm Tris buffer, pH 8.0. The electrophoresis was run at 100 V for 30 min. Resulting separation was observed under UV light and acquired using a CDD camera. Optical density of the bands was measured using NIH image software, and the results were expressed as the ratio of phosphorylated over nonphosphorylated peptide.
Electrophoresis and immunoblotting
Protein electrophoresis was performed as described previously (Galas et al., 2000). Briefly, cells were homogenized in lysis buffer (M-PER, Pierce) containing a protease inhibitor mixture (Complete, Roche Molecular Biochemicals) (6 × 106 cells in 500 μl). Samples were stored at −20°C until they were analyzed. Protein concentration was determined using MicroBCA Protein Assay (Pierce). Equal amounts of proteins (10 μg) were denaturated in 2× Laemmli buffer at 100°C for 5 min and then separated on 10% SDS-polyacrylamide gels. Proteins were transferred to nitrocellulose (Bio-Rad, Hercules, CA) at 250 mA for 90 min at 4°C. The membrane was blocked with 5% BSA, 0.1% Tween 20 in PBS buffer containing 2% goat serum and then incubated with the primary antibody (anti-A1 receptor, A1R11-A, 1:1000; Alpha Diagnostic, San Antonio, TX) overnight at 4°C. After washing in PBS buffer containing 0.1% Tween 20, the membrane was incubated with the HRP-labeled secondary antibody (goat anti-rabbit IgG; DuPont NEN, Boston, MA) at a concentration of 0.1 μg/ml for 60 min at room temperature. Immunoreactive bands were visualized by chemiluminescent ECL Plus Western blotting detection reagents (Amersham, Buckinghamshire, UK). A control experiment performed without the primary antibody demonstrated the absence of signal at the molecular weight corresponding to A1 receptor (data not shown).
Analysis and statistics
Results were expressed as means ± SEM. Depending on the parameter studied, comparisons among groups were made using either Mann–Whitney or Kruskal–Wallis/Dunn nonparametric tests, the unpairedt test, the Fisher test with Yates correction, or one-way ANOVA followed by a Newman–Keuls post hoc test. Electrophysiological data were analyzed by a paired t test (GraphPad Software).
RESULTS
Effects of ADAC on 3NP-induced motor deficits
Chronic treatment
The first 2 d after minipump implantation, animals did not present any behavioral changes (Fig.3A). Disabilities began the third day, and the evolution of motor symptomatology was similar in the vehicle/3NP and the ADAC−8/5/3NP groups (Fig.3A). At day 3, three of seven and four of seven rats were clinically affected in the vehicle/3NP and the ADAC−8/5/3NP groups, respectively, with animals presenting slight intermittent dystonia of one hindlimb and gait abnormalities. The following day (day 4), motor impairments worsened. In both groups, all animals were clinically affected and shared gait abnormalities. Rats presented either pronounced dystonia of one hindlimb or intermittent dystonia of both hindlimbs. After 5 d of subcutaneous infusion of 3NP, all of the rats were alive and homogeneously clinically affected with strong impairment of locomotor activity (Fig.3A, Table 1). Permanent dystonia of both hindlimbs was observed in six of seven or five of seven rats in the vehicle/3NP or the ADAC−8/5/3NP groups, respectively. In both groups, inability to remain on the platform for a short time was observed in six of seven rats; among them, two were recumbent. Neurological symptoms were accompanied with pronounced weight loss, reaching 17–18% at day 5 (Table 1). Therefore, motor disabilities as well as weight loss were similar in the 3NP group of rats chronically treated or not with ADAC (Fig. 3A, Table 1). The neurological score of the sham animals as well as the rats chronically treated with ADAC alone was always of zero.
Fig. 3.
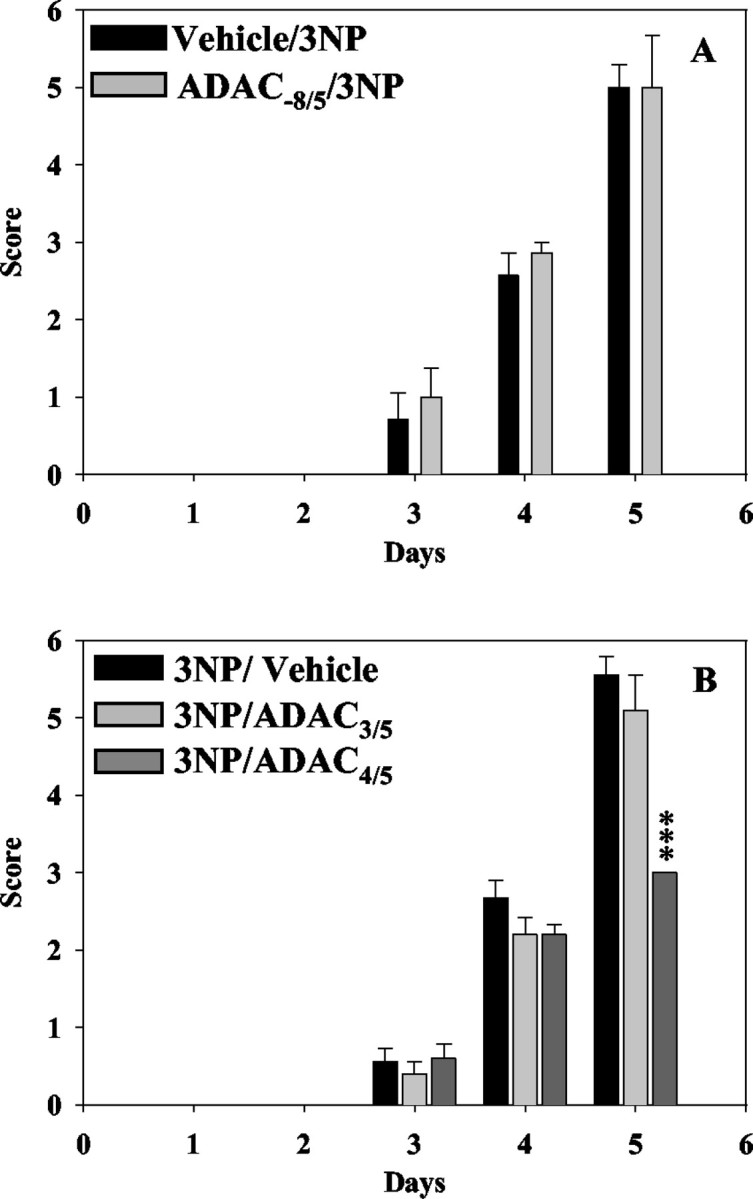
Evolution of neurological score in rats treated with 3NP alone or with ADAC and 3NP. A, Behavioral changes for both 3NP and ADAC−8/5/3NP rats in the chronic experiment. B, Behavioral changes of rats treated with either 3NP alone or 3NP with acute administration of ADAC (3NP/ADAC3/5 and 3NP/ADAC4/5 groups). ***p < 0.001; Kruskal–Wallis/Dunn test versus rats treated with 3NP alone.
Table 1.
Behavioral and weight changes observed after 5 d in rats treated by 3NP alone or in animals cotreated with chronic or acute ADAC administration
| Score at day 5 | Permanent dystonia of two hindlimbs | Recumbency | % of weight loss | |
|---|---|---|---|---|
| Vehicle/3NP | 5.0 ± 0.3 | 6 /7 | 2 /7 | 17.9 ± 1.1 |
| ADAC−8/5/3NP | 5.0 ± 0.7 | 5 /7 | 2 /7 | 16.9 ± 2.1 |
| 3NP/Vehicle | 5.5 ± 0.2 | 9 /9 | 7 /9 | 18.8 ± 0.7 |
| 3NP/ADAC3/5 | 5.1 ± 0.5 | 6 /10 | 6 /10 | 15.8 ± 0.81-160 |
| 3NP/ADAC4/5 | 3.0 ± 01-245 | 0 /101-173 | 0 /101-173 | 13.4 ± 2.1***1-246 |
F1-245: p < 0.001 using non-parametric Kruskall–Wallis/Dunn test versus 3NP-treated rats.
F1-173: p < 0.001 using Fisher–Yates correction test versus 3NP-treated rats.
F1-160: p < 0.01,
F1-165: p< 0.001 using Newman-Keuls ANOVA post hoc test versus 3NP-treated rats.
F1-246: p < 0.05 using Newman-Keuls ANOVA post hoc test versus 3NP/ADAC3/5-treated rats.
Acute treatments
Behavioral changes observed in the 3NP/vehicle-treated rats were essentially similar to those described above (Fig. 3B, Table1). At day 3, five of nine rats were clinically affected, with animals presenting gait abnormalities. At day 4, all animals were clinically affected and shared gait abnormalities. Seven of nine rats presented intermittent dystonia of both hindlimbs. At day 5, all rats (nine of nine) were alive (Fig. 3B, Table 1). Permanent dystonia of both hindlimbs and inability to remain on the platform were observed in all rats, and seven of nine were recumbent (Table 1). 3NP rats acutely treated by ADAC 4 and 5 d after minipump implantation were relatively spared (3NP/ADAC4/5 group) (Fig.3B, Table 1). Indeed, although evolution of motor symptomatology was similar to that of 3NP/vehicle group from days 1–4, at day 5 all of these rats (10 of 10) had a neurological score of 3 (Fig. 3B, Table 1), and none of them (0 of 10) presented either permanent dystonia of both hindlimbs or recumbency (Table 1). They were all (10 of 10) able to remain on the platform for >10 sec, and their motor impairment consisted only of intermittent dystonia of both hindlimbs. Additionally, the mean weight loss of 3NP/ADAC4/5 group was lower than for the 3NP/vehicle animals (Table 1). Conversely, although only 6 of 10 animals presented permanent dystonia of both hindlimbs and the weight loss was slightly lower than for 3NP/vehicle animals, behavioral alterations observed at day 5 in the 3NP/ADAC3/5group were not significantly different from the 3NP/vehicle group (Fig.3B, Table 1). As in the first experiment, ADAC alone did not alter body weight or motor behavior (data not shown).
Effects of ADAC on 3NP-induced striatal lesion
Chronic treatment
In the 3NP-treated rats of the first experiment, macroscopic analysis of hematoxylin-stained sections confirmed the presence of a large striatal pale area topologically restricted to the lateral striatum (surface of 8.2 ± 0.6 mm2at the stereotaxic plane approximatively +0.48 mm) (Fig.4A,C). The surface of this striatal lesion (7.95 ± 0.45 mm2) was not modified in rats chronically treated with ADAC (ADAC−8/5/3NP group) (Fig.4A,C).
Fig. 4.

Histological evaluation of the neuroprotection induced by ADAC in 3NP-treated rats. A, Typical hematoxylin staining of section for a rat treated by 3NP alone or after a chronic treatment with ADAC (ADAC−8/5/3NP group).B, Typical hematoxylin staining of a section from a rat treated with 3NP alone or after acute treatments with ADAC (3NP/ADAC3/5 and 3NP/ADAC4/5 groups).C, D, Quantification of the striatal lesion size at the +0.48 mm stereotaxic plane represented inA and B, respectively. **p < 0.01 using Newman–Keuls ANOVA post hoc test versus 3NP-treated rats.▴▴▴ p < 0.001 using Newman–Keuls ANOVA post hoc test versus 3NP/ADAC3/5-treated rats.
Acute treatments
In the 3NP-treated rats of the second experiment, the surface of the striatal lesion was 11.3 ± 0.6 mm2 at the stereotaxic plane approximatively +0.48 mm (Fig.4B,D). The surface of the lesion core was significantly reduced by 41.2 ± 7.1% in the 3NP/ADAC4/5 group (Fig.4B,D). Conversely, it was not modified in the 3NP/ADAC3/5 group (Fig.4B,D). Neither chronic nor acute injections of ADAC alone induced striatal histological alterations (data not shown). It is noteworthy that in the protected group, the lesion core was less extended to the ventral part of the striatum when compared with 3NP animals (Fig. 4B). Similar results were found when we measured the volume of the lesion core in the anterior part of the striatum (bregma approximately +1.4 mm to bregma approximately −0.2 mm using Cavalieri's principle on sections separated by 320 μm intervals). Indeed, we found a value of 15.2 ± 0.5 mm3 for the 3NP-treated rats. The volume of the lesion core was significantly reduced by 30 ± 6% in the 3NP/ADAC4/5 group (10.6 ± 1 mm3; p < 0.05; Newman–Keuls post hoc test vs 3NP group). Conversely, it was not modified in the 3NP/ADAC3/5 group (17.3 ± 1.7 mm3; NS; Newman–Keulspost hoc test vs 3NP group).
Effects of ADAC on 3NP-induced succinate dehydrogenase inhibition
Because SDH is a well known irreversible target for 3NP (Brouillet et al., 1998, 1999), we aimed to determine whether ADAC treatment could interfere with the 3NP-induced complex II inhibition. Semiquantitative histochemical measurements showed that 3NP greatly decreased SDH activity in both the striatum (ROI 1) (Fig. 2) and the outer cortical layers (ROI 5) (Fig. 2), with the higher alteration in the former (Fig.5A,B). In the 3NP/ADAC4/5 group, striatal enzymatic activity was slightly but significantly increased when compared with 3NP/vehicle animals, whereas 3NP-induced SDH inhibition was similar in the cortex (Fig. 5B). Striatal and cortical SDH activity impairment was not modified by a chronic treatment with ADAC or within the 3NP/ADAC3/5 group (Fig.5A,B). It should also be noted that ADAC alone did not modify basal SDH activity in any of the conditions tested (data not shown).
Fig. 5.

Determination of succinate dehydrogenase activity in the striatum and the cortex of 3NP-treated rats and animals receiving chronic (A) or acute (B) treatment with ADAC. ***p< 0.001, **p < 0.01 using Newman–Keuls ANOVApost hoc test versus sham rats.▴▴▴ p < 0.001 using Newman–Keuls ANOVA post hoc test versus 3NP/Vehicle or 3NP/ADAC3/5-treated rats.
Striatal and cortical effects of ADAC on the density of A1 receptor binding sites
To determine whether the lack of striatal protection observed after chronic administration or in the 3NP/ADAC3/5 group was caused by downregulation of the adenosine A1 receptors, binding experiments were performed in rats treated with the agonist alone (ADAC−8/5, ADAC3/5, and ADAC4/5 groups). As shown in Figure6, chronic ADAC treatment dramatically decreased the density of [3H]-DPCPX binding sites, reaching 60.9 ± 10.6 and 73.9 ± 4.9% in the striatum (ROI 1) and the overlying cortex (ROI 6), respectively (Fig.6A,B). It is noteworthy that the [3H]-DPCPX binding signal observed in the ADAC−8/5 group is higher than the nonspecific binding level, itself indistinguishable from the film background (Fig. 6A). In opposition to chronic treatment, in acute conditions ADAC did not significantly modify the density of [3H]-DPCPX binding sites (Fig. 6C,D).
Fig. 6.

Striatal and cortical density of A1receptor binding sites in vehicle- or ADAC-treated rats.A, [3H]-DPCPX binding in rats chronically treated with ADAC. C, [3H]-DPCPX binding in rats acutely treated with ADAC. B, D, Quantification of autoradiograms represented in A and C. ***p < 0.001 using unpaired t test versus vehicle rats.
To determine whether the lack of protective effects of ADAC in the 3NP/ADAC3–5 group was not caused by a functional desensitization of the A1 receptor, we have tested the efficacy of A1 receptor activation to inhibit the forskolin-induced activation of PKA in brain homogenates from rats treated with vehicle, with ADAC for 3 d, or, as a control of desensitization, with ADAC for 13 d. We found that in vehicle rats, forskolin induced PKA activation to 155.2 ± 9.4% of the control value (p < 0.05 vs control; Newman–Keuls post hoc test). This increase was reduced to 100.4 ± 18.6% of the control value in the presence of ADAC (p < 0.05 vs forskolin and NS vs control; Newman–Keuls post hoc test), although the agonist alone did not share a significant effect by itself in the absence of forskolin (104.9 ± 5% of the control; NS; Newman–Keuls post hoc test). In accordance with the binding experiment, after a chronic treatment with the agonist, we found that ADAC was unable to reduce the forkolin-induced PKA activation (data not shown). Similar results were found in rats treated with ADAC for 3 d. Indeed, in these rats, although forskolin induced PKA activity by 179 ± 38% (p < 0.01 vs control; Newman–Keuls post hoc test), in the presence of the agonist, PKA activity did not return to the basal level because it was still activated by 199 ± 23% as compared with the control condition (NS vs forskolin condition; Newman–Keuls post hoc test). Consequently, it appears that a 3 d treatment with ADAC is sufficient to induce a functional desensitization of the A1 receptor.
Histological and functional effects of acute ADAC4/5treatment on the striatal alterations induced by 3NP
NeuN immunoreactivity and TUNEL staining
Using NeuN immunohistochemistry, we observed neuronal loss after 3NP treatment within both the dorsolateral and ventrolateral striatum (ROIs 2 and 4) (Fig.7C,D). In accordance with the limited extension of the lesion core toward the ventrolateral striatum (Fig. 4B), the neurons of 3NP/ADAC4/5-treated rats located within this area appeared healthy (Fig. 7F). Using TUNEL staining, we aimed to determine whether the remaining cells located within the lesion core (ROI 2) (Fig. 2) underwent ongoing DNA damage. In 3NP animals, the density of TUNEL-positive cells was measured as 640 ± 46 cells per millimeters squared. This density was significantly reduced by 29.7 ± 7.7% in the lesion core of the 3NP/ADAC4/5 group (p < 0.01 vs 3NP group; unpaired t test) (Fig.8). ADAC is thus able to reduce not only the lesion size but also the ongoing striatal degeneration.
Fig. 7.
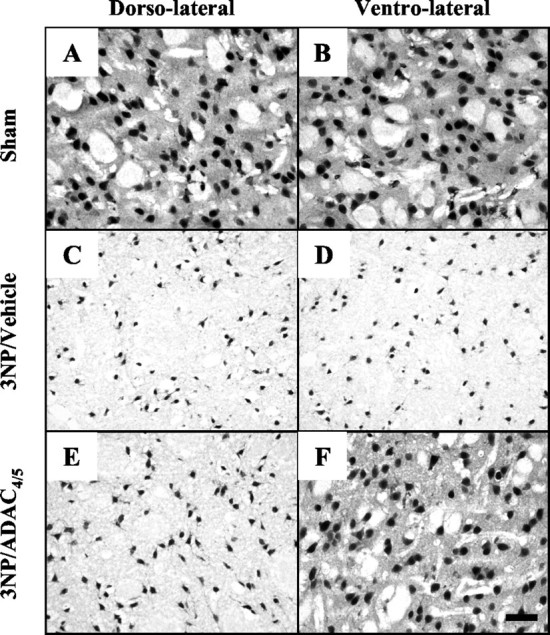
Micrographs showing NeuN immunoreactivity in the dorsolateral (A, C, E) and ventrolateral (B, D,F) striatum of sham rats (A–B) in animals treated with 3NP (C, D) or in animals from the 3NP/ADAC4/5 group (E,F). Scale bar, 25 μm.
Fig. 8.
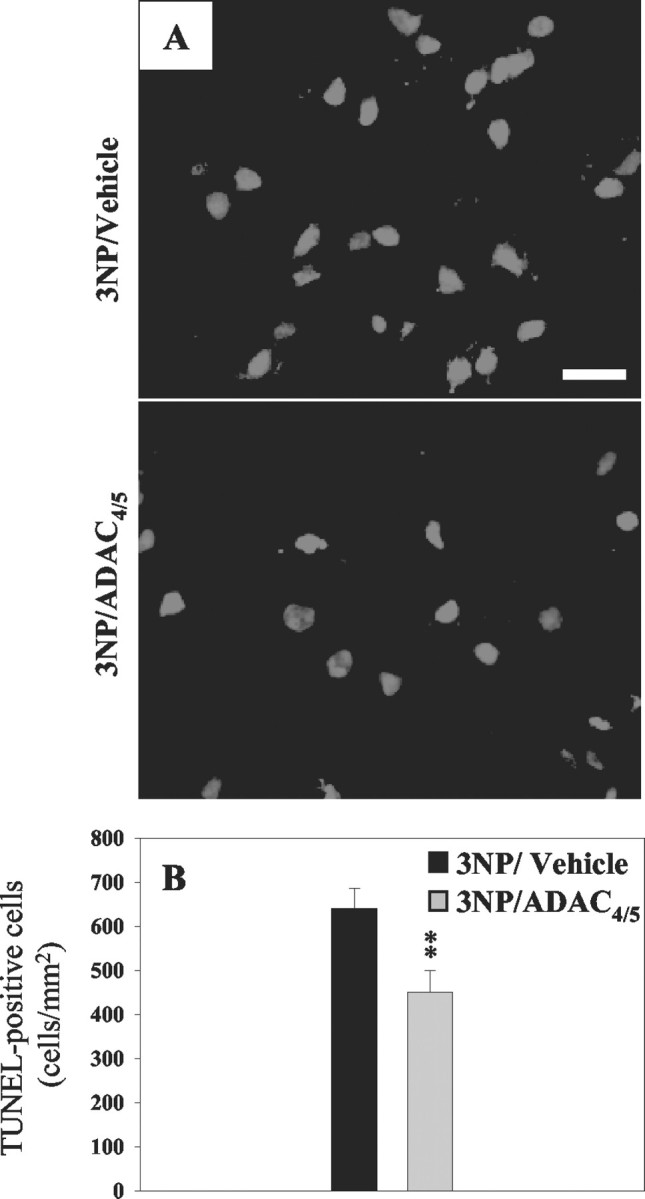
A, Micrographs showing TUNEL labeling in the dorsolateral striatum of animals treated with 3NP or in rats from the 3NP/ADAC4/5 group. B, Quantification of the density of TUNEL-positive cells represented inA. **p < 0.001 unpairedt test versus 3NP rats. Scale bar, 25 μm.
Enkephalin mRNA expression
Using in situ hybridization, we also quantified the expression of enkephalin mRNA, which is expressed by a subpopulation of the affected spiny neurons. As shown in Figure9, ADAC4/5 reduced the loss of enkephalin mRNA expression observed after the 3NP treatment (Fig. 9A,B). Indeed, 3NP alone decreased the enkephalin mRNA level by 65 ± 5.4%, whereas this reduction was lowered to 42 ± 5.7% in the 3NP/ADAC4/5 group (Fig. 9B). It is noteworthy that the striatal surface presenting enkephalin mRNA depletion was strongly reduced in the 3NP/ADAC4/5group (stereotaxic plane approximatively +0.48 mm) as compared with the 3NP rats and that ADAC alone did not modify basal enkephalin mRNA expression (data not shown).
Fig. 9.

A, Striatal enkephalin mRNA expression in sham rats, after a 5 d subcutaneous infusion of 3NP or in 3NP/ADAC4/5 animals. B, Quantification of the mRNA expression represented in A. ***p < 0.001, **p < 0.01, *p < 0.05 using Newman–Keuls ANOVA post hoc test versus sham rats.▴▴ p < 0.01 using Newman–Keuls ANOVA post hoc test versus 3NP/vehicle-treated rats.
Effect of ADAC on corticostriatal synaptic transmission
Because glutamate was suggested to be, at least in part, a component of striatal vulnerability to 3NP (Beal et al., 1993; Guyot et al., 1997b), we aimed to determine whether ADAC, which has never before been evaluated by electrophysiology, would be able to modify glutamatergic corticostriatal transmission. In agreement with previous results obtained with other A1 receptor agonists (Flagmeyer et al., 1997; D'Alcantara et al., 2001), the fEPSP amplitude was markedly decreased after addition of ADAC (33 ± 6% of the control value; n = 5; p < 0.001) (Fig. 10), and this effect was totally reversed by washing out this compound from the external solution.
Fig. 10.

Effect of A1 receptor activation on the corticostriatal fEPSP amplitude in rat brain slices.Inset, Superimposed fEPSP traces obtained before (○) and 30 min after (●) ADAC addition. Calibration: horizontal bar, 5 msec; vertical bar, 0.1 mV.
Effects of ADAC on 3NP-induced striatal cell deathin vitro
We finally aimed to determine whether the protective effect of ADAC could be related to an intrinsic effect involving A1 receptors located on striatal cells. As shown in Figure 11A, striatal neurons from primary cultures express A1receptors, confirming RT-PCR experiments (data no shown). Treatment of striatal cells with 75 μm 3NP led to a significant reduction of neuronal viability (Fig.11B). The 3NP concentration was chosen according to the dose–effect experiments that we performed, and we gave 75 μm as the ED50 in our cellular system (data not shown). ADAC alone did not significantly alter cellular viability (data not show), and this A1 agonist did not modify the 3NP-induced cell death whatever the concentration (Fig. 11B).
Fig. 11.

A, Western blotting analysis of A1 receptor expression (79 kDa) in a primary culture of striatal cells. B, Effect of various concentrations of ADAC against deleterious effects of 3NP (75 μm) on striatal cells in vitro. ADAC was added 60 min before 3NP treatment. Cell viability was determined 3 d thereafter. Each value represents mean ± SEM of four measurements from a representative experiment. Similar results were obtained in three separate experiments. ***p < 0.001 versus untreated control, Newman–Keuls post hoc test.
Because it was reported previously that in immature rat brain the lack of a neuroprotective effect of ADAC may be caused by a functional uncoupling of the A1 receptor (Aden et al., 2001), we measured the functional state of the latter in our culture system. We found that A1 receptors were functionally and negatively coupled to adenylyl cyclase because ADAC, by itself, reduced basal PKA activation by 31.7 ± 4.2% (p < 0.001 vs untreated control; Newman–Keulspost hoc test). In the presence of forskolin, PKA activity was increased to 228.4 ± 10.8% of the untreated control (p < 0.05 vs untreated control; Newman–Keulspost hoc test), and this activation was reduced by 52.71 ± 9.8% in the presence of ADAC (p< 0.001 vs forskolin-activated cells; Newman–Keuls post hoc test).
DISCUSSION
At present, there are no pharmacological treatments for Huntington's disease despite first attempts with compounds such as riluzole or coenzyme Q10 (Beal et al., 1994; Guyot et al., 1997b; Palfi et al., 1997; The Huntington's Disease Study Group, 2001) and more recent promising studies evaluating dichloroacetate and creatine (Andreassen et al., 2001a,b; Tarnopolsky and Beal, 2001). The most striking experimental and preclinical advances in the treatment of this neurodegenerative disorder have been obtained recently using cellular and gene therapy strategies (Hantraye et al., 1992; Emerich et al., 1997; Palfi et al., 1998; Bachoud-Levi et al., 2000; Mittoux et al., 2000, 2002; De Almeida et al., 2001). However, considering the technical complexity of such approaches, it remains important to test new drugs susceptible to display neuroprotective properties in this neurodegenerative disease.
In the present study, we report that treatment with a low dose of the A1 receptor agonist ADAC protects Lewis rats from 3NP-induced striatal degeneration and motor disabilities. In particular, our results show that acute administration of ADAC on the day preceding the lesion (day 4) (Ouary et al., 2000; Blum et al., 2002) not only significantly decreases the size of the striatal lesion but also delays the ongoing cellular degeneration that occurs in the remaining lesion core. Also of great interest, ADAC efficiently slowed the worsening of motor disturbances, notably dystonia. To our knowledge, this is the first report suggesting the potential use of an A1 receptor agonist in a model of HD, opening new possibilities for adenosinergic compounds.
The beneficial potential of adenosine, and particularly of A1 receptor agonists, in neurodegenerative disorders has already been suggested, especially for ischemia and epilepsy (Connick and Stone, 1989; Rudolphi et al., 1992; De Mendonca et al., 2000; Dunwiddie and Masino, 2001; Huber et al., 2001). Nevertheless, their clinical implementation was mitigated by cardiovascular and hypotensive side effects (Williams, 1993; White et al., 1996). Recently, new classes of A1 agonists, devoid of such disturbing issues, have been disclosed (Knutsen et al., 1995; Bischofberger et al., 1997). Among them, ADAC has been studied extensively as a potential candidate for the treatment of ischemia (Von Lubitz et al., 1996a,b, 1999).
Our electophysiological data clearly demonstrate that ADAC powerfully inhibits striatal fEPSPs generated by the stimulation of cortical afferent fibers. This is in agreement with previous findings showing that presynaptic activation of A1 receptors interferes with glutamate neurotransmission, thereby reducing AMPA- and NMDA-mediated field potentials (Malenka and Kocsis, 1988; De Mendonca et al., 1995; Flagmeyer et al., 1997; D'Alcantara et al., 2001). It can thus be suggested that ADAC likely prevents striatal excitotoxicity generated by 3NP (Brouillet et al., 1999), without generating any behavioral side effects. This hypothesis is supported by several studies demonstrating the protective potency of NMDA antagonists against striatal neuropathology induced by quinolinic acid, malonate, or 3NP (Beal et al., 1993; Schulz et al., 1995, 1996; Jenkins et al., 1996). At a cellular level, ADAC could then indirectly counteract the NMDA-mediated neuronal calcium overload induced by 3NP (Brouillet et al., 1999), thereby depressing the deleterious activation of detrimental enzymes such as nitric oxide synthase or calpain (Nishino et al., 1996; Bizat et al., 2001). Activation of neuronal postsynaptic A1 receptors could also directly (Mogul et al., 1993) and indirectly (Trussell and Jackson, 1985) depress calcium influx. However, the lack of protection provided by ADAC against 3NP-induced striatal cell death in vitro, despite the presence of functional A1 receptors, argues against this latter possibility. It has to be mentioned that electrophysiological and cell culture experiments have been performed on Wistar instead of Lewis rats. Although very unlikely, an effect of the strain difference therefore may not be ruled out.
Striatal 3NP toxicity also involves dopaminergic neurotransmission. Indeed, this neurotoxin increases the striatal dopamine overflow (Nishino et al., 1997; Johnson et al., 2000) that is responsible, at least in part, for the rise in intracellular calcium (Nishino et al., 1997). Enhancing striatal dopamine release using methamphetamine or sulpiride potentiates 3NP neurotoxicity (Reynolds et al., 1998; Nishino et al., 2000), whereas nigral lesions prevent it (Reynolds et al., 1998). Given that stimulation of presynaptic A1receptors results in an inhibition of striatal dopamine release (Wood et al., 1989; Zetterstrom and Fillenz, 1990), ADAC-mediated neuroprotection could also be attributed to A1receptor regulation at the nigrostriatal terminals.
A possible interaction between ADAC and 3NP limiting the access of the toxin to SDH was ruled out because our results indicate that both chronic and acute treatments with ADAC do not alleviate the 3NP-induced SDH inhibition in either the striatum or the cortex. The slight increase in SDH activity observed within the whole striatum in the 3NP/ADAC4/5 group is likely attributable to the reduction of the lesion size rather than to a direct interaction between ADAC and 3NP, because we showed previously that the inhibition of SDH activity was greater within the lesion core after a 5 d subcutaneous infusion of 3NP (Blum et al., 2001, 2002).
We did not observe any neuroprotective effects of ADAC in the chronic ADAC−8/5/3NP or acute 3NP/ADAC3/5 groups. It appears that a sustained administration of the A1 receptor agonist (chronic condition) but also a 3 d treatment (ADAC3–5) leads to a functional desensitization of the A1 receptors. This phenomenon has already been described after chronic exposure to various A1 receptor agonists such as R-PIA and N6-cyclopentyladenosine (Abbracchio et al., 1992; Lee et al., 1993). Additionally, such stimulations can also result in a desensitization characterized by a loss of Giα-protein expression (Longabaugh et al., 1989). Surprisingly, Von Lubitz et al. (1999)reported a protective effect of ADAC against ischemia in gerbils after chronic treatment for 60 d. This suggests that under their conditions, ADAC did not downregulate or desensitize the A1 receptors. The reasons for this discrepancy with our data are elusive. It remains possible, however, that the pharmacokinetics of ADAC or the mechanisms of A1receptor desensitization may be species dependent. Nevertheless, despite profound A1 receptor downregulation, the chronic treatment with ADAC did not increase the neurotoxic effects of 3NP. Therefore, ADAC does not produce any regimen-dependent inversion of the beneficial effect as reported previously for other A1 agonists (Von Lubitz et al., 1994a,b; Jacobson et al., 1996).
The fact that the first injection of ADAC in the 3NP/ADAC3–5 group did not reduce the neurological score of the animals as compared with the 3NP/vehicle group suggests also that the development of motor symptoms 4 d after minipump implantation is probably not caused by a modulation of glutamate or dopamine release within the striatum. It could likely reflect intrinsic, striatal, metabolic alterations as suggested by the early decrease in striatal N-acetylaspartate level and zif-268 mRNA expression induced by 3NP (Dautry et al., 2000; Blum et al., 2002).
In conclusion, our results provide the first demonstration that A1 receptor activation is able to delay the development of striatal lesions as well as the worsening of motor disabilities in a rat model of Huntington's disease. This is of particular interest given the lack of pharmacological therapy for this neurological affection. Although the absence of a beneficial effect of ADAC after more than two injections precludes a direct application at the therapeutic level, the present strategy clearly deserve further evaluation to assess whether (1) post-lesional administration of ADAC or spaced short treatments at different times after the onset of degeneration would also be beneficial and (2) ADAC or related compounds may have functional effects in transgenic mice models of HD.
D.G. and M.-C.G. contributed equally to this work.
This work was supported by the Queen Elisabeth Medical Foundation (FMRE-Neurobiology 99-01 and 02-04), Fund for Medical Scientific Research (FRSM-Belgium 3.4551.98/3.4507.02), and the Fondation Alice et David Van Buuren. D.B. is supported by the Fondation Simone et Cino Del Duca and the Fonds National pour la Recherche Scientifique (FNRS) (Belgium). D.G. is a post-doctoral researcher of the FNRS (Belgium). M.C.G. is a researcher of the Centre National de la Recherche Scientifique (France) and is supported by the FNRS (Belgium). K.B. is supported by a Televie grant. We thank Dr. Emmanuel Brouillet for his help and continuous support. We are grateful to Drs. Raphaël Hourez, Nathalie Lambeng, Serge Pinto, and Patrick Van Bogaert for their helpful comments. We thank Fiona Hemming for English review and Laetitia Cuvelier and Huy Nguyen-Tran for their very valuable technical support.
Correspondence should be addressed to David Blum, Laboratoire de Neurophysiologie, Université Libre de Bruxelles-Erasme, CP601, 808 route de Lennik, 1070 Brussels, Belgium. E-mail:David.Blum@ulb.ac.be.
REFERENCES
- 1.Abbracchio MP, Fogliatto G, Paoletti AM, Rovati GE, Cattabeni F. Prolonged in vitro exposure of rat brain slices to adenosine analogues: selective desensitization of adenosine A1 but not A2 receptors. Eur J Pharmacol. 1992;227:317–324. doi: 10.1016/0922-4106(92)90010-s. [DOI] [PubMed] [Google Scholar]
- 2.Aden U, Leverin AL, Hagberg H, Fredholm BB. Adenosine A(1) receptor agonism in the immature rat brain and heart. Eur J Pharmacol. 2001;426:185–192. doi: 10.1016/s0014-2999(01)01220-1. [DOI] [PubMed] [Google Scholar]
- 3.Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF. Creatine increases survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis. 2001a;8:479–491. doi: 10.1006/nbdi.2001.0406. [DOI] [PubMed] [Google Scholar]
- 4.Andreassen OA, Ferrante RJ, Huang HM, Dedeoglu A, Park L, Ferrante KL, Kwon J, Borchelt DR, Ross CA, Gibson GE, Beal MF. Dichloroacetate exerts therapeutic effects in transgenic mouse models of Huntington's disease. Ann Neurol. 2001b;50:112–117. doi: 10.1002/ana.1085. [DOI] [PubMed] [Google Scholar]
- 5.Bachoud-Levi AC, Remy P, Nguyen JP, Brugieres P, Lefaucheur JP, Bourdet C, Baudic S, Gaura V, Maison P, Haddad B, Boisse MF, Grandmougin T, Jeny R, Bartolomeo P, Dalla BG, Degos JD, Lisovoski F, Ergis AM, Pailhous E, Cesaro P, Hantraye P, Peschanski M. Motor and cognitive improvements in patients with Huntington's disease after neural transplantation. Lancet. 2000;356:1975–1979. doi: 10.1016/s0140-6736(00)03310-9. [DOI] [PubMed] [Google Scholar]
- 6.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beal MF, Henshaw DR, Jenkins BG, Rosen BR, Schulz JB. Coenzyme Q10 and nicotinamide block striatal lesions produced by the mitochondrial toxin malonate. Ann Neurol. 1994;36:882–888. doi: 10.1002/ana.410360613. [DOI] [PubMed] [Google Scholar]
- 8.Bischofberger N, Jacobson KA, Von Lubitz DK. Adenosine A1 receptor agonists as clinically viable agents for treatment of ischemic brain disorders. Ann NY Acad Sci. 1997;825:23–29. doi: 10.1111/j.1749-6632.1997.tb48411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bizat C, Créminon JM, Hermel F, Condé F, Boyer S, Ouary S, Krajewski JC, Reed P, Hantraye P, Brouillet E. In vivo study of the apoptotic machinery in the preferential neurodegeneration of the striatum due to chronic energy compromise. Soc Neurosci Abstr. 2001;31:432.6. [Google Scholar]
- 10.Blum D, Gall D, Cuvelier L, Schiffmann SN. Topological analysis of striatal lesions induced by 3-nitropropionic acid in the Lewis rat. NeuroReport. 2001;12:1769–1772. doi: 10.1097/00001756-200106130-00050. [DOI] [PubMed] [Google Scholar]
- 11.Blum D, Galas MC, Gall D, Cuvelier L, Schiffmann SN. Striatal and cortical neurochemical changes induced by a chronic metabolic compromise in the 3-nitropropionic model of Huntington's disease. Neurobiol Dis. 2002;10:410–426. doi: 10.1006/nbdi.2002.0512. [DOI] [PubMed] [Google Scholar]
- 12.Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brouillet E, Guyot MC, Mittoux V, Altairac S, Conde F, Palfi S, Hantraye P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem. 1998;70:794–805. doi: 10.1046/j.1471-4159.1998.70020794.x. [DOI] [PubMed] [Google Scholar]
- 14.Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington's disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427–468. doi: 10.1016/s0301-0082(99)00005-2. [DOI] [PubMed] [Google Scholar]
- 15.Connick JH, Stone TW. Quinolinic acid neurotoxicity: protection by intracerebral phenylisopropyladenosine (PIA) and potentiation by hypotension. Neurosci Lett. 1989;101:191–196. doi: 10.1016/0304-3940(89)90529-6. [DOI] [PubMed] [Google Scholar]
- 16.D'Alcantara P, Ledent C, Swillens S, Schiffmann SN. Inactivation of adenosine A2A receptor impairs long term potentiation in the accumbens nucleus without altering basal synaptic transmission. Neuroscience. 2001;107:455–464. doi: 10.1016/s0306-4522(01)00372-4. [DOI] [PubMed] [Google Scholar]
- 17.Dassesse D, Vanderwinden JM, Goldberg I, Vanderhaeghen JJ, Schiffmann SN. Caffeine-mediated induction of c-fos, zif-268 and arc expression through A1 receptors in the striatum: different interactions with the dopaminergic system. Eur J Neurosci. 1999;11:3101–3114. doi: 10.1046/j.1460-9568.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- 18.Dassesse D, Massie A, Ferrari R, Ledent C, Parmentier M, Arckens L, Zoli M, Schiffmann SN. Functional striatal hypodopaminergic activity in mice lacking adenosine A(2A) receptors. J Neurochem. 2001;78:183–198. doi: 10.1046/j.1471-4159.2001.00389.x. [DOI] [PubMed] [Google Scholar]
- 19.Dautry C, Conde F, Brouillet E, Mittoux V, Beal MF, Bloch G, Hantraye P. Serial 1H-NMR spectroscopy study of metabolic impairment in primates chronically treated with the succinate dehydrogenase inhibitor 3-nitropropionic acid. Neurobiol Dis. 1999;6:259–268. doi: 10.1006/nbdi.1999.0244. [DOI] [PubMed] [Google Scholar]
- 20.Dautry C, Vaufrey F, Brouillet E, Bizat N, Henry PG, Conde F, Bloch G, Hantraye P. Early N-acetylaspartate depletion is a marker of neuronal dysfunction in rats and primates chronically treated with the mitochondrial toxin 3-nitropropionic acid. J Cereb Blood Flow Metab. 2000;20:789–799. doi: 10.1097/00004647-200005000-00005. [DOI] [PubMed] [Google Scholar]
- 21.De Almeida LP, Zala D, Aebischer P, Deglon N. Neuroprotective effect of a CNTF-expressing lentiviral vector in the quinolinic acid rat model of Huntington's disease. Neurobiol Dis. 2001;8:433–446. doi: 10.1006/nbdi.2001.0388. [DOI] [PubMed] [Google Scholar]
- 22.De Mendonca A, Sebastiao AM, Ribeiro JA. Inhibition of NMDA receptor-mediated currents in isolated rat hippocampal neurones by adenosine A1 receptor activation. NeuroReport. 1995;6:1097–1100. doi: 10.1097/00001756-199505300-00006. [DOI] [PubMed] [Google Scholar]
- 23.De Mendonca A, Sebastiao AM, Ribeiro JA. Adenosine: does it have a neuroprotective role after all? Brain Res Brain Res Rev. 2000;33:258–274. doi: 10.1016/s0165-0173(00)00033-3. [DOI] [PubMed] [Google Scholar]
- 24.De Sarro G, Donato DP, Falconi U, Ferreri G, De Sarro A. Repeated treatment with adenosine A1 receptor agonist and antagonist modifies the anticonvulsant properties of CPPene. Eur J Pharmacol. 1996;317:239–245. doi: 10.1016/s0014-2999(96)00746-7. [DOI] [PubMed] [Google Scholar]
- 25.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 26.Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- 27.El Massioui N, Ouary S, Cheruel F, Hantraye P, Brouillet E. Perseverative behavior underlying attentional set shifting deficit in rats chronically treated with the neurotoxin 3-nitropropionic acid. Exp Neurol. 2001;172:172–181. doi: 10.1006/exnr.2001.7766. [DOI] [PubMed] [Google Scholar]
- 28.Emerich DF, Winn SR, Hantraye PM, Peschanski M, Chen EY, Chu Y, McDermott P, Baetge EE, Kordower JH. Protective effect of encapsulated cells producing neurotrophic factor CNTF in a monkey model of Huntington's disease. Nature. 1997;386:395–399. doi: 10.1038/386395a0. [DOI] [PubMed] [Google Scholar]
- 29.Flagmeyer I, Haas HL, Stevens DR. Adenosine A1 receptor-mediated depression of corticostriatal and thalamostriatal glutamatergic synaptic potentials in vitro. Brain Res. 1997;778:178–185. doi: 10.1016/s0006-8993(97)01060-3. [DOI] [PubMed] [Google Scholar]
- 30.Fredholm BB, Dunwiddie TV. How does adenosine inhibit transmitter release? Trends Pharmacol Sci. 1988;9:130–134. doi: 10.1016/0165-6147(88)90194-0. [DOI] [PubMed] [Google Scholar]
- 31.Galas MC, Chasserot-Golaz S, Dirrig-Grosch S, Bader MF. Presence of dynamin-syntaxin complexes associated with secretory granules in adrenal chromaffin cells. J Neurochem. 2000;75:1511–1519. doi: 10.1046/j.1471-4159.2000.0751511.x. [DOI] [PubMed] [Google Scholar]
- 32.Guyot MC, Hantraye P, Dolan R, Palfi S, Maziere M, Brouillet E. Quantifiable bradykinesia, gait abnormalities and Huntington's disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience. 1997a;79:45–56. doi: 10.1016/s0306-4522(96)00602-1. [DOI] [PubMed] [Google Scholar]
- 33.Guyot MC, Palfi S, Stutzmann JM, Maziere M, Hantraye P, Brouillet E. Riluzole protects from motor deficits and striatal degeneration produced by systemic 3-nitropropionic acid intoxication in rats. Neuroscience. 1997b;81:141–149. doi: 10.1016/s0306-4522(97)00192-9. [DOI] [PubMed] [Google Scholar]
- 34.Hantraye P, Riche D, Maziere M, Isacson O. Intrastriatal transplantation of cross-species fetal striatal cells reduces abnormal movements in a primate model of Huntington disease. Proc Natl Acad Sci USA. 1992;89:4187–4191. doi: 10.1073/pnas.89.9.4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huber A, Padrun V, Deglon N, Aebischer P, Mohler H, Boison D. Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc Natl Acad Sci USA. 2001;98:7611–7616. doi: 10.1073/pnas.131102898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobson KA, Von Lubitz DK, Daly JW, Fredholm BB. Adenosine receptor ligands: differences with acute versus chronic treatment. Trends Pharmacol Sci. 1996;17:108–113. doi: 10.1016/0165-6147(96)10002-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenkins BG, Brouillet E, Chen YC, Storey E, Schulz JB, Kirschner P, Beal MF, Rosen BR. Non-invasive neurochemical analysis of focal excitotoxic lesions in models of neurodegenerative illness using spectroscopic imaging. J Cereb Blood Flow Metab. 1996;16:450–461. doi: 10.1097/00004647-199605000-00011. [DOI] [PubMed] [Google Scholar]
- 38.Johnson JR, Robinson BL, Ali SF, Binienda Z. Dopamine toxicity following long term exposure to low doses of 3-nitropropionic acid (3-NPA) in rats. Toxicol Lett. 2000;116:113–118. doi: 10.1016/s0378-4274(00)00214-9. [DOI] [PubMed] [Google Scholar]
- 39.Knutsen LJS, Lau J, Sheardown MJ, Eskesen K, Thomsen C, Weis JU, Judge ME, Klitgaard H. Anticonvulsivant actions of novel and reference adenosine agonists. In: Belardinelli L, Pelleg A, editors. Adenine and adenosine nucleotides: From molecular biology to integrative physiology. Kluwer; Boston: 1995. pp. 479–488. [Google Scholar]
- 40.Lee HT, Thompson CI, Hernandez A, Lewy JL, Belloni FL. Cardiac desensitization to adenosine analogues after prolonged R-PIA infusion in vivo. Am J Physiol. 1993;265:H1916–H1927. doi: 10.1152/ajpheart.1993.265.6.H1916. [DOI] [PubMed] [Google Scholar]
- 41.Longabaugh JP, Didsbury J, Spiegel A, Stiles GL. Modification of the rat adipocyte A1 adenosine receptor-adenylate cyclase system during chronic exposure to an A1 adenosine receptor agonist: alterations in the quantity of GS alpha and Gi alpha are not associated with changes in their mRNAs. Mol Pharmacol. 1989;36:681–688. [PubMed] [Google Scholar]
- 42.Malenka RC, Kocsis JD. Presynaptic actions of carbachol and adenosine on corticostriatal synaptic transmission studied in vitro. J Neurosci. 1988;8:3750–3756. doi: 10.1523/JNEUROSCI.08-10-03750.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mittoux V, Joseph JM, Conde F, Palfi S, Dautry C, Poyot T, Bloch J, Deglon N, Ouary S, Nimchinsky EA, Brouillet E, Hof PR, Peschanski M, Aebischer P, Hantraye P. Restoration of cognitive and motor functions by ciliary neurotrophic factor in a primate model of Huntington's disease. Hum Gene Ther. 2000;11:1177–1187. doi: 10.1089/10430340050015220. [DOI] [PubMed] [Google Scholar]
- 44.Mittoux V, Ouary S, Monville C, Lisovoski F, Poyot T, Conde F, Escartin C, Robichon R, Brouillet E, Peschanski M, Hantraye P. Corticostriatopallidal neuroprotection by adenovirus-mediated ciliary neurotrophic factor gene transfer in a rat model of progressive striatal degeneration. J Neurosci. 2002;22:4478–4486. doi: 10.1523/JNEUROSCI.22-11-04478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mogul DJ, Adams ME, Fox AP. Differential activation of adenosine receptors decreases N-type but potentiates P-type Ca2+ current in hippocampal CA3 neurons. Neuron. 1993;10:327–334. doi: 10.1016/0896-6273(93)90322-i. [DOI] [PubMed] [Google Scholar]
- 46.Nishino H, Fujimoto I, Shimano Y, Hida H, Kumazaki M, Fukuda A. 3-Nitropropionic acid produces striatum selective lesions accompanied by iNOS expression. J Chem Neuroanat. 1996;10:209–212. doi: 10.1016/0891-0618(96)00134-2. [DOI] [PubMed] [Google Scholar]
- 47.Nishino H, Kumazaki M, Fukuda A, Fujimoto I, Shimano Y, Hida H, Sakurai T, Deshpande SB, Shimizu H, Morikawa S, Inubushi T. Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood-brain barrier: involvement of dopamine toxicity. Neurosci Res. 1997;27:343–355. doi: 10.1016/s0168-0102(97)01170-x. [DOI] [PubMed] [Google Scholar]
- 48.Nishino H, Hida H, Kumazaki M, Shimano Y, Nakajima K, Shimizu H, Ooiwa T, Baba H. The striatum is the most vulnerable region in the brain to mitochondrial energy compromise: a hypothesis to explain its specific vulnerability. J Neurotrauma. 2000;17:251–260. doi: 10.1089/neu.2000.17.251. [DOI] [PubMed] [Google Scholar]
- 49.Ouary S, Bizat N, Altairac S, Menetrat H, Mittoux V, Conde F, Hantraye P, Brouillet E. Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: implications for neuroprotection studies. Neuroscience. 2000;97:521–530. doi: 10.1016/s0306-4522(00)00020-8. [DOI] [PubMed] [Google Scholar]
- 50.Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyot MC, Peschanski M, Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington's disease. J Neurosci. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palfi S, Riche D, Brouillet E, Guyot MC, Mary V, Wahl F, Peschanski M, Stutzmann JM, Hantraye P. Riluzole reduces incidence of abnormal movements but not striatal cell death in a primate model of progressive striatal degeneration. Exp Neurol. 1997;146:135–141. doi: 10.1006/exnr.1997.6520. [DOI] [PubMed] [Google Scholar]
- 52.Palfi S, Conde F, Riche D, Brouillet E, Dautry C, Mittoux V, Chibois A, Peschanski M, Hantraye P. Fetal striatal allografts reverse cognitive deficits in a primate model of Huntington disease. Nat Med. 1998;4:963–966. doi: 10.1038/nm0898-963. [DOI] [PubMed] [Google Scholar]
- 53.Palmer TM, Stiles GL. Adenosine receptors. Neuropharmacology. 1995;34:683–694. doi: 10.1016/0028-3908(95)00044-7. [DOI] [PubMed] [Google Scholar]
- 54.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1990. [DOI] [PubMed] [Google Scholar]
- 55.Reynolds DS, Carter RJ, Morton AJ. Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington's disease. J Neurosci. 1998;18:10116–10127. doi: 10.1523/JNEUROSCI.18-23-10116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-r. [DOI] [PubMed] [Google Scholar]
- 57.Schiffmann SN, Vanderhaeghen JJ. Adenosine A2 receptors regulate the gene expression of striatopallidal and striatonigral neurons. J Neurosci. 1993;13:1080–1087. doi: 10.1523/JNEUROSCI.13-03-01080.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schiffmann SN, Desdouits F, Menu R, Greengard P, Vincent JD, Vanderhaeghen JJ, Girault JA. Modulation of the voltage-gated sodium current in rat striatal neurons by DARPP-32, an inhibitor of protein phosphatase. Eur J Neurosci. 1998;10:1312–1320. doi: 10.1046/j.1460-9568.1998.00142.x. [DOI] [PubMed] [Google Scholar]
- 59.Schulz JB, Matthews RT, Jenkins BG, Ferrante RJ, Siwek D, Henshaw DR, Cipolloni PB, Mecocci P, Kowall NW, Rosen BR, Beal MF. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schulz JB, Matthews RT, Henshaw DR, Beal MF. Neuroprotective strategies for treatment of lesions produced by mitochondrial toxins: implications for neurodegenerative diseases. Neuroscience. 1996;71:1043–1048. doi: 10.1016/0306-4522(95)00527-7. [DOI] [PubMed] [Google Scholar]
- 61.Sebastiao AM, Ribeiro JA. Fine-tuning neuromodulation by adenosine. Trends Pharmacol Sci. 2000;21:341–346. doi: 10.1016/s0165-6147(00)01517-0. [DOI] [PubMed] [Google Scholar]
- 62.Sieradzan KA, Mann DM. The selective vulnerability of nerve cells in Huntington's disease. Neuropathol Appl Neurobiol. 2001;27:1–21. doi: 10.1046/j.0305-1846.2001.00299.x. [DOI] [PubMed] [Google Scholar]
- 63.Tarnopolsky MA, Beal MF. Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Ann Neurol. 2001;49:561–574. [PubMed] [Google Scholar]
- 64.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 65.The Huntington's Disease Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington's disease. Neurology. 2001;57:397–404. doi: 10.1212/wnl.57.3.397. [DOI] [PubMed] [Google Scholar]
- 66.Trussell LO, Jackson MB. Adenosine-activated potassium conductance in cultured striatal neurons. Proc Natl Acad Sci USA. 1985;82:4857–4861. doi: 10.1073/pnas.82.14.4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vis JC, Verbeek MM, De Waal RM, Ten Donkelaar HJ, Kremer HP. 3-Nitropropionic acid induces a spectrum of Huntington's disease-like neuropathology in rat striatum. Neuropathol Appl Neurobiol. 1999;25:513–521. doi: 10.1046/j.1365-2990.1999.00212.x. [DOI] [PubMed] [Google Scholar]
- 68.Von Lubitz DK, Paul IA, Ji XD, Carter M, Jacobson KA. Chronic adenosine A1 receptor agonist and antagonist: effect on receptor density and N-methyl-d-aspartate induced seizures in mice. Eur J Pharmacol. 1994a;253:95–99. doi: 10.1016/0014-2999(94)90762-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Von Lubitz DK, Lin RC, Melman N, Ji XD, Carter MF, Jacobson KA. Chronic administration of selective adenosine A1 receptor agonist or antagonist in cerebral ischemia. Eur J Pharmacol. 1994b;256:161–167. doi: 10.1016/0014-2999(94)90241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Von Lubitz DK, Beenhakker M, Lin RC, Carter MF, Paul IA, Bischofberger N, Jacobson KA. Reduction of postischemic brain damage and memory deficits following treatment with the selective adenosine A1 receptor agonist. Eur J Pharmacol. 1996a;302:43–48. doi: 10.1016/0014-2999(96)00101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Von Lubitz DK, Lin RC, Paul IA, Beenhakker M, Boyd M, Bischofberger N, Jacobson KA. Postischemic administration of adenosine amine congener (ADAC): analysis of recovery in gerbils. Eur J Pharmacol. 1996b;316:171–179. doi: 10.1016/s0014-2999(96)00667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Von Lubitz DK, Lin RC, Bischofberger N, Beenhakker M, Boyd M, Lipartowska R, Jacobson KA. Protection against ischemic damage by adenosine amine congener, a potent and selective adenosine A1 receptor agonist. Eur J Pharmacol. 1999;369:313–317. doi: 10.1016/s0014-2999(99)00073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 74.White PJ, Rose'Meyer RB, Hope W. Functional characterization of adenosine receptors in the nucleus tractus solitarius mediating hypotensive responses in the rat. Br J Pharmacol. 1996;117:305–308. doi: 10.1111/j.1476-5381.1996.tb15191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams M. Purinergic drugs: opportunities in the 1990s. Drug Res Dev. 1993;28:438–441. [Google Scholar]
- 76.Wood PL, Kim HS, Boyar WC, Hutchison A. Inhibition of nigrostriatal release of dopamine in the rat by adenosine receptor agonists: A1 receptor mediation. Neuropharmacology. 1989;28:21–25. doi: 10.1016/0028-3908(89)90062-2. [DOI] [PubMed] [Google Scholar]
- 77.Zetterstrom T, Fillenz M. Adenosine agonists can both inhibit and enhance in vivo striatal dopamine release. Eur J Pharmacol. 1990;180:137–143. doi: 10.1016/0014-2999(90)90601-2. [DOI] [PubMed] [Google Scholar]