

Abstract
Huntington's disease (HD) is a neurodegenerative disorder caused by a CAG expansion that results in elongation of the polyglutamine tract at the N terminus of huntingtin (Htt). Abnormal proteolytic processing of mutant Htt has been implicated as a critical step in the initiation of HD. The protease(s) involved in this process has not been fully characterized. Here we report that activated calpain was detected in the caudate of human HD tissue but not in age-matched controls. In addition, one of the major N-terminal Htt proteolytic fragments found in human HD tissue appears to be derived from calpain cleavage. Htt fragments in HD lysates were similar in size to those produced by exposure of in vitro-translated Htt to exogenous calpain. Incubation of in vitro-translated Htt with calpain generated a cascade of cleavage events with an initial intermediate cleavage product at 72 kDa and a final cleavage product at 47 kDa. The rate of cleavage of Htt by calpain was polyglutamine-length-dependent. These results suggest that cleavage of Htt in human HD tissue is mediated in part by the Ca2+-activated neutral protease, calpain.
Keywords: huntingtin, Huntington's disease, calpain, proteases, triplet repeat disease, neurodegeneration
Huntington's disease (HD) is an autosomal-dominant neurodegenerative disease caused by a CAG expansion in the huntingtin gene (htt) (Huntington's Disease Collaborative Research Group, 1993). The essential neuropathological characteristic of HD is the loss of medium spiny neurons in the caudate nucleus and the cortical projection neurons in layers V and VI (Cudkowicz and Kowall, 1990; Hedreen et al., 1991; Albin, 1995). Expression of truncated forms of mutant huntingtin protein (Htt) and not the full-length protein induces cell death by apoptosis (Martindale et al., 1998). This led to the hypothesis that toxic protein fragments derived from full-length mutant Htt are required for disease initiation (DiFiglia et al., 1997; Li and Li, 1998; Hackam et al., 1999; Miyashita et al., 1999; Ona et al., 1999; Peters et al., 1999; Sanchez et al., 1999; Wellington et al., 2000). One family of proteases that promote the cleavage of Htt and other polyglutamine expansion disease proteins is the cell death proteases, caspases (Goldberg et al., 1996;Martindale et al., 1998; Wellington et al., 1998, 2000; Ellerby et al., 1999a,b). However, one of the principal pathways of neurotoxicity in the mammalian brain is glutamate excitotoxicity, which depends on excessive Ca2+ influx into the cell. In neurons, this pathway is often accompanied by the activation of cysteine proteases from both the caspase and calpain family (Wang, 2000). Multiple lines of evidence suggest that alterations in intracellular Ca2+ levels play a role in HD pathogenesis.
In an HD mouse model expressing full-length expanded human Htt, resting Ca2+ levels are increased by almost twofold in CA1 pyramidal neurons (Hodgson et al., 1999). Transgenic mice expressing full-length mutant Htt show significantly reduced synaptic vesicular uptake of glutamate (Li et al., 2000). NMDA receptor currents are also enhanced in in vitro and in vivo HD models (Chen et al., 1999). Because the NMDA receptor is glutamate-sensitive, Ca2+-permeable, and expressed in the medium spiny neostriatal neurons targeted in HD, it follows that intracellular Ca2+ levels may increase through increased NMDA receptor-mediated signaling. A third line of evidence linking Ca2+dysregulation to HD is that the levels of proteins involved in Ca2+ regulation are altered in HD patients and mouse models (Hodgson et al., 1999; Luthi-Carter et al., 2000). Given these multiple lines of evidence linking HD with alterations in Ca2+ homeostasis, we investigated whether the Ca2+ responsive protease, calpain, plays a role in the cleavage of Htt in HD.
Calpains are a family of Ca2+-dependent intracellular cysteine proteases, including the ubiquitously expressed μ- and m-calpains. μ-Calpain requires micromolar levels of Ca2+, whereas m-calpain requires millimolar levels of Ca2+ for activation. Both μ- and m-calpains are heterodimeric and consist of a distinct large 80 kDa catalytic subunit and a common small 28 kDa regulatory subunit. The addition of Ca2+ results in the autolytic processing of the catalytic subunit of μ-calpain from an 80 kDa protein to a 76 kDa protein, whereas activation of the m-calpain catalytic subunit results in 20 amino acids being removed from the 80 kDa protein N terminus. The small calpain regulatory subunit is converted from a 28 kDa protein to a 21 kDa polypeptide with increased Ca2+ levels. The physiological roles and possible functional distinctions of μ- and m-calpains remain unclear, but suggested functions include participation in cell division and migration (Huttenlocher et al., 1997), integrin-mediated signal transduction, and apoptosis (Kulkarni et al., 1999).
In vitro, we have shown that Htt is cleaved by caspases at three sites, yielding N-terminal fragments of 70, 75, and 80 kDa (Wellington et al., 2000). These fragments are also generated when Htt is incubated with apoptotic extracts or cells (Hackam et al., 1998;Martindale et al., 1998). Mutation of the caspase sites in Htt prevented the accumulation of these fragments during apoptotic challenge (Wellington et al., 2000). However, these initial studies did not evaluate whether altered Ca2+ homeostasis would influence the generation of toxic fragments. In many forms of cell death, both caspases and calpains are activated (Nakagawa and Yuan, 2000; Wang, 2000; Blomgren et al., 2001). Furthermore, despite the difference in cleavage-site specificity, an increasing number of cellular proteins are found to be dually susceptible to caspases and calpains, including α- and β-spectrin, calmodulin-dependent protein kinases, and tau (Wang, 2000). Here, we report that Htt is a substrate of caspases and calpains. We show that cleavage of Htt by calpain is polyglutamine repeat-dependent, with increasing length of the tract correlating with increased susceptibility of Htt to cleavage. The Htt fragments generated from calpain cleavage are smaller than those generated from caspase cleavage and therefore are more toxic to cells (Hackam et al., 1998). Calpain activation is detected in human HD tissue but not in age-matched controls. The total levels of both active and inactive calpains are increased in HD patients when compared with age-matched controls. Some of the cleavage products in HD tissue are similar in size to those generated by recombinant calpains.
MATERIALS AND METHODS
Cell culture. Superfect reagent (Qiagen, Valencia, CA) was used for transient transfections in human embryonic kidney 293T cells with plasmids described previously (Goldberg et al., 1996; Martindale et al., 1998; Wellington et al., 2000) and included wild-type and expanded full-length huntingtin (Htt15 and Htt44, respectively), caspase-resistant huntingtin (Htt15 D513A, D552A, D586A), and the wild-type and expanded N-terminal fragment of Htt (Htt 3949-15 and Htt 3949-138). Thapsigargin (2.5 μm; Sigma, St. Louis, MO) was added 24 hr after transfection, and cells were lysed and collected at 48 hr.
Western blot analysis. Cell pellets or human brain tissue (Harvard Brain Tissue Resource Center, Belmont, MA) were homogenized in NP-40 lysis buffer (0.1% NP-40, 50 mm HEPES, pH 7.4, 250 mmNaCl, and 5 mm EDTA) or RIPA (10 mmTris, pH 8.0, 150 mm NaCl, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, and 5 mm EDTA) with protease inhibitors (complete mini, Roche, Mannheim, Germany; Z-VAD, Sigma) (Ellerby et al., 1999a,b). Controls were prepared by treating Htt-transfected lysates in lysis buffer with 5 μm DTT, 10 mmCaCl2, and 3 μm m-calpain (Sigma) for 5 min at 30°C. Lysate proteins were resolved on a 12% polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and probed with monoclonal Htt 2166 (3.5 μg/ml; Chemicon, Temecula, CA), calpain small subunit 3083 (9 μg/ml; Chemicon), and μ-calpain 3104 (3.5 μg/ml; Chemicon) antibodies. Immunoblots were developed with a peroxidase-conjugated secondary antibody and enhanced chemiluminescence.
In vitro protein synthesis and cleavage. The Htt 1955-15, Htt 3949-15, and Htt 3949-138 constructs were translated with a TnT-coupled kit (Promega, Madison, WI) and the products were incubated with μ-calpain (Calbiochem, La Jolla, CA) or m-calpain (Sigma) in NP-40 lysis buffer with 5 μm DTT and no added 3 μm or 10 mmCaCl2 at 30°C for 5 min or for the time indicated in Figure 2C. Calpain inhibitor I (Bachem, Bubendorf, Switzerland) was added where indicated. Reactions were terminated by addition of EDTA, SDS sample buffer, and boiling. Control caspase-2, caspase-3, and caspase-6 cleavage products were produced as described previously (Ellerby et al., 1999a,b).
Fig. 2.

The length of the polyglutamine tract modulates calpain cleavage of Htt. A, Cleavage of in vitro-translated Htt 3949-15 with m-calpain produced three distinct cleavage products, whereas the cleavage of Htt 3949-138 produced only a small amount of the largest calpain repeat-dependent cleavage product and barely detectable levels of the two smaller repeat-dependent cleavage products. B, Incubatingin vitro-translated Htt (Htt 3949-15 and Htt 3949-138) with increasing concentrations of m-calpain demonstrated that the 92 kDa cleavage product derived from mutant Htt is more rapidly produced and degraded at lower enzyme concentrations than the corresponding 72 kDa wild-type Htt cleavage product (n = 3).C, In the presence of m-calpain (30 nm), expanded Htt (Htt 3949-138) is preferentially cleaved relative to the normal protein (Htt 3949-15) (n = 3).
Immunocytochemistry. Formalin-fixed human caudate tissue (Harvard Brain Tissue Resource Center) was embedded in paraffin, sectioned, and deparaffinized with xylene. Antigen retrieval was performed by microwaving sections in 10 mm citrate buffer, pH 6.0, for 5 min. Primary antibodies were as follows: monoclonal calpain regulatory subunit [14 μg/ml; monoclonal antibody (mAb) 3083; Chemicon]; polyclonal m-calpain catalytic subunit (2 μg/ml; sc-7533; Santa Cruz Biotechnology, Santa Cruz, CA); monoclonal μ-calpain catalytic subunit (7 μg/ml; mAb 3104; Chemicon); and monoclonal Htt (7 μg/ml; mAb 2166; Chemicon). Rabbit IgG (2 μg/ml) or goat IgG (2 μg/ml) was used as a negative control. Biotinylated secondary antibody (6 μg/ml; Vector Laboratories, Burlingame, CA) was incubated for 1 hr at 37°C followed by signal amplification with biotin/avidin and diaminobenzidine visualization. Mayers hematoxylin (American MasterTech, Lodi, CA) was used as a counter-stain.
RESULTS
Htt is cleaved by calpains into three major N-terminal cleavage products
We have shown previously that caspases cleave Htt to produce N-terminal fragments that are toxic to cells (Hackam et al., 1998;Martindale et al., 1998; Wellington et al., 2000). Caspases cleave Htt in a 76 aa region to produce 70, 75, and 80 kDa N-terminal fragments (Wellington et al., 2000). Here we show that Htt is also cleavedin vitro by μ-calpain and m-calpain (Fig.1) and that some of the N-terminal fragments produced are smaller in length than those derived from caspase cleavage and therefore more toxic (Hackam et al., 1999). We initially evaluated calpain cleavage of Htt by treating the in vitro-translated, [35S]-labeled, N-terminal Htt fragment (Htt 1955-15) with either μ-calpain or m-calpain. Lower concentrations of calpains produce cleavage fragments at 67 and 62 kDa, whereas higher concentrations of calpains produce a cleavage product at 47 kDa (Fig. 1A,B). μ-Calpain and m-calpain cleaved Htt at the same sites (Fig.1A,B). μ-Calpain cleaved Htt in the presence of low (3 μm) and high (10 mm) Ca2+, m-calpain cleaved Htt only in the presence of high Ca2+ (10 mm), and preincubation with calpain inhibitor 1 (30 μm) completely blocked cleavage of Htt by both calpains (Fig. 1C).
Fig. 1.

Htt is cleaved by calpains into four major cleavage products. A, Treatment of the in vitro-translated N-terminal Htt construct Htt 1955-15 with μ- and m-calpain produced three products. The cleavage is dependent on enzyme concentrations with low levels (L) of μ- and m-calpain (0.1 and 0.3 μm, respectively) producing 67 and 62 kDa fragments and higher levels (H) of μ- and m-calpain (0.3 and 3.0 μm, respectively) producing a single 47 kDa fragment. B, The two larger Htt fragments produced by μ- and m-calpain at lower (L) enzyme concentrations are better visualized with longer exposure times. C, Calpain cleavage ofin vitro-translated Htt (1955-15) is also dependent on Ca2+ concentration and is inhibited by the calpain inhibitor known as calpain inhibitor 1. D, Cleavage ofin vitro-translated Htt 3949-15 with caspase-3, m-calpain, caspase-2, and caspase-6. E, Table of potential calpain cleavage sites. Caspase cleavage sites in Htt are highlighted in red. Caspase-3 cleaves at 513 and 552, caspase-2 cleaves at 552, and caspase-6 cleaves at 586.a, Molecular weight of a fragment on the gel (MWG, in kilodaltons; a, b andc–f values from two separate experiments);b, predicted molecular weight determined from the relative molecular weight of the caspase-3 cleavage product [MWP, in kilodaltons; MWP = MWG × MWC (caspase-3)/MWG(caspase-3)]; c, calculated molecular weight (MWC) of a fragment based on predicted cleavage site(s) (in kilodaltons); d, P1 amino acid number and sequence for predicted calpain cleavage site(s) and known caspase cleavage sites; e, predicted amino acid sequence recognized by calpains (P2, P1, and P1′) or the amino acid sequence recognized by caspases; f, amino acid sequence of a calpain cleavage site in protein kinase C; g, amino acid sequence of a calpain cleavage site in caspase-12.F, Htt amino acid sequence from 437 to 586. Caspase sites are highlighted in red, and potential calpain sites are highlighted in blue.
To further evaluate the calpain cleavage sites of Htt with respect to caspase cleavage, we used a larger Htt construct encoding a 1211 aa fragment because it contains all three previously mapped caspase sites (Wellington et al., 2000). Treatment of in vitro-translated, [35S]-labeled, N-terminal Htt 3949-15 with calpain produced three fragments of 72, 67, and 62 kDa (Fig.1D). Previous work with recombinant caspases shows that caspase-3 cleaves Htt at D513 and D552, caspase-2 cleaves at D552, and caspase-6 cleaves at D586 (Fig. 1E). As shown in Figure 1D, calpain generated a 72 kDa fragment that migrates between the caspase-3 Htt fragment at 70 kDa and the caspase-2 Htt fragment at 75 kDa. Therefore, one of the calpain sites lies between amino acids 513 and 552 of Htt. Additional experiments with the expanded forms of in vitro-translated Htt showed that three of the calpain cleavage products (72, 67, and 62 kDa) contain the polyglutamine tract (Fig. 2), whereas the smallest fragment (47 kDa) does not (data not shown). In summary, calpain treatment of Htt produces three N-terminal cleavage products (72, 67, and 62 kDa) and one C-terminal cleavage fragment (47 kDa) derived from the N-terminal portion of Htt.
Predicted calpain cleavage sites
Previous studies have mapped the caspase sites in Htt (Wellington et al., 2000), which are shown in Figure 1E. Using this information and known substrate cleavage sites for calpain, we can predict likely calpain cleavage sites in Htt for the N-terminal cleavage products. Based on amino acid sequence, the caspase-3 Htt fragment has a molecular weight of 55 kDa (observed 70 kDa). Therefore, the predicted size of the calpain-derived N-terminal Htt fragments were ∼46, 50, and 58 kDa (Fig. 1E, MWP) (Croall and DeMartino, 1991). Using this information, we were able to determine potential calpain cleavage sites in Htt (Fig. 1E), which are based on the fact that calpain cleaves preferentially in the P2 position at a Val, Leu, or Ile and on sequenced calpain cleavage sites in calpain-susceptible proteins. Our predictions show that two cleavage sites in Htt are identical in sequence to known calpain substrates, most notably, a Leu-Thr-Ala motif at position 469 and a Ser-Ser-Ser motif at position 536, which is the calpain cleavage site in caspase-12 (Nakagawa and Yuan, 2000) and protein kinase C (Croall and DeMartino, 1991), respectively. Both of these sites are in close proximity to our predicted cleavage sites for the largest calpain-derived Htt fragments. As shown in Figure 1F, a Ser-Ser-Ser motif lies within the 76 aa sequence in which the caspase cleavage sites reside in Htt.
The length of the polyglutamine tract modulates calpain cleavage of Htt
Because production of N-terminal cleavage products plays an important role in HD pathogenesis (DiFiglia et al., 1997), we compared the rate of cleavage of normal and expanded Htt by calpain. The disease form of Htt was more readily cleaved by calpain (Fig. 2). Cleavage of normal Htt (Htt 3949-15) with m-calpain produced three calpain-derived N-terminal Htt fragments (62, 67, and 72 kDa) (Fig.2A). Calpain cleavage of expanded Htt (Htt 3949-138) under identical conditions produced only small quantities of the repeat-dependent Htt fragments (72, 77, and 92 kDa) (Fig.2A). The results were similar using either μ-calpain or m-calpain.
To further evaluate the repeat-dependent cleavage of Htt, we incubatedin vitro-translated normal and expanded Htt with increasing concentrations of m-calpain. The 92 kDa fragment derived from expanded Htt was more rapidly produced and degraded than the corresponding 72 kDa fragment of wild-type Htt (Fig. 2B) (n = 3). Furthermore, incubating normal and expanded Htt with low levels of m-calpain (30 nm) demonstrated that expanded Htt is more sensitive to calpain degradation (Fig. 2C) (n = 3). After a 120 min incubation at 30°C with m-calpain, a major portion of the in vitro-translated expanded Htt was cleaved (41%), whereas normal repeat Htt remained fully intact. Interestingly, previous work using the same in vitro-translated constructs demonstrated that cleavage of Htt by caspases is repeat-independent (Wellington et al., 1998).
Increased calpain expression and Htt fragmentation in thapsigargin-treated 293T cells
Given our initial in vitro experiments showing cleavage of Htt by calpains, we investigated whether Htt is cleaved by calpains in intact cells under conditions of altered Ca2+ homeostasis. We treated 293T cells overexpressing full-length Htt (Htt15) with thapsigargin, a proapoptotic agent that increases intracellular Ca2+ levels through inhibition of the endoplasmic reticulum Ca2+/Mg2+ATPase. Treatment increased levels of the 28 kDa calpain regulatory subunit and converted calpain to a 21 kDa polypeptide consistent with calpain activation (Fig. 3A). Thapsigargin treatment resulted in a twofold increase in activated calpain (Fig. 3A). Thapsigargin-treated cells generated additional cleavage products of Htt when compared with untreated cells (Fig. 3B). These cleavage products are identical in size to those generated by treating full-length Htt15-transfected cell lysates with recombinant calpains (Fig. 3B, lane 5). In addition, treatment of 293T cells overexpressing the caspase-resistant form of full-length Htt15 (D513A, D552A, D586A) with thapsigargin produced increased levels of calpain-derived Htt fragments, demonstrating that the Htt fragments are not attributable to caspase cleavage (Fig. 3B).
Fig. 3.
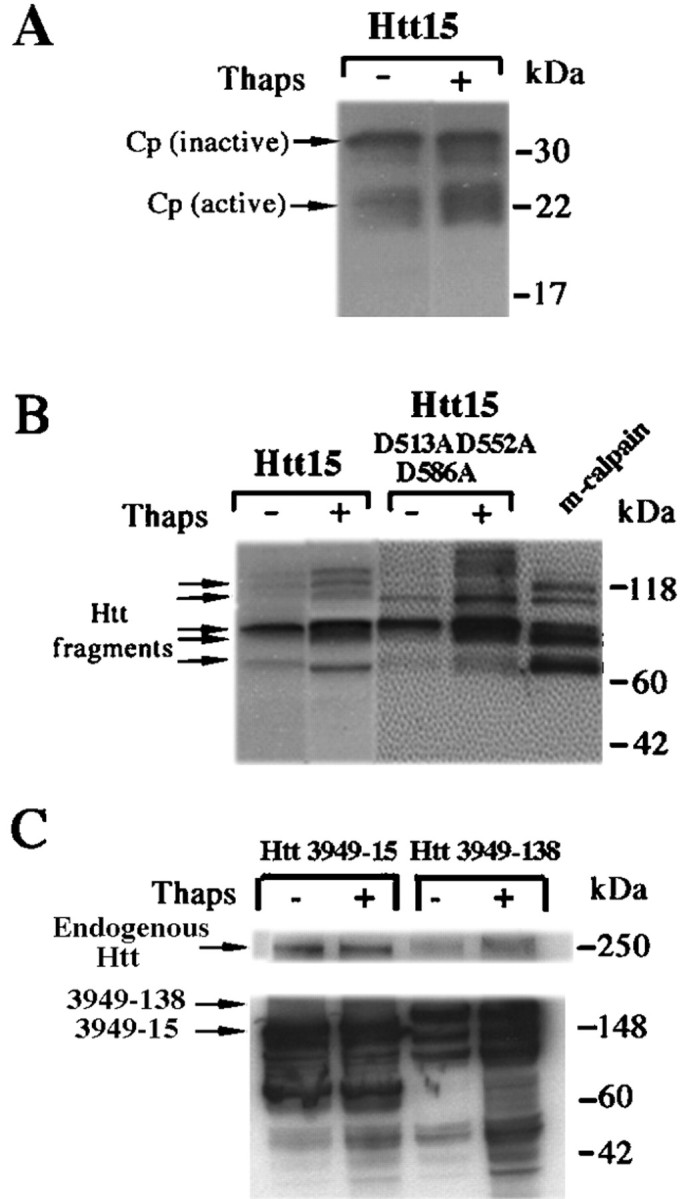
According to Western blot analysis, calpain activation and Htt fragmentation are observed in thapsigargin-treated 293T cells overexpressing Htt. A, 293T cells overexpressing full-length Htt15 treated with thapsigargin (Thaps) and probed with calpain (Cp) regulatory subunit antibody. B, 293T cells overexpressing full-length Htt15 and caspase-resistant full-length Htt15 D513A, D552A, D586A treated with thapsigargin (Thaps) and probed with anti-Htt antibody 2166. The controls in B are lysates from 293T cells overexpressing full-length Htt15, which are subsequently treated with m-calpain.C, 293T cells overexpressing normal Htt 3949-15 and expanded Htt 3949-138 treated with thapsigargin (Thaps) and probed with anti-Htt antibody 2166.
We subsequently investigated whether differential cleavage of mutant Htt would be observed in a cell culture model by treating 293T cells overexpressing normal and expanded Htt (3949-15 and 3949-138) with thapsigargin. Strikingly, we found increased levels of expanded Htt cleavage products relative to wild-type (Fig. 3C). Interestingly, the expression of mutant Htt resulted in depletion of endogenous full-length normal Htt (Fig. 3C). A 30% reduction in endogenous Htt was observed in the presence of the mutant Htt fragment when compared with the normal Htt fragment (Fig.3C).
Increased calpain expression and Htt fragmentation in the human HD caudate
Increased calpain activation and Htt fragmentation was observed in the caudate of human HD patients (Fig. 4) (n = 3). The age, sex, and postmortem interval of the HD and control caudate tissue used for these studies are shown in Table1. The 21 kDa active form of calpain was detected in HD tissue and not in controls (Fig. 4A) (n = 3). Interestingly, both the catalytically inactive precursor of the calpain regulatory subunit and the activated forms are increased in the HD patients relative to controls. Also noteworthy was the finding that a patient with earlier disease onset has higher levels of calpain expression (Fig. 4A,B). The expression of the 21 kDa active form of calpain regulatory subunit was on average eightfold higher in the HD group when compared with controls, and the total calpain levels were 2.5-fold higher in the HD group relative to controls (Fig. 4B) (n = 3). Furthermore, total levels of the large subunit of μ-calpain were increased in HD relative to control tissue (Fig. 4C) (n = 3).
Fig. 4.
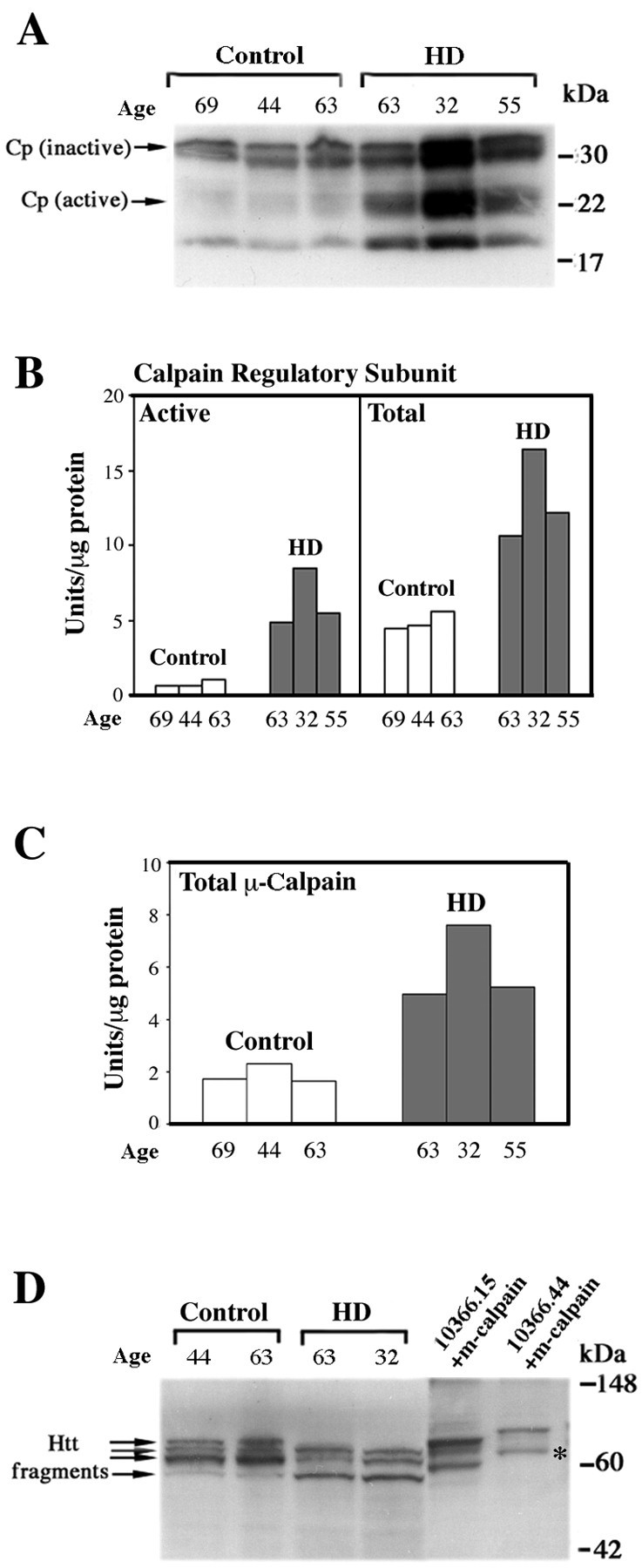
According to Western blot analysis, calpain activation and Htt fragmentation are observed in the HD caudate.A, Human HD and control caudate lysates probed with calpain (Cp) regulatory subunit antibody (n = 3). B, Quantification of calpain regulatory subunit expression in the human HD and control caudate (n = 3). C, Quantification of μ-calpain large subunit expression in the HD and control caudate (n = 3). D, Human HD and control caudate lysates probed with anti-Htt antibody 2166 (n = 3). The controls are lysates from 293T cells overexpressing full-length Htt15 and Htt44, which are subsequently treated with m-calpain. The cleavage product labeled with anasterisk represents an Htt44 N-terminal calpain cleavage product equivalent in size to the largest N-terminal cleavage product found in HD lysates.
Table 1.
Human caudate tissue samples used for Western blot analysis and immunohistochemistry
| Tissue type Age | Sex | PMI (hr) | Use |
|---|---|---|---|
| Control | |||
| 69 | F | 20.7 | WB |
| 44 | F | 10.7 | WB |
| 63 | M | 21.5 | WB |
| 73 | M | 12.0 | IHC |
| 71 | M | 11.0 | IHC |
| 71 | M | 24.0 | IHC |
| 70 | M | 17.3 | IHC |
| HD (grade 3): | |||
| 63 | F | 16.0 | WB |
| 32 | F | 13.4 | WB |
| 55 | F | 24.4 | WB |
| 66 | M | 22.0 | IHC |
| 48 | M | 19.2 | IHC |
| 74 | F | 21.0 | IHC |
PMI, Postmortem interval; WB, Western blot; IHC, immunohistochemistry; F, female; M, male.
HD patients also had a decrease in full-length Htt (data not shown), along with a degradation of Htt into smaller fragments, despite the increase in length of the polyglutamine tract (Fig.4D) (n = 3) when compared with age-matched controls. The different pattern of fragmentation observed is likely attributable to activation of distinct proteolytic pathways. Because calpains are so dramatically activated in the HD caudate, it follows that this pathway contributes to the cleavage pattern observed in HD and not in control tissue. Additional experiments demonstrate that all three of the Htt cleavage products in the human HD caudate contain the N terminus (data not shown). Cleavage of full-length Htt15 and Htt44 (approximate size of repeat in HD patients) with m-calpain showed that the largest N-terminal cleavage product in HD patients is identical in size to one of the calpain-derived N-terminal Htt fragments (Fig. 4D, see asterisk), suggesting that at least one of the HD fragments may be calpain-derived.
Increased expression of calpain and altered subcellular localization in the caudate of HD patients
Given our finding that Htt is a substrate for calpains, immunohistochemical analysis was performed on human HD caudate and age-matched controls (Fig. 5) (n = 3). The antibodies used were specific to the large catalytic subunits of μ-calpain and m-calpain as well as to the small calpain regulatory subunit, as shown by Western blot analysis (data not shown). In HD patients, immunoreactivity to the calpain regulatory subunit was detected in perinuclear vesicular structures (Fig.5C–F), whereas age-matched controls showed much lower levels of calpain staining (Fig. 5A,B). In addition, we saw a number of cells in deeper layers of the HD caudate exhibiting intense perinuclear vesicular staining, including medium spiny neurons (Fig. 5D,F). The enhanced immunoreactivity of calpains in the HD caudate correlates with the higher levels of calpain found by Western blot analysis (Fig. 4). The pattern of calpain staining is distinct from the diffuse pattern of calpain staining reported in other studies. The intensity of staining in HD tissue is not only attributable to increased calpain levels in HD tissue but also to the concentration of calpains in Htt-containing vesicular structures.
Fig. 5.

Increased expression of the calpain regulatory subunit in the HD caudate. Immunohistochemical analysis shows that high levels of calpain are localized to perinuclear vesicles in cells near the lateral ventricle (A, C, E) as well as to medium spiny neurons deeper within the caudate (B, D, F) in HD patients relative to controls (n = 3). In addition, calpain is expressed at high levels in the Lewy bodies of HD patients (C). Arrowheads indicate perinuclear vesicles. Lewy bodies are to the right of the asterisk. In A, C, andE, the lateral ventricle is toward thebottom of the picture. Insets inC and D represent boundaries of the 100× image in E and F, respectively.
Immunohistochemical analysis with antibodies to μ-calpain and m-calpain showed intense staining in a large number of cells surrounding the ventricle; this staining appeared to colocalize with Htt in aggregates in cytosolic and possibly nuclear compartments (Fig.6). Based on staining with specific cell-type markers, it was determined that these cells are both neurons and glia (Figs. 5C,E and Fig.6B,C,E,F,H,I) (data not shown). Calpain and Htt aggregation was observed in age-matched control caudate, although at greatly reduced levels in all regions (Fig.6A,D,G). Interestingly, the two calpain isoforms were differentially expressed in Lewy bodies within the HD caudate. Although the μ-calpain catalytic subunit and calpain regulatory subunit were expressed at high levels with Htt within Lewy body structures, expression of the m-calpain catalytic subunit was not detected in these structures (Fig. 5C).
Fig. 6.

Both μ-calpain and m-calpain are upregulated in the HD caudate. Immunohistochemical analysis shows increased expression of the μ- and m-calpain catalytic subunit, as well as Htt, in the caudate from HD patients (A–I). Intense perinuclear vesicular staining of all three proteins is observed in HD caudate tissue surrounding the lateral ventricle (A–I) as well as in cells deeper within the caudate, including medium spiny neurons. Staining with calpain isoform-specific antibodies also indicates that only μ-calpain and Htt proteins (not m-calpain) are associated with Lewy bodies (data not shown). Arrowheads indicate perinuclear vesicles, and intranuclear staining is indicated by an asterisk. In A–I, the lateral ventricle is toward thebottom of the picture. Insets inB, E, and H represent boundaries of the 100× image in C, F, and I, respectively.
DISCUSSION
One pathological mechanism proposed for HD is that the production of a toxic fragment(s) containing the polyglutamine tract amplifies pathways leading to neuronal dysfunction and cell death (DiFiglia et al., 1997; Li and Li, 1998; Hackam et al., 1999; Miyashita et al., 1999; Ona et al., 1999; Peters et al., 1999; Sanchez et al., 1999;Wellington et al., 2000). Caspases represent one class of proteases that may initiate the cleavage of Htt in HD (Goldberg et al., 1996). Evidence for caspase activation has been observed in the HD brain, and expression of the expanded polyglutamine form of Htt in cell culture promotes cell death (Goldberg et al., 1996). These initial results do not exclude the possibility that other proteases may contribute to the initiation or further truncation of Htt. To continue our investigation of the proteolytic pathways that contribute to the generation of toxic fragments in HD, we investigated the role of calpains in the cleavage of Htt. In the present work, we demonstrate that Htt is cleaved by calpains, but more importantly that the cleavage is modulated by CAG repeat length. In addition, cleavage of Htt by calpains occurs under conditions that modulate Ca2+homeostasis and not necessarily all conditions that induce cell death (Wellington et al., 2000).
It is interesting to note that although potential calpain sites in Htt encompass a range of locations within the Htt protein, one site is tightly clustered in the region containing the caspase cleavage sites. In some cases, calpains play an upstream role in activating caspases, whereas in other cases, they act in parallel with caspases to promote cell death or shuttle the cell toward a necrotic death by rendering caspases inactive (Pike et al., 1998; Chua et al., 2000;Lankiewicz et al., 2000; Wang, 2000; Blomgren et al., 2001). Our current work demonstrates that Htt is cleaved by calpains independently of caspases. Future work will address how these two families of cysteine proteases interact in HD pathogenesis.
One particularly important finding is that some of the fragments generated by calpains are small enough to diffuse into the nucleus. Larger N-terminal Htt fragments form strictly perinuclear aggregates, whereas smaller Htt fragments (<60 kDa) can also translocate to the nucleus (Hackam et al., 1998). In a number of transgenic mouse models expressing full-length Htt, the N-terminal fragments redistribute to the nucleus and cleavage of Htt is believed to precede entry of Htt into the nucleus (Hodgson et al., 1999). The sizes of caspase cleavage products range from 70 to 80 kDa and therefore are found in perinuclear aggregates in the cytoplasm. In addition, caspases produce single cleavage products that are not further truncated. In contrast, calpains generate a cascade of fragments and intermediates derived from full-length Htt. One might predict from our results that truncation by calpains may contribute to the redistribution of Htt to the nucleus.
Uncontrolled calpain activity or activation may contribute to ischemic brain injury, Alzheimer's disease, multiple sclerosis, and Parkinson's disease (for review, see Wang, 2000). In models of ischemia, calpain activation serves as a link between initial ionic disturbances and apoptotic pathways. We have shown that calpains are aberrantly activated in the HD caudate. Saito et al. (1993) previously have shown activation of the large subunit of μ-calpain with no net increase in total levels of calpain in the HD putamen. Our results demonstrate a dramatic increase in the levels of both the precursor and active forms of calpain in the caudate, which is more severely affected in HD.
Currently, the physiological function(s) of calpains is unknown. However, it should be noted that calpain activity is essential for a number of cellular functions unrelated to cell death. Therefore, cleavage of Htt by calpains under normal physiological conditions may modulate important cellular events. Unlike many cysteine proteases, calpains tend to cleave substrates at interdomain boundaries, thereby modulating the function of their substrates rather than inactivating them. It is possible that calpain cleavage of normal Htt modulates events related to the potential role of Htt in vesicular trafficking (DiFiglia et al., 1995) and/or the control of BDNF levels by Htt (Zuccato et al., 2001). This would be in contrast to the cleavage of expanded Htt by calpain, which generates products that are toxic to the cell. Furthermore, increased levels of Htt fragments lead to the depletion of normal full-length Htt, which could also impede normal cell function.
Our work suggests that calpain cleavage of Htt may play an important role in the pathogenesis of HD and compliments a recent report that Htt can be cleaved by calpains (Kim et al., 2001). Further work will be directed at evaluating the relative contribution of caspase and calpain cleavage in the natural history of disease pathology and progression in the HD transgenic mouse models (Hodgson et al., 1999; Lin et al., 2001).
Footnotes
This work was supported by National Institutes of Health Grant NS40251 (L.M.E.), the Huntington's Disease Society of America, and the Multiple Dystrophy Association. The human brain tissue was provided by the Harvard Brain Tissue Resource Center (United States Public Health Service Grant MN/NS31862). We thank Drs. Greenberg, Hermel, LaFevre-Bernt, and Sarah Lamson for critical comments and Dr. Michael Hayden for the htt vectors.
Correspondence should be addressed to Dr. Lisa M. Ellerby, Buck Institute for Age Research, 8001 Redwood Boulevard, Novato, CA 94945. E-mail: lellerby@buckinstitute.org.
REFERENCES
- 1.Albin RL. Selective neurodegeneration in Huntington's disease. Ann Neurol. 1995;38:835–836. doi: 10.1002/ana.410380602. [DOI] [PubMed] [Google Scholar]
- 2.Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- 3.Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J Neurochem. 1999;72:1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x. [DOI] [PubMed] [Google Scholar]
- 4.Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem. 2000;275:5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- 5.Croall DE, DeMartino GN. Calcium-activated neutral protease (calpain) system: structure, function, and regulation. Physiol Rev. 1991;71:813–847. doi: 10.1152/physrev.1991.71.3.813. [DOI] [PubMed] [Google Scholar]
- 6.Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol. 1990;27:200–204. doi: 10.1002/ana.410270217. [DOI] [PubMed] [Google Scholar]
- 7.DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, Boyce FM, Aronin N. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 8.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 9.Ellerby LM, Andrusiak RL, Wellington CL, Hackam AS, Propp SS, Wood JD, Sharp AH, Margolis RL, Ross CA, Salvesen GS, Hayden MR, Bredesen DE. Cleavage of atrophin-1 at caspase site aspartic acid 109 modulates cytotoxicity. J Biol Chem. 1999a;274:8730–8736. doi: 10.1074/jbc.274.13.8730. [DOI] [PubMed] [Google Scholar]
- 10.Ellerby LM, Hackam AS, Propp SS, Ellerby HM, Rabizadeh S, Cashman NR, Trifiro MA, Pinsky L, Wellington CL, Salvesen GS, Hayden MR, Bredesen DE. Kennedy's disease: caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J Neurochem. 1999b;72:185–195. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- 12.Hackam AS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden MR. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hackam AS, Singaraja R, Zhang T, Gan L, Hayden MR. In vitro evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington's disease. Hum Mol Genet. 1999;8:25–33. doi: 10.1093/hmg/8.1.25. [DOI] [PubMed] [Google Scholar]
- 14.Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntington's disease. Neurosci Lett. 1991;133:257–261. doi: 10.1016/0304-3940(91)90583-f. [DOI] [PubMed] [Google Scholar]
- 15.Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 16.Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 17.Huttenlocher A, Palecek SP, Lu Q, Zhang W, Mellgren RL, Lauffenburger DA, Ginsberg MH, Horwitz AF. Regulation of cell migration by the calcium-dependent protease calpain. J Biol Chem. 1997;272:32719–32722. doi: 10.1074/jbc.272.52.32719. [DOI] [PubMed] [Google Scholar]
- 18.Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, Qin Z-H, Aronin N, DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci USA. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulkarni S, Saido TC, Suzuki K, Fox JE. Calpain mediates integrin-induced signaling at a point upstream of Rho family members. J Biol Chem. 1999;274:21265–21275. doi: 10.1074/jbc.274.30.21265. [DOI] [PubMed] [Google Scholar]
- 20.Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275:17064–17071. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- 22.Li SH, Li XJ. Aggregation of N-terminal huntingtin is dependent on the length of its glutamine repeats. Hum Mol Genet. 1998;7:777–782. doi: 10.1093/hmg/7.5.777. [DOI] [PubMed] [Google Scholar]
- 23.Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 24.Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 25.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 26.Miyashita T, Matsui J, Ohtsuka Y, Mami U, Fujishima S, Okamura-Oho Y, Inoue T, Yamada M. Expression of extended polyglutamine sequentially activates initiator and effector caspases. Biochem Biophys Res Commun. 1999;257:724–730. doi: 10.1006/bbrc.1999.0447. [DOI] [PubMed] [Google Scholar]
- 27.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 29.Peters MF, Nucifora FC, Jr, Kushi J, Seaman HC, Cooper JK, Herring WJ, Dawson VL, Dawson TM, Ross CA. Nuclear targeting of mutant huntingtin increases toxicity. Mol Cell Neurosci. 1999;14:121–128. doi: 10.1006/mcne.1999.0773. [DOI] [PubMed] [Google Scholar]
- 30.Pike BR, Zhao X, Newcomb JK, Posmantur RM, Wang KK, Hayes RL. Regional calpain and caspase-3 proteolysis of α-spectrin after traumatic brain injury. NeuroReport. 1998;9:2437–2442. doi: 10.1097/00001756-199808030-00002. [DOI] [PubMed] [Google Scholar]
- 31.Saito K-I, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 33.Wang KK. Calpain and caspase: can you tell the difference? Trends Neurosci. 2000;23:20–26. doi: 10.1016/s0166-2236(99)01479-4. [DOI] [PubMed] [Google Scholar]
- 34.Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–9167. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- 35.Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, Cattaneo E, Hackam A, Sharp A, Thornberry N, Nicholson DW, Bredesen DE, Hayden MR. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–19838. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- 36.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]