

Abstract
Long-term potentiation (LTP) at sensory input synapses to the lateral amygdala (LA) is a candidate mechanism for memory storage during fear conditioning. We evaluated the effect of L-type voltage-gated calcium channel (VGCC) and NMDA receptor (NMDAR) blockade in LA on LTP at thalamic input synapses induced by two different protocols in vitro and on fear memoryin vivo. When induced in vitro by pairing weak presynaptic stimulation with strong (spike eliciting) postsynaptic depolarization, LTP was dependent on VGCCs and not on NMDARs, but, when induced by a form of tetanic stimulation that produced prolonged postsynaptic depolarization (but not spikes), LTP was dependent on NMDARs and not on VGCCs. In behavioral studies, bilateral infusions of NMDAR antagonists into the LA impaired both short-term and long-term memory of fear conditioning, whereas VGCC blockade selectively impaired long-term memory formation. Collectively, the results suggest that two pharmacologically distinct forms of LTP can be isolated in the LAin vitro and that a combination of both contribute to the formation of fear memories in vivo at the cellular level.
Keywords: fear conditioning, APV, ifenprodil, verapamil, LTP, amygdala
Neural plasticity in the lateral amygdala (LA) is believed to underlie the acquisition and retention of Pavlovian fear conditioning, including auditory fear conditioning in which a tone [conditioned stimulus (CS)] is paired with foot shock [unconditioned stimulus (US)] (Davis, 1992; Fanselow and LeDoux, 1999; LeDoux, 2000). The LA receives sensory information from both the auditory thalamus and cortex, and CS and US inputs converge onto individual cells in the LA (Romanski et al., 1993). Furthermore, pairing of CS and US inputs during fear conditioning leads to long-lasting alterations in synaptic transmission and neuronal activity in the LA (McKernan and Shinnick-Gallagher, 1997; Quirk et al., 1995,1997; Rogan et al., 1997; Repa et al., 2001).
One candidate mechanism for the plasticity seen in the LA during fear conditioning is associative long-term potentiation (LTP), an experience-dependent form of synaptic plasticity widely believed to underlie many learning processes (Malenka and Nicoll, 1993, 1999;Milner et al., 1998). In support of this hypothesis, LTP has been demonstrated in each of the major sensory input pathways to the LA (Clugnet and LeDoux, 1990; Chapman and Bellavance, 1992; Rogan and LeDoux, 1995), and neural activity in the LA is altered during auditory fear conditioning in a manner that is similar to artificial LTP induction (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). Moreover, LTP in the LA and fear conditioning are sensitive to the same stimulus contingencies (Bauer et al., 2001) and share similar biochemical mechanisms (Huang et al., 2000; Schafe and LeDoux, 2000;Schafe et al., 2000).
Associative LTP requires calcium entry into postsynaptic cells, which triggers a cascade of intracellular processes and, if sufficiently strong, ultimately leads to gene expression and protein synthesis (Milner et al., 1998; Malenka and Nicoll, 1999; Sweatt, 1999). In the CA1 region of the hippocampus, in which LTP has been studied most extensively, calcium can enter the postsynaptic cell through several channels, including NMDA receptors (NMDARs) and voltage-gated calcium channels (VGCCs), and LTP can be induced in such a way as to involve either or both channels. For example, LTP induction protocols that involve high-frequency tetanization (>200 Hz) or pairing of presynaptic and postsynaptic inputs require VGCCs (Grover and Teyler, 1990; Magee and Johnston, 1997). Conversely, those that use lower-frequency tetanus protocols (≤30 Hz) require NMDARs but not VGCCs (Cavus and Teyler, 1996), whereas those tetanus protocols that fall in the middle (100–200 Hz) can involve both types of channel (Cavus and Teyler, 1996; Morgan and Teyler, 1999).
In the LA, calcium influx through L-type VGCCs is necessary for LTP at thalamic input synapses in the LA induced in vitro by pairing presynaptic stimulation with postsynaptic depolarizations (Weisskopf et al., 1999). This same LTP is insensitive to blockade of NMDARs (Weisskopf et al., 1999). However, calcium entry through NMDARs is required for LTP induced by tetanization of the cortical pathway to LA (Huang and Kandel, 1998). Furthermore, behavioral studies have shown that blockade of NMDARs in the LA impairs fear conditioning, suggesting that an NMDAR-dependent plasticity in the LA underlies fear memory formation (Miserendino et al., 1990; Maren et al., 1996; Lee and Kim, 1998; Rodrigues et al., 2001). The contribution of VGCCs to fear memory, however, has not been tested.
In the present study, we examined the role of L-type VGCCs and NMDARs in thalamo-LA LTP and fear memory formation. We first compared the effects of VGCC and NMDAR blockade on LTP induced by protocols tailored to recruit either VGCCs (pairing presynaptic stimulation with strong postsynaptic depolarization) or NMDARs (a tetanus). We then compared the effects of NMDAR and VGCC blockade in LA on the acquisition and formation of auditory fear conditioning. Collectively, the results suggest that both NMDARs and L-type VGCCs contribute to the synaptic plasticity that underlies fear memory formation but in unique ways.
MATERIALS AND METHODS
Slice electrophysiology. Electrophysiological experiments in amygdala slices were conducted as documented previously (for a detailed description, see Weisskopf et al., 1999). Briefly, male Sprague Dawley rats (3–5 weeks old) were deeply anesthetized with halothane, and the brain was rapidly removed and transferred to ice-cold artificial CSF (ACSF) containing (in mm): 115 NaCl, 3.3 KCl, 1 MgSO4, 2 CaCl2, 25.5 NaHCO3, 1.2 NaH2PO4, 5 lactic acid, and 25 glucose (equilibrated with 95% O2–5% CO2). Coronal slices (400-μm-thick) containing the amygdala were cut and recovered in a holding chamber at 32–34°C for 30 min and were then allowed to return to room temperature for at least another 30 min before recording. An upright microscope equipped with infrared differential interference contrast optics (OlympusOptical, Tokyo, Japan) was used to perform whole-cell patch recordings under visual guidance. Electrodes were filled with (in mm): 130 K-gluconate, 0.6 EGTA, 2 MgCl2, 5 KCl, 10 HEPES, 2 Mg-ATP, and 0.3 Na3-GTP. The electrodes typically had resistances of 4–8 MΩ. All cells were allowed to remain at their resting potentials. Whole-cell recordings were made from 71 neurons in the LA. All of these showed various degrees of spike frequency adaptation and had relatively broad action potentials, typical of excitatory cells in the amygdala (Rainnie et al., 1991; McDonald 1992; Paré et al., 1995). The average ± SD resting membrane potential, input resistance, and membrane time constant were −69.4 ± 2.7 mV, 138 ± 45.4 MΩ, and 16.1 ± 4.8 msec, respectively.
For in vitro experiments, stimuli (150 μsec duration) were delivered through bipolar stainless steel electrodes placed in the ventral striatum, just medial to LA (see Fig. 1A,left). This stimulating protocol activates fibers that originate, at least in part, in the auditory thalamus (Weisskopf et al., 1999). For each cell, we measured the initial slope of the EPSP during the first 2–3 msec after the onset of the response. Confounds introduced by polysynaptic responses were controlled for by keeping the stimulation intensity at a minimum to produce a reliable EPSP without also recruiting polysynaptic responses or spiking, by computing percentage of increase of the initial slope of the EPSP and by excluding any data that demonstrated a change in EPSP latency after LTP induction. Baseline responses were monitored at 0.1 Hz. After stabilization of baseline responses, LTP was induced by one of two protocols. The first consisted of pairing trains of 10 stimuli at 30 Hz with 1 nA, 5 msec depolarizations given 5–10 msec after the onset of each EPSP in the train. This pattern of stimulation yields an action potential at the peak of each EPSP of the train. This pairing was given 15 times at 10 sec intervals. The second induction protocol consisted of a 30 Hz tetanus (100 stimuli, given twice with a 20 sec interval). For each cell, the stimulation intensity for LTP induction was the same as that used to elicit baseline EPSPs.
Fig. 1.

Pairing-induced LTP at thalamic input synapses in the LA is L-type VGCC dependent and NMDAR independent.A, Left, Schematic of the amygdala slice preparation, showing placement of stimulating and recording electrodes. Afferent fibers from the auditory thalamus enter the LA medially, coursing through the ventral most part of the striatum just above the central nucleus. Recordings were made just below the site of termination of auditory thalamic fibers terminating in the LA.CE, Central nucleus of amygdala; B, basal nucleus of amygdala; IC, internal capsule;OT, optic tract; EC, external capsule.Right, Example of the response of one cell to 10 stimuli delivered at 30 Hz paired with 1 nA, 5 msec depolarizations. This pairing was given 15 times at 10 sec intervals to induce LTP.B, Mean ± SE percentage of EPSP slope (relative to baseline) in cells treated with ACSF vehicle (n = 6; squares) or 50 μm verapamil (n = 5; triangles) before and after LTP induction by pairing at time 0. Traces (averages of 10 responses) from individual experiments before and 30 min after LTP induction are shown below. C, Mean ± SE percentage of EPSP slope (n = 5; squares) and amplitude (n = 4; triangles) relative to baseline before and after treatment with 50 μm verapamil at time 0. Traces (averages of 10 responses) are taken from an individual experiment before and 20 min after verapamil application. Inset, Mean ± SE integral under evoked EPSP (AUC) relative to baseline before and 20 min after verapamil application (n = 5).D, Mean ± SE percentage of EPSP slope (relative to baseline) in cells treated with ACSF vehicle (n = 8; squares) or 50 μm APV (n = 8; triangles) before and after LTP induction by pairing at time 0. Traces (averages of 10 responses) from individual experiments before and 30 min after LTP induction are shown below.
Picrotoxin (75 μm) was included in the bath in all experiments to block fast GABAergic transmission but was not observed to produce epileptiform bursting in the amygdala. Drugs were made up in DMSO stock solution and diluted 1000 times into the superfusing ACSF, yielding a final concentration of 50 μm verapamil ord-2-amino-5-phosphonopentanoic acid (APV) and 10 μm ifenprodil. Drugs used were d-APV, methoxy-(−)-verapamil, picrotoxin, and ifenprodil (all from Sigma, St. Louis, MO). Drugs were washed onto the slice between 10 and 30 min before LTP induction and washed off the slices 10–15 min after LTP induction in each experiment.
After LTP induction, test stimuli were presented once every 10 sec (0.1 Hz) for a total of 30 min. For analysis, values were binned into 1 min intervals and expressed as a percentage of baseline. The values recorded during the last 5 min of the recording session (minutes 25–30) were averaged into a single score for each cell. The amount of potentiation was analyzed by comparing preinduction values (5 min before induction) with those collected 25–30 min after LTP induction. Significance of potentiation relative to baseline was tested with a paired Student's t test. Comparison of the amount of potentiation between groups (drug treatment vs vehicle) was tested with a two-tailed, independent Student's t test. Differences were considered significant if p < 0.05.
Behavioral procedures. Behavioral procedures were conducted as described previously (Schafe and LeDoux, 2000; Schafe et al., 2000). Under Nembutal anesthesia (45 mg/kg, i.p.), rats were first implanted bilaterally with 26 gauge stainless steel guide cannulas aimed at the LA. The guide cannulas were fixed to screws in the skull using a mixture of acrylic and dental cement, and a 33 gauge dummy cannula was inserted into each guide to prevent clogging. Rats were given at least 5 d to recover before experimental procedures. All surgical procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Experimental Animalsand were approved by the New York University Animal Care and Use Committee.
In the first experiment, we evaluated the effects of different doses of the L-type VGCC blocker verapamil on auditory fear conditioning. Rats were habituated to dummy cannula removal and to the conditioning chamber for a minimum of 10–15 min. On the following day, rats were given an intra-LA infusion of either 0.5 μl of dH2O (vehicle) or one of three different doses of verapamil-HCl (0.04, 0.4, or 4.0 μg/side in 0.5 μl of dH2O; 0.25 μl/min). Distilled water was used as a vehicle in these experiments because we were unable to dissolve verapamil in ACSF or NaCl at the highest concentration (8.0 μg/μl). Injectors remained in the cannulas for 1 min after drug infusion to allow diffusion of the drug from the tip. Ten to 15 min after drug infusions, rats were trained with two conditioning trials consisting of a 20 sec, 5 kHz, 75 dB tone that coterminated with a 0.5 sec, 1.0 mA foot shock (intertrial interval of 120 sec). Testing for auditory fear conditioning took place ∼24 hr later. For this test, rats were placed in a distinctive environment (for details, see Schafe et al., 2000) and exposed to eight CS tones (5 kHz, 75 dB, 30 sec) without foot shocks. Total seconds of freezing during each CS presentation was scored for each rat, and this number was expressed as a percentage of the total CS presentation time. For analysis, scores from each trial were averaged and treated as a single score. All data were analyzed with ANOVA and Tukey's honestly significant difference post hoc t tests. Differences were considered significant if p < 0.05.
In the second experiment, we evaluated the effects of intra-LA infusion of verapamil on memory consolidation of auditory fear conditioning. Rats were habituated as before. On the conditioning day, rats were given intra-LA infusion of 0.5 μl of dH2O (vehicle), 4.0 μg of verapamil, or 5.0 μg of the NMDAR antagonist APV (0.5 μl/side; 0.25 μl/min). Ten to 15 min after drug infusions, rats were trained with three conditioning trials (20 sec, 5 kHz, 75 dB tone paired with 1.0 mA, 0.5 sec foot shock). After conditioning, rats were tested for auditory fear conditioning at 1, 3, 6, and 24 hr after conditioning (three tones for each test; 5 kHz, 20 sec, 75 dB). Freezing scores across trials were averaged into a single score and analyzed using ANOVA as in the first experiment.
At the end of each behavioral experiment, rats were killed by an overdose of chloral hydrate (600 mg/kg) and perfused with 10% buffered formalin. Nissl staining and light microscopy were used to verify the location of the cannula tips within the amygdala.
RESULTS
Pairing induces a VGCC-dependent, NMDAR-independent LTP at thalamic input synapses in the LA
In the LA, LTP can be induced at thalamic input synapses by using a pairing protocol in which weak presynaptic stimulation of thalamo-amygdala afferents is presented concurrently with brief depolarization of the postsynaptic cell by current injection (Weisskopf et al., 1999; Schafe et al., 2000; Bauer et al., 2001). This type of LTP induction protocol produces action potentials in the soma of the recorded neuron, which backpropagates into the dendrites (Magee and Johnston, 1997; Markram et al., 1997; Nishiyama et al., 2000). It has been proposed that these backpropagating action potentials (BPAPs) invade the dendrites during pairing and interact with EPSPs, leading to calcium entry through VGCCs (Magee and Johnston, 1997; Stuart et al., 1997; Johnston et al., 1999; Stuart and Hausser, 2001). Consistent with this notion, we showed that application of nifedipine, an L-type VGCC blocker, prevents the induction of LTP induced by pairing (Weisskopf et al., 1999). However, nifedipine is poorly suited for use in behavioral studies of fear conditioning because it cannot be dissolved into a suitable concentration in aqueous solution for infusion into the LA. Verapamil-HCl is a more soluble L-type VGCC blocker that can be used in behavioral studies, but the effects of verapamil on LTP in the LA have not been studied previously. We therefore examined the ability of verapamil to block pairing-induced LTP in amygdala brain slices. Specifically, we induced LTP by pairing trains of 10 stimuli at 30 Hz delivered to the thalamic inputs to LA with 1nA, 5 msec depolarizations of the postsynaptic LA cell (Fig.1A, right). This pairing was given 15 times at 10 sec intervals.
VGCCs and pairing-induced LTP
Application of 50 μm verapamil blocked the induction of LTP induced by pairing (Fig. 1B). The control group showed 166 ± 23% potentiation, which was significantly different from baseline (t(5) = 2.83; p < 0.05). The verapamil group showed 104 ± 16% potentiation, which was not significantly different from baseline (p> 0.05), but was significantly different from vehicle controls (t(9) = 2.31; p < 0.05).
To determine whether verapamil affects baseline synaptic transmission in the LA, we next examined the effects of 50 μmverapamil on the initial slope and maximum amplitude of EPSPs induced by thalamic stimulation (Fig. 1C). Verapamil was added to the superfusing ACSF after a baseline period of at least 10 min. An analysis of the size of the initial slope and maximum amplitude of the EPSPs 15–20 min after verapamil application showed no significant effects of the drug (slope, 95 ± 1.8%; amplitude, 96 ± 2.0%; p > 0.05 for both). Additionally, there was no change in resting membrane potential (−71.4 ± 0.55 mV predrug vs −69 ± 1.15 mV postdrug; p > 0.05). Finally, as a more general measure of the effects of verapamil on EPSP kinetics, we calculated the integral under the evoked EPSP [area under the curve (AUC)] (Fig. 1C, inset) and found no significant effect (110 ± 20.1% of predrug baseline; p > 0.05).
NMDARs and pairing-induced LTP
Next, we examined the ability of the NMDAR antagonist APV to block the induction of LTP by the pairing protocol (Fig.1D). In contrast to cells treated with verapamil, cells treated with 50 μm APV did not show significantly different levels of LTP. The APV-treated cells showed 147 ± 21% potentiation, which was significantly different from baseline (t(7) = 2.49; p < 0.05) but not from vehicle controls (148 ± 21%;p > 0.05).
Thus, blockade of L-type VGCCs, but not NMDARs, impairs pairing-induced LTP at thalamic input synapses in the LA without affecting routine synaptic transmission. These data are consistent with previous findings from our laboratory showing that LTP induced by pairing at thalamic input synapses to the LA is VGCC dependent and NMDAR independent (Weisskopf et al., 1999).
A 30 Hz tetanus induces an NMDAR-dependent, L-type VGCC-independent form of LTP at thalamic input synapses in the LA
Although the pairing protocol induces an NMDAR-independent form of LTP, previous studies using tetanus protocols have demonstrated an NMDAR-dependent LTP in the LA, particularly in the cortical input pathway (Huang and Kandel, 1998). Moreover, NMDARs are necessary for auditory fear conditioning (Lee and Kim, 1998; Rodrigues et al., 2001). Because the pairing protocol described above triggers BPAPs that may open dendritic VGCCs, we next used a second induction protocol that did not trigger action potentials but rather produced a long depolarization of the postsynaptic cell. This protocol, inspired by those used to induce NMDAR-dependent LTP in the hippocampus (Cavus and Teyler, 1996), consisted of 100 stimuli at 30 Hz delivered to the thalamic afferents (Fig. 2A). This tetanus was given twice, with a 20 sec interval between presentations.
Fig. 2.
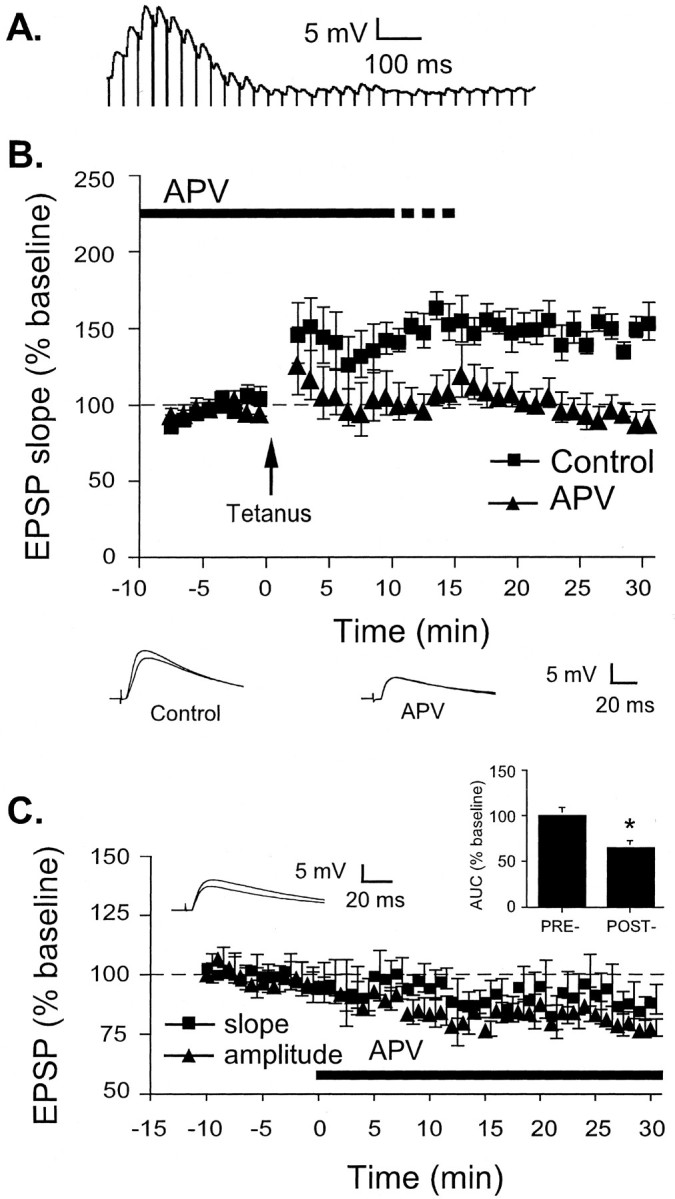
Both tetanus-induced LTP and synaptic transmission at thalamic input synapses to LA are impaired by APV. A, Example of the response of one cell to 30 stimuli at 30 Hz. A tetanus of 100 stimuli at 30 Hz given twice was used to induce LTP.B, Mean ± SE percentage of EPSP slope (relative to baseline) in cells treated with ACSF vehicle (n = 7; squares) or 50 μm APV (n = 6; triangles) before and after LTP induction by a 30 Hz tetanus at time 0. Traces(averages of 10 responses) from individual experiments before and 30 min after LTP induction are shown below. C, Mean ± SE percentage of EPSP slope (n = 6;squares) and amplitude (n = 6;triangles) relative to baseline before and after treatment with 50 μm APV at time 0. Traces(averages of 10 responses) are taken from an individual experiment before and 30 min after APV application. Inset, Mean ± SE AUC (relative to baseline) before and 30 min after APV application (n = 6). *p < 0.05 relative to predrug AUC.
NMDARs and tetanus-induced LTP
Treatment of cells with 50 μm APV blocked LTP induced by the tetanus relative to control cells (Fig.2B). The control group showed 149 ± 5.3% potentiation, which was significantly different from baseline (t(6) = 9.81; p < 0.05). The APV-treated group showed 94 ± 5.0% potentiation, which was not significantly different from baseline (p > 0.05) but was significantly different from vehicle controls (t(11) = 8.24,p < 0.05).
To determine whether APV affects baseline synaptic transmission in the LA, we next examined the effects of 50 μm APV on the initial slope and maximum amplitude of EPSPs (Fig. 2C). An analysis of the size of the initial slope of the EPSPs 25–30 min after APV application showed no significant effects of the drug (slope, 90 ± 5.3%; p > 0.05). There was also no change in resting membrane potential (−67.4 ± 1.2 mV predrug vs −67.3 ± 1.8 mV postdrug; p > 0.05). However, there was a significant decrease in the maximum amplitude of the EPSPs (79 ± 3.4%; t(6) = 6.38;p < 0.05). There was also a significant decrease in the AUC of the EPSP (Fig. 2C, inset) (64 ± 8.2% of predrug baseline; t(5) = 5.02; p < 0.05). Thus, consistent with the results of previous experiments, APV appeared to impair routine synaptic transmission at thalamic input synapses (Li et al., 1995, 1996;Weisskopf and LeDoux, 1999). As a result, it is not possible to unambiguously conclude that the effect of APV on tetanus-induced LTP in this pathway is a result of an impairment of synaptic plasticity.
The NR2B subunit and tetanus-induced LTP
APV is an NMDAR antagonist that disrupts the entire NMDA receptor complex, including the NR1 subunits, which are critical for channel function, and the NR2 subunits, which appear to regulate channel function (Monyer et al., 1992). Thus, it may be possible to circumvent the effects of APV on synaptic transmission in the thalamic input pathway by selectively blocking individual subunits of the NMDA receptor complex. In vitro studies have shown that the NR1–NR2B complex exhibits longer EPSPs than the NR1–NR2A complex (Monyer et al., 1994), suggesting that NR2B might be particularly well suited for coincidence detection and plasticity. Indeed, tyrosine phosphorylation of NR2B has been correlated with synaptic plasticity in the hippocampus (Rostas et al., 1996) and with taste learning in the insular cortex (Rosenblum et al., 1996, 1997). Overexpression of this subunit enhances LTP in the hippocampus, as well as learning in several tasks, including fear conditioning (Tang et al., 1999). Furthermore, we showed recently that intra-LA blockade of the NR2B subunit by the selective antagonist ifenprodil disrupts the acquisition but not the expression of fear conditioning (Rodrigues et al., 2001). This suggests that NMDARs incorporating the NR2B subunit may be involved specifically in synaptic plasticity, but not synaptic transmission, in the LA. In the present study, we tested this hypothesis by examining whether application of ifenprodil to amygdala brain slices impairs tetanus-induced LTP at thalamic input synapses independently of an effect on baseline synaptic transmission. Although, in cortical preparations, ifenprodil has been shown to paradoxically increase NMDA currents at concentrations ranging from 3 to 10 μm (Zhang et al., 2000), its effects have not been examined on the electrophysiological properties of amygdala neurons.
Bath application of 10 μm ifenprodil blocked LTP relative to control cells (Fig.3A). The control group showed 143 ± 13.1% potentiation, which was significantly different from baseline (t(6) = 3.31;p < 0.05). The ifenprodil group showed 103 ± 9.5% potentiation, which was not significantly different from baseline (p > 0.05) but was significantly different from vehicle controls (t(11) = 2.56;p < 0.05). Unlike APV, however, ifenprodil had no significant effects on EPSP kinetics (Fig. 3B). There was no significant effect of ifenprodil on the size of the initial slope of the EPSP (99 ± 11% at 20–25 min relative to baseline;p > 0.05) or the maximum amplitude (95 ± 5%;p > 0.05). Additionally, there was no change in the resting membrane potential (−69.3 ± 0.8 mV predrug vs −69.4 ± 0.9 postdrug; p > 0.05) or in the AUC (111 ± 16.6% of predrug baseline; p > 0.05) (Fig. 3B, inset). Thus, bath application of ifenprodil in our preparation impaired LTP induced by tetanus but had no effect on different aspects of routine synaptic transmission.
Fig. 3.
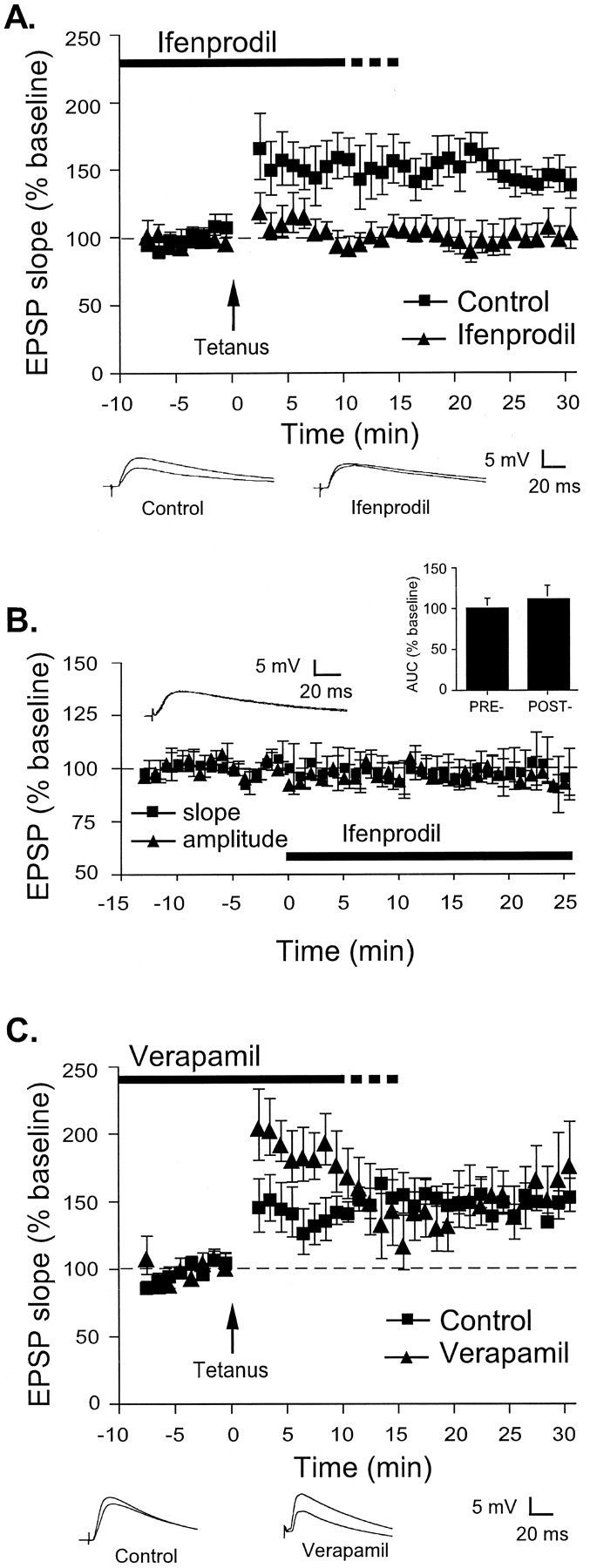
Tetanus-induced LTP at thalamic input synapses is impaired by selective NR2B blockade but not by an L-type VGCC antagonist. A, Mean ± SE percentage of EPSP slope (relative to baseline) in cells treated with ACSF vehicle (n = 7; squares) or 10 μm of the selective NR2B antagonist ifenprodil (n = 6;triangles) before and after LTP induction by a tetanus at time 0. Traces (averages of 10 responses) from individual experiments before and 30 min after LTP induction are shown below. B, Mean ± SE percentage of EPSP slope (n = 6; squares) or amplitude (n = 6; triangles) relative to baseline before and after treatment with 10 μm ifenprodil at time 0. Traces (averages of 10 responses) are taken from an individual experiment before and 25 min after ifenprodil application. Inset, Mean ± SE AUC relative to baseline before and 25 min after ifenprodil application (n = 6). C, Mean ± SE percentage of EPSP slope (relative to baseline) in cells treated with ACSF vehicle (n = 7; squares) or 50 μm verapamil (n = 7;triangles) before and after LTP induction by a tetanus at time 0. Traces (averages of 10 responses) from individual experiments before and 30 min after LTP induction are shown below.
VGCCs and tetanus-induced LTP
Finally, we examined the effect of VGCC blockade in the LA on tetanus-induced LTP (Fig. 3C). Cells treated with 50 μm verapamil showed significant potentiation from baseline (t(6) = 2.58;p < 0.05) but did not show significantly different levels of LTP relative to vehicle controls (p > 0.05). The control group showed 150 ± 5% potentiation, whereas the verapamil group showed 164 ± 25% potentiation. The apparent enhancement observed in the first 10 min after the tetanus is consistent with the findings of previous reports that have examined L-type VGCC-independent LTP in the hippocampus (Cavus and Teyler, 1996) and may be similar to the previously documented depolarization-dependent excitatory effects of dihydropyridine compounds (O'Regan et al., 1990).
In summary, LTP induced by tetanus at thalamic input synapses in the LA is impaired by NMDAR antagonists, including a selective NR2B antagonist, but not by blockade of VGCCs. Together with the results of the pairing protocol experiments, these findings suggest that, as in the hippocampus, two pharmacologically distinct forms of LTP can be induced at thalamic input synapses to the LA depending on the induction protocol used. Furthermore, in each of our LTP experiments, the drugs (verapamil, APV, and infenprodil) were washed off of the slice 10–15 min after LTP induction, whereas the impairment in LTP was observed to last out to at least 30 min (Figs. 1-3). Additionally, in many experiments, we held cells out to 1 hr after LTP induction, and the impairment, where evident, was long lasting in each case (data not shown). Thus, we believe that the most likely explanation for our results is that we impaired LTP induction rather than expression with these compounds.
Intra-LA blockade of L-type VGCCs and NMDARs impairs memory formation of auditory fear conditioning but in different ways
Whereas the role of NMDA receptors in fear conditioning has been studied extensively (Miserendino et al., 1990; Kim et al., 1991; Maren et al., 1996; Walker and Davis, 2000; Rodrigues et al., 2001), no study has to date examined the role of VGCCs in fear conditioning. In the present experiments, we therefore gave rats intra-LA infusions of different doses of the selective L-type VGCC blocker verapamil before fear conditioning.
Blockade of VGCCs dose-dependently impairs fear memory formation
In the first experiment, rats were infused with vehicle (dH2O) or different doses of verapamil (0.04, 0.4, or 4.0 μg/side) before conditioning and tested for retention of auditory fear conditioning ∼24 hr later (Fig.4A). Infusion of dH2O as a vehicle did not appear to affect the amount of freezing in controls. Vehicle-treated rats froze 75–80% during retention tests, which is comparable with the level of freezing that we observed in previous studies in which controls have been infused with NaCl or ACSF (Wilensky et al., 1999; Schafe and LeDoux, 2000). Cannula tips were observed to lie throughout the rostrocaudal extent of the amygdala (Fig. 4B). Only rats with cannula tips at or within the borders of the LA or the basal amygdala were included in the data analysis.
Fig. 4.

Intra-LA infusion of an L-type VGCC antagonist dose-dependently impairs auditory fear conditioning. A, Schematic of behavioral protocol. B, Cannula tip placements from rats infused with dH2O (black squares), 0.04 μg of verapamil (white squares), 0.4 μg of verapamil (white circles), or 4 μg of verapamil (white triangles). Adapted fromPaxinos and Watson (1997). B, Basal nucleus of amygdala;CE, central nucleus of amygdala. C, Mean ± SE postshock freezing after the two conditioning trials in rats given intra-LA infusions of dH2O (vehicle;n = 10): 0.04 μg (n = 6), 0.4 μg (n = 8), or 4 μg (n = 9). D, Mean ± SE auditory fear memory 24 after conditioning in the same rats. *p < 0.05 relative to vehicle controls.
Infusion of verapamil had no effect on postshock freezing at any dose (Fig. 4C), suggesting that foot shock sensitivity and US processing in the LA were not disrupted by the drug. The ANOVA (drug dose by trial) for postshock freezing scores showed an effect only for trials (F(1,29) = 78.27;p < 0.01). The effect for dose and the interaction were not significant (p > 0.05). Twenty-four hours later, however, rats treated with verapamil showed a dose-dependent impairment of long-term memory (LTM) to the tone (Fig.4D). The ANOVA for LTM scores revealed a significant effect for group (drug dose) (F(3,29)= 16.08; p < 0.01). Tukey's post hoc t tests revealed that significant differences existed between vehicle controls and the two highest doses of verapamil (0.4 and 4 μg; p < 0.05). No difference was detected between the vehicle group and the lowest dose group (0.04 μg;p > 0.05). Thus, long-term retention of Pavlovian fear memory is dose-dependently disrupted by intra-LA infusion of verapamil.
Blockade of VGCCs selectively impairs LTM of fear conditioning
In the second experiment, we evaluated the role of L-type VGCCs in fear memory consolidation. That is, rats were infused with the highest dose of verapamil (4 μg/side), conditioned, and tested for auditory fear conditioning at 1, 3, 6, and 24 hr after conditioning (Fig.5A). For comparison with previous studies, in this latter experiment, we also included a group that was infused with 5 μg/side APV, a dose that has been shown recently to block both short-term memory (STM) and LTM of fear conditioning (Walker and Davis, 2000). Cannula tips were observed to lie throughout the rostrocaudal extent of the amygdala (Fig.5B).
Fig. 5.

Blockade of L-type VGCCs or NMDARs in the LA impairs memory formation of auditory fear conditioning in different ways. A, Schematic of behavioral protocol.B, Cannula tip placements from rats infused with dH2O (black squares), 4 μg of verapamil (white triangles), or 5 μg of APV (white circles). Adapted from Paxinos and Watson (1997).C, Mean ± SE postshock freezing between conditioning trials in rats given intra-LA infusions of dH2O (vehicle; n = 11), 4 μg of verapamil (n = 11), or 5 μg of APV (n = 8). D, Mean ± SE auditory fear memory assessed at 1 hr after conditioning in the rats fromC. E, Mean ± SE auditory fear memory assessed at 3 hr after conditioning in the rats fromC. F, Mean ± SE auditory fear memory assessed at 6 hr after conditioning in the rats fromC. G, Mean ± SE auditory fear memory assessed at 24 hr after conditioning in the rats fromC. Freezing during the 24 hr test is also expressed as a percentage of that during the 1 hr test (% of STM) for each rat (inset). *p < 0.05 relative to vehicle controls.
As in the first experiment, intra-LA infusion of 4 μg of verapamil had no effect on postshock freezing (Fig. 5C). However, consistent with previous studies (Lee and Kim, 1998), APV (5 μg) was observed to have a significant effect. The ANOVA (drug by trials) for postshock freezing scores showed a significant effect of drug (F(2,27) = 137.01; p< 0.01), a significant effect of trials (F(2,54) = 48.09; p < 0.01), and a significant drug by trials interaction (F(4,54) = 8.71; p < 0.01). Tukey's post hoc t tests showed that significant differences existed between the vehicle–verapamil groups and the APV group on each trial (p < 0.05). No significant differences were observed between the vehicle group and the verapamil group after any of the training trials (p > 0.05).
Figure 5D–F depicts retention of auditory fear at 1, 3, and 6 hr after conditioning. Although APV-infused rats were observed to have impaired fear memory at every time point, no significant differences were observed between the vehicle and 4 μg of verapamil-treated groups. The ANOVAs for the 1, 3, and 6 hr tests revealed a significant effect for drug (1 hr,F(2,27) = 57.45, p < 0.01; 3 hr, F(2,27) = 13.92,p < 0.01; 6 hr,F(2,27) = 10.35, p < 0.01). Tukey's post hoc t tests revealed that, for each test, the APV-treated group was significantly different from the vehicle–verapamil-treated groups (p < 0.05). No significant difference was detected between the vehicle and verapamil-treated groups at any time point (p > 0.05). Thus, postshock freezing, as well as STM, was not affected by intra-LA infusions of verapamil, which indicates that verapamil had no significant effect on either shock or tone sensitivity in the LA.
Twenty-four hours after conditioning, however, verapamil-treated rats showed substantial impairments in fear retention (Fig. 5G). The ANOVA for the 24 hr test revealed a significant effect for drug (F(2,27) = 8.86; p < 0.01). Tukey's post hoc t tests showed that the both the APV- and verapamil-treated groups were significantly different from the vehicle group (p < 0.05). No difference existed between verapamil-treated rats and those receiving APV (p > 0.05). To further analyze the memory impairment in verapamil-treated rats at 24 hr, we examined 24 hr freezing scores as a percentage of freezing scores for the 1 hr test in rats from the vehicle- and verapamil-treated groups (Fig.5G, inset). Once again, a significant difference was observed (t(20) = 3.32;p < 0.01).
Thus, intra-LA blockade of L-type VGCCs with verapamil impaired long-term memory formation of auditory fear conditioning (>24 hr) but had no effect on memory shortly after training. On the other hand, consistent with the results of previous studies (Walker and Davis, 2000; Rodrigues et al., 2001), blockade of NMDARs impaired both STM and LTM. Together, these findings suggest that NMDARs and L-type VGCCs contribute to fear memory formation in qualitatively different ways.
The effect of verapamil on fear memory formation is not altered when dissolved in saline vehicle
In our behavioral experiments, we dissolved the different doses of verapamil in dH20 because the highest dose (4 μg/side) was not readily soluble in ionic solution. Thus, it could be argued that our lack of an effect on acquisition and STM is attributable to either (1) less than optimal performance of our vehicle controls as a result of osmotic changes in the amygdala, bringing them down to the level of verapamil-treated rats and thus masking a difference in acquisition and STM, or (2) the drug temporarily coming out of solution during infusion into brain, which would make it unavailable at sufficient concentrations to affect acquisition or STM. To address these two possibilities, we performed an additional behavioral experiment using saline as a vehicle and a dose of verapamil (1 μg/side) that is soluble in saline. Rats were infused with either 0.9% NaCl (0.5 μl/side; n = 8) or verapamil (1 μg/side in 0.9% NaCl; n = 7) 10–15 min before receiving three conditioning trials. One hour after conditioning, rats were given an STM test consisting of presentation of five tones, followed ∼24 hr later by an LTM test consisting of 10 tones. For analysis, freezing scores across trials for both the STM and LTM tests were averaged into a single score.
Results (data not shown) showed that both groups of rats had equivalent STM at 1 hr (t(13) = 0.48;p = 0.63). The NaCl-infused rats exhibited 77 ± 4.7% freezing, whereas the verapamil-treated rats exhibited 80 ± 3.8% freezing. Twenty-four hours later, however, a significant difference was observed between groups (t(13) = 2.65; p = 0.01). The NaCl-infused rats exhibited 54.25 ± 6.8% freezing, whereas the verapamil-treated rats exhibited 29.07 ± 6.3% freezing. This difference was even more pronounced when LTM was expressed as a percentage of STM for each rat (t(13) = 3.04; p = 0.009; NaCl, 69.9 ± 6.6%; verapamil, 23.1 ± 8.7%). Thus, as in our previous experiments in which dH20 was used as a vehicle, verapamil impaired LTM of auditory fear conditioning, whereas STM was left intact.
DISCUSSION
The present series of experiments evaluated the roles of VGCCs and NMDARs in both the induction of LTP at thalamic input synapses in LAin vitro and the acquisition and memory formation of auditory fear conditioning. The findings indicate that two pharmacologically distinct forms of LTP in this pathway can be distinguished depending on the induction protocol used. Using a pairing protocol, in which presynaptic stimulation of thalamic input fibers is paired with brief postsynaptic depolarizations, we were able to induce a VGCC-dependent, NMDAR-independent form of LTP. Using a tetanus that produced prolonged depolarization of the postsynaptic cell but not spiking, we induced a VGCC-independent LTP that was blocked by the NMDAR antagonist APV, as well as the more selective NR2B subunit antagonist ifenprodil. In behavioral experiments, blockade of NMDARs in the LA before training produced deficits in both STM, as assessed at 1 hr, and in LTM, as assessed 24 hr later. Blockade of VGCCs, on the other hand, produced an impairment only at 24 hr, suggesting that VGCCs are selectively involved in LTM formation.
Two pharmacologically distinct forms of LTP at thalamic input synapses in the LA
Recent experiments in our laboratory have shown that LTP at thalamic input synapses to the LA is L-type VGCC dependent and NMDAR independent (Weisskopf et al., 1999). Those experiments used a pairing protocol in which subthreshold presynaptic stimulation of auditory afferents was paired with brief postsynaptic depolarizations (Magee and Johnston, 1997; Markram et al., 1997; Johnston et al., 1999; Nishiyama et al., 2000). In this protocol, BPAPs originating in the soma are thought to invade the dendrites and interact with EPSPs, leading to calcium influx through VGCCs (Magee and Johnston, 1997; Stuart et al., 1997; Johnston et al., 1999). Accordingly, LTP induced by pairing in the thalamic pathway is blocked by application of nifedipine, an L-type VGCC blocker (Weisskopf et al., 1999). In the present study, a second L-type VGCC blocker, verapamil, similarly blocked LTP induced by pairing, whereas the NMDAR antagonist APV had no significant effect. Thus, when EPSPs and brief depolarizations are induced repeatedly in a synchronous train, they appear to cause enough calcium influx through VGCCs to induce LTP independently of NMDARs (Weisskopf et al., 1999;Blair et al., 2001).
It is clear, however, that NMDARs are essential for fear conditioning (Miserendino et al., 1990; Kim et al., 1991; Maren et al., 1996; Walker and Davis, 2000; Rodrigues et al., 2001) and that an NMDAR-independent form of LTP in the LA is not adequate to account for fear conditioning at the cellular level. In the LA, NMDAR-dependent LTP has been induced by tetanic stimulation in the cortical input pathway (Huang and Kandel, 1998). However, no study has to date demonstrated the involvement of NMDARs in LTP induced in thalamic input synapses to the LA. In fact, previous studies have shown that NMDARs contribute to transmission in the LA (Rainnie et al., 1991; Danober and Pape, 1998;Mahanty and Sah, 1999; Weisskopf and LeDoux, 1999) and that application of APV can reduce synaptic transmission in the LA (Li et al., 1995, 1996; Weisskopf and LeDoux, 1999). In the present study, we used a tetanus protocol to induce LTP in the thalamic pathway, a protocol that, unlike the pairing protocol, does not trigger action potentials but instead produces a sustained depolarization of the postsynaptic cell. This type of LTP was effectively blocked by the NMDAR antagonist APV, as well as the more selective NR2B subunit antagonist ifenprodil, but spared by application of VGCC blockers. Moreover, whereas APV was observed, as in previous studies, to have effects on routine transmission in this pathway, ifenprodil had no effect on synaptic transmission. Thus, consistent with behavioral studies that have demonstrated a role for NMDARs (Miserendino et al., 1990; Kim et al., 1991; Maren et al., 1996; Walker and Davis, 2000) and the NR2B subunit (Rodrigues et al., 2001) in fear memory, NMDAR-dependent LTP exists at thalamic input synapses to the LA. As in the hippocampus, however, the involvement of NMDARs appears to be sensitive to the LTP induction protocol.
Although the present experiments show that it is possible to artificially induce LTP in the LA that selectively requires either NMDARs or VGCCs, this should not be taken to indicate that amygdala LTP must always depend on one of these mechanisms and not the other. In fact, as discussed previously, some forms of hippocampal LTP appear to require both types of receptor (Huber et al., 1995; Cavus and Teyler, 1996; Magee and Johnston, 1997; Morgan and Teyler, 1999). Furthermore, a recent study found that LTP in the LA at cortical input synapses is attenuated, but not completely blocked, by NMDAR antagonists (Huang and Kandel, 1998). That study used a protocol that involved pairing of low-frequency (0.2 Hz) presynaptic stimulation with prolonged depolarization of the postsynaptic cell by current injection. In contrast to our pairing protocol in which brief depolarizations of the postsynaptic cell were used, this prolonged depolarization protocol appeared to be sufficient to open the channel of NMDARs. Furthermore, although not explicitly tested, the present data would suggest that the NMDAR-independent component of this type of LTP might depend on VGCCs (Huang and Kandel, 1998).
Implications for fear memory formation
The LTP findings in the present study indicate that both L-type VGCCs and NMDARs are essential to synaptic plasticity in the LA but that their involvement depends on the LTP induction protocol used. In the behavioral studies, we showed that both NMDARs and L-type VGCCs are necessary for fear memory formation in the LA but in qualitatively different ways. Consistent with previous studies (Walker and Davis, 2000; Rodrigues et al., 2001), intra-LA blockade of NMDARs resulted in impaired STM and LTM. Blockade of L-type VGCCs, however, selectively impaired the formation of LTM. This latter finding is consistent with that of a recent report that demonstrated the involvement of L-type VGCCs in the maintenance of spatial memory over long, but not shorter, time periods (Borroni et al., 2000).
Because fear conditioning in the LA requires both NMDARs and L-type VGCCs, this suggests that a combination of the two in vitroLTP models is necessary to explain fear memory formation at the cellular level (Blair et al., 2001). Specifically, we propose that pairing of CS and US inputs during fear conditioning leads to calcium entry through both NMDARs and L-type VGCCs in LA principal cells. The increase in intracellular calcium through both types of channels ultimately, however, results in the formation of different kinds of memory. NMDARs, in particular the NR2B subunit, play an essential role in STM formation (Walker and Davis, 2000; Rodrigues et al., 2001), possibly by activation of local protein kinases that can phosphorylate existing membrane proteins (Soderling and Derkach, 2000). L-type VGCCs, on the other hand, play a selective role in LTM, possibly by activation of different protein kinases that can translocate to the nucleus and initiate gene expression and the synthesis of new proteins that are required for LTM formation (Blair et al., 2001; Schafe et al., 2001). In support of this hypothesis, a number of studies have implicated the involvement of L-type VGCCs in cAMP response element-binding protein (CREB) phosphorylation and CRE-driven gene expression in hippocampal neurons (Bading et al., 1993; Deisseroth et al., 1998; Mermelstein et al., 2000; Deisseroth and Tsien, 2002). More recently, it has been shown that stimulation of L-type VGCCs, but not NMDARs, leads to sustained nuclear CREB phosphorylation and CREB binding protein-driven gene expression via the Ca2+/calmodulin-dependent kinase IV (CaMKIV) signaling pathway (Hardingham et al., 2001). Furthermore, Greenberg and colleagues have demonstrated recently a selective role for L-type VGCCs in signaling to the nucleus to initiate CRE-mediated transcription via the extracellular signal-regulated kinase/mitogen-activated protein (ERK/MAP) kinase signaling pathway (Dolmetsch et al., 2001). Importantly, ERK/MAP kinase, CaMKIV, and CREB have all been shown to be necessary for the formation of long-term, but not short-term, fear memories (Bourtchuladze et al., 1994; Schafe et al., 2000; Josselyn et al., 2001; Kang et al., 2001). Additional experiments will be necessary to determine the contribution of L-type VGCCs to activation of protein kinases and CRE-driven gene expression in the LA after fear conditioning.
As in previous studies that have directly examined the relationship between LTP and memory in the amygdala, LTP in our verapamil-treated slices appears to decay with a much faster time course than fear memory after the same manipulation (Figs. 1, 5). This is not a novel finding but is in fact quite common in the LTP–behavior literature and one that we wrote about recently (Schafe et al., 2001). Many compounds that impair long-term but not short-term memory have been shown to block LTP within minutes after induction. In the in vitro amygdala preparation, for example, bath application of inhibitors of protein synthesis, PKA or ERK/MAPK activity begins to impair LTP immediately after induction (Huang et al., 2000), whereas intra-amygdala infusion of the same compounds in behavioral experiments results in memory impairment only at 24 hr after conditioning (Schafe and LeDoux, 2000;Schafe et al., 2000). It is not immediately clear what this pattern of findings could mean. However, it does not appear to be unique to pharmacological manipulations. The same pattern of findings is observed in the molecular genetic literature, in which genetic manipulation of the Ras/ERK/Rsk pathway results in complete decay of LTP in the amygdala with 30 min after induction, whereas fear memory in the same mice is intact for at least 1 hr after training (Brambilla et al., 1997). One likely possibility is that it is simply a quantitative difference created by the in vitro slice preparation (Schafe et al., 2001). Future experiments using in vivoelectrophysiological methods will be necessary to examine this question further.
The findings of the present study clearly suggest that NMDARs and L-type VGCCs make unique contributions to fear memory formation in the LA. However, many important questions remain to be addressed. For example, how do CS and US inputs engage these different channels during fear learning? How are these channels differentially involved in the activation of signal transduction pathways that promote short-term and long-term memory formation? Additional experiments will be required to address these questions. However, our ability to isolate two pharmacologically distinct types of LTP at thalamic input synapses in the LA and explicitly test hypotheses generated from those findings in behavioral studies of fear conditioning has distinct advantages. Not only does it allow us to address biophysical questions of cellular and synaptic activity as they relate to memory storage, but it may also allow us to determine the relative contribution of VGCCs and NMDARs in initiating second-messenger systems and/or gene expression that underlie different types of memory formation in the LA.
E.P.B. and G.E.S. contributed equally to this work.
This research was supported in part by National Institute of Mental Health Grants MH 46516, MH00956, MH 39774, and MH 11902 and a National Science Foundation Graduate Fellowship to E.P.B. The work was also supported by a grant from the W. M. Keck Foundation to New York University. We thank Kathryn Johnson and Annemieke Schoute for technical assistance. We also thank Hugh T. Blair for helpful comments on this manuscript.
Correspondence should be addressed to Elizabeth P. Bauer, Center for Neural Science, 4 Washington Place, Room 809, New York, NY 10003. E-mail: bauer@cns.nyu.edu.
REFERENCES
- 1.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 2.Bauer EP, LeDoux JE, Nader K. Fear conditioning and LTP in the lateral amygdala are sensitive to the same stimulus contingencies. Nat Neurosci. 2001;4:687–688. doi: 10.1038/89465. [DOI] [PubMed] [Google Scholar]
- 3.Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem. 2001;8:229–242. doi: 10.1101/lm.30901. [DOI] [PubMed] [Google Scholar]
- 4.Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ. Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J Neurosci. 2000;20:9272–9276. doi: 10.1523/JNEUROSCI.20-24-09272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourtchuladze R, Frenguelli B, Blendy J, Cioff D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 6.Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer VC, Cestari V, Rossi-Arnaud C, Grant SE, Chapman PF, Lipp H-P, Sturani E, Klein R. A role for the Ras signaling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- 7.Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- 8.Chapman PF, Bellavance LL. Induction of long-term potentiation in the basolateral amygdala does not depend on NMDA receptor activation. Synapse. 1992;11:310–318. doi: 10.1002/syn.890110406. [DOI] [PubMed] [Google Scholar]
- 9.Clugnet MC, LeDoux JE. Synaptic plasticity in fear conditioning circuits: induction of LTP in the lateral nucleus of the amygdala by stimulation of the medial geniculate body. J Neurosci. 1990;10:2818–2824. doi: 10.1523/JNEUROSCI.10-08-02818.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danober L, Pape H-C. Mechanisms and functional significance of a slow inhibitory potential in neurons of the lateral amygdala. Eur J Neurosci. 1998;10:853–867. doi: 10.1046/j.1460-9568.1998.00092.x. [DOI] [PubMed] [Google Scholar]
- 11.Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 12.Deisseroth K, Tsien RW. Dynamic multiphosphorylation passwords for activity-dependent gene expression. Neuron. 2002;34:179–182. doi: 10.1016/s0896-6273(02)00664-5. [DOI] [PubMed] [Google Scholar]
- 13.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 14.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 15.Fanselow MS, LeDoux JE. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron. 1999;23:229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- 16.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 17.Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- 18.Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- 19.Huang YY, Martin KC, Kandel ER. Both protein kinase A, mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci. 2000;20:6317–6325. doi: 10.1523/JNEUROSCI.20-17-06317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huber KM, Mauk MD, Kelly PT. Distinct LTP induction mechanisms: contribution of NMDA receptors and voltage-dependent calcium channels. J Neurophysiol. 1995;73:270–279. doi: 10.1152/jn.1995.73.1.270. [DOI] [PubMed] [Google Scholar]
- 21.Johnston D, Hoffman DA, Colbert CM, Magee JC. Regulation of back-propagating action potentials in hippocampal neurons. Curr Opin Neurobiol. 1999;9:288–292. doi: 10.1016/s0959-4388(99)80042-7. [DOI] [PubMed] [Google Scholar]
- 22.Josselyn SA, Shi C, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang H, Sun LD, Atkins CM, Soderling TR, Wilson MA, Tonegawa S. An important role of neural activity-dependent CaMKIV signaling in the consolidation of long-term memory. Cell. 2001;106:771–783. doi: 10.1016/s0092-8674(01)00497-4. [DOI] [PubMed] [Google Scholar]
- 24.Kim JJ, DeCola JP, Landeira-Fernandez J, Fanselow MS. N-methyl-d-aspartate receptor antagonist APV blocks acquisition but not expression of fear conditioning. Behav Neurosci. 1991;105:126–133. doi: 10.1037//0735-7044.105.1.126. [DOI] [PubMed] [Google Scholar]
- 25.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 26.Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci. 1998;18:8444–8454. doi: 10.1523/JNEUROSCI.18-20-08444.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XF, Philips R, LeDoux JE. NMDA and non-NMDA receptors contribute to synaptic transmission between the medial geniculate body and the lateral nucleus of the amygdala. Exp Brain Res. 1995;105:87–100. doi: 10.1007/BF00242185. [DOI] [PubMed] [Google Scholar]
- 28.Li XF, Stutzmann GE, LeDoux JE. Convergent but temporally separated inputs to lateral amygdala neurons from the auditory thalamus and auditory cortex use different postsynaptic receptors: in vivo intracellular and extracellular recordings in fear conditioning pathways. Learn Mem. 1996;3:229–242. doi: 10.1101/lm.3.2-3.229. [DOI] [PubMed] [Google Scholar]
- 29.Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- 30.Mahanty NK, Sah P. Excitatory synaptic inputs to pyramidal neurons of the lateral amygdala. Eur J Neurosci. 1999;11:1217–1222. doi: 10.1046/j.1460-9568.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- 31.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 32.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 33.Maren S, Aharonov G, Stote DL, Fanselow MS. N-methyl-d-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 34.Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs, EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 35.McDonald AJ. Cell types and intrinsic connections of the amygdala. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotion, memory, and mental dysfunction. Wiley; New York: 1992. pp. 67–96. [Google Scholar]
- 36.McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 37.Mermelstein PG, Bito H, Deisseroth K, Tsien RW. Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci. 2000;20:266–273. doi: 10.1523/JNEUROSCI.20-01-00266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- 39.Miserendino MJ, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 40.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 41.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 42.Morgan SL, Teyler TJ. VDCCs and NMDARs underlie two forms of LTP in CA1 hippocampus in vivo. J Neurophysiol. 1999;82:736–740. doi: 10.1152/jn.1999.82.2.736. [DOI] [PubMed] [Google Scholar]
- 43.Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- 44.O'Regan MH, Kocsis DJ, Waxman SG. Depolarization-dependent actions of dihydropyridines on synaptic transmission in the in vitro rat hippocampus. Brain Res. 1990;527:181–191. doi: 10.1016/0006-8993(90)91136-5. [DOI] [PubMed] [Google Scholar]
- 45.Paré D, Pape HC, Dong J. Bursting and oscillating neurons of the cat basolateral amygdaloid complex in vivo: electrophysiological properties and morphological features. J Neurophysiol. 1995;74:1179–1191. doi: 10.1152/jn.1995.74.3.1179. [DOI] [PubMed] [Google Scholar]
- 46.Paxinos G, Watson C. The rat brain in stereotaxic coordinates: computer graphics files, Ed 3. Academic; San Diego: 1997. [Google Scholar]
- 47.Quirk GJ, Repa C, LeDoux JE. Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron. 1995;15:1029–1039. doi: 10.1016/0896-6273(95)90092-6. [DOI] [PubMed] [Google Scholar]
- 48.Quirk GJ, Armony JL, LeDoux JE. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron. 1997;19:613–624. doi: 10.1016/s0896-6273(00)80375-x. [DOI] [PubMed] [Google Scholar]
- 49.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Excitatory transmission in the basolateral amygdala. J Neurophysiol. 1991;66:986–998. doi: 10.1152/jn.1991.66.3.986. [DOI] [PubMed] [Google Scholar]
- 50.Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE. Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci. 2001;4:724–731. doi: 10.1038/89512. [DOI] [PubMed] [Google Scholar]
- 51.Rodrigues SM, Schafe GE, LeDoux JE. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci. 2001;21:6889–6896. doi: 10.1523/JNEUROSCI.21-17-06889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15:127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 53.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 54.Romanski LM, Clugnet MC, Bordi F, LeDoux JE. Somatosensory and auditory convergence in the lateral nucleus of the amygdala. Behav Neurosci. 1993;107:444–450. doi: 10.1037//0735-7044.107.3.444. [DOI] [PubMed] [Google Scholar]
- 55.Rosenblum K, Dudai Y, Richter-Levin G. Long-term potentiation increases tyrosine phosphorylation of the N-methyl-d-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc Natl Acad Sci USA. 1996;93:10457–10460. doi: 10.1073/pnas.93.19.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenblum K, Berman DE, Hazvi S, Lamprecht R, Dudai Y. NMDA receptor and the tyrosine phosphorylation of its 2B subunit in taste learning in the rat insular cortex. J Neurosci. 1997;17:5129–5135. doi: 10.1523/JNEUROSCI.17-13-05129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rostas JA, Brent VA, Voss K, Errington ML, Bliss TV, Gurd JW. Enhanced tyrosine phosphorylation of the 2B subunit of the N-methyl-d-aspartate receptor in long-term potentiation. Proc Natl Acad Sci USA. 1996;93:10452–10456. doi: 10.1073/pnas.93.19.10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20 2000. RC96(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schafe GE, Nader K, Blair HT, LeDoux JE. Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 2001;24:540–546. doi: 10.1016/s0166-2236(00)01969-x. [DOI] [PubMed] [Google Scholar]
- 61.Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- 62.Stuart G, Spruston N, Sakmann B, Hausser M. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci. 1997;20:125–131. doi: 10.1016/s0166-2236(96)10075-8. [DOI] [PubMed] [Google Scholar]
- 63.Stuart GJ, Hausser M. Dendritic coincidence detection of EPSPs and action potentials. Nat Neurosci. 2001;4:63–71. doi: 10.1038/82910. [DOI] [PubMed] [Google Scholar]
- 64.Sweatt JD. Toward a molecular explanation for long-term potentiation. Learn Mem. 1999;6:399–416. doi: 10.1101/lm.6.5.399. [DOI] [PubMed] [Google Scholar]
- 65.Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 66.Walker DL, Davis M. Involvement of NMDA receptors within the amygdala in short- versus long-term memory for fear conditioning as assessed with fear-potentiated startle. Behav Neurosci. 2000;114:1019–1033. [PubMed] [Google Scholar]
- 67.Weisskopf MG, LeDoux JE. Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol. 1999;81:930–934. doi: 10.1152/jn.1999.81.2.930. [DOI] [PubMed] [Google Scholar]
- 68.Weisskopf MG, Bauer EP, LeDoux JE. L-Type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci. 1999;19:10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wilensky AE, Schafe GE, LeDoux JE. Functional inactivation of amygdala before but not after auditory fear conditioning disrupts memory formation. J Neurosci 19 1999. RC48(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang XX, Bunney BS, Shi WX. Enhancement of NMDA-induced current by the putative NR2B selective antagonist ifenprodil. Synapse. 2000;37:56–63. doi: 10.1002/(SICI)1098-2396(200007)37:1<56::AID-SYN6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]