

Abstract
Aggregated and oligomeric amyloid β-protein (Aβ) is known to exhibit neurotoxicity. However, the action of Aβ monomers on neurons is not fully understood. We have studied aggregation state-dependent actions of Aβ and found an oligomer-specific effect of Aβ on lipid metabolism in neurons (Michikawa et al., 2001). Here, we show a novel function of monomeric Aβ1–40, which is the major species found in physiological fluid, as a natural antioxidant molecule that prevents neuronal death caused by transition metal-induced oxidative damage. Monomeric Aβ1–40, which is demonstrated by SDS-PAGE after treatment with glutaraldehyde, protects neurons cultured in a medium containing 1.5 μm Fe(II) without antioxidant molecules. Metal ion chelators such as EDTA, CDTA (trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid), and DTPA (diethylenetriamine-N,N,N′,N",N"-penta-acetic acid, an iron-binding protein, transferrin, and antioxidant scavengers such as catalase, glutathione, and vitamin E also inhibit neuronal death under the same conditions. Monomeric Aβ1–40 inhibits neuronal death caused by Cu(II), Fe(II), and Fe(III) but does not protect neurons against H2O2-induced damage. Monomeric Aβ1–40 inhibits the reduction of Fe(III) induced by vitamin C and the generation of superoxides and prevents lipid peroxidation induced by Fe(II). Aβ1–42 remaining as a monomer also exhibits antioxidant and neuroprotective effects. In contrast, oligomeric and aggregated Aβ1–40 and Aβ1–42 lose their neuroprotective activity. These results indicate that monomeric Aβ protects neurons by quenching metal-inducible oxygen radical generation and thereby inhibiting neurotoxicity. Because aggregated Aβ is known to be an oxygen radical generator, our results provide a novel concept that the aggregation-dependent biological effects of Aβ are dualistic, being either an oxygen radical generator or its inhibitor.
Keywords: Alzheimer's disease, amyloid β-protein, transition metals, oxygen radicals, antioxidant, neuronal death
One of the neuropathological hallmarks of Alzheimer's disease (AD) is the formation of extracellular amyloid deposits (Selkoe, 1994). The major component of the amyloid deposits is the 39–42 amino acid peptide of the amyloid β-protein (Aβ) (Glenner and Wong, 1984; Masters et al., 1985). One of the Aβ species, ending with a C terminus at residue 40 (Aβ1–40), is the predominant soluble species in biological fluids (Vigo-Pelfrey et al., 1993; Ida et al., 1996). The longer form of Aβ, ending at residue 42 (Aβ1–42), accumulates initially and predominantly in parenchymal plaques (Roher et al., 1993; Iwatsubo et al., 1994). Aβ1–42 is normally produced and secreted by cells in much lower quantities than Aβ1–40, which represents ∼90% of the total secreted Aβ. It is believed that aggregated Aβ exerts neurotoxicity and initiates the progressive pathophysiology of AD (Mattson et al., 1993; Pike et al., 1993; Lorenzo and Yankner, 1994;Hartley et al., 1999). However, the function of monomeric Aβ on neurons is not yet fully understood.
It has been reported that the levels of metals such as zinc, iron, and copper are significantly concentrated in senile plaques (Smith et al., 1997; Lovell et al., 1998b). These observations followed original reports showing that these metals promote Aβ aggregation (Bush et al., 1994a,b; Huang et al., 1997; Atwood et al., 1998), which is reversed by treatment with chelators in vitro (Huang et al., 1997) and in vivo (Cherny et al., 2001). In support of these findings, a recent study has clearly demonstrated that zinc and copper induce non-β-sheeted Aβ aggregation but inhibit β-sheeted aggregation and fibril formation (Yoshiike et al., 2001). Other studies have suggested that accumulated metals support the AD pathology as a possible source of reactive oxygen radicals (Smith et al., 1997; Lovell et al., 1998b; Sayre et al., 2000).
Recent studies showed that the surrounding regions of Aβ deposits in brains of patients with AD and Down's syndrome have no damage (Nunomura et al., 2000, 2001) and that there is an inverse correlation between Aβ burden and the levels of oxidized nucleic acids in the AD brain (Cuajungco et al., 2000b). Although aggregated Aβ is reported to generate free radicals (Hensley et al., 1994; Schubert and Chevion, 1995; Kay, 1997; Huang et al., 1999a; Monji et al., 2001b), these lines of evidence imply a new function of Aβ other than that of a radical generator. A previous report has suggested its antioxidant activity for lipoproteins (Kontush et al., 2001); however, no explanation has been provided as to the mechanism behind the disparate results from different laboratories regarding Aβ-induced oxidative stress versus others suggesting antioxidant properties.
In light of the above, we have studied the aggregation state-dependent actions of Aβ on neurons (Michikawa et al., 2001; Gong et al., 2002). Here, we show that monomeric Aβ1–40 and also Aβ1–42 serve as antioxidant molecules protecting neurons from oxygen radicals generated in a metal-dependent manner, providing new insights into the strategy for developing a therapy for patients with AD.
MATERIALS AND METHODS
Reagents and preparation. Synthetic human Aβ1–40 was purchased from Peptide Institute Inc. (Osaka, Japan; lot numbers 510116 and 501001) and Bachem (Bubendorf, Switzerland; lot number 0538913). Aβ40–1 (lot number D539530) was purchased from Bachem, and Aβ1–42 (lot number 510523), Aβ1–16 (lot number 490704), and Aβ25–35 (lot number 500701) were purchased from Peptide Institute Inc. Aβ1–40, Aβ1–42, and Aβ25–35 were dissolved in DMSO at 2 mm and then diluted with distilled water to a concentration of 200 μm. Although the solution was clear, it is known that an Aβ solution contains short fibrils (Naiki et al., 1998). To remove short fibrils, Aβ solutions were centrifuged at 100,000 × g for 1 hr at 4°C, using a Beckman Optima TLX table ultracentrifuge and a Beckman TLA-120.2 fixed-angle rotor. Aβ1–16 was directly dissolved in water to a concentration of 200 μm. Oligomeric Aβ1–40 was prepared as described previously (Michikawa et al., 2001). Transferrin, insulin, progesterone, putrescine, selenite, superoxide dismutase (SOD), catalase, glutathione, vitamin E, and vitamin E acetate were obtained from Sigma (St. Louis, MO). The B27 supplement and B27 minus antioxidants (B27-AO) were purchased from Invitrogen (Grand Island, NY). EDTA was purchased from Eastman Kodak Company (Rochester, NY).trans-1,2-diaminocyclohexane-N,N,N′,N′-tetra-acetic acid (CDTA), diethylenetriamine-N,N,N′,N",N"-penta-acetic acid (DTPA), iron sulfate heptahydrate, iron nitrate nonahydrate, and copper sulfate pentahydrate were obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Monoclonal antibody, namely, anti-4-hydroxy-2-nonenal (4-HNE) antibody, which recognizes oxidized 4-HNE, was purchased from NOF Corporation (Tokyo, Japan).
Cell culture. All experiments were performed in compliance with existing laws and the institutional guidelines. Cerebral cortical neuronal cultures were prepared from Sprague Dawley rats at embryonic day 17 as described previously (Michikawa and Yanagisawa, 1998). The dissociated single cells were suspended in a feeding medium and plated onto poly-d-lysine-coated 12-well plates at a cell density of 5 × 10−5. The feeding medium consisted of DMEM/F12 containing 0.1% bovine albumin fraction V solution (Invitrogen) and N2 (Bottenstein and Sato, 1979), B27, or B27-AO supplements.
Quantification of neuron survival. For assessment of cell viability of cultured neurons, phase-contrast photomicrographs were taken before treatment and at various time points after treatment. The number of viable neurons on each micrograph was determined in premarked microscope fields (10× objective). Viable neurons were identified on the basis of morphological criteria. Neurons with intact neurites with uniform diameter and soma with a smooth round appearance were considered viable, whereas neurons with fragmented neurites and shrunken cell bodies were considered nonviable. In a pilot study, cell viability was confirmed by testing cell membrane permeability using propidium iodide (PI) or by staining with a viable cell-specific marker, calcein AM, as described previously (Michikawa and Yanagisawa, 1998). Neurons were stained with Hoechst 33342 (bis-benzamide; 2.5 μg/ml), to visualize their nuclear morphology. Most neurons died during culturing in DMEM/F12 medium supplemented with B27-AO (B27-AO medium) or DMEM/F12 medium supplemented with N2 and FeSO4, CuSO4, or Fe(NO3)3. For each determination of cell viability, 1000–1400 cells were counted.
Thioflavin-T binding assay for aggregated Aβ. Determination of the aggregated state of Aβ in solution was performed on the basis of a previously established method (LeVine, 1995;LeVine, 1999). The conditioned media, in which the cultures were incubated with Aβ, were collected. Each well contained 50 μl of each medium in 1 ml per well of 5 μmthioflavin-T in 50 mm glycin-NaOH, pH 8.5. Steady-state fluorescence intensities for each sample were determined in 48-well plates with a multiplate reader (Fluoroskan Ascent, Labsystems Inc., Franklin, MA) (excitation 446 nm, emission 490 nm). The culture media to which Aβ was not added were used as the background.
Cross-linking of Aβ with glutaraldehyde. SDS-PAGE of cross-linked fAβ1–40 and iAβ1–40 was performed as described previously (Levine, 1995). Briefly, 8 μg of each peptide in a stock solution (200 μm) was diluted to 35 μl with H2O. One-tenth volume of glutaraldehyde (3.5 μl of 0.625% diluted from a 25% stock solution) was added to each solution followed by the addition of an excess amount of NaBH4 (10 μl of 0.175m, 6.6 mg/ml, in 0.1 mNaOH). After 10 min of incubation, 15 μl of the SDS-PAGE sample buffer containing 100 mm dithiothreitol and 20% sucrose was added. Boiling of the amyloid peptides in the sample buffer was avoided, because SDS-resistant multimeric complexes are formed from non-cross-linked peptides during heating in SDS (LeVine, 1995). Then, 20 μl of the mixture was subjected to SDS-PAGE using a 4–20% gradient gel as described previously (Michikawa et al., 2001). The gel was then visualized by silver staining. To compare the aggregation status of Aβ1–40 and Aβ1–42 in 8 mm sodium phosphate, pH 7.4, or DMEM/F12 medium, freshly dissolved Aβ1–40 and Aβ1–42 were incubated at 20 μm for 3 hr at 37°C in each solution. The protein concentration of each solution of Aβ1–40 and Aβ1–42 was determined, 2.8 μg of each peptide was subjected to cross-linking, and the peptides were subjected to electrophoresis and silver staining.
Iron reduction assays. Assays were performed according to a previously reported method (Huang, 1999a). Aβ1–40 (10 μm), Aβ1–42 (10 μm), DTPA (10 and 300 μm) or Fe(III) (25 μm), and 3-(2-pyridyl)-5,6-bis(4-sulfo-phenyl)-1,2,4-triazine (PDT) (250 μm) were added to 1 ml of 8 mm sodium phosphate, pH 7.4, and rotated at 25°C for 6 hr. Vitamin C (10 μm) was then added, and the mixture was further incubated at 37°C for 14 hr. A solution containing Fe(III) and Aβ1–40 at the same concentrations in the absence of the indicator PDT was used to determine the background levels of this assay system. The absorbance at 562 nm indicates the amount of reduced iron ion, Fe(II).
Measurement of superoxide levels. The levels of intracellular superoxide anion radicals were measured using hydroethidium (HE), which is oxidized to a fluorescent ethidium cation by superoxides, using methods similar to those described previously (Bindokas et al., 1996). In brief, cells were incubated for 30 min in the presence of 5 μm HE (Molecular Probes, Eugene, OR) at 37°C in 5% CO2 atmosphere, and confocal images of cell-associated HE fluorescence were acquired (excitation = 488 nm and emission >560 nm).
Western blot analysis for determination of lipid peroxidation. Cerebral cortices of Sprague Dawley rats at embryonic day 17 were isolated and minced with a cutter and incubated in PBS in the presence of 3 or 5 μm Fe(II) with or without fAβ1–40 at 10 μm for 4 hr at 37°C. The fragments were then homogenized in RIPA buffer (150 mm NaCl, 10 mm Tris-HCl, pH 7.5, 1% Nonidet P-40, 0.1% SDS, and 0.25% sodium deoxycholate) and centrifuged at 10,000 × g for 10 min at 4°C, and the supernatants were recovered. Protein concentrations of the supernatants were determined by the BCA method (Pierce, Rockford, IL). Western blot analysis was performed according to the methods described previously (Michikawa et al., 2001). In brief, 24 μg of each protein was separated by 4–20% gradient SDS-PAGE and electrotransferred onto nitrocellulose membranes. The membranes were incubated with monoclonal primary antibody, anti-4-HNE antibody, at 2 μg/ml overnight at 4°C. The membranes were then washed in PBS containing 0.05% Tween 20 (PBS-T) three times, followed by incubation with HRP-conjugated goat anti-mouse IgG (1:5000 dilution) for 1 hr at room temperature. The membranes were washed four times in PBS-T and visualized with an ECL kit (Amersham Pharmacia Biotech, Buckinghamshire, UK).
RESULTS
Freshly dissolved Aβ1–40 protects neurons against death induced by antioxidant-depleted medium
We studied the effect of Aβ1–40 on neuronal viability. The medium used was DMEM nutrient mixture (DMEM/F12, 50:50) containing B27-AO. When neurons were incubated in the B27-AO medium, cultured neurons appeared healthy 40 hr after plating; however, neuronal death was induced 48 hr after plating, and most of the cells were dead 64 hr after plating (Fig.1a–c). In contrast, neuronal death was inhibited in the presence of freshly dissolved Aβ1–40 (fAβ1–40) at a concentration of 5 μm 64 hr after plating (Fig. 1d). Addition of DMSO at a final concentration of 1% DMSO to the B27-AO medium did not prevent or accelerate neuronal death (Fig.1e). Figure 1e shows the time-dependent curves of neuronal viability of the cultures treated with fAβ1–40 at various concentrations. The neuronal death induced by incubation in the B27-AO medium was inhibited by fAβ1–40 in a dose-dependent manner. Neuronal viability was maintained at the initial levels when the cultures were treated with fAβ1–40 at concentrations of 10 and 20 μm until 4 d after the commencement of the treatment (Fig. 1e). The neurons at each time point were stained with Hoechst 33342 and PI. The viable neurons at culture day 2 had larger swollen cell bodies (Fig. 1b), and the nuclei of dead neurons were shrunken as demonstrated by Hoechst 22336 and PI staining (Figs. 1f,h). The effect of freshly prepared Aβ1–42 on neuronal viability was also examined. fAβ1–42 could not inhibit neurotoxicity but rather promoted neuronal death (Fig. 1e).
Fig. 1.

fAβ1–40 inhibits neuronal death induced in an antioxidant-depleted medium. Rat cortical neurons were cultured in the B27-AO medium without Aβ1–40 treatment: a, 40 hr culture; b, 48 hr culture;c, 64 hr culture; d, with fAβ1–40 (5 μm) treatment 4 hr after plating, 64 hr culture.a–c are the same microscope field.e, Cell viability in the cultures treated with Aβ: none (●); DMSO vehicle (■); Aβ1–40 at 2 μm (▪), 10 μm (▵), and 20 μm (○); and Aβ1–42 at 10 μm (▴). Data represent means ± SE;n = 6 each. Six independent experiments showed similar results. Representative Hoechst staining showing the nuclear morphology of cortical neurons with or without Aβ1–40 treatment is shown in f, g. Cell membrane permeability is indicated by PI staining in h,i.
Monomeric Aβ1–40, but not oligomeric Aβ1–40, has an ability to protect neurons in the B27-AO medium
We examined the effect of incubated Aβ1–40 (iAβ1–40), which was filtered and the protein concentration of which was determined before addition, on neuronal viability cultured in the B27-AO medium. As shown in Figure 2a, neuronal death occurred 72 hr after the commencement of the treatment, which was completely inhibited by fAβ1–40, but not by iAβ1–40, at a concentration of 5 μm. Results of the quantitative analysis of neuronal viability within 72 hr of incubation are shown in Figure 1b, showing that fAβ1–40 at concentrations of 5 and 10 μm completely inhibited neuronal death, whereas iAβ1–40 inhibited neuronal death at 10 μm but not at 5 μm. To determine the oligomeric state of Aβ, the reaction of the conditioned medium of each culture with thioflavin-T was determined. As shown in Figure 2c, the value of the conditioned medium of the cultures treated with iAβ1–40 was significantly higher than that treated with fAβ1–40, indicating that iAβ1–40 contains highly oligomerized Aβ. To determine more directly that the amount of monomeric Aβ was decreased and that of oligomeric Aβ was increased, a cross-linking study of each Aβ sample was performed. As shown in Figure 2d, fAβ1–40 contains mostly monomers, whereas iAβ1–40 contains many oligomers, including dimers, trimers, and tetramers, in addition to decreased levels of monomers. These results indicate that Aβ monomers have a neuroprotective activity and that the lack of neuroprotective activity of iAβ1–40 at 5 μm is not caused by its toxic effect on neurons but rather by the low levels of monomers.
Fig. 2.

fAβ1–40 but not iAβ1–40 protects neurons in the B27-AO medium. Neurons were treated with fAβ1–40 or iAβ1–40 24 hr after plating. Phase-contrast photomicrographs were taken (a), and the cell viability was determined (b) 48 hr after the commencement of each treatment. a, p < 0.0001 versus fAβ1–40 (5 μm), iAβ1–40 (10 μm), and fAβ1–40 (10 μm). c, Thioflavin-T fluorescence with the conditioned medium of each cultured neuron treated with fAβ1–40 or iAβ1–40 for 3 d. b,p < 0.01 versus iAβ1–40. d, Detection of oligomeric Aβ in fAβ and iAβ samples by cross-linking with glutaraldehyde. fAβ1–40 or iAβ1–40 (2.5 μg) was cross-linked with glutaraldehyde and subjected to a 4–20% SDS-PAGE. The gel was then visualized by silver staining.
Metal-binding protein and metal chelators inhibit neuronal death in the B27-AO medium
Because neurotoxicity was induced in the media deficient of antioxidant reagents, it is reasonable to assume that the Aβ-mediated neuronal protection may possibly be explained by an antioxidant action of Aβ. Antioxidant actions include a direct antioxidant effect, and the indirect actions of fAβ1–40 include quenching of metal ions to inhibit secondary generation of free radicals. Thus, we examined the effect of molecules that have antioxidant activities. As shown in Figure 3a, catalase, glutathione, vitamin E acetate, and vitamin E inhibited neuronal death at culture day 2. Catalase and vitamin E acetate and vitamin E partially and completely inhibited cell death at culture day 4, respectively; however, SOD did not show any neuroprotective activity. Because we have observed that the N2 supplements (Bottenstein and Sato, 1979) inhibited neuronal death induced by incubation in the B27-AO medium, we examined the inhibitory effect of each component of N2 supplements. Figure 3b shows that among the components examined, only transferrin successfully inhibited neuronal death. Because transferrin is known to bind iron, inhibiting cell death by quenching the iron-dependent generation of reactive oxygen radicals (Halliwell and Gutteridge, 1989), it is reasonable to postulate that iron in DMEM/F12 plays a critical role in neuronal death in the B27-AO medium. Thus, we next examined the effect of various iron chelators on neuronal death under these conditions. EDTA (400 μm), CDTA (40 μm), and DTPA (8 μm) protected neurons against toxicity induced in the B27-AO medium at culture day 4 (Table1).
Fig. 3.

Transferrin and antioxidant scavengers inhibit neuronal death that occurred in the B27-AO medium. a, SOD (1500 U/ml), catalase (21,600 U/ml), glutathione (450 μg/ml), vitamin E acetate (1 μg/ml), or vitamin E (1 μg/ml) was added to neuronal cultures maintained in the B27-AO medium 4 hr after plating.b, N2 supplements or each component of N2 supplements, transferrin (100 μg/ml), insulin (5 μg/ml), progesterone (0.0063 μg/ml), putrescine (16.11 μg/ml), and selenite (0.0052 μg/ml) was added to neuronal cultures maintained in the B27-AO medium 4 hr after plating. Neuronal viability was determined as described in Materials and Methods at culture days 1, 2, and 4 (a) or 1, 2, and 7 (b).
Table 1.
Metal chelators inhibit neuronal death induced in the B27-AO medium
| Chelators | Concentration (μm) | Viability (% of control) |
|---|---|---|
| None | 0 | 0 |
| EDTA | 400 | 60 ± 3 |
| CDTA | 40 | 99 ± 4 |
| DTPA | 8 | 93 ± 7 |
Primary cortical neurons were cultured in the B27-AO medium. Four hours after plating, the cultures were incubated with metal chelators. Cell viability was determined 48 h after the start of treatment. The data represent means ± SE. n = 6 each. Three independent experiments showed similar results.
Monomeric Aβ1–40 protects neurons against iron- and copper-mediated neuronal toxicity
Because the culture medium, DMEM/F12 supplemented with B27-AO, contains 1.5 μm Fe (II), 124 nm Fe(III), and 5.2 nm Cu(II), our findings that neuronal death induced in the B27-AO medium is prevented by antioxidant scavengers, metal chelators, and transferrin indicate that neurotoxicity is induced by oxygen radicals generated in an Fe(II)-mediated manner. Thus, we next determined whether transition metal ions, such as iron and copper ions, and H2O2 exhibit neurotoxicity on cultured neurons, and whether fAβ1–40 has the ability to prevent this toxicity. Twenty-four hours after plating, neuronal cultures were treated with 1.5 μmCuSO4, 3.0 μmFeSO4, 25 μmFe(NO3)3, and 30 μm H2O2 in the presence or absence of 5 μm fAβ1–40 in the N2 medium. After 24 hr incubation, photographs were taken, and the neuronal viability was determined. As shown in Figure4, Cu(II), Fe(II), Fe(III), and H2O2 caused cell death (a, c, e, and g, respectively). fAβ1–40 at a concentration of 5 μm inhibited cell death caused by these metals (Fig. 4b,d,f) but did not inhibit cell death caused by H2O2 (Fig. 4h), indicating that protection of neurons by fAβ1–40 is not caused by a direct antioxidant activity but by an indirect one via interaction with metal ions. The quantitative analysis of these experiments is shown in Figure 4i. We performed additional experiments to determine the effect of transferrin on neuronal toxicity induced by these metals. We found that 3.8 μm transferrin inhibited 3.0 μm Fe(II)- and 25 μmFe(III)-mediated neurotoxicity, but even 13 μmtransferrin did not inhibit 1.5 μmCu(II)-mediated neurotoxicity (data not shown), supporting the idea that Fe(II) but not Cu(II) is responsible for the generation of oxygen radicals and induces toxicity. This is supported by the fact that the B27-AO medium contains 1.5 μm Fe(II), which is sufficiently high to induce cell toxicity, whereas it contains much lower concentrations of Cu(II) and Fe(III).
Fig. 4.
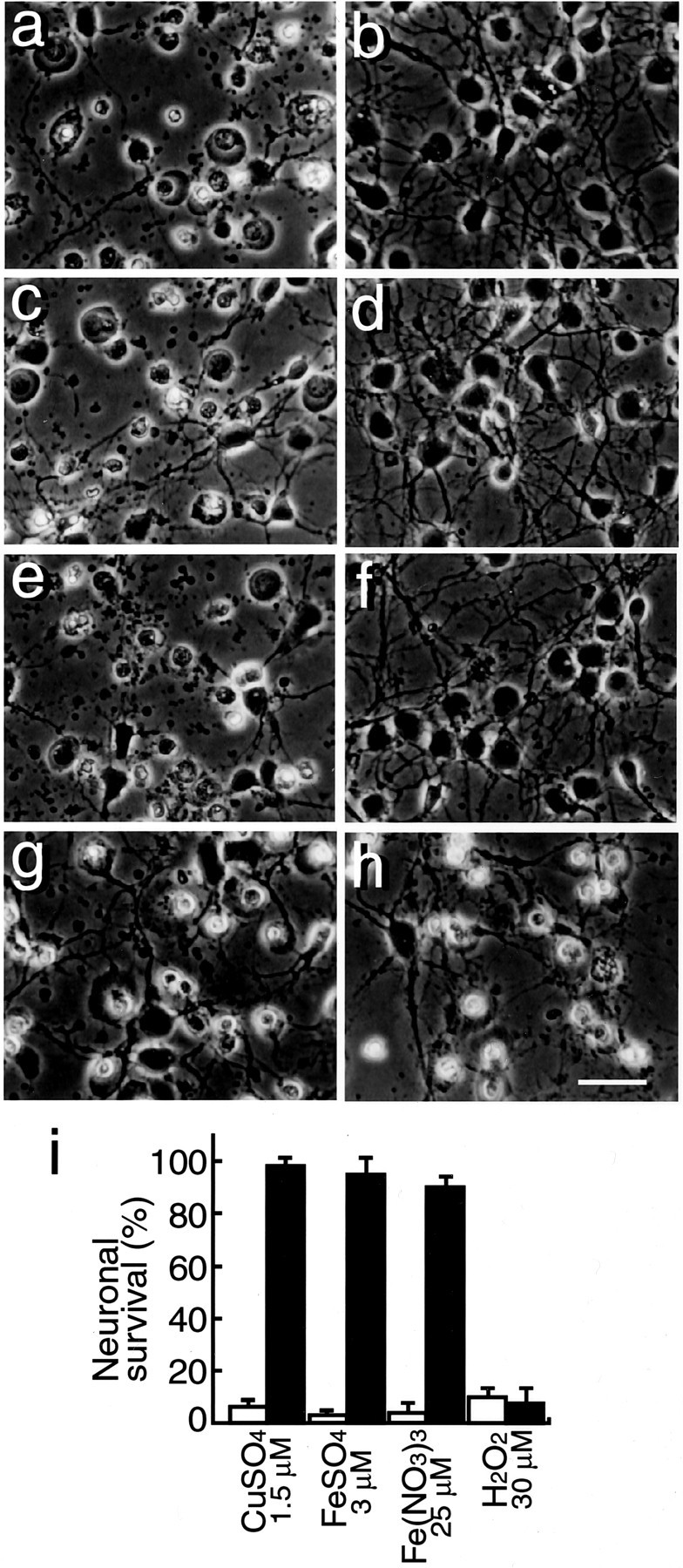
f Aβ1–40 inhibits neuronal death induced by transition-metal ions. Neurons were cultured in N2 medium for 24 hr, followed by treatment with 1.5 μm CuSO4(a, b), 3.0 μm Fe SO4 (c, d), 25 μm Fe(NO3)3, (e, f), and 30 μmH2O2 (g,h), and incubated for another 24 hr; photographs were taken to determine cell viability. a, c,e, and g represent control cultures, andb, d, f, andh represent neurons treated with fAβ1–40 (5 μm) in addition to metal ions. i, Quantitative analysis of these treatments 24 hr after the commencement of the metal treatment. Open and closed bars indicate cell viability in the cultures in the absence or presence, respectively, of fAβ1–40 (5 μm).
Monomeric Aβ1–40 inhibits vitamin C-mediated reduction of Fe(III)
Because reduced metal ions are known to generate oxygen radicals that initiate subsequent reactions of radical productions (Halliwell and Gutteridge, 1984), we determined whether fAβ1–40 has any effect on Fe(III) reduction. To examine the inhibitory effect of fAβ1–40 on Fe(III) reduction, a vitamin C-mediated metal reduction system was used. As shown in Figure 5a, fAβ1–40 inhibited Fe(III) reduction mediated by vitamin C. In addition to fAβ1–40, fAβ1–42, Aβ1–16, Aβ25–35, and a metal ion chelator, DTPA, also inhibited Fe(III) reduction. Because the action of Aβ is known to depend on the state of aggregation of the peptides, we next determined the aggregation states of Aβ used in this study by cross-linking of peptides with glutaraldehyde and subsequent silver staining. As shown in Figure 5b, most of both Aβ1–40 and Aβ1–42 incubated in 8 mmsodium phosphate buffer and Aβ1–40 incubated in DMEM/F12 for 3 hr were found to be monomers, whereas fAβ1–42 incubated in DMEM/F12 for 3 hr was found to form aggregation (Fig. 5b, ∗), and the amount of monomeric Aβ1–42 was significantly decreased (Fig.5b).
Fig. 5.

Inhibitory effect of Aβ peptides on vitamin C-mediated Fe(III) reduction and the aggregation state of Aβ peptides in PB and DMEM/F12. a, Aβ peptides were incubated with Fe(III) (25 μm) and PDT (250 μm), followed by the addition of vitamin C (10 μm) and subsequent incubation for 14 hr at 37°C. The effects of freshly dissolved Aβ peptides (10 μm) (fAβ1–40, fAβ1–42, fAβ1–16, and fAβ25–35, all of which were dissolved in distilled water to make a stock solution at 200 μm) and DTPA (10 and 300 μm) on the reduction of Fe(III) were determined. The amount of reduced iron ions was determined by measuring the absorbance at 562 nm. Data represent means ± SE; n = 5 replicate wells. p < 0.001 (a) and p < 0.0001 (b) versus control. b, Freshly dissolved Aβ1–40 and Aβ1–42 peptides at 20 μm were incubated for 3 hr at 37°C in 8 mm sodium phosphate buffer, pH 7.4 (lanes 1, 3), or in DMEM/F12 medium (lanes 2, 4). The aggregation state of Aβ1–40 (lanes 1,2) and Aβ1–42 (lane 3,4) was visualized by SDS-PAGE and silver staining after cross-linking as described in Materials and Methods. Note that Aβ1–42 aggregated immediately in DMEM/F12 (∗), but the majority of both peptides, Aβ1–40 in both solutions and Aβ1–42 in PB, remained as a monomer.
Generation of superoxides in the B27-AO medium and Fe(II)-induced lipid peroxidation are inhibited by monomeric Aβ1–40
Using HE dye, we examined whether the generation of oxygen radicals is enhanced in the B27-AO medium and whether this enhancement is inhibited by fAβ1–40. As shown in Figure6a, strong signals of oxidized ethidium dye were observed in some viable neurons with swollen cell bodies (Fig. 6a, arrows) and dead neurons with shrunken cell bodies (Fig. 6c), whereas the signals with ethidium dye in the cultures treated with fAβ1–40 (5 μm) were not detected (Fig. 6b). We then examined the effect of fAβ1–40 on Fe(II)-induced lipid peroxidation in rat brain cortices by investigating the production of 4-HNE-modified proteins, a product of lipid peroxidation in rat brains, using a monoclonal antibody against 4-HNE-modified proteins. As shown in Figure 6e (∗), the amount of 4-HNE-modified proteins increased in brains incubated in PBS in the presence of 3 and 5 μm Fe(II), whereas treatment with 10 μm fAβ1–40 attenuated this increase. This result indicates that fAβ1–40 prevented lipid peroxidation of the brain tissues induced by oxygen radicals generated by the Fenton reaction.
Fig. 6.
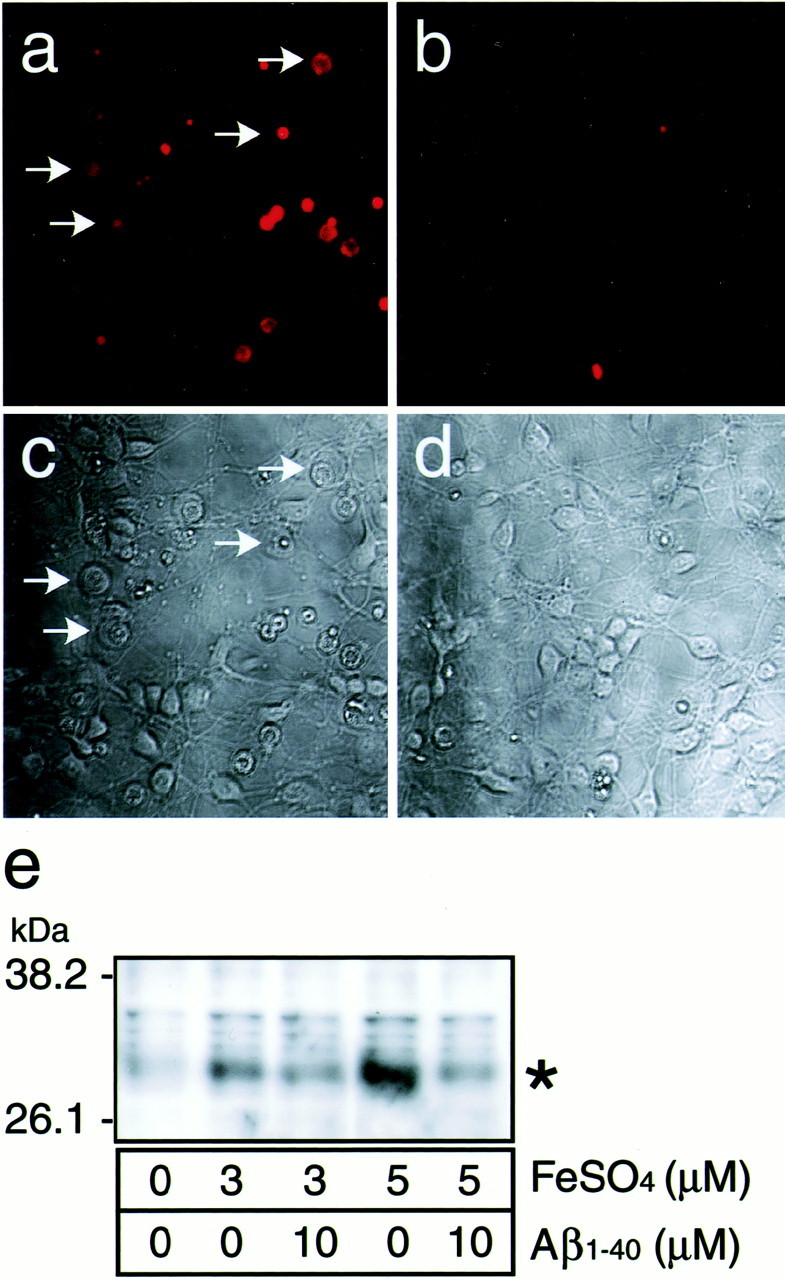
fAβ1–40 inhibits the generation of superoxide and lipid peroxidation. Neurons were treated with (a,c) or without (b, d) 5 μm fAβ1–40 4 hr after plating and were cultured for 48 hr in the B27-AO medium. The cultures were then incubated with 5 μm HE fluorescence (a, b) for 30 min, and transmissive light micrographs of these cultures were taken. Note that the increased signal of superoxides was observed in swollen neurons (arrows) as well as in shrunken neurons in cultures without fAβ (a). e, The effect of Aβ on the production of lipid peroxidation in rat cerebral cortices in the presence of Fe(II). Cerebral cortices were isolated, minced with a cutter, and incubated in PBS in the presence of 3 and 5 μm Fe(II) with or without fAβ1–40 at 10 μm for 4 hr at 37°C. The fragments were then homogenized in RIPA buffer and centrifuged at 10,000 ×g for 10 min at 4°C. The supernatant of the homogenate was subjected to Western blot analysis using anti-4-HNE antibody as the primary antibody.
Comparison of effects of various kinds of Aβ species on neuronal protection
We examined the neuroprotective effect of Aβ1–42, Aβ1–16, and Aβ25–35, in addition to Aβ1–40, on cultured cells incubated in the B27-AO medium. Treatment of Aβ1–40 at a concentration of 5 μm inhibited neuronal death at a percentage of 95 ± 5, whereas Aβ1–42 at concentrations of 0.01, 0.1, 1, 2, 5, 10, and 20 μm, Aβ1–16 at concentrations of 1, 2, 5, 10, 20, and 40, or Aβ25–35 at concentrations of 1, 2, 5, 10, 20, and 40 did not prevent neuronal death 48 hr after the commencement of incubation (Table 2). In the case of Aβ1–42, the thioflavin-T value of the culture medium at the end of treatment significantly increased compared with that of the culture medium treated with Aβ1–40, Aβ1–16, or Aβ25–35, indicating that Aβ1–42 becomes highly aggregated in a culture medium. However, because Aβ1–40, when it remains as a monomer, has an antioxidant effect in the in vitro assay system (Fig. 5a), we next performed an experiment to determine whether monomeric Aβ1–42 at a concentration of 5 μm has a neuroprotective effect on neurons. Because Congo red is known to inhibit oligomerization of Aβ by stabilizing Aβ monomer (Podlisny et al., 1995, 1998), we used Congo red to maintain Aβ1–42 as a monomer. As shown in Table 2, concurrent treatment of 100 μm Congo red with 5 μmAβ1–42 inhibited neuronal death, whereas treatment with 100 μm Congo red alone did not. The thioflavin-T value of these conditioned media was not determined, because Congo red affects the thioflavin-T assay system. These data indicate that monomeric Aβ, regardless of its species, Aβ1–40 or Aβ1–42, rescues neurons.
Table 2.
Effect of various kinds of Aβ peptides on neuronal viability in the B27-AO medium
| Peptides | Concentration Aβ (μm) | Viability (% of control) | Thioflavin-T arbitrary unit (Aβ, 5 μm) |
|---|---|---|---|
| None | 0 | 0 | 0 |
| Aβ1–40 | 5 | 95 ± 5 | 0.11 ± 0.12 |
| Aβ1–42 | 0.01, 0.1, 1, 2, 5, 10, 20 | 0 | 0.51 ± 0.12* |
| Aβ1–16 | 1, 2, 5, 10, 20, 40 | 0 | 0.02 ± 0.01 |
| Aβ25–35 | 1, 2, 5, 10, 20, 40 | 0 | 0.02 ± 0.04 |
| Aβ40-1 | 1, 2, 5, 10, 20, 40 | 0 | ND |
| Aβ1–42 + CR (100 μm) | 5 | 96 ± 4 | ND |
| CR (100 μm) | 0 | 0 | ND |
Primary cortical neurons were cultured in the B27-AO medium. Four hours after plating the cultures were incubated with various kinds of Aβ peptides. In the case of Congo red (CR) treatment, the cultures were incubated with CR (100 μm), with or without Aβ1–42 (5 μm). Cell viability was determined 48 hr after the start of treatment. The data represent means ± SE.n = 6 each. Three independent experiments showed similar results.
p < 0.0001 versus Aβ1–40, Aβ1–16, and Aβ25–35. ND, Not determined.
Effect of tachykinin neuropeptides on monomeric Aβ1–40-mediated neuroprotection
Because a previous study has demonstrated the neurotrophic effects of Aβ1–40, which can be reversed by tachykinin neuropeptides (Yankner et al., 1990), we further examined whether the neuroprotective effect of monomeric Aβ1–40 is inhibited by tachykinin neuropeptides. In our culture system, takykinine neuropeptides such as substance P, physalaemin, eledoisin, neurokinin A, and neurokinin B at 1, 2, 5, 10, and 20 μm did not inhibit neuronal death. Moreover, substance P and physalaemin did not inhibit the neuroprotective effect of Aβ1–40 (Table 3). These results indicate that the mechanism underlying the neurotrophic effects of Aβ1–40 is different from that underlying the antioxidant functions of monomeric Aβ.
Table 3.
Effect of substance P and physalaemin on the neuroprotective effect of monomeric Aβ1–40 in the B27-AO medium
| Treatment | Viability (% of control) |
|---|---|
| None | 0 |
| Aβ1–40 (10 μm) | 95 ± 5 |
| Aβ1–40 (10 μm) + substance P (20 μm) | 101 ± 12 |
| Aβ1–40 (10 μm) + physalaemin (20 μm) | 94 ± 6 |
| Substance P (1, 2, 5, 10, 20 μm) | 0 |
| Physalaemin (1, 2, 5, 10, 20 μm) | 0 |
| Eledoisin (1, 2, 5, 10, 20 μm) | 0 |
| Neurokinin A (1, 2, 5, 10, 20 μm) | 0 |
| Neurokinin B (1, 2, 5, 10, 20 μm) | 0 |
Primary cortical neurons were cultured in the B27-AO medium. Four hours after plating the cultures were incubated with 10 μm freshly dissolved Aβ1–40 in the presence or absence of substance P (20 μm) or physalaemin (20 μm). The cultures were also treated with substance P, physalaemin, eledoisin, neurokinin A, and neurokinin B at various concentrations. Cell viability was determined 48 hr after the start of treatment. The data represent means ± SE. n = 6 each.
DISCUSSION
In this study, we demonstrated a novel function of monomeric Aβ1–40 as an antioxidant molecule on cultured neurons. Monomeric Aβ1–40 exhibits a neuroprotective effect on neurons by quenching transition metal-mediated oxygen radical generation; however, oligomeric Aβ1–40 loses its neuroprotective activity. Monomeric Aβ1–42 also exhibits a neuroprotective effect; however, when monomeric Aβ1–42 is incubated in the culture medium, it rapidly aggregates and exhibits neurotoxicity, whereas monomeric Aβ1–40 remains as a monomer under the same conditions and protects neurons. These findings indicate a novel concept that the biological action of Aβ is dualistic. Aβ, as a monomer, functions as an antioxidant molecule, preventing the generation of oxygen radicals, whereas oligomerized or aggregated Aβ not only loses its antioxidant activity but also contributes to the generation of oxygen radicals (Kay, 1997; Monji et al., 2001a,b), disrupts lipid homeostasis (Michikawa et al., 2001; Gong et al., 2002), and eventually exhibits neurotoxicity (Mattson et al., 1993; Pike et al., 1993; Lorenzo and Yankner, 1994).
Neuronal death induced in the B27-AO medium is inhibited by the addition of radical scavengers, indicating that neuronal toxicity is caused by oxygen radicals. The B27-AO medium contains 1.5 μm Fe(II), 124 nm Fe(III), and 5.2 nm Cu(II), and redox-active transition metals such as iron and copper are known to stimulate oxygen radical chain reactions (Halliwell and Gutteridge, 1984). Because 1.5 μm or higher concentrations of Fe(II) and Cu(II) induce neuronal death in culture [our unpublished data and previous reports (White et al., 1999; Wang and Cynader, 2001)], Fe(II) is most likely responsible for inducing neurotoxicity by generating oxygen radicals in the B27-AO medium. Furthermore, the facts that transferrin successively protects neurons in the B27-AO medium (Fig. 3b) and inhibits Fe(II)-mediated neuronal death in N2 medium, whereas it does not prevent Cu(II)-induced neuronal death (data not shown), strongly support this notion. Thus, it is possible that fAβ1–40 protects neurons from oxygen radicals generated in an Fe(II)-mediated manner.
Antioxidant actions include a direct antioxidant action such as that of scavengers and indirect actions including the quenching metal ions to inhibit secondary generation of free radicals. The neuroprotective activity of monomeric Aβ1–40 includes an inhibitory effect on the generation of superoxides in cultured neurons and lipid peroxidation in brains (Fig. 6). Furthermore, the direct inhibitory effects of monomeric Aβ1–40 on metal reduction induced by vitamin C are also demonstrated (Fig. 5). These findings, together with the result showing that monomeric Aβ1–40 does not serve as a radical scavenger (Fig.4), indicate that the neuroprotective activity of Aβ1–40 is not caused by a direct antioxidant effect but rather by an indirect effect of this peptide, probably the sequestration of metal ions leading to the quenching of the secondary generation of oxygen radicals as other metal-binding proteins do (Halliwell and Gutteridge, 1989).
Free-radical involvement in AD pathogenesis is a well established hypothesis (Lovell et al., 1998a; Markesbery and Lovell, 1998). Aβ is widely believed to serve as a neurotoxic molecule by producing oxygen radicals leading to cell dysfunction and death (Behl et al., 1994;Hensley et al., 1994). The oxygen radicals generated by the interaction of Aβ with redox-active metal ions are suggested to be the possible source of Aβ neurotoxicity, which is suppressed by the redox-inactive form of zinc or metal ion chelators (Huang et al., 1999a,b; Cuajungco et al., 2000a). These lines of evidence seem to contradict our present results that monomeric Aβ1–40 is a potent antioxidant molecule. This discrepancy can be explained by the notion that the action of Aβ is aggregation state-dependent. We show that monomeric Aβ1–40 protects neurons from metal-induced neurotoxicity, whereas iAβ1–40 contains fewer Aβ monomers but more oligomers (Fig. 2d), which could be the reason for the loss of its neuroprotective ability. This is also the case for Aβ1–42, because we have found that Aβ1–42, remaining as a monomer in PB, inhibits the reduction of Fe(III) caused by vitamin C as does Aβ1–40 (Fig. 5a), indicating that monomeric Aβ1–42 also functions as an antioxidant molecule. In addition, the finding that Aβ1–42, when it is maintained as a monomer by coincubation with Congo red in DMEM/F12 medium, exhibits neuroprotective activity (Table 2) strongly supports this notion. However, when fAβ1–42 is incubated in DMEM/F12 medium that contains salt, it aggregates rapidly (Fig. 5b, Table 2) and exhibits neurotoxicity (Fig. 1e, Table 2), whereas fAβ1–40 remaining as a monomer under the same conditions protects neurons (Figs. 1e, 5b). Thus, under physiological conditions, Aβ1–42, a highly amyloidogenic peptide, rapidly aggregates, loses its neuroprotective activity, generates free radicals, and subsequently exhibits neurotoxicity (Pike et al., 1993;Lorenzo and Yankner, 1994; Roher et al., 1996; Kay, 1997; Huang et al., 1999b; Cuajungco et al., 2000b; Monji et al., 2001a,b). These lines of evidence suggest that it may not the differences in Aβ species, Aβ1–40 or Aβ1–42, but those in the state of aggregation, monomers, or other states of aggregation such as oligomers or fibrils, that determine whether the action of Aβ is either neuroprotective or neurotoxic.
Another possible explanation for the discrepancy between the effect of Aβ1–40 and that of Aβ1–42 on neuronal survival may be that at low iron/Aβ binding ratios, iron is captured by Aβ and sequestered from inducing oxygen radical generation, but at higher iron/Aβ ratios, the interaction of Aβ and iron promotes oxygen radical generation (Huang et al., 1999a). Because Aβ1–42 is suggested to bind iron with greater affinity than Aβ1–40 (Atwood et al., 2000), it may be possible to postulate that Aβ1–42 may acquire gain of adverse action at lower concentrations than Aβ1–40.
One may say that because a previous study has demonstrated that aggregated Aβ does not lose the stoichiometry of copper binding (Atwood et al., 2000), an increased amount of oligomerized Aβ may undergo oxidization, reduce metal ions, and serve as an oxygen radical generator (Huang et al., 1999a), leading to neuronal death. Actually, at present we have no evidence indicating that oligomeric Aβ has lesser binding affinity to iron than monomeric Aβ. This may be the case for Aβ1–42, because Aβ1–42 that rapidly aggregates in the culture medium not only loses its neuroprotective activity but also exhibits neurotoxicity (Fig. 1e); however, this may not be the case for Aβ1–40. Our findings that 5 μmiAβ1–40 loses its neuroprotective effect on neurons, whereas 10 μm iAβ1–40 protects neurons (Fig.2b), do not favor the idea that the loss of neuroprotective function is caused by oxidized oligomeric Aβ but favor the notion that monomeric but not oligomeric Aβ1–40 can serve as an antioxidant molecule.
The last question to be addressed is that the neuroprotective effects of monomeric Aβ1–40 shown in our present study are the same as the previously reported neurotrophic effects of Aβ1–40, which can be reversed by tachykinin neuropeptides (Yankner et al., 1990). However, monomeric Aβ1–40 has a neuroprotective effect even on mature neurons at high concentrations, whereas takykinin neuropeptides including substance P and physalaemin at 10 and 20 μm did not inhibit neuronal death in our culture system. Moreover, substance P and physalaemin did not reverse the neuroprotective effect of Aβ1–40 (Table 3), indicating that the mechanism underlying the neurotrophic effects of Aβ1–40 is different from that underlying the antioxidant functions of monomeric Aβ.
The notion that monomeric Aβ1–40 functions as an antioxidant is supported by previous studies showing that the surrounding regions of Aβ deposits in the brains of patients with AD and Down's syndrome have no damage (Nunomura et al., 2000, 2001) and that the inverse correlation is found between Aβ burden and levels of oxidized nucleic acids in AD brain (Cuajungco et al., 2000b). Interestingly, a recent report suggests that brain oxidative damage occurs before Aβ accumulation in the brains of a model mouse of AD amyloidosis (Pratico et al., 2001). Previous reports have shown that Aβ1–42 accumulates with aging, whereas Aβ1–40 does not but accumulates in AD brains (Funato et al., 1998), and that oxidative stress promotes amyloidogenesis (Misonou et al., 2000). These lines of evidence may allow us to assume that oxygen radicals generated in an age-dependent manner enhance generation of Aβ, which may protect neurons from oxygen radical toxicity generated by metal-dependent chain reactions. However, with the increasing amount of Aβ serving as an antioxidant, Aβ aggregates with longer incubation periods in extracellular local fluid and, in turn, exhibits neurotoxicity.
On the basis of our findings, we envisage that Aβ may serve dual actions both by being involved in mechanisms attempting to quench oxidative stress and neurotoxicity probably by sequestrating metal ions when Aβ is in a monomeric state and by exhibiting neurotoxicity when Aβ is highly oligomerized and aggregated by generating oxygen radicals in a metal-mediated manner. Hence, although the toxic actions of Aβ have been exaggerated to date, our observations may provide a new insight into the strategies for development of AD therapy that not only reduction of the amount of Aβ but also inhibition of Aβ aggregation could be the pivotal target for AD therapy.
Footnotes
This study was supported by a research grant for Longevity Sciences (H11-001), Research on Brain Science from the Ministry of Health and Welfare, and by Core Research for Evolutional Sciences and Technology, Japan.
Correspondence should be addressed to Dr. Makoto Michikawa, Department of Dementia Research, National Institute for Longevity Sciences, 36-3 Gengo, Morioka, Obu, Aichi, 474-8522, Japan. E-mail:michi@nils.go.jp.
REFERENCES
- 1.Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, Romano DM, Hartshorn MA, Tanzi RE, Bush AI. Dramatic aggregation of Alzheimer Aβ by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 2.Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI. Characterization of copper interactions with alzheimer amyloid β peptides: identification of an attomolar-affinity copper binding site on amyloid β1–42. J Neurochem. 2000;75:1219–1233. doi: 10.1046/j.1471-4159.2000.0751219.x. [DOI] [PubMed] [Google Scholar]
- 3.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β-protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 4.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bush AI, Pettingell WH, Jr, Paradis MD, Tanzi RE. Modulation of Aβ adhesiveness and secretase site cleavage by zinc. J Biol Chem. 1994a;269:12152–12158. [PubMed] [Google Scholar]
- 7.Bush AI, Pettingell WH, Multhaup G, Paradis MD, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science. 1994b;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 8.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 9.Cuajungco MP, Faget KY, Huang X, Tanzi RE, Bush AI. Metal chelation as a potential therapy for Alzheimer's disease. Ann NY Acad Sci. 2000a;920:292–304. doi: 10.1111/j.1749-6632.2000.tb06938.x. [DOI] [PubMed] [Google Scholar]
- 10.Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI. Evidence that the β-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of Aβ by zinc. J Biol Chem. 2000b;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- 11.Funato H, Yoshimura M, Kusui K, Tamaoka A, Ishikawa K, Ohkoshi N, Namekata K, Okeda R, Ihara Y. Quantitation of amyloid β-protein (Aβ) in the cortex during aging and in Alzheimer's disease. Am J Pathol. 1998;152:1633–1640. [PMC free article] [PubMed] [Google Scholar]
- 12.Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 13.Gong JS, Sawamura N, Zou K, Sakai J, Yanagisawa K, Michikawa M (2002) Amyloid β-protein affects cholesterol metabolism in cultured neurons: implications for pivotal role of cholesterol in the amyloid cascade. J Neurosci Res, in press. [DOI] [PubMed]
- 14.Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine, Ed 2. Oxford UP; New York: 1989. Protection by sequestration of metal ions. pp. 131–133. [Google Scholar]
- 16.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang X, Atwood CS, Moir RD, Hartshorn MA, Vonsattel JP, Tanzi RE, Bush AI. Zinc-induced Alzheimer's Aβ1–40 aggregation is mediated by conformational factors. J Biol Chem. 1997;272:26464–26470. doi: 10.1074/jbc.272.42.26464. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999a;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 20.Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. Cu(II) potentiation of Alzheimer Aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999b;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 21.Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters CL, Beyreuther K. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 22.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 23.Kay CJ. Mechanochemical mechanism for peptidyl free radical generation by amyloid fibrils. FEBS Lett. 1997;403:230–235. doi: 10.1016/s0014-5793(97)00076-8. [DOI] [PubMed] [Google Scholar]
- 24.Kontush A, Berndt C, Weber W, Akopyan V, Arlt S, Schippling S, Beisiegel U. Amyloid-β is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic Biol Med. 2001;30:119–128. doi: 10.1016/s0891-5849(00)00458-5. [DOI] [PubMed] [Google Scholar]
- 25.LeVine H., 3rd Soluble multimeric Alzheimer β(1–40) pre-amyloid complexes in dilute solution. Neurobiol Aging. 1995;16:755–764. doi: 10.1016/0197-4580(95)00052-g. [DOI] [PubMed] [Google Scholar]
- 26.LeVine H., 3rd Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 27.Lorenzo A, Yankner BA. β-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lovell MA, Xie C, Markesbery WR. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer's disease. Neurology. 1998a;51:1562–1566. doi: 10.1212/wnl.51.6.1562. [DOI] [PubMed] [Google Scholar]
- 29.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. J Neurol Sci. 1998b;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 30.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 31.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattson MP, Tomaselli KJ, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated β-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621:35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- 33.Michikawa M, Yanagisawa K. Apolipoprotein E4 induces neuronal cell death under conditions of suppressed de novo cholesterol synthesis. J Neurosci Res. 1998;54:58–67. doi: 10.1002/(SICI)1097-4547(19981001)54:1<58::AID-JNR7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 34.Michikawa M, Gong JS, Fan QW, Sawamura N, Yanagisawa K. A novel action of Alzheimer's β-protein (Aβ): oligomeric Aβ promotes lipid release. J Neurosci. 2001;21:7226–7235. doi: 10.1523/JNEUROSCI.21-18-07226.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misonou H, Morishima-Kawashima M, Ihara Y. Oxidative stress induces intracellular accumulation of amyloid β-protein (Aβ) in human neuroblastoma cells. Biochemistry. 2000;39:6951–6959. doi: 10.1021/bi000169p. [DOI] [PubMed] [Google Scholar]
- 36.Monji A, Utsumi H, Yoshida I, Hashioka S, Tashiro K, Tashiro N. The relationship between Aβ-associated free radical generation and Aβ fibril formation revealed by negative stain electron microscopy and thioflavin-T fluorometric assay. Neurosci Lett. 2001a;304:65–68. doi: 10.1016/s0304-3940(01)01756-6. [DOI] [PubMed] [Google Scholar]
- 37.Monji A, Utsumi H, Ueda T, Imoto T, Yoshida I, Hashioka S, Tashiro K, Tashiro N. The relationship between the aggregational state of the amyloid-β peptides and free radical generation by the peptides. J Neurochem. 2001b;77:1425–1432. doi: 10.1046/j.1471-4159.2001.00392.x. [DOI] [PubMed] [Google Scholar]
- 38.Naiki H, Hasegawa K, Yamaguchi I, Nakamura H, Gejyo F, Nakakuki K. Apolipoprotein E and antioxidants have different mechanisms of inhibiting Alzheimer's β-amyloid fibril formation in vitro. Biochemistry. 1998;37:17882–17889. doi: 10.1021/bi980550y. [DOI] [PubMed] [Google Scholar]
- 39.Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S, Smith MA. Neuronal oxidative stress precedes amyloid-β deposition in Down syndrome. J Neuropathol Exp Neurol. 2000;59:1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- 40.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 41.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid β-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 43.Podlisny MB, Walsh DM, Amarante P, Ostaszewski BL, Stimson ER, Maggio JE, Teplow DB, Selkoe DJ. Oligomerization of endogenous and synthetic amyloid β-protein at nanomolar levels in cell culture and stabilization of monomer by Congo red. Biochemistry. 1998;37:3602–3611. doi: 10.1021/bi972029u. [DOI] [PubMed] [Google Scholar]
- 44.Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ. β-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:10836–10840. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roher AE, Chaney MO, Kuo YM, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, Bonnell BS, Emmerling MR. Morphology and toxicity of Aβ-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J Biol Chem. 1996;271:20631–20635. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- 47.Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals. J Neurochem. 2000;74:270–279. doi: 10.1046/j.1471-4159.2000.0740270.x. [DOI] [PubMed] [Google Scholar]
- 48.Schubert D, Chevion M. The role of iron in β-amyloid toxicity. Biochem Biophys Res Commun. 1995;216:702–707. doi: 10.1006/bbrc.1995.2678. [DOI] [PubMed] [Google Scholar]
- 49.Selkoe DJ. Alzheimer's disease: a central role for amyloid. J Neuropathol Exp Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 50.Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci USA. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vigo-Pelfrey C, Lee D, Keim P, Lieberburg I, Schenk DB. Characterization of β-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang XF, Cynader MS. Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity. J Neurosci. 2001;21:3322–3331. doi: 10.1523/JNEUROSCI.21-10-03322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White AR, Multhaup G, Maher F, Bellingham S, Camakaris J, Zheng H, Bush AI, Beyreuther K, Masters CL, Cappai R. The Alzheimer's disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J Neurosci. 1999;19:9170–9179. doi: 10.1523/JNEUROSCI.19-21-09170.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 55.Yoshiike Y, Tanemura K, Murayama O, Akagi T, Murayama M, Sato S, Sun X, Tanaka N, Takashima A. New insights on how metals disrupt amyloid β-aggregation and their effects on amyloid-β cytotoxicity. J Biol Chem. 2001;276:32293–32299. doi: 10.1074/jbc.M010706200. [DOI] [PubMed] [Google Scholar]