

Abstract
The L1 adhesion molecule regulates axon growth and is mutated in the X-linked mental retardation syndrome CRASH (acronym for corpus callosum agenesis, retardation, aphasia, spastic paraplegia, hydrocephalus). A novel role for L1 as a potentiator of neuronal cell migration to extracellular matrix proteins through β1 integrins and intracellular signaling to mitogen-activated protein (MAP) kinase was identified. L1 potentiated haptotactic migration of B35 neuroblastoma cells toward fibronectin, vitronectin, and laminin through the signaling intermediates c-Src, phosphatidylinositol-3 kinase, and MAP kinase. L1 potentiated migration toward fibronectin through α5β1 integrin in human embryonic kidney 293 cells and depended on determinants of L1 endocytosis: dynamin I, c-Src, and the AP2/clathrin binding site (Arg-Ser-Leu-Glu) in the neuronal splice form of L1. L1 clustering on the cell surface enhanced the internalization of activated β1 integrins and L1 into distinct endocytic vesicles. L1-potentiated migration, enhancement of β1 integrin endocytosis, and activation of MAP kinase were coordinately inhibited by mutation of an RGD sequence in the sixth immunoglobulin-like domain of L1. Moreover, three CRASH mutations in the L1 cytoplasmic domain (1194L, S1224L, Y1229H), two of which interfere with ankyrin association, inhibited L1-potentiated migration and MAP kinase activation. Function-blocking antibodies to L1 and β1 integrin retarded the migration of 5-bromo-2′-deoxyuridine-labeled mouse cerebellar granule cells in slice cultures, underscoring the potential physiological relevance of these findings. These studies suggest that L1 functionally interacts with β1 integrins to potentiate neuronal migration toward extracellular matrix proteins through endocytosis and MAP kinase signaling, and that impairment of this function by L1 cytoplasmic domain mutations may contribute to neurological deficits in CRASH.
Keywords: L1, integrin, cell migration, endocytosis, MAP kinase, mental retardation
The L1 cell adhesion molecule is an integral membrane protein that promotes axon growth and guidance critical to nervous system development (for review, see Schmid and Maness, 2001). L1 belongs to a family of Ig-like cell recognition molecules including L1, CHL1, NrCAM, NgCAM, neurofascin, andDrosophila neuroglian, which share common structural elements and the ability to promote axon growth. The L1 gene (on chromosome Xq28) is the target for mutations in the human mental retardation syndrome CRASH (acronym for corpus callosum agenesis, retardation, aphasia, spastic paraplegia, hydrocephalus) (Kenwrick et al., 2000). L1 null mutant mice display aspects of the CRASH phenotype including axon guidance errors in the corticospinal tract and corpus callosum, cortical dendrite abnormalities, reduced numbers of hippocampal neurons, defects in spatial memory, and enlarged ventricles (Cohen et al., 1997; Dahme et al., 1997; Fransen et al., 1998;Demyanenko et al., 1999).
The L1 extracellular region is composed of six Ig-like and five fibronectin type III domains that engage in L1–L1 homophilic binding and heterophilic interactions with other Ig-class cell recognition molecules including axonin 1/TAG1, F11/F3/contactin, and NCAM (Kadmon et al., 1990; Kuhn et al., 1991; Brummendorf et al., 1993). L1 has been shown to interact functionally with some integrins, including the fibronectin receptor α5β1 and vitronectin receptor αvβ3, for adhesion or neurite growth on L1 substrates (Moos et al., 1988, Ruppert et al., 1995; Ebeling et al., 1996; Montgomery et al., 1996;Felding-Habermann et al., 1997). The L1 intracellular domain bears a binding site for the cytoskeletal linker protein ankyrin (Bennett and Chen, 2001) and a neuron-specific sequence Arg-Ser-Leu-Glu (RSLE) arising from alternative splicing, which together with a preceding tyrosine residue, comprises a motif for axon targeting and clathrin- and dynamin-mediated endocytosis (Kamiguchi and Lemmon, 1998; Kamiguchi et al., 1998). Although originally identified as a neural cell adhesion molecule, L1 is also expressed in Schwann cells, melanocytes, hematopoietic cells of lymphoid and myelomonocytic lineages, and epithelial cells (Takeda et al., 1996; Pancook et al., 1997; Kadmon et al., 1998; Nolte et al., 1999). Potential involvement of L1 in tumor development and metastasis is further suggested by its expression on many tumor cell lines including neuroblastoma, melanoma, and carcinomas (Linnemann et al., 1989; Reid and Hemperly, 1992; Pancook et al., 1997).
In addition to the known function of L1 in axon growth, there is evidence that L1 can participate in the migration of neuronal precursors. L1 is expressed in dopaminergic neuronal precursors during their tangential migration on L1-positive fiber tracts (Ohyama et al., 1998), and their final location is altered in L1 knock-out mice (Demyanenko et al., 2001). L1 is also expressed transiently in migrating neuronal precursors within the developing cerebellum and neocortex (Fushiki and Schachner, 1986; Beasley and Stallcup, 1987;Chung et al., 1991; Demyanenko et al., 1999), and function-blocking L1 antibodies have been shown to perturb the inward migration of granule cell precursors in cerebellar explant cultures (Lindner et al., 1986;Chuong et al., 1987; Crossin et al., 1990). In addition, L1 can promote haptotactic migration of several types of culture cells toward purified L1 substrates (Montgomery et al., 1996; Gutwein et al., 2000).
L1 promotes neurite outgrowth in cell cultures through the central signal integrator MAP kinase (Schaefer et al., 1999; Schmid et al., 2000) by a pathway that includes the nonreceptor tyrosine kinase c-Src (Ignelzi et al., 1994), phosphatidylinositol-3 (PI3) kinase, Rac1 GTPase (Schmid et al., 2000), and the guanine nucleotide exchange factor Vav-2 (R.-S. Schmid and P. F. Maness, unpublished results). This pathway is closely related to an early-acting signaling pathway induced by integrin activation that promotes membrane ruffling, lamellipodia, and migration of non-neural cells (Clark et al., 1998; Meng and Lowell, 1998; Miranti et al., 1998). The common features of signaling by L1 and integrins and evidence that L1 may participate in regulating neuronal precursor migration lead us to speculate that L1 might interact functionally with integrins to stimulate neuronal cell migration on extracellular matrix molecules. In this study, we report a new role for L1 as a potentiator of neuronal migration to extracellular matrix proteins through β1 integrin and intracellular signaling and show that CRASH mutations in the L1 cytoplasmic domain impair this function.
MATERIALS AND METHODS
Reagents. Plasmids subcloned into pcDNA3 included human L1(+/−RSLE) (John Hemperly, BD Technologies, Research Triangle Park, NC), hemagglutinin (HA)-tagged extracellular signal regulated kinase (ERK)2 (Melanie Cobb, University of Texas Southwestern Medical School, Dallas, TX), dynamin I (K44A) (Marc Caron, Duke University, Durham, NC), Src (K295M) (Sara Courtneidge, Sugen, San Francisco, CA), and enhanced cyan fluorescence protein (Invitrogen, San Diego, CA). All L1 mutants used here have been described in our previous work (Needham et al., 2001). Mouse monoclonal antibody against human L1 (Neuro4) and anti-L1 polyclonal rabbit antibody 6096 were gifts of John Hemperly (BD Technologies). The following antibodies were obtained commercially: polyclonal anti-Active MAP kinase antibody (pTEpY) (Promega, Madison, WI), antibody against MAP kinase protein (Santa Cruz Biotechnology, Santa Cruz, CA), anti-human β1 integrin monoclonal antibody (clone P4C10) against the RGD binding site (Invitrogen, Gaithersburg, MD), anti-human α5 integrin (clone P1D6) against the PHSRN synergy site (Invitrogen), anti-human αvβ3 integrin monoclonal antibody 1976Z (Chemicon, Temecula, CA), and monoclonal antibody MAB 2000 that activates human β1 integrin (Chemicon). Human vitronectin, human fibronectin, murine laminin, and peptides G RGDSP and G RGESP were from Invitrogen or Peninsula laboratories; murine collagen type IV was from Sigma (St. Louis, MO). Recombinant reelin secreted from reelin-transfected human embryonic kidney (HEK) 293T cells and control supernatant from untransfected cells were a gift from Eva Anton (University of North Carolina, Chapel Hill, NC). The Src inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine (PP2) (Hanke et al., 1996) and inactive analog 4-amino-7-phenylpyrazolo[3,4-d]pyrimidine (PP3) were from Calbiochem (La Jolla, CA). MAP kinase kinase (MEK) inhibitor U0126 (Promega), PI3 kinase inhibitor Ly294002 (Calbiochem), PP2, and PP3 were dissolved in dimethylsulfoxide (DMSO).
Cell culture procedures. Rat B35 neuroblastoma cell lines stably expressing human L1(+RSLE) or L1 point mutants S1194L, S1224L, and S1229H in pcDNA3 have been described (Needham et al., 2001). B35 cell lines were maintained in DMEM, 10% fetal bovine serum, and gentamycin/kanamycin, 250 μg/ml G418. HEK293 cells were maintained in the same medium without G418 and transfected for transient expression of pcDNA3 plasmids (10 μg/100 mm dish) using lipofectamine with transfection efficiency of ∼70% as described (Needham et al., 2001).
Haptotactic cell migration assay. Haptotactic cell migration assays were performed in modified Boyden chambers (Transwells; Corning/Costar 3422) in serum-free DMEM containing 0.4 mmMnCl2·4H2O, 50 μg/ml gentamicin, and 200 μg/ml kanamycin. Transwell chambers had polycarbonate filters with 8.0-μm-diameter pores, the bottom sides of which were coated with extracellular matrix proteins or bovine serum albumin (BSA; fatty acid free, Sigma). Proteins in PBS were applied to the bottom side of filters (3 μg of protein per filter) and air dried. The bottom surfaces of filters were washed and blocked in 2% BSA. Cells were detached using PBS/Na-EDTA (5 mm) and 10,000–20,000 cells plated per chamber. Some cells were preincubated with antibodies (2 μg/100 μl medium) for 10 min at 4°C or peptides (2 mm) for 1 hr at 4°C in serum-free medium.
To score B35 cells, cells from the top side of the filter were removed, and cells on the bottom side of the filter were stained for 10–30 min with Gill's formula hematoxylin (Vector Laboratories, Burlingame, CA), and at least 150 cells were scored within a 10 × 10 mm grid from nine or more randomly selected fields using a 20× microscope objective. The mean number of cells per field was multiplied by a factor based on the number and size of fields scored and on a filter diameter of 6.5 mm to obtain the total number of cells migrated. Experiments were performed in duplicate or triplicate, and results were averaged. The means of experimental and control samples and SEM were compared for significant differences using Student's ttest. L1-expressing HEK293 cells were visualized by indirect immunofluorescence staining with anti-L1 monoclonal antibody Neuro4 and FITC-labeled goat anti-mouse IgG and scored on both top and bottom surfaces of filters under epifluorescence illumination. The percentage of L1-expressing cells that transmigrated was calculated by determining the ratio of L1-positive cells on the bottom of the filter compared with the total number of L1-positive cells on both sides of the filter.
MAP kinase assay. B35 cells were transfected for transient expression with HA-tagged ERK2 plasmid together with L1 plasmids, and phosphorylation of immunoprecipitated HA-ERK2 protein was measured by Western blotting with anti-Active MAPK antibody against dually phosphorylated, activated ERKs as described (Schmid et al., 2000). At 36–40 hr after transfection, the medium was replaced with OptiMEM-I, and 8 hr later cells were treated with preformed complexes of either nonimmune mouse IgG or L1 antibody Neuro4 and F(ab′)2 fragments of secondary antibodies against Fc fragments of mouse IgG (Jackson ImmunoResearch, West Grove, PA) for 10 min at 37°C. Cells were lysed in buffer containing 1% NP-40, 0.25% Na-deoxycholate, 50 mm HEPES, pH 7.4, 137 mm NaCl, 1 mm Na-EDTA, 10 mm phenylmethylsulfonyl fluoride, 10 mm β-glycerophosphate, 10 μg/ml leupeptin, 0.1 TIU/ml aprotinin, 1 μg/ml pepstatin, 2 nmcalyculin A, and 10% glycerol. HA-tagged ERK2 was immunoprecipitated from lysates with anti-HA antibody and protein G-Sepharose. Immune complexes were analyzed by immunoblotting with anti-Active MAPK antibody using enhanced chemiluminescence. Blots were stripped and reprobed with antibodies against total ERK protein. Bands were quantified by densitometric scanning, and levels of phosphorylated ERK2 were normalized to ERK2 protein. Each cell line was assayed in duplicate, and results were averaged. L1-induced ERK2 phosphorylation was expressed as the fold-activation relative to normal IgG control. Experiments were repeated 3–10 times, and mean ERK2 phosphorylation relative to normal IgG and SEM was determined for each cell line. Some amount of nonspecific ERK2 phosphorylation was elicited by nonimmune IgG in cells expressing mutant or wild-type L1, presumably attributable to binding of residual uncomplexed IgG to endogenous Fc receptors (Schmid et al., 2000), but little variation in the normal IgG control was observed for each cell line. Means for each L1 mutant or splice form were compared with L1(+RSLE), and significant differences were determined by the t test.
Immunofluorescence staining for endocytosis of L1 and β1 integrins. HEK cells were plated at 20,000 cells per well onto LabTek II chamber slides (Nunc, Naperville, IL) coated with poly-d-lysine (0.01%) and human fibronectin (5 μg/ml) and transfected with plasmid encoding human L1(+RSLE). After 16–18 hr cells were washed and incubated for 5–60 min at 37°C with polyclonal rabbit antibody 6096 against L1 (50 μg/ml) and MAB2000, an activating monoclonal antibody against human β1 integrin (40 μg/ml). Cells were fixed in 2% paraformaldehyde for 30 min and then permeabilized with 0.05% Triton X-100 in PBS, 10% goat serum for 1 hr. Cells were incubated with FITC-conjugated goat anti-mouse IgG (750 μg/ml) in PBS, 10% goat serum for 1 hr to label surface and internalized integrins. After washing, cells were incubated with tetramethylrhodamine isothiocyanate-conjugated goat anti-rabbit IgG (750 μg/ml) in PBS containing 10% goat serum for 30 min to label surface and internalized L1. Slides were mounted with Vectashield, and cells were analyzed using a Zeiss LSM10 confocal microscope with an argon laser. Images of 0.5 μm optical thickness through the middle of the cells were captured at the University of North Carolina Microscopy Services Facility, Department of Pathology (Dr. Robert Bagnell, Director). L1 labeling was recorded using the 514 nm excitation filter with an emission range of 575–645 nm. Integrin labeling was recorded using a narrow band filter with 488 nm excitation range and 515–545 nm emission range. NIH Image J software was used for pseudocolor. The mean fluorescent pixel density of internalized integrins was measured in 18–28 randomly selected cells using Scion imaging for three experimental groups: HEK293, L1(+RSLE)-HEK293, and mutant L1(KGE)-HEK293 cells treated for 5 min at 37°C with L1 and β1 integrin antibodies as described above. The number of fluorescent pixels inside each cell was measured and divided by the total number of pixels inside the cell to obtain the fluorescent pixel density of each cell. The mean fluorescent pixel density was calculated for all cells within each experimental group. The mean fluorescent pixel densities of L1 and L1(KGE)-expressing cells in three different experiments were normalized to the corresponding mean fluorescent pixel densities of HEK293 cells (no L1) in the same experiment and averaged (see Fig. 9). Significant differences in means were determined by the ttest (one-tailed; p < 0.05).
Fig. 9.
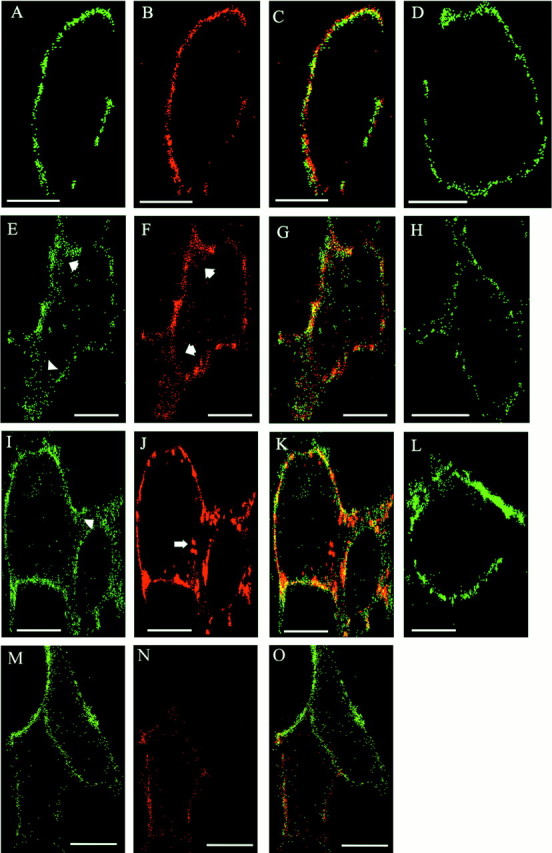
Antibody-induced endocytosis of L1 and β1 integrins in L1-expressing HEK cells. HEK293 cells were transfected for transient expression with L1(+RSLE) (A–C,E–G,I–K) or mutant L1(KGE) (M–O) plasmids. Confocal microscopy images of cells are labeled green for β1 integrin (A, E, I,M), red for wild-type or mutant L1 (B, F, G), andyellow for colocalized β1 integrin and L1 (C, G, K,O). Nontransfected cells were labeledgreen for β1 integrin alone (D,H, L). Live HEK cells were incubated with antibodies against L1 and β1 integrin for 0 min (A–D), 5 min (E–H,M–O), or 60 min (I–L). After 5 min, increased internal staining of β1 integrins was seen in L1-transfected cells (E,arrowheads) compared with nontransfected cells (H). Long arrow inJ indicated larger endocytotic vesicles of wild-type L1 resembling multivesicular bodies. L1(KGE) mutant cells (M) did not show increased β1 integrin internalization after 5 min. Images show representative cells from multiple experiments. Scale bars, 10 μm.
Neuronal migration in cerebellar slices. Newly generated neurons were labeled with 5-bromo-2′-deoxyuridine (BrdU), and the extent of their radial migration away from the external granular layer (EGL) was measured after 21 hr in culture. Mice (C57BL/6) at postnatal day 4 were injected intraperitoneally with BrdU (10 mg/kg body weight) in saline (Boehringer Mannheim, Indianapolis, IN). After 45 min brains were removed, cerebella were dissected out, and 200 μm coronal slices were made. Adjacent slices were cultured on Millicell-CM 0.4 μm membranes (Millipore, Milford, MA) in MEM supplemented with 10% horse serum, 40 mm glucose, 1.8 mm glutamine, 24 mmNa-bicarbonate, and 90 U/ml penicillin–streptomycin. In some samples, medium with adjacent sections of cerebellar slices from different mice was supplemented with purified antibodies; rabbit polyclonal antibody 6096 against L1 (1 mg/ml), hamster anti-rat CD29 monoclonal antibody against β1 integrin (0.1 mg/ml) (BD PharMingen), or nonimmune rabbit IgG (1.1 mg/ml). Slices were removed at 0 or 21 hr of incubation in culture at 37°C in 5% CO2 and processed for BrdU by immunofluorescence staining. Slices were fixed in 70% ethanol for 12 hr at 4°C. After three rinses in PBS, slices were fixed in 2N HCl for 1 hr at room temperature, washed in PBS, and incubated for 4 hr with BrdU monoclonal antibody (1:75 in PBS, 0.5% Tween 20) for 4 hr (Becton Dickinson, Research Triangle Park, NC). Slices were washed in PBS and incubated with anti-mouse IgG/M conjugated to Cy3 (1:100;Jackson ImmunoResearch) for 2 hr. Bis-benzimide (10 μm) was added 5 min before the end of the incubation. Filters were rinsed in PBS, and slices were cut out and mounted onto glass slides with Vectashield mounting medium. Slices were visualized with a Zeiss confocal microscope using rhodamine or UV filter sets. Images of ∼3 μm optical sections were collected and stored on a CD and then analyzed for cell migration using Scion Image software or assembled into the panel of figures presented using Adobe Photoshop.
To determine the extent of migration of BrdU-labeled cells, the shortest distance between the BrdU-labeled cells and the pial surface was measured within at least four different slices at different locations, and the average distance was calculated for at least 150 cells per experimental condition. The average distance was then expressed relative to the respective thickness of the cerebellar cortex within the same location measured from bis-benzimide images to yield the migration index. Only cells with brightly labeled nuclei were scored. Bis-benzimide labeling of nuclei demarcated the different regions of the cerebellar cortex and white matter. The location of the EGL, molecular layer (ML), and internal granular layer (IGL) were confirmed by staining with hematoxylin-eosin. After such demarcation, the number of BrdU-labeled cells within three randomly selected regions of the cerebellar cortex from the pial surface to the bottom of the IGL was compared with the number of labeled cells within the EGL of the corresponding region for each experimental condition to estimate the percentage of BrdU-labeled cells in the EGL. Statistical differences between experimental groups was tested by Student's t test (p < 0.05; one-tailed).
RESULTS
L1 potentiates haptotactic cell migration to extracellular matrix molecules through integrins
L1 signaling and endocytosis have been analyzed previously in the CNS-derived B35 neuroblastoma cell line that stably expresses the neuronal form of human L1(+RSLE) (Schmid et al., 2000; Needham et al., 2001). Haptotactic migration of L1(+RSLE)-B35 cells and parental B35 cells, which lack L1, was studied in Transwell assays using modified Boyden chambers in which cells migrated from top to bottom chambers through filters coated on the bottom side with extracellular matrix protein. Parental B35 neuroblastoma cells displayed greater migration in 16 hr toward fibronectin compared with their random migration toward BSA (Fig. 1). Migration of L1(+RSLE)-B35 cells toward fibronectin was strongly stimulated (three- to fourfold) compared with parental B35 cells. Migration of L1(+RSLE)-B35 cells toward laminin and vitronectin was also stimulated compared with B35 cells not expressing L1. Neither B35 cells nor L1(+RSLE)-B35 cells displayed significant migration toward collagen type IV or reelin, an extracellular matrix-like molecule that regulates cortical neuron migration on radial glia (Dulabon et al., 2000). The slight increase in migration of L1(+RSLE)-B35 cells to control and reelin-containing supernatants was probably caused by small amounts of fibronectin in the conditioned medium.
Fig. 1.

L1 potentiates migration of B35 neuroblastoma cells to extracellular matrix proteins. Haptotactic cell migration was measured in B35 cells (20,000 cells per chamber) expressing no L1 or L1(+RSLE) toward extracellular matrix proteins or BSA for 16 hr. Each sample was assayed in triplicate, and experiments were repeated at least twice with similar results.
To determine whether L1-potentiated migration to fibronectin was mediated through an integrin-dependent mechanism, HEK293 cells were used, because unlike B35 cells, they express defined integrins for which function-blocking antibodies are available. HEK293 cells express α5β1 integrin, which functions as an RGD-dependent fibronectin receptor, and αvβ1 integrin, which functions as a fibronectin or vitronectin receptor (Bodary and McLean, 1990), but they lack αvβ3 or αvβ5 integrins (Simon et al., 1997) or L1 (Needham et al., 2001). HEK293 cells were transfected for transient expression with a plasmid encoding human L1(+RSLE) or the control pcDNA3 plasmid, and haptotactic migration was assayed toward fibronectin for 4 hr. L1(+RSLE)-HEK293 cells exhibited increased migration to fibronectin compared with nonexpressing cells (Fig.2). The migration of HEK293 cells after 4 hr was greater than that of B35 cells after 16 hr. Treatment of L1-expressing cells with antibodies against the extracellular region of L1 (anti-L1; Neuro4), at concentrations that did not induce clustering of L1, reduced migration to the levels of untransfected cells, whereas an equivalent amount of nonimmune IgG did not affect migration (Fig.2). Antibody P1D6 specific for the α5 integrin subunit (anti-α5) inhibited haptotactic migration of both L1-expressing and nonexpressing cells toward fibronectin (Fig. 2). This antibody maps to the binding site for the fibronectin synergy sequence (PHSRN) in α5 integrin (Burrows et al., 1999). Antibody P4C10 against the β1 integrin RGD binding site also inhibited migration toward fibronectin, as did a mixture of α5 and β1 integrin antibodies (Fig. 2). Monoclonal antibodies specific for αvβ3 integrin had no effect on migration. These results demonstrated that in the absence and presence of L1, haptotactic migration toward fibronectin occurred primarily through α5β1 integrins in HEK293 cells.
Fig. 2.

L1 potentiates migration of HEK293 cells through integrins. Haptotactic migration of HEK293 (10,000 cells per chamber) expressing no L1 or L1(+RSLE) toward fibronectin was measured for 4 hr. Cells were pretreated as follows: none (None), nonimmune mouse IgG (NIgG), anti-L1 monoclonal antibody Neuro4 (Anti-L1), anti-integrin α5 antibody (Anti-α5), anti-integrin β1 antibody (Anti-β1), a combination of antibodies against α5 and β1 integrins (Anti-α5+β1), or anti-integrin αvβ3 (Anti-αvβ3) monoclonal antibody (each at 20 μg/ml). *Statistically significant differences in means of treated and untreated (None) samples using the t test (p < 0.05).
An RGD sequence within the cell binding domain of fibronectin is the major binding site for α5β1 integrin. To investigate the requirement for RGD binding in L1-potentiated migration to fibronectin, HEK293 cells were preincubated with the RGD-containing hexapeptide GRGDSP (2 mm) or an RGE-containing control peptide GRGESP that does not bind to integrins and assayed for haptotactic migration to fibronectin. Migration of both L1-expressing and nonexpressing HEK293 cells was completely inhibited by RGD peptides but not by RGE peptides, consistent with an α5β1 integrin mechanism (Fig.3A). Haptotactic migration of both L1-expressing and nonexpressing B35 neuroblastoma cells toward fibronectin was also inhibited by RGD but not RGE peptides (Fig.3B). However, RGD peptides did not fully block migration of L1-B35 cells, suggesting the minor contribution of an RGD-independent mechanism. As an example, L1 might increase α5β1 integrin affinity for the fibronectin synergy sequence, or alternatively L1 might activate a non-RGD-binding integrin, such as α4β1 integrin, which binds the Glu-Ile-Leu-Asp-Val (EILDV) sequence in fibronectin. In any case, adhesion of cells was not significantly altered by RGD peptides, because there was no decrease in total cell number on the top and bottom sides of the filters.
Fig. 3.
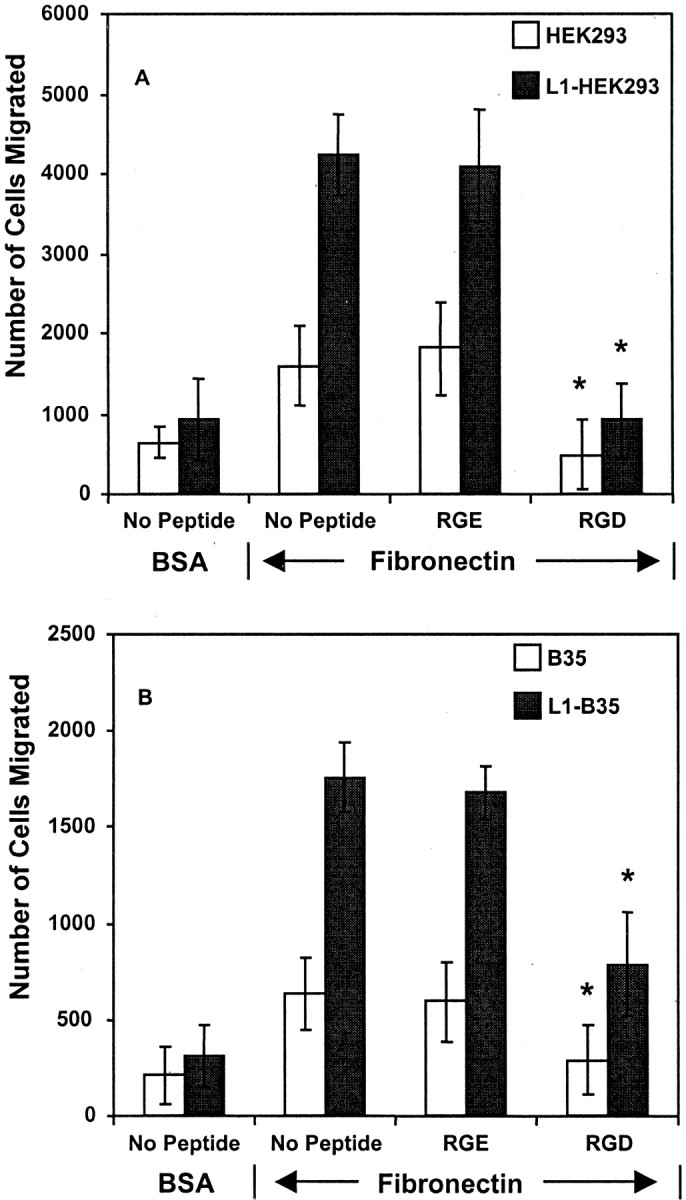
RGD peptides abrogate L1-potentiated migration toward fibronectin. HEK293 (10,000 cells per chamber) (A) or B35 cells (20,000 cells/chamber) expressing no L1 or L1(+RSLE) (B) were incubated with or without hexapeptide GRGDS (RGD) or inactive GRGES (RGE) control peptides (2 mm) for 1 hr on ice and then assayed for haptotactic migration toward fibronectin or BSA for 4 hr. Each sample was assayed in triplicate, and experiments were repeated at least twice with similar results. *Statistically significant differences in means of treated and untreated (No Peptide) samples using the t test (p < 0.05).
An RGD in the L1 Ig6 domain is required for potentiating migration
There is only one RGD sequence in the Ig6 domain of human L1 (Hlavin and Lemmon, 1991). Conservative mutation of the L1 RGD to Lys-Gly-Glu (KGE) reduced L1-potentiated migration of HEK293 cells toward fibronectin to the level of HEK293 cells not expressing L1 (Fig.4). This effect was highly specific for the RGD→KGE mutation, because L1-potentiated migration was not adversely affected by a number of different CRASH point mutations in other extracellular domains of L1 (R184Q in Ig2, H210Q in Ig2, E309K in Ig3, Y784C in FN2, Y1070C in FN5) (Fig. 4). Each of these mutants is expressed at levels equivalent to wild type on the surface of HEK293 cells and is able to recruit ankyrin normally to the plasma membrane (Needham et al., 2001). Surface expression of some of these (R184Q, E309K, Y784C) has been reported to be impaired (29–72%) in Chinese hamster ovary (CHO) cells (DeAngelis et al., 2002) and thus the altered surface expression appears to be cell-type dependent. The R184Q and E309K mutations are known to interfere with L1 binding to heterophilic partners F3/F11/contactin and axonin-1/Tag-1 (DeAngelis et al., 1999); thus the ability of wild-type L1 to stimulate migration to fibronectin was unlikely to be mediated through these molecules. In addition, the R184Q mutation as well as the H210Q mutation perturb homophilic L1–L1 binding (DeAngelis et al., 1999) and neurite outgrowth on L1 substrates (Zhao and Siu, 1996), suggesting that the mechanism of L1-potentiated migration to extracellular matrix proteins also did not involve homophilic L1–L1 interactions. These results indicated that the RGD sequence in the Ig6 domain of L1 was a critical determinant of L1-potentiated cell migration to extracellular matrix.
Fig. 4.

L1-potentiated migration to fibronectin requires RGD and RSLE sequences in L1. HEK293 cells were transfected for transient expression of L1(+RSLE), L1(−RSLE), or L1(+RSLE) with RGD→KGE or extracellular CRASH mutations. Cells were assayed for haptotactic migration toward fibronectin for 4 hr. Each sample was assayed in duplicate or triplicate, and experiments were repeated at least twice with similar results. *Statistically significant differences in means of L1 mutants compared with L1(+RSLE) using thet test (p < 0.05).
Neuronal L1 enhances migration through dynamin I, c-Src, PI3 kinase, and MAP kinase
The neuronal splice form L1(+RSLE) is endocytosed rapidly by clathrin and dynamin-mediated endocytosis required for MAP kinase signaling (Kamiguchi et al., 1998; Schaefer et al., 1999; Schmid et al., 2000), whereas the non-neuronal form lacking RSLE is endocytosed more slowly by a clathrin-independent mechanism (Long et al., 2001). Haptotactic migration of HEK293 cells toward fibronectin was potentiated after 4 hr by L1(+RSLE), which binds the AP2/clathrin adapter (Kamiguchi et al., 1998), and not by the non-neuronal form of L1 lacking RSLE (Fig. 4). Expression of the dominant negative dynamin I (K44A) mutant, which impairs receptor-mediated endocytosis, abolished the migration-potentiating effect of L1(+RSLE) (Fig. 5). This mutant also produced a small but significant inhibitory effect on migration of HEK cells to fibronectin in accord with evidence that dynamin-dependent internalization of β1 integrins plays a role in cell motility (Ng et al., 1999; Pierini et al., 2000). These results suggested that L1-potentiated migration to fibronectin in HEK293 cells after 4 hr occurred through a mechanism involving clathrin- and dynamin-mediated endocytosis. Cells expressing L1(−RSLE) might exhibit potentiated migration after longer migration times because this form is internalized more slowly (Long et al., 2001); however, HEK293 cells have a rapid rate of migration that precluded examining this possibility.
Fig. 5.

L1-potentiated migration is regulated by dynamin-1 and c-Src. HEK293 cells were transfected with or without human L1(+RSLE) and one of the following plasmids: pcDNA3 (Control), dominant negative dynamin-1(K44A) (DN-Dynamin), or dominant negative c-Src(K295M) (DN-Src). Cells were assayed for haptotactic migration toward fibronectin or random migration to BSA for 2 hr. Each sample was assayed in triplicate and repeated at least twice with similar results. *Statistically significant differences in means of mutant-expressing and control cells using the t test (p < 0.05).
The nonreceptor tyrosine kinase c-Src is required for endocytosis of L1 (Schmid et al., 2000), regulation of neurite outgrowth on L1 (Ignelzi et al., 1994), and L1-induced MAP kinase activation (Schmid et al., 2000). Expression of the dominant negative Src (K295M) mutant in L1(+RSLE)-transfected HEK293 cells decreased L1-potentiated migration to the level of untransfected cells (Fig. 5). To confirm the role of c-Src in L1-potentiated haptotactic migration in neuronal cells, B35 neuroblastoma cells stably expressing L1(+RSLE) and parental B35 cells lacking L1 were treated with PP2, a pyrazolopyrimidine inhibitor selective for Src family kinases, under conditions that inhibit Src in cell culture (Hanke et al., 1996). PP2 substantially inhibited the haptotactic migration of L1(+RSLE)-B35 cells to fibronectin but did not significantly inhibit migration of parental B35 cells (Fig.6). PP3, an inactive structural analog, did not affect the migration of either L1(+RSLE)-expressing or parental B35 cells, and the inhibitor solvent DMSO (0.05%) had no significant effect. PP2 did not completely inhibit migration of L1-expressing cells, suggesting that there was either an Src-independent component of L1-potentiated migration or that sufficient levels of PP2 were not sustained over the duration of the assay.
Fig. 6.

Src kinase, MAP kinase, and PI3 kinase are required for L1-potentiated migration of B35 cells. Migration of B35 cells expressing no L1 or L1(+RSLE) was measured toward fibronectin for 16 hr. Cells were untreated or treated with 0.05% DMSO alone or with Src kinase inhibitor PP2 (1 μm), inactive analog PP3 (1 μm), PI3 kinase inhibitor Ly294002 (25 μm), or MEK inhibitor UO126 (10 or 20 μm). Each sample was assayed in triplicate, and experiments were repeated at least twice with similar results. *Statistically significant differences in means of treated and untreated cells using the t test (p < 0.05).
MAP kinase and PI3 kinase are phosphorylated and activated during L1 endocytosis through Src (Schmid et al., 2000). To determine whether MAP kinase was important for haptotactic cell migration to fibronectin, B35 cells and L1(+RSLE)-B35 cells were treated with an inhibitor (U0126) of the dual specificity kinase MEK, which phosphorylates and activates MAP kinase. The inhibitor was used under conditions that produce maximal inhibition of L1-promoted neurite growth and phosphorylation of the MAP kinases ERK1 and ERK2 in B35 cells (Schmid et al., 2000). The MEK inhibitor effectively suppressed migration of both parental and L1-expressing B35 cells to fibronectin, indicating that MAP kinase was required for haptotactic migration to fibronectin (Fig. 6). To assess the involvement of PI3 kinase, B35 cells and L1(+RSLE)-B35 cells were treated with the PI3 kinase inhibitor LY294002 under conditions that inhibit ERK1,2 phosphorylation in L1-B35 cells (Schmid et al., 2000). Ly294002 significantly inhibited migration of both L1-expressing and parental B35 cells to fibronectin, indicating that PI3 kinase also played a role in haptotactic migration to extracellular matrix proteins.
MAP kinase activation and haptotactic migration are impaired by L1 CRASH mutations
Three known mutations in the intracellular domain of L1 occurring in the CRASH syndrome were assessed for adverse effects on potentiating migration to fibronectin. Established B35 cell lines were studied that expressed L1(S1194L), L1(S1224L), or L1(Y1229H) at levels equivalent to wild-type L1 in L1(+RSLE)-B35 cells (Needham et al., 2001). All three mutants displayed reduced ability to potentiate haptotactic migration to fibronectin (Fig. 7). Similar results were observed in HEK293 cells transiently expressing these mutants (data not shown). Attachment of B35 or HEK293 cells to the filters was unaffected by expression of L1 mutant proteins, because the total number of cells on both sides of the filters was the same for mutant and wild-type L1-expressing cells after the migration assay.
Fig. 7.

CRASH mutations in the L1 cytoplasmic domain suppress L1-potentiated migration to fibronectin. B35 cell lines stably expressing no L1 (B35), wild-type L1(+RSLE), or L1(+RSLE) with point mutations S1194L, S1224L, or Y1229H were assayed for haptotactic migration to fibronectin for 24 hr. Each sample was assayed in triplicate, and experiments were repeated at least twice with similar results. *Statistically significant differences in means compared with L1(+RSLE) using the t test (p< 0.05).
To determine whether the intracellular L1 CRASH mutants were coordinately impaired for intracellular signaling to MAP kinase, ERK2 phosphorylation was assayed in B35 cells expressing L1(+RSLE) or L1 CRASH mutants. L1 was clustered on the B35 cell surface with complexes of Neuro4 monoclonal antibodies against the L1 extracellular domain, followed by immunoprecipitation and immunoblotting for dually phosphorylated, activated ERK2 as described (Schmid et al., 2000). This antibody recognizes all L1 mutants equivalently as shown by immunoblotting and immunolabeling (Needham et al., 2001). L1(+RSLE)-B35 cells displayed levels of ERK2 phosphorylation approximately threefold greater than parental B35 cells as expected (Schaefer et al., 1999;Schmid et al., 2000), whereas B35 cells expressing L1 mutants S1194L, S1224L, or Y1229H exhibited significantly lower levels of ERK2 phosphorylation (Fig. 8). The neuronal L1 RSLE sequence in the cytoplasmic domain was also required for maximal MAP kinase activation in B35 cells, as shown by the substantial reduction in ERK2 phosphorylation in B35 cells expressing L1(−RSLE) (Fig. 8) in accord with studies using other cell types (Schaefer et al., 1999). To address whether the RGD sequence in the L1 Ig6 domain was required for MAP kinase activation induced by L1 clustering, B35 neuroblastoma cells expressing the RGD→KGE mutation were analyzed similarly for ERK2 phosphorylation. Cells expressing the L1(KGE) mutant displayed a significantly reduced level of ERK2 phosphorylation compared with wild-type L1(+RSLE) (Fig.9). Thus effective MAP kinase activation and potentiated migration required determinants of the L1 cytoplasmic domain, including the RSLE motif necessary for L1 internalization through the clathrin pathway, and additionally the extracellular RGD sequence in the L1 Ig6 domain.
Fig. 8.

MAP kinase activation is impaired by L1 CRASH mutations. Parental B35 cells were cotransfected for transient expression of HA-tagged ERK2 and plasmids encoding L1(+ or −RSLE) or indicated L1 mutants. Cells were stimulated for 10 min with NIgG or L1 monoclonal antibody Neuro4 complexed with F(ab′)2 fragments of anti-mouse IgG (Fc-specific). HA-tagged ERK2 was immunoprecipitated with anti-HA antibody and blotted with anti-Active MAPK antibodies (p ERK2), and then the amount of immunoprecipitated ERK2 protein was analyzed by stripping and reprobing blots with ERK2 antibodies (ERK2) as shown in a representative experiment in the panels below. Densitometric scanning determined the amount of ERK2 phosphorylation relative to ERK2 protein in L1 antibody-treated and NIgG-treated cells. Results of multiple experiments were averaged to obtain the mean values with SEM shown in the panel above. ERK2 phosphorylation is expressed relative to basal levels of ERK2 phosphorylation in nonimmune IgG-treated B35 cells (NIg). *Statistically significant differences in the mean ERK2 phosphorylation of L1 mutant-expressing B35 cells compared with L1(+RSLE)-B35 cells were evaluated by the t test (p < 0.02).
Endocytosis of L1 and β1 integrins
The results above raised the possibility that L1 might act by stimulating the endocytosis of β1 integrins. To test this, L1(+RSLE)-transfected and nontransfected HEK293 cells were treated with L1 antibody 6096 under conditions sufficient to cluster cell surface L1 and also with the β1 integrin-activating antibody MAB2000, and then internalization was followed by double-label indirect immunofluorescence. As seen by confocal microscopy, L1 and β1 integrins were initially present on the surface of L1-transfected cells (Fig. 9A,B) (0 min). A similar distribution of β1 integrin was seen in untransfected cells (Fig.9D). In image overlays, L1 and β1 integrins appeared to represent primarily distinct molecular populations on the cell surface (Fig. 9C). After 5 min of antibody treatment, β1 integrins and L1 were observed in small cytoplasmic vesicles (Fig.9E,F, arrows), which were probably early endosomes as judged from their size, location, and time of appearance (Ng et al., 1999). L1 and β1 integrins did not colocalize to a major degree within these endocytic vesicles (Fig.9G). Interestingly, substantially more β1 integrin-containing vesicles were seen after 5 min in L1-transfected cells (Fig. 9E) compared with nontransfected cells (Fig.9H). Image analysis revealed that ∼2.5-fold more β1 integrin accumulated in internalized vesicles in L1(+RSLE)-expressing cells than in untransfected HEK cells (no L1) (Fig. 10). After 60 min, L1 antibody complexes accumulated in larger endocytotic vesicles with the appearance of multivesicular bodies (Fig. 9J, long arrow) in accord with previous results (Kamiguchi et al., 1998; Needham et al., 2001). At this time β1 integrins were still present in the small endocytic vesicles and few colocalized with L1 (Fig. 9I,K). Nontransfected cells displayed less prominent internalization of β1 integrin than L1 transfected cells at 60 min (Fig. 9L) or 40 min (data not shown). These results were consistent with the interpretation that L1 clustering induced an accumulation of β1 integrins in early endocytic vesicles.
Fig. 10.

Mean fluorescent pixel density of internalized β1 integrin in confocal micrographs from experiments of Figure 9. After 5 min at 37°C there was a statistically significant increase in internalized β1 integrins in L1(+RSLE)-HEK293 cells compared with HEK293 cells not expressing L1 (∗) and a decrease in internalized β1 integrins in L1(KGE)-expressing cells compared with L1(+RSLE)-HEK293 cells (**) by the t test (one-tailed;p < 0.05).
This increase was dependent on the RGD sequence in the L1 Ig6 domain, because clustering of the L1(KGE) mutant expressed in HEK293 cells did not elicit enhanced β1 integrin accumulation at 5 min of incubation (Fig. 9M), although the L1 mutant appeared to be internalized (Fig. 9N). Image analysis revealed that L1 (KGE)-expressing cells internalized approximately the same amount of β1 integrin as untransfected HEK293 cells (no L1) (Fig. 10). It should be noted that a physical association between L1 and β1 integrin was also not detected by co-immunoprecipitation from 1% Triton X-100 extracts of L1(+RSLE)-transfected HEK293 cells plated on fibronectin with or without antibody-induced L1 clustering or from extracts of postnatal day 7 mouse cerebelli (data not shown).
L1 and β1 antibodies perturb neuronal migration in acute cerebellar slices
To evaluate the significance of L1 and β1 integrins in neuronal cell migration in a bioassay, we tested the ability of function blocking L1 and β1 integrin antibodies to perturb migration of cerebellar granule neurons in mouse cerebellar slice cultures. L1 is expressed on postmitotic premigratory and migrating cerebellar granule neurons (Persohn and Schachner, 1987), and L1 antibodies have been reported to impede granule cell migration in explants (Lindner et al., 1986; Chuong et al., 1987; Crossin et al., 1990). Extracellular matrix proteins such as laminin, thrombospondin, and tenascin are also present in the developing cerebellum and participate in aspects of cell migration (O'Shea et al., 1990; Husmann et al., 1992; Liesi et al., 1992; Fishman and Hatten, 1993). To ask whether L1 and β1 integrins could cooperate in modulating granule cell migration under conditions reflective of an in vivo environment, mouse cerebellar neurons generated on postnatal day 4 were pulse labeled with BrdU in vivo as described (Anton et al., 1996). Labeled cerebella were sectioned coronally into 200 μm slices and incubated in culture for 21 hr in the presence and absence of L1 and β1 integrin antibodies. After 21 hr the extent of radial migration of BrdU-labeled neurons from the EGL into the IGL was assessed after fixation by immunofluorescence staining with BrdU antibodies. At time 0, labeled cells were present almost exclusively in the EGL (Fig.11A). After 21 hr, many labeled cells had migrated into the IGL with some remaining in the EGL (Fig. 11B). Nonimmune IgG had no effect on granule cell migration (Fig. 11C). In the presence of either polyclonal L1 antibodies (Fig. 11D) or monoclonal β1 integrin antibodies (E), there was little visible retardation in granule cell migration. However, in the presence of both L1 and β1 integrin antibodies, migration was strikingly inhibited, as manifested in an accumulation of labeled neurons in the EGL (Fig. 11F). Cell migration was not inhibited completely, because labeled cells were also present in the top part of the IGL, indicating the involvement of other factors. Quantitative evaluation of the effect of antibodies on migration of labeled neurons was made by measurement of the migration index; i.e., the radial distance (perpendicular to the pial surface) that BrdU-labeled granule cells migrated relative to the width of the cerebellar cortex in the same region. L1 or β1 integrin antibodies alone produced a small but significant decrease in the migration index. The inhibition of migration by our L1 antibody (17%) after 21 hr in vitro was in general accord with the inhibition (33%) reported by Lindner et al. (1986) for slices from P10 mice using a different L1 antibody after 3 d in vitro. A combination of the two antibodies decreased the migration index to a greater than additive extent (Table1). The percentage of BrdU-labeled cells in the EGL was also measured and was found to increase to a greater extent in the presence of both antibodies compared with either antibody alone (Table 1). By this measurement the β1 integrin antibody but not the L1 antibody significantly reduced migration when used alone. Differences in age of mice, antibodies, labeling, and culture conditions may have contributed to the smaller inhibition of migration produced by L1 antibodies in our experiment compared with Lindner et al. (1983). The results support a potential role for L1 and β1 integrins in coordinate regulation of radial migration of cerebellar granule cells.
Fig. 11.
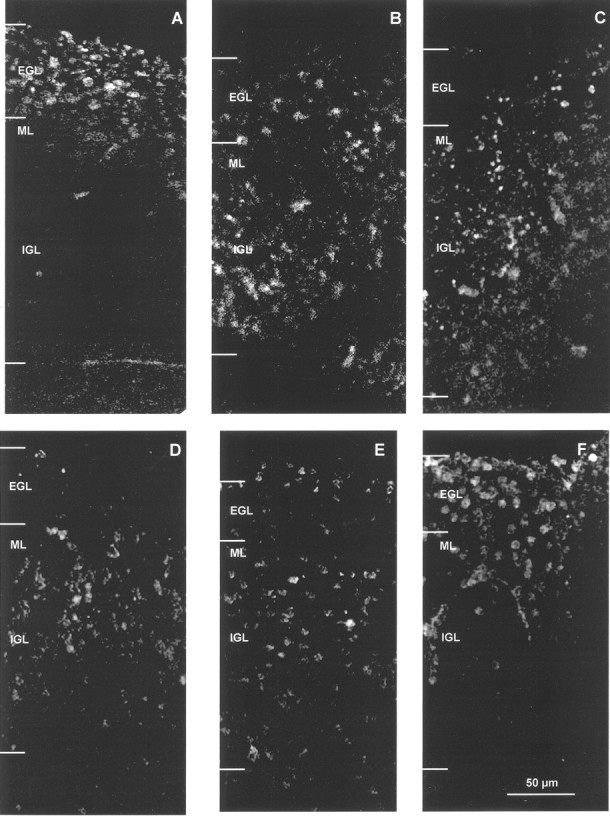
Effect of L1 and β1 integrin antibodies on migration of newly generated BrdU-labeled neurons in cerebellar slice cultures. BrdU-labeled cell distribution in postnatal day 4 mouse cerebellar slices after 0 hr in culture (A), after 21 hr in culture (B), after 21 hr in culture with nonimmune IgG (C), after 21 hr in culture with L1 polyclonal antibody 6096 (D), after 21 hr in culture with β1 integrin monoclonal antibody CD29 (E), and after 21 hr in culture with both L1 antibody 6096 and β1 integrin antibody CD29 (F). At 0 hr, BrdU-labeled cells were restricted primarily to the EGL (A). In the next 21 hr, newly generated BrdU-positive neurons migrated into the IGL under control conditions (B) or in the presence of nonimmune IgG (C). In the presence of both L1 and β1 integrin antibodies (F), migration was impeded, and neurons did not migrate as far as neurons in the presence of only L1 antibody (D) or β1 integrin antibody (E). EGL, External granular layer;ML, molecular layer; IGL, internal granular layer. The borders of the Purkinje cell layer and ML/IGL were indistinct and thus were not delineated in this figure. Scale bar (shown in F forA–F), 50 μm.
Table 1.
Effect of L1 and β1 integrins on migration of BrdU-labeled neurons in P4 mouse cerebellar slices1-a
| Treatment | Migration index (relative depth of labeled cells in the cerebellar cortex) | Labeled neurons in EGL (%) |
|---|---|---|
| None (0 hr) | 0.18 ± 0.01 | 94 |
| None (21 hr) | 0.59 ± 0.03 | 11 |
| Nonimmune IgG (21 hr) | 0.54 ± 0.02 | 6 |
| L1 antibody (21 hr) | 0.49 ± 0.02* | 7 |
| β1 integrin antibody (21 hr) | 0.44 ± 0.02* | 14* |
| L1 + β1 integrin antibodies (21 hr) | 0.34 ± 0.02* | 27* |
Significantly different compared with nonimmune IgG control (21 hr) by the t test (one-tailed; p< 0.05).
See Materials and Methods for assay details.
DISCUSSION
This study describes a novel function for L1 as a potentiator of neuronal cell migration toward extracellular matrix proteins through β1 integrins by a MAP kinase signaling pathway dependent on receptor-mediated endocytosis. L1 potentiated haptotactic migration through the fibronectin-specific integrin α5β1 in HEK293 cells but appeared capable of activating other integrin subclasses, because migration of B35 neuroblastoma cells to laminin and vitronectin was also stimulated by L1. Potentiated migration depended on determinants of L1 endocytosis: dynamin I, c-Src, and the AP2/clathrin binding sequence (RSLE) in the neuronal form of L1. Determinants within the cytoplasmic domain of L1 that are mutated in the CRASH syndrome and the RGD sequence in the extracellular Ig6 domain of L1 were essential for potentiating cell migration. Surprisingly, clustering of cell surface L1 caused enhanced β1 integrin accumulation into endocytic vesicles in addition to L1 internalization. Results were consistent with a model in which L1 potentiated neuronal cell migration toward extracellular matrix proteins through L1 and β1 integrin endocytosis leading to MAP kinase activation. Function-blocking L1 and β1 integrin antibodies cooperatively retarded the migration of granule neurons in early postnatal mouse cerebellar slices, underscoring the potential for a functional L1 integrin interaction in neuronal migration in vivo.
Several lines of evidence support the interpretation that potentiation of integrin-mediated haptotactic migration by L1 derived from its ability to activate MAP kinase through endocytosis of L1. (1) Dominant negative dynamin I (K44A), which suppresses L1 endocytosis and MAP kinase activation (Schaefer et al., 1999; Schmid et al., 2000), inhibited L1-potentiated migration to fibronectin. (2) The L1 RSLE sequence, which mediates clathrin-dependent rapid L1 endocytosis (Schaefer et al., 1999; Long et al., 2001), was required for L1-potentiated migration as well as MAP kinase activation. Because the ERK inhibitor PD98059 does not block L1 internalization, MAP kinase activation occurs subsequent to L1 endocytosis (Schmid et al., 2000). Because the L1(-RSLE) form recycles more slowly than L1(+RSLE), it remains to be determined whether this form would stimulate migration and MAP kinase activity at longer times than were evaluated in our assays. (3) Inhibition of c-Src kinase by PP2 or the kinase-inactive Src (K295M) mutant blocked L1-potentiated migration. c-Src is required for dynamin-mediated internalization of L1 (Schmid et al., 2000) and other receptors (Carpenter, 2000). Interestingly, c-Src can phosphorylate tyrosine residues in components of the endocytic machinery, because β2 adrenergic stimulation causes c-Src-induced tyrosine phosphorylation of dynamin (Ahn et al., 1999; Luttrell et al., 1999), and epidermal growth factor stimulation induces c-Src-induced tyrosine phosphorylation and membrane recruitment of clathrin (Wilde et al., 1999).
L1-potentiated migration and MAP kinase activation may also depend on its ability to stimulate the internalization of β1 integrins as observed in this study. One model consistent with all of the evidence is that transient association of L1 with β1 integrin through the L1 RGD sequence induces endocytosis of both L1 and β1 integrins, leading to activation of MAP kinase through β1 integrin signaling to potentiate migration toward extracellular matrix proteins. In accord with this model, mutation of the L1 RGD sequence coordinately abolished its ability to stimulate the accumulation of β1 integrin into endocytic vesicles and to activate MAP kinase. Furthermore, the time of maximal ERK phosphorylation induced by L1 (10 min) (Schmid et al., 2000) closely followed the time of maximal β1 integrin accumulation into endocytic vesicles (5 min). β1 integrins have been shown to internalize with similar kinetics into early endosomes in non-neuronal cells by a dynamin-dependent pathway (Ng et al., 1999). The possibility that L1 signals to MAP kinase through β1 integrins is consistent with the common intermediates shared by L1 and early integrin signaling pathways (Src, PI3 kinase, Rac1, Vav-2, ERK; see introductory remarks). Indeed PI3 kinase was shown here to be required for haptotactic migration of B35 cells to fibronectin. PI3 kinase may contribute to migration through its ability to influence integrin endocytosis or recycling (Siddhanta et al., 1998; Ng et al., 1999), because its product PI-3,4,5 phosphate can bind the plextrin homology domain of dynamin I and stimulate dynamin GTPase activity in vitro(Barylko et al., 1998); however, its exact function in haptotactic migration has not been determined.
L1 also potentiated migration of B35 cells toward laminin, which can bind in a non-RGD manner to certain integrins such as α6β1. An interesting possibility is that interactions between L1 and an RGD binding integrin might stimulate migration by inside-out signaling to increase the affinity of non-RGD binding integrins such as α6β1 for extracellular matrix. Such cross-talk among integrin subclasses to increase integrin affinity is established (Simon et al., 1997; Blystone et al., 1999) and can be mediated by signaling from Ig-like cell adhesion molecules such as PECAM (Chiba et al., 1999). Alternatively, L1 may regulate non-RGD-binding integrins through its ability to interact with integrins through its third fibronectin III domain (Silletti et al., 2000) or through interaction with other cell surface molecules such as the tetraspan cell surface molecule CD9, which can enhance laminin-induced cell migration and neurite growth through α6β1 integrin (Schmidt et al., 1996).
Current and previous findings do not preclude a mechanism in which internalization of only L1 and not β1 integrins is the critical factor in MAP kinase activation. Nonetheless β1 integrins are clearly required for L1-potentiated migration as shown by antibody perturbation experiments, but they could perform a function other than signaling to MAP kinase. For example, integrin endocytosis at the rear of cells may facilitate detachment from the substrate necessary for forward migration, and together with integrin recycling to the leading edge, endocytosis may help establish an adhesive gradient needed for directional cell motility (Bretscher, 1996; Lauffenburger and Horwitz, 1996; Condic and Letourneau, 1997).
There is increasing evidence that signaling to MAP kinase can occur from endosomal compartments (Ceresa and Schmid, 2000). Elevated levels of internalized L1 or β1 integrins could prolong signaling from endocytic compartments or place receptor complexes in an appropriate cellular location to interact with downstream signaling molecules necessary for stimulating motility. The substrates of MAP kinase important for L1-potentiated haptotactic cell migration have not been identified but might include cytoplasmic targets such as myosin light chain kinase, the phosphorylation of which by ERK1,2 has been shown to be required for collagen-dependent FG carcinoma cell motility (Klemke et al., 1997), or nuclear transcription factors modulating expression of genes important for cell motility such as those encoding cytoskeletal proteins.
The RGD sequence in the L1 Ig6 domain was necessary for potentiating haptotactic migration to fibronectin through α5β1 integrin, for stimulating β1 integrin accumulation in endocytic vesicles, and for MAP kinase activation. The L1 RGD sequence may directly interact with RGD-binding integrins either in cis or trans, or it may simply stabilize a conformation necessary for the signaling properties of L1. However, structural modeling predicts that the L1 RGD sequence is located at the molecular surface within a β turn, where it would be available for binding to integrins (Drescher et al., 1996). Because we did not observe colocalization of L1(+RSLE) and β1 integrins in internalized vesicles and did not detect a stable association between these proteins by co-immunoprecipitation from transfected HEK293 cells or cerebellar extracts, such an association within the plasma membrane might be transient or low in affinity. Acis-interaction of the L1 and integrins α5β1, αvβ3, and α9β1 mediating adhesion in M21 melanoma cells that requires two dibasic sequences in the third fibronectin III domain of L1 but could also involve the RGD sequence has been described previously (Silletti et al., 2000). The third fibronectin III domain was shown to be critical for neurite outgrowth by cerebellar neurons in vitro (Stallcup, 2000). A potential cis-interaction between L1 and integrins was implicated previously in neurite growth of sensory neurons (Felsenfeld et al., 1994) and PC12 cells (Yip and Siu, 2001). Through its RGD sequence, L1 has also been shown to interact functionally, although not physically, with several RGD-binding integrins (α5β1, αvβ1, α4β1, αvβ3, and αIIbβ3) to mediate adhesion and neurite outgrowth on L1 substrates (Moos et al., 1988; Ruppert et al., 1995; Ebeling et al., 1996; Montgomery et al., 1996; Felding-Habermann et al., 1997; Yip et al., 1998). In addition to enhancing β1 integrin internalization, potential binding of the L1-RGD sequence to integrins might have the effect of increasing integrin affinity for ligands, inasmuch as low concentrations of RGD peptides superactivate integrin affinity for RGD-containing ligands (Legler et al., 2001).
While our studies were in progress, it was reported that ectodomain cleavage of L1 by ADAMs (A Disintegrin and Metalloproteinase) can promote migration of transfected CHO cells through αvβ5 integrin independent of the L1 cytoplasmic domain (Mechtersheimer et al., 2001). Our experiments do not rule out an involvement of metalloprotease cleavage of L1 in the mechanism of haptotoactic migration. However, in our studies with B35 neuroblastoma cells and HEK293 cells, the cytoplasmic domain of L1 plays an essential role in stimulating cell migration by providing linkage to AP2/clathrin necessary for signaling and perhaps also to the actin cytoskeleton through ankyrin as shown by impairment of potentiated migration by particular CRASH mutations in the L1 cytoplasmic domain.
Three mutations in the intracellular domain of L1 (S1194L, S1224L, Y1229H) associated with the CRASH syndrome coordinately impaired L1-potentiated haptotactic migration and MAP kinase activation. Each of these mutants is capable of antibody-induced internalization (Needham et al., 2001); thus their reduced capacity to potentiate haptotactic migration cannot be ascribed to defective L1 internalization. Instead these L1 mutants might be compromised in their ability to associate with critical intracellular components important for cytoskeletal association. In particular the L1(S1224L) and L1(Y1229H) mutants fail to recruit the actin adapter protein ankyrin in cellular assays (Needham et al., 2001). On the other hand, the L1(S1194L) mutant recruits ankyrin normally (Needham et al., 2001), but it may interfere with the function of juxtamembrane residues in the L1 cytoplasmic domain that have been shown to mediate association with the actin cytoskeleton through a molecule other than ankyrin (Dahlin-Huppe et al., 1997). The inabilities of the L1(S1194L) mutation to potentiate haptotactic migration and to activate MAP kinase represent the first defective cellular responses to be reported for this CRASH mutation. Because extracellular CRASH mutants (R184Q, H210Q, E309K, Y784C, and Y1070C) potentiated migration of HEK293 cells normally, impaired integrin-mediated functions would not appear to contribute to the pathology associated with these mutations.
The capacity of L1 to potentiate haptotactic migration toward extracellular matrix proteins through integrins in these in vitro studies may reflect a physiological role for L1 in integrin-dependent migration or guidance of neuronal precursors during nervous system development. Neuronal migration and axon guidance are influenced by extracellular matrix proteins in the embyronic brain (Reichardt and Tomaselli, 1991; Liesi et al., 1992; Sheppard et al., 1995), and integrin knock-out mice reveal critical functions for β1 integrins in neuronal migration and laminar organization of the cerebral cortex (Anton et al., 1999; Dulabon et al., 2000). The reduction of cerebellar neuron migration in acute slice preparations by a combination of function-blocking L1 and β1 integrin antibodies may reflect alterations in glial-dependent or glial-independent stages of neuronal migration, which differ in mode, rate, and mechanism (Komuro and Rakic, 1998; Komuro et al., 2001). It should be noted that the combined inhibition by L1 and β1 integrin antibodies, although greater than additive, did not completely prevent neuronal migration and thus may not reflect a cooperative mechanism for cerebellar granule cell migration in vivo. Clearly not all cerebellar neurons were affected by the antibodies, and the conditions used for acute slice preparations could affect neuronal migration mechanisms. Nonetheless, the reduction in migration of labeled neurons through the IGL in the presence of L1 antibodies alone was in general accord with other studies using L1 antibodies to perturb granule cell migration in mouse cerebellar slices (Lindner et al., 1983, 1986) and Ng-CAM antibodies in chick cerebellar slices (Chuong et al., 1987; Crossin et al., 1990). Although β1 integrin antibodies alone produced a small inhibition of granule cell migration in our system, greater inhibition was seen in combination with L1 antibodies, predicting that the relatively unimpaired migration of cerebellar neurons in conditional β1 integrin knock-out mice (Graus-Porta et al., 2001) might be more strongly perturbed in L1–β1 integrin double mutants. Other L1 family members may share the ability to promote cell migration or axon growth through integrins. Indeed, NrCAM cooperates with β1 integrins in retinal ganglion cells to promote fibronectin-induced neurite outgrowth (Treubert and Brummendorf, 1998). In conjunction with β1 integrins, L1 may contribute to normal neurodevelopmental processes, whereas mutation of critical residues in the L1 cytoplasmic domain may give rise to abnormal aspects of neuronal migration that result in pathological features of brain development or mental function in the CRASH syndrome.
K.T., V.K., and A.K.P. contributed equally to this work.
This work was supported by National Institutes of Health Grants NS26620 and HD35170. We thank Eva Anton for providing reagents and assistance with the cerebellar slice bioassay. The contribution of Galina Demyanenko in preparation of cerebellar slices is gratefully acknowledged. We thank John Hemperly for providing reagents, Lee Needham for discussion, and Dr. Robert Bagnell for assistance in confocal microscopy.
Correspondence should be addressed to Patricia F. Maness, Department of Biochemistry and Biophysics, 505 Mary Ellen Jones Building, CB#7260, University of North Carolina School of Medicine, Chapel Hill, NC 27599-7260. E-mail: srclab@med.unc.edu.
REFERENCES
- 1.Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. Src-mediated tyrosine phosphorylation of dynamin is required for β2-adrenergic receptor internalization and mitogen-activated protein kinase signaling. J Biol Chem. 1999;274:1185–1188. doi: 10.1074/jbc.274.3.1185. [DOI] [PubMed] [Google Scholar]
- 2.Anton ES, Cameron RS, Rakic P. Role of neuron–glial junctional domain proteins in the maintenance and termination of neuronal migration across the embryonic cerebral wall. J Neurosci. 1996;16:2283–2293. doi: 10.1523/JNEUROSCI.16-07-02283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anton ES, Kreidberg JA, Rakic P. Distinct functions of α3 and α(v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron. 1999;22:277–289. doi: 10.1016/s0896-6273(00)81089-2. [DOI] [PubMed] [Google Scholar]
- 4.Barylko B, Binns D, Lin KM, Atkinson MA, Jameson DM, Yin HL, Albanesi JP. Synergistic activation of dynamin GTPase by Grb2 and phosphoinositides. J Biol Chem. 1998;273:3791–3797. doi: 10.1074/jbc.273.6.3791. [DOI] [PubMed] [Google Scholar]
- 5.Beasley L, Stallcup WB. The nerve growth factor-inducible large external (NILE) glycoprotein and neural cell adhesion molecule (N-CAM) have distinct patterns of expression in the developing rat CNS. J Neurosci. 1987;7:708–715. doi: 10.1523/JNEUROSCI.07-03-00708.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett V, Chen L. Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol. 2001;13:61–67. doi: 10.1016/s0955-0674(00)00175-7. [DOI] [PubMed] [Google Scholar]
- 7.Blystone SD, Slater SE, Williams MP, Crow MT, Brown EJ. A molecular mechanism of integrin cross-talk: αvβ3 suppression of calcium/calmodulin-dependent protein kinase II regulates α5β1 function. J Cell Biol. 1999;145:889–897. doi: 10.1083/jcb.145.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bodary SC, McLean JW. The integrin β1 subunit associates with the vitronectin receptor αv subunit to form a novel vitronectin receptor in a human embryonic kidney cell line. J Biol Chem. 1990;265:5938–5941. [PubMed] [Google Scholar]
- 9.Bretscher MS. Moving membrane up to the front of migrating cells. Cell. 1996;85:465–467. doi: 10.1016/s0092-8674(00)81246-5. [DOI] [PubMed] [Google Scholar]
- 10.Brummendorf T, Hubert M, Treubert U, Leuschner R, Tarnok A, Rathjen FG. The axonal recognition molecule F11 is a multifunctional protein: specific domains mediate interactions with Ng-CAM and restrictin. Neuron. 1993;10:711–727. doi: 10.1016/0896-6273(93)90172-n. [DOI] [PubMed] [Google Scholar]
- 11.Burrows L, Clark K, Mould AP, Humphries MJ. Fine mapping of inhibitory anti-α5 monoclonal antibody epitopes that differentially affect integrin-ligand binding. Biochem J. 1999;344:527–533. [PMC free article] [PubMed] [Google Scholar]
- 12.Carpenter G. The EGF receptor: a nexus for trafficking and signaling. BioEssays. 2000;22:697–707. doi: 10.1002/1521-1878(200008)22:8<697::AID-BIES3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 13.Ceresa BP, Schmid SL. Regulation of signal transduction by endocytosis. Curr Opin Cell Biol. 2000;12:204–210. doi: 10.1016/s0955-0674(99)00077-0. [DOI] [PubMed] [Google Scholar]
- 14.Chiba R, Nakagawa N, Kurasawa K, Tanaka Y, Saito Y, Iwamoto I. Ligation of CD31 (PECAM-1) on endothelial cells increases adhesive function of αvβ3 integrin and enhances β1 integrin-mediated adhesion of eosinophils to endothelial cells. Blood. 1999;94:1319–1329. [PubMed] [Google Scholar]
- 15.Chung WW, Lagenaur CF, Yan YM, Lund JS. Developmental expression of neural cell adhesion molecules in the mouse neocortex and olfactory bulb. J Comp Neurol. 1991;314:290–305. doi: 10.1002/cne.903140207. [DOI] [PubMed] [Google Scholar]
- 16.Chuong CM, Crossin KL, Edelman GM. Sequential expression and differential function of multiple adhesion molecules during formation of cerebellar cortical layers. J Cell Biol. 1987;104:331–342. doi: 10.1083/jcb.104.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark EA, King WG, Brugge JS, Symons M, Hynes RO. Integrin-mediated signals regulated by members of the rho family of GTPases. J Cell Biol. 1998;142:573–586. doi: 10.1083/jcb.142.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen NR, Taylor JSH, Scott LB, Guillery RW, Soriano P, Furley AJW. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol. 1997;8:26–33. doi: 10.1016/s0960-9822(98)70017-x. [DOI] [PubMed] [Google Scholar]
- 19.Condic ML, Letourneau PC. Ligand-induced changes in integrin expression regulate neuronal adhesion and neurite outgrowth. Nature. 1997;389:852–856. doi: 10.1038/39878. [DOI] [PubMed] [Google Scholar]
- 20.Crossin KL, Prieto AL, Hoffman S, Jones FS, Friedlander DR. Expression of adhesion molecules and the establishment of boundaries during embryonic and neural development. Exp Neurol. 1990;109:6–18. doi: 10.1016/s0014-4886(05)80004-4. [DOI] [PubMed] [Google Scholar]
- 21.Dahlin-Huppe K, Berglund EO, Ranscht B, Stallcup WB. Mutational analysis of the L1 neuronal cell adhesion molecule identifies membrane-proximal amino acids of the cytoplasmic domain that are required for cytoskeletal anchorage. Mol Cell Neurosci. 1997;9:144–156. doi: 10.1006/mcne.1997.0608. [DOI] [PubMed] [Google Scholar]
- 22.Dahme M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat Genet. 1997;17:346–349. doi: 10.1038/ng1197-346. [DOI] [PubMed] [Google Scholar]
- 23.DeAngelis E, MacFarlane J, Du JS, Yeo G, Hicks R, Rathjen FG, Kenwrick S, Brummendorf T. Pathological missense mutations of neural cell adhesion molecule L1 affect homophilic and heterophilic binding activities. EMBO J. 1999;18:4744–4753. doi: 10.1093/emboj/18.17.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeAngelis E, Watkins A, Schaefer M, Brümmendorf T, Kenwrick S. Disease-associated mutations in L1 CAM interfere with ligand interactions and cell-surface expression. Hum Mol Genet. 2002;11:1–12. doi: 10.1093/hmg/11.1.1. [DOI] [PubMed] [Google Scholar]
- 25.Demyanenko G, Tsai A, Maness PF. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knock-out mice. J Neurosci. 1999;19:4907–4920. doi: 10.1523/JNEUROSCI.19-12-04907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Demyanenko GP, Shibata Y, Maness PF. Altered distribution of dopaminergic neurons in the brain of L1 null mice. Brain Res Dev Brain Res. 2001;126:21–30. doi: 10.1016/s0165-3806(00)00129-2. [DOI] [PubMed] [Google Scholar]
- 27.Drescher B, Spiess E, Schachner M, Probstmeier R. Structural analysis of the murine cell adhesion molecule L1 by electron microscopy and computer-assisted modelling. Eur J Neurosci. 1996;8:2467–2478. doi: 10.1111/j.1460-9568.1996.tb01541.x. [DOI] [PubMed] [Google Scholar]
- 28.Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds α3β1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 29.Ebeling O, Duczmal A, Aigner S, Geiger C, Schollhammer S, Kemshead JT, Moller P, Schwartz-Albiez R, Altevogt P. L1 adhesion molecule on human lymphocytes and monocytes: expression and involvement in binding to αvβ3 integrin. Eur J Immunol. 1996;26:2508–2516. doi: 10.1002/eji.1830261035. [DOI] [PubMed] [Google Scholar]
- 30.Felding-Habermann B, Silletti S, Mei F, Siu CH, Yip PM, Brooks PC, Cheresh DA, O'Toole TE, Ginsberg MH, Montgomery AM. A single immunoglobulin-like domain of the human neural cell adhesion molecule L1 supports adhesion by multiple vascular and platelet integrins. J Cell Biol. 1997;139:1567–1581. doi: 10.1083/jcb.139.6.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Felsenfeld DP, Hynes MA, Skoler KM, Furley AJ, Jessell TM. TAG-1 can mediate homophilic binding, but neurite outgrowth on TAG-1 requires an L1-like molecule and β1 integrins. Neuron. 1994;12:675–690. doi: 10.1016/0896-6273(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 32.Fishman RB, Hatten ME. Multiple receptor systems promote CNS neural migration. J Neurosci. 1993;13:3485–3495. doi: 10.1523/JNEUROSCI.13-08-03485.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fransen E, D'Hooge R, Van Camp G, Verhoye M, Sijbers J, Reyniers E, Soriano P, Kamiguchi H, Willemsen R, Koekkoek SKE, De Zeeuw CI, De Deyn PP, Van der Linden A, Lemmon V, Kooy RF, Willems PJ. L1 knockout mice show dilated ventricles, vermis hypoplasia, and impaired exploration patterns. Hum Mol Genet. 1998;7:999–1009. doi: 10.1093/hmg/7.6.999. [DOI] [PubMed] [Google Scholar]
- 34.Fushiki S, Schachner M. Immunocytological localization of cell adhesion molecules L1 and N-CAM and the shared carbohydrate epitope L2 during development of the mouse neocortex. Brain Res. 1986;389:153–167. doi: 10.1016/0165-3806(86)90183-5. [DOI] [PubMed] [Google Scholar]
- 35.Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta 1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 36.Gutwein P, Oleszewski M, Mechtersheimer S, Agmon-Levin N, Krauss K, Altevogt P. Role of Src kinases in the ADAM-mediated release of L1 adhesion molecule from human tumor cells. J Biol Chem. 2000;275:15490–15497. doi: 10.1074/jbc.275.20.15490. [DOI] [PubMed] [Google Scholar]
- 37.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 38.Hlavin ML, Lemmon V. Molecular structure and functional testing of human L1CAM: an interspecies comparison. Genomics. 1991;11:416–423. doi: 10.1016/0888-7543(91)90150-d. [DOI] [PubMed] [Google Scholar]
- 39.Husmann K, Faissner A, Schachner M. Tenascin promotes cerebellar granule cell migration and neurite outgrowth by different domains in the fibronectin type III repeats. J Cell Biol. 1992;116:1475–1486. doi: 10.1083/jcb.116.6.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ignelzi MA, Miller DR, Soriano P, Maness PF. Impaired neurite outgrowth of src-minus cerebellar neurons on the cell adhesion molecule L1. Neuron. 1994;12:873–884. doi: 10.1016/0896-6273(94)90339-5. [DOI] [PubMed] [Google Scholar]
- 41.Kadmon G, Kowitz A, Altevogt P, Schachner M. The neural cell adhesion molecule N-CAM enhances L1-dependent cell-cell interactions. J Cell Biol. 1990;110:193–208. doi: 10.1083/jcb.110.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kadmon G, Montgomery AM, Altevogt P. L1 makes immunological progress by expanding its relations. Dev Immunol. 1998;6:205–213. doi: 10.1155/1998/23451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamiguchi H, Lemmon V. A neuronal form of the cell adhesion molecule L1 contains a tyrosine-based signal required for sorting to the axonal growth cone. J Neurosci. 1998;18:3749–3756. doi: 10.1523/JNEUROSCI.18-10-03749.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamiguchi H, Long KE, Pendergast M, Schaefer AW, Rapoport I, Kirchhausen T, Lemmon V. The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J Neurosci. 1998;18:5311–5321. doi: 10.1523/JNEUROSCI.18-14-05311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kenwrick S, Watkins A, Angelis ED. Neural cell recognition molecule L1: relating biological complexity to human disease mutations. Hum Mol Genet. 2000;9:879–886. doi: 10.1093/hmg/9.6.879. [DOI] [PubMed] [Google Scholar]
- 46.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Komuro H, Rakic P. Distinct models of neuronal migration in different domains of developing cerebellar cortex. J Neurosci. 1998;18:1478–1490. doi: 10.1523/JNEUROSCI.18-04-01478.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komuro H, Yacubova E, Yacubova E, Rakic P. Mode and tempo of tangential cell migration in the cerebellar external granular layer. J Neurosci. 2001;21:527–540. doi: 10.1523/JNEUROSCI.21-02-00527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuhn TB, Stoeckli ET, Condrau MA, Rathjen FG, Sonderegger P. Neurite outgrowth on immobilized axonin-1 is mediated by a heterophilic interaction with L1(G4). J Cell Biol. 1991;115:1113–1126. doi: 10.1083/jcb.115.4.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 51.Legler DF, Wiedle G, Ross FP, Imhof BA. Superactivation of integrin αvβ3 by low antagonist concentrations. J Cell Sci. 2001;114:1545–1553. doi: 10.1242/jcs.114.8.1545. [DOI] [PubMed] [Google Scholar]
- 52.Liesi P, Seppala I, Trenkner E. Neuronal migration in cerebellar microcultures is inhibited by antibodies against a neurite outgrowth domain of laminin. J Neurosci Res. 1992;33:170–176. doi: 10.1002/jnr.490330122. [DOI] [PubMed] [Google Scholar]
- 53.Lindner J, Rathjen FG, Schachner M. L1 mono- and polyclonal antibodies modify cell migration in early postnatal mouse cerebellum. Nature. 1983;305:427–430. doi: 10.1038/305427a0. [DOI] [PubMed] [Google Scholar]
- 54.Lindner J, Zinser G, Werz W, Goridis C, Bizzini B, Schachner M. Experimental modification of postnatal cerebellar granule cell migration in vitro. Brain Res. 1986;377:298–304. doi: 10.1016/0006-8993(86)90872-3. [DOI] [PubMed] [Google Scholar]
- 55.Linnemann D, Raz A, Bock E. Differential expression of cell adhesion molecules in variants of K1735 melanoma cells differing in metastatic capacity. Int J Cancer. 1989;43:709–712. doi: 10.1002/ijc.2910430428. [DOI] [PubMed] [Google Scholar]
- 56.Long KE, Asou H, Snider MD, Lemmon V. The role of endocytosis in regulating L1-mediated adhesion. J Biol Chem. 2001;276:1285–1290. doi: 10.1074/jbc.M006658200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 58.Mechtersheimer S, Gutwein P, Agmon-Levin N, Stoeck A, Oleszewski M, Riedle S, Fogel F, Lemmon V, Altevogt P. Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J Cell Biol. 2001;155:661–673. doi: 10.1083/jcb.200101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meng F, Lowell CA. A β1 integrin signaling pathway involving Src-family kinases Cbl and PI-3 kinase is required for macrophage spreading and migration. EMBO J. 1998;17:4391–4403. doi: 10.1093/emboj/17.15.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miranti CK, Leng L, Maschberger P, Brugge JS, Shattil SJ. Identification of a novel integrin signaling pathway involving the kinase Syk and the guanine nucleotide exchange factor Vav1. Curr Biol. 1998;8:1289–1299. doi: 10.1016/s0960-9822(07)00559-3. [DOI] [PubMed] [Google Scholar]
- 61.Montgomery AM, Becker JC, Siu CH, Lemmon VP, Cheresh DA, Pancook JD, Zhao X, Reisfeld RA. Human neural cell adhesion molecule L1 and rat homologue NILE are ligands for integrin αvβ3. J Cell Biol. 1996;132:475–485. doi: 10.1083/jcb.132.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moos M, Tacke R, Scherer H, Teplow D, Früh K, Schachner M. Neural adhesion molecule L1 as a member of the immunoglobulin superfamily with binding domains similar to fibronectin. Nature. 1988;334:701–703. doi: 10.1038/334701a0. [DOI] [PubMed] [Google Scholar]
- 63.Needham LK, Thelen K, Maness PF. Cytoplasmic domain mutations of the L1 cell adhesion molecule reduce L1-ankyrin interactions. J Neurosci. 2001;21:1490–1500. doi: 10.1523/JNEUROSCI.21-05-01490.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ. PKCα regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 1999;18:3909–3923. doi: 10.1093/emboj/18.14.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nolte C, Moos M, Schachner M. Immunolocalization of the neural cell adhesion molecule L1 in epithelia of rodents. Cell Tissue Res. 1999;298:261–273. doi: 10.1007/s004419900063. [DOI] [PubMed] [Google Scholar]
- 66.Ohyama K, Kawano H, Asou H, Fukuda T, Oohira A, Uyemura K, Kawamura K. Coordinate expression of L1 and 6B4 proteoglycan/phosphacan is correlated with the migration of mesencephalic dopaminergic neurons in mice. Brain Res Dev Brain Res. 1998;107:219–226. doi: 10.1016/s0165-3806(97)00220-4. [DOI] [PubMed] [Google Scholar]
- 67.O'Shea KS, Rheinheimer JS, Dixit VM. Deposition and role of thrombospondin in the histogenesis of the cerebellar cortex. J Cell Biol. 1990;110:1275–1283. doi: 10.1083/jcb.110.4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pancook JD, Reisfeld RA, Varki N, Vitiello A, Fox RI, Montgomery AM. Expression and regulation of the neural cell adhesion molecule L1 on human cells of myelomonocytic and lymphoid origin. J Immunol. 1997;158:4413–4421. [PubMed] [Google Scholar]
- 69.Persohn E, Schachner M. Immunoelectron microscopic localization of the neural cell adhesion molecules L1 and N-CAM during postnatal development of the mouse cerebellum. J Cell Biol. 1987;105:569–576. doi: 10.1083/jcb.105.1.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pierini LM, Lawson MA, Eddy RJ, Hendey B, Maxfield FR. Oriented endocytic recycling of α5β1 in motile neutrophils. Blood. 2000;95:2471–2480. [PubMed] [Google Scholar]
- 71.Reichardt LF, Tomaselli KJ. Extracellular matrix molecules and their receptors: functions in neural development. Annu Rev Neurosci. 1991;14:531–570. doi: 10.1146/annurev.ne.14.030191.002531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reid RA, Hemperly JJ. Variants of human L1 cell adhesion molecule arise through alternate splicing of RNA. J Mol Neurosci. 1992;3:127–135. doi: 10.1007/BF02919404. [DOI] [PubMed] [Google Scholar]
- 73.Ruppert M, Aigner S, Hubbe M, Yagita H, Altevogt P. The L1 adhesion molecule is a cellular ligand for VLA-5. J Cell Biol. 1995;131:1881–1891. doi: 10.1083/jcb.131.6.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schaefer AW, Kamiguchi H, Wong EV, Beach CM, Landreth G, Lemmon V. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37965–37973. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- 75.Schmid RS, Maness PF. Cell recognition molecules and disorders of neurodevelopment. In: Kalverboer AF, Gramsbergen A, editors. International handbook on brain and behaviour in human development. Kluwer; Groningen, The Netherlands: 2001. pp. 199–218. [Google Scholar]
- 76.Schmid RS, Pruitt WM, Maness PF. A MAP kinase signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. J Neurosci. 2000;11:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmidt C, Kunemund V, Wintergerst ES, Schmitz B, Schachner M. CD9 of mouse brain is implicated in neurite outgrowth and cell migration in vitro and is associated with the α6/β1 integrin and the neural adhesion molecule L1. J Neurosci Res. 1996;43:12–31. doi: 10.1002/jnr.490430103. [DOI] [PubMed] [Google Scholar]
- 78.Sheppard AM, Brunstrom JE, Thornton TN, Gerfen RW, Broekelmann TJ, McDonald JA, Pearlman AL. Neuronal production of fibronectin in the cerebral cortex during migration and layer formation is unique to specific cortical domains. Dev Biol. 1995;172:504–518. doi: 10.1006/dbio.1995.8034. [DOI] [PubMed] [Google Scholar]
- 79.Siddhanta U, McIlroy J, Shah A, Zhang Y, Backer JM. Distinct roles for the p110α and hVPS34 phosphatidylinositol 3′-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol. 1998;143:1647–1659. doi: 10.1083/jcb.143.6.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Silletti S, Mei F, Sheppard D, Montgomery AM. Plasmin-sensitivedibasic sequences in the third fibronectin-like domain of L1-cell adhesion molecule (CAM) facilitate homomultimerization and concomitant integrin recruitment. J Cell Biol. 2000;149:1485–1502. doi: 10.1083/jcb.149.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simon KO, Nutt EM, Abraham DG, Rodan GA, Duong LT. The αvβ3 integrin regulates α5β1-mediated cell migration toward fibronectin. J Biol Chem. 1997;272:29380–29389. doi: 10.1074/jbc.272.46.29380. [DOI] [PubMed] [Google Scholar]
- 82.Stallcup WB. The third fibronectin type III repeat is required for L1 to serve as an optimal substratum for neurite extension. J Neurosci Res. 2000;61:33–43. doi: 10.1002/1097-4547(20000701)61:1<33::AID-JNR4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 83.Takeda Y, Asou H, Murakami Y, Miura M, Kobayashi M, Uyemura K. A nonneuronal isoform of cell adhesion molecule L1: tissue-specific expression and functional analysis. J Neurochem. 1996;66:2338–2349. doi: 10.1046/j.1471-4159.1996.66062338.x. [DOI] [PubMed] [Google Scholar]
- 84.Treubert U, Brummendorf T. Functional cooperation of β1 integrins and members of the Ig superfamily in neurite outgrowth induction. J Neurosci. 1998;18:1795–1805. doi: 10.1523/JNEUROSCI.18-05-01795.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC, Soriano P, Brodsky FM. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677–687. doi: 10.1016/s0092-8674(00)80578-4. [DOI] [PubMed] [Google Scholar]
- 86.Yip PM, Siu CH. PC12 cells utilize the homophilic binding site of L1 for cell-cell adhesion but L1-αvβ3 interaction for neurite outgrowth. J Neurochem. 2001;76:1552–1564. doi: 10.1046/j.1471-4159.2001.00152.x. [DOI] [PubMed] [Google Scholar]
- 87.Yip PM, Zhao X, Montgomery AM, Siu CH. The Arg-Gly-Asp motif in the cell adhesion molecule L1 promotes neurite outgrowth via interaction with the αvβ3 integrin. Mol Biol Cell. 1998;9:277–290. doi: 10.1091/mbc.9.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao X, Siu CH. Differential effects of two hydrocephalus/MASA syndrome-related mutations on the homophilic binding and neuritogenic activities of the cell adhesion molecule L1. J Biol Chem. 1996;271:6563–6566. doi: 10.1074/jbc.271.12.6563. [DOI] [PubMed] [Google Scholar]