

Abstract
We examined the role of excitatory amino acids (EAAs) in the activation of midbrain dopaminergic (DA) neurons evoked by stimulation of the ventromedial and ventrolateral (subcommissural) bed nucleus of the stria terminalis (vBNST). Using anesthetized rats and extracellular recording techniques, we found that 84.8% of ventral tegmental area (VTA) DA neurons were activated synaptically by single-pulse electrical stimulation of the vBNST. In contrast, similar stimulation did not affect the activity of presumed GABA neurons in the VTA. Three characteristic responses were observed in VTA DA neurons: short latency activation (<25 msec; 55.1% of cells), long latency activation (>65 msec; 56% of cells), and inhibition (61.8% of cells, usually followed by long latency excitation). Microinfusion of antagonists of EAA receptors (3 mm kynurenic acid, 100 μm AP-5, or 50 μm CNQX) from a micropipette adjacent to the recording electrode significantly reduced both short and long latency activations evoked in DA neurons by vBNST stimulation. Specific responses were attenuated similarly by AP-5 alone, CNQX alone, or a cocktail of AP-5+CNQX, indicating that joint activation of NMDA plus non-NMDA receptors was required. Stimulation of the vBNST by local microinfusion of glutamate increased the firing and bursting activity of VTA DA neurons. Similar microinfusion of GABA decreased bursting of VTA DA neurons without altering their firing rate. Retrograde and anterograde labeling and antidromic activation of vBNST neurons by VTA stimulation confirmed a direct projection from the vBNST to the VTA. These results reveal that inputs from the vBNST exert a strong excitatory influence on VTA DA neurons mediated by both NMDA and non-NMDA receptors.
Keywords: dopamine neurons, ventral tegmental area, bed nucleus of the stria terminalis, NMDA receptors, non-NMDA receptors, extracellular recording
The amino acidl-glutamate (Glu) is recognized as the major excitatory neurotransmitter in the CNS (for review, see Danbolt, 2001). Anatomical studies have provided evidence for glutamatergic afferents to midbrain DA neurons as well as glutamatergic receptors on dopaminergic (DA) neurons (for review, see Meltzer et al., 1997). The frontal cortex (Carter, 1982; Kornhuber et al., 1984; Sesack and Pickel, 1992), subthalamic nucleus (Kita and Kitai, 1987; Rinvik and Ottersen, 1993), and pedunculopontine nucleus (Tokuno et al., 1988) are regions from which glutamatergic afferents to midbrain DA neurons originate. Recent studies revealed an important role of the ventromedial and ventrolateral bed nucleus of the stria terminalis (vBNST) in opiate abuse (Walker et al., 2000) and opiate withdrawal (Delfs et al., 2000). DA neurons in the ventral tegmental area (VTA) also have long been linked to motivation and drug abuse (Koob and Le Moal, 2001). Recently, we found that stimulation of the vBNST potently and consistently activated VTA DA neurons (Georges and Aston-Jones, 2001). However, the mechanism for activation of VTA DA neurons by vBNST stimulation has not been elucidated. Here we combined neuropharmacological and anatomical methods to identify the neurotransmitter and pathway used in this response. We found that excitatory amino acid (EAA) receptor antagonists locally microinfused into the VTA eliminated or reduced the response of DA neurons to stimulation of the vBNST, indicating that an EAA transmitter likely is involved. Coactivation of both NMDA and non-NMDA receptors is required for one response, revealing a novel EAA mechanism in the VTA. Using tract tracing and antidromic activation, we also identified projections from the vBNST to the VTA, demonstrating a direct anatomic connection. Excitatory synaptic inputs mediated by Glu are a key component of the regulation of dopamine cell excitability and play an important role in the actions of many drugs of abuse (Kalivas and Stewart, 1991; Kalivas and Alesdatter, 1993). These findings reveal a previously unknown EAA pathway in the CNS and characterize a major component in neuronal circuits that control the VTA dopaminergic system.
MATERIALS AND METHODS
Animals. Forty-six Sprague Dawley rats (200–225 gm; Taconic Farms, Germantown, NY) were used. Rats were housed three or four per cage under controlled conditions (22–23°C, 40% relative humidity; 12 hr light/dark illumination cycle; lights on from 07:00 A.M. to 7:00 P.M.) and were allowed free access to commercial chow and tap water.
Surgery. Animals were anesthetized initially with 3% halothane in air that was administered through a facemask. A tracheotomy was performed, and 2% halothane was delivered through a tracheal cannula via spontaneous respiration for surgical procedures. During recording experiments the concentration of halothane was kept at 1.0–1.2%. Animals were placed in a stereotaxic frame, and body temperature was maintained at 36–38°C, using a thermistor-controlled electric heating pad. All incision points were infiltrated with a long-lasting anesthetic (0.5% Sensorcaine). The skull was exposed, and a hole was drilled above the VTA (5.3 mm caudal to bregma; 0.8 mm lateral to the midline). Another hole was drilled to expose the surface of the brain above the BNST (0.3 mm caudal to bregma; 1.5 mm lateral to the midline).
Electrical stimulation of the ventral BNST. Bipolar electrical stimulation of the vBNST was conducted with a concentric electrode (250 μm in diameter overall; 100 μm diameter for inner electrode, which extended 100 μm beyond the outer electrode; FHC, Bowdoinham, ME). This electrode was inserted into the vBNST (see Fig.1A,B; coordinates in millimeters relative to bregma, with the skull flat: anteroposterior, −0.3; mediolateral, 1.5; dorsoventral, 7.2). Electrical stimulation (1.0–5.0 mA, 0.5 Hz, 0.5 msec duration pulses) was administered by using a square pulse stimulator (Master 8; A.M.P.I., Alomone Labs, Jerusalem, Israel) and stimulus isolator (ISO-Flex, A.M.P.I., Alomone Labs).
Fig. 1.

Stimulation and recording sites. A, B, Photomicrographs of coronal sections through two levels of the BNST. The sections were counterstained with DBH immunohistochemistry (dark staining) to delineate the region of dense noradrenergic innervation in the vBNST. White crosses show effective sites of electrical stimulation.Black crosses show the locations of three ineffective electrical stimulation sites in the caudate putamen or in the lateral preoptic area. White and graycircles show locations of effective Glu and GABA microinjections, respectively. Blackcircles depict ineffective microinjections of GABA in the lateral hypothalamus or in the substantia innominata. Ineffective Glu injections in the lateral preoptic area and ventral pallidum are plotted in our recent publication (Georges and Aston-Jones, 2001).C, An electrical stimulation site in the DBH-positive area of the vBNST, marked by passing current through the stimulation electrode (dark lesioned area, arrow).D, Iontophoretic injection of pontamine sky blue (dark spot, arrow; coronal section) marks the recording location for a VTA DA neuron (neutral red stain). Scale bars: A, B, D, 0.5 mm; C, 1.0 mm.
Local chemical manipulation of the ventral BNST. An injection pipette (tip, <50 μm in diameter) was filled with Glu [10 or 50 mm in artificial CSF (aCSF)] or GABA (1m in aCSF) and was lowered into the vBNST. Glu or GABA was microinjected into the vBNST, using brief pulses of pneumatic pressure (Picospritzer, General Valve, Fairfield, NJ). In all experiments a total volume of 60 nl was infused over 30 sec for each injection. Two injections at a single vBNST site typically were given at an interval >30 min. All injection sites were marked by iontophoretic ejection of pontamine sky blue dye from an adjacent micropipette barrel. Data concerning the chemical stimulation of the vBNST with Glu have been reported elsewhere (Georges and Aston-Jones, 2001).
Ventral tegmental area recordings. A glass micropipette (tip diameter, 2–3 μm; 4–6 MΩ) filled with a 2% pontamine sky blue solution in 0.5 m sodium acetate was lowered into the VTA. DA neurons were identified according to well established electrophysiological features (Guyenet and Aghajanian, 1978; Grace and Bunney, 1983, 1984; Tepper et al., 1984; Chiodo, 1988). These included (1) action potential with biphasic or triphasic waveform >2.5 msec in duration, (2) slow spontaneous firing rate (2–9 impulses/sec), (3) single and burst spontaneous firing patterns (characterized by spike–amplitude decrement), and (4) inhibition of spontaneous activity by DA receptor agonists and subsequent reversal by DA receptor antagonists. VTA GABA neurons were identified according to electrophysiological features established by Steffensen et al. (1998). These included (1) action potential with biphasic waveform <500 μsec in duration, (2) rapid spontaneous firing rate, and (3) uninterrupted phasic activity characterized by alternating 0.5–2.0 sec ON/OFF periods. Signals were amplified and filtered (0.1–5 kHz bandpass) by using conventional electronics. Spikes of single neurons were discriminated, and digital pulses were led to a computer for on-line data collection with the use of a laboratory interface and software (CED 1401, SPIKE 2; Cambridge Electronic Design).
After we isolated a single VTA neuron, we recorded prestimulation spontaneous activity to establish baseline activity for at least 5 min. Subsequently, in electrical stimulation experiments single pulses were delivered to the BNST every 2 sec. At least 100 trials were administered per cell. In experiments with chemical manipulation of the vBNST, microinfusions were delivered for 30 sec after at least 5 min of baseline activity from a VTA DA neuron were recorded. Only one microinfusion was made per cell, and no more than eight microinfusions were made in the same site.
Ventral tegmental area recordings and microinfusion of NMDA and non-NMDA antagonists. A double-barrel pipette assembly similar to that described previously (Akaoka and Aston-Jones, 1991) was used for recording VTA spike activity with simultaneous local microinjection of drugs. The recording tip (2–3 μm diameter) protruded ∼130 μm beyond the injection tip (<50 μm diameter). The recording pipette was filled with a 2% pontamine sky blue solution in 0.5 msodium acetate. The injection pipette was filled with one of the following: the broad-spectrum EAA antagonist kynurenic acid (3 mm), the specific NMDA antagonist amino-5-phosphonopentanoic acid (AP-5; 100 μm), the specific non-NMDA antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 50 μm), a mixture of AP-5 (100 μm) plus CNQX (50 μm), or aCSF. All drugs were dissolved in aCSF. Signals were amplified and filtered as described above. After isolating a single VTA neuron, we recorded spontaneous activity for at least 3 min before stimulation tests to establish baseline activity. Subsequently, 100 single electrical pulses were delivered to the vBNST at a rate of 0.5/sec, and VTA responses were recorded. At least 5 min after this control stimulation, an EAA antagonist or aCSF was applied at a rate of 60 nl/min simultaneously with the electrical stimulation to determine the role of EAAs in evoked responses of VTA neurons. At least 15 min after the microinjection, electrical stimulation was applied again to measure the recovery of evoked responses. One hundred trials of single-pulse stimulation were administered in each of the control, drug infusion, and recovery periods. For chemical stimulation of the vBNST an injection pipette (tip, <50 μm in diameter) was filled with Glu (50 mm in aCSF) and was lowered into position. Glu was microinjected into the vBNST by using brief pulses of pneumatic pressure (Picospritzer, General Valve). In all experiments a total volume of 60 nl was infused over 30 sec for each injection. At least 5 min after this control stimulation, AP-5 plus CNQX was applied in the VTA at a rate of 60 nl/min for 2 min before and throughout the BNST stimulation by Glu microinjection to determine the role of EAAs in evoked responses of VTA neurons.
Intravenous drug injection. The jugular vein was cannulated for intravenous administration of pharmacological agents. The DA receptor agonist apomorphine (0.1 mg/kg) and the D2 DA receptor antagonist eticlopride (0.1 mg/kg) were prepared in 0.9% sterile NaCl and were used to characterize VTA DA neurons pharmacologically.
Antidromic activation. Electrical stimulation of the VTA was used to test for antidromic activation of vBNST neurons by using the high-frequency following and collision methods, as described previously (Aston-Jones et al., 1985). A bipolar concentric stimulation electrode was inserted into the VTA, and stimulation of the VTA and recordings in the vBNST used the methods described above. Driven impulses were considered antidromic if they met the following criteria: (1) constant latency driving at 1.5 times threshold stimulation intensity, (2) driving by each of paired stimulus pulses at frequencies of 100 Hz or greater, and (3) collision of driven spikes by spontaneous impulses occurring within a critical interval approximately equal to the sum of the refractory period plus the driving latency. A measure of the axonal refractory period was obtained by determining the minimum interval between paired stimuli producing two action potentials 100% of the time. The delay at which the second response disappeared corresponded to the absolute refractory period of that particular unit. Absolute refractory period measurement and collision of driven spikes with spontaneous impulses were conducted with stimulation amplitudes of 1.5–2.0 times the threshold for driving.
Histology. At the end of each recording penetration the electrode placement was marked with an iontophoretic deposit of pontamine sky blue dye (−20 μA, continuous current for 12–15 min) (see Fig. 1D). In experiments with chemical manipulations of the BNST the dye was deposited similarly to mark injection sites in the BNST. To mark electrical stimulation sites, we passed 10 μA of positive current through the stimulation electrode for 1 min to deposit iron at the electrode tips (see Fig.1C). After the experimental procedures the animals were anesthetized deeply with halothane (5%) and perfused with 4% formaldehyde in 0.1 m phosphate buffer (PB) containing 5% potassium ferrocyanide. The ferrocyanide in this perfusate yields a visible Prussian blue reaction product at the site of the iron deposit (see Fig. 1C). Brains were removed and snap-frozen in a solution of isopentane at −70°C.
There are no sharp cytoarchitectural boundaries differentiating subaspects of the BNST. However, the vBNST is well defined by a dense noradrenergic innervation (Delfs et al., 2000). Thus the vBNST was identified by immunohistochemical staining for dopamine-β-hydroxylase (DBH) to reveal noradrenergic fibers. Brains were cut (40-μm-thick sections) on a cryostat. Sections through the BNST were collected on gelatin-coated slides and were processed for DBH immunohistochemical staining as described previously (Delfs et al., 1998). The sections were incubated overnight at room temperature (25°C) in rabbit anti-DBH primary antibody (1:2000; Eugene Tech, Ridgefield, NJ). After washes, the sections were placed sequentially for 90 min at room temperature in biotinylated donkey anti-rabbit IgG (1:1000; Jackson ImmunoResearch, West Grove, PA) and avidin-biotin-peroxidase complex (ABC; 1:1000; Elite ABC Kit, Vector Laboratories, Burlingame, CA). Then the sections were reacted with DAB (0.025%) containing 0.006% H2O2 in PB. All sections were dried, dehydrated, and coverslipped with Deppex.
Tract tracing methods. Cholera toxin B subunit (CTb; List Biological, Campbell, CA) was prepared for iontophoretic ejection as described previously (Luppi et al., 1995). In brief, 1 mg of lyophilized CTb was reconstituted with 1 ml of 0.1 m PB, pH 6.0, and then desalted; the buffer was exchanged and concentrated to 1% by two repeated 1 hr and 30 min ultrafiltrations (from 1 to 0.1 ml) at 7000 rpm with a Centricon 10 microconcentrator (Amicon, Beverly, MA).
Rats were anesthetized with pentobarbital (50 mg/kg, i.p.) and placed in a stereotaxic apparatus. A scalp incision was made, a hole was drilled in the skull overlaying the VTA or the BNST, and the dura was reflected. Micropipettes (8–10 μm tip diameter) were back-filled with the 1% solution of CTb in 0.1 m PB, pH 6.0, and lowered stereotaxically into the VTA (5.3 mm caudal to bregma; 0.8 mm lateral and 8.0 mm ventral to skull surface) or the BNST (0.3 mm caudal to bregma; 1.5 mm lateral and 7.2 mm ventral to skull surface). To eject CTb, we applied a pulsed positive current (7 sec on, 7 sec off; 1 μA, 12 min) (model number 51413 Precision Current Source, Stoelting, Wood Dale, IL). After the injection the pipette was left in place for 15 min to prevent leakage of the tracer along the pipette track. Animals were allowed to survive 7 d and then were perfused through the heart with 250 ml of an ice-cold fixative in 0.1 m PB containing 4% paraformaldehyde (4% PFA) for 10 min. Brains were postfixed for 2 hr by immersion in 4% PFA and then immersed overnight in 20% sucrose/0.1 m PB. Afterward, these specimens were frozen rapidly with CO2, and coronal 40-μm-thick sections were cut on a cryostat.
Immunocytochemical methods. Immunohistochemical detection of CTb was performed by sequential incubations of free-floating sections that used a standard ABC technique as described previously (Luppi et al., 1995). Briefly, sections were incubated in 2% normal donkey serum (NDS; Jackson ImmunoResearch) containing 0.2% Triton X-100 in PBS (PBST) for 2 hr and then were incubated overnight at room temperature in goat anti-CTb primary antibody (1:20,000; List Biological). After washes, the sections were placed sequentially for 90 min at room temperature in biotinylated donkey anti-goat IgG (1:1000; Jackson ImmunoResearch) and ABC (1:1000; Elite ABC Kit, Vector). Then the sections were reacted with DAB (0.025%) containing 0.006% H2O2 and 0.6% nickel ammonium sulfate in 0.05 m Tris-HCl buffer, pH 7.6, for 10–15 min at room temperature. For double labeling the CTb-stained sections were incubated overnight at room temperature in rabbit anti-DBH primary antibody (1:2000; Eugene Tech) or in rabbit anti-TH primary antibody (1:10,000; Institut Jacques Boy). After washes, the sections were placed sequentially for 90 min at room temperature in biotinylated donkey anti-rabbit IgG (1:1000; Jackson ImmunoResearch) and ABC (1:1000; Elite ABC Kit, Vector). The sections were reacted with DAB (0.025%) containing 0.006% H2O2 in PB. After this procedure the CTb retrogradely labeled cell bodies were blue-black, whereas the cytoplasm of dopaminergic neurons in the VTA or the noradrenergic terminals in the vBNST was brown. Then all sections were mounted on gelatin-coated glass slides, dried, dehydrated, and coverslipped with Deppex.
Data analysis. During chemical inhibition and stimulation of the vBNST two parameters of VTA impulse activity were studied: the basal firing rate and the proportion of spikes that occurred in bursts. The onset of a burst was defined as the occurrence of two spikes with an interspike interval <80 msec (Grace and Bunney, 1983). The percentage of spikes in bursts was calculated by dividing the number of spikes occurring in bursts by the total number of spikes occurring in the same period of time. Results are expressed throughout as mean ± SEM. These values were subjected to a one-way ANOVA, followed bypost hoc Student's t tests.
During electrical stimulation of the vBNST, cumulative peristimulus time histograms (PSTHs; 5 msec bin width) of VTA activity were generated for each neuron that was recorded. PSTHs were analyzed to determine excitatory and inhibitory epochs as described previously (Jodo et al., 1998). In brief, the mean and SD of counts per bin were determined for a baseline period, defined as the 500 msec epoch preceding stimulation. The onset of excitation was defined as the first of 5 bins for which the mean value exceeded mean baseline activity by 2 SD, and response offset was determined as the time at which activity had returned to be consistently within 2 SD of baseline. Excitatory and inhibitory response magnitudes (Rmags) were normalized for different levels of baseline impulse activity, allowing for comparison of drug effects on evoked responses independent of effects on baseline activity. Rmags for excitation were calculated with the following equation: ExcitationRmag = (counts in excitatory epoch) − (mean counts per baseline bin × number of bins in excitatory epoch).
Inhibition was defined as an epoch of at least 15 bins in which the mean count per bin was at least 35% less than that during baseline. To distinguish postactivation auto-inhibition (Aghajanian and Bunney, 1977; Shepard and Bunney, 1988; Nirenberg et al., 1996) from inhibitory responses mediated by extrinsic afferents, we have considered here only inhibitory epochs not preceded by an excitatory response. Response magnitude for inhibition was calculated as follows: InhibitionRmag = (counts in inhibitory epoch) − (mean counts per baseline bin × number of bins in inhibitory epoch).
Results are expressed throughout as mean ± SEM. These values were subjected to a one-way ANOVA, followed by post hocStudent's t test.
RESULTS
Electrophysiological study
Data are reported for 198 histologically verified VTA neurons (Fig. 1D), which were identified as dopaminergic according to well established electrophysiological features (see Materials and Methods). Spontaneously discharging nonburst- and burst-firing DA cells (Grace and Bunney, 1984) typically fired in a slow, irregular pattern at an average rate of 4.2 ± 0.02 Hz. Action potentials of these cells had biphasic or triphasic waveforms with an average duration of 3.02 ± 0.05 msec. The DA receptor agonist apomorphine was administered for the last DA neuron recorded in each experiment (0.1 mg/kg, i.v.; n = 15). As reported previously (Grace and Bunney, 1983, 1984), this drug consistently inhibited spontaneous impulse activity of DA neurons within 30 sec of injection. A subsequent injection of the D2 DA receptor antagonist eticlopride (0.1 mg/kg, i.v.) consistently restored impulse activity (data not shown).
VTA DA neurons were activated by electrical stimulation of the vBNST
Electrical stimulation of the vBNST typically produced synaptically driven activation of VTA DA neurons (Fig.2). Properties of VTA DA neuronal responses evoked by stimulation of the vBNST are summarized in Table1. Overall, 84.8% of VTA DA neurons (100 of the 118 tested) were activated synaptically by single-pulse stimulation of the vBNST. Three characteristic responses were observed in VTA DA neurons with vBNST stimulation: 55.1% (65 of 118 cells) were activated synaptically with a short onset latency (<25 msec) (Fig.2A,C), 56.0% (66 of 118 cells) were activated synaptically with a long onset latency (>65 msec) (Fig.2B,D), and 24.6% (29 of 118 cells) exhibited an initial inhibition, often followed by a long latency excitation (75.0% of inhibitory responses preceded a late excitatory response) (Fig.2D). Short and long onset latency responses exhibited two distinct distributions (Fig. 2E). Short onset latencies ranged from 5 to 25 msec, whereas long onset latencies ranged from 65 to 270 msec (Table 1). Most of the long onset latency excitatory responses (66.6%) were preceded by an inhibition with an onset latency of 10.4 ± 5.9 msec and a duration of 104.3 ± 8.5 msec (Table 1) (Fig. 2D). Notably, 26.3% of the VTA DA neurons activated by stimulation of the vBNST exhibited both short and long latency activations after single-pulse stimulation (31 of 118) (Fig. 2C). No VTA neurons were driven antidromically by vBNST stimulation.
Fig. 2.

Typical peristimulus time histograms (PSTHs) showing vBNST-evoked excitation and inhibition in four VTA DA neurons. Three characteristic responses were observed after single-pulse stimulation of the vBNST: activation with short latency (<25 msec; A, C), activation with long latency (>65 msec; B, D), or inhibition often associated with a long latency excitation (D). For all PSTHs here and in subsequent figures, stimulation was at time 0; bin width was 10 msec, and 100 sweeps were collected. E, Histogram of the distribution of onset latencies for initial excitatory responses of VTA DA neuronal responses driven by single-pulse stimulation of the vBNST. Note that electrical stimulation of the vBNST produced two distinct excitatory responses: activation with a short or long onset latency.
Table 1.
Summary of onset latencies and durations of excitatory and inhibitory responses of VTA DA neurons to vBNST stimulation
| Short latency excitation | Inhibition | Long latency excitation | |
|---|---|---|---|
| Response onset latency (msec) | 16.4 ± 1.3 | 10.4 ± 5.9 | 160.5 ± 6.7 |
| Response duration (msec) | 16.2 ± 1.9 | 104.3 ± 8.5 | 93.3 ± 6.8 |
| Response magnitude (spikes) | +39.7 ± 3.3 | −54.5 ± 5.7 | +107.5 ± 8.4 |
| Baseline firing rate (spikes/sec) | 4.0 ± 0.3 | 4.5 ± 0.3 | 4.2 ± 0.2 |
All values are means ± SEM. Figures for response magnitudes are the numbers of spikes relative to baseline activity during the response interval integrated over 100 trials. Response magnitudes were calculated as described in Materials and Methods.
VTA GABA neurons were unaffected by electrical stimulation of the vBNST
Putative GABA neurons in the VTA were distinguished from VTA DA neurons according to criteria described by Steffensen et al. (1998)(see Materials and Methods). These neurons were located in a cluster between 200 and 500 μm dorsal to DA neurons (as reported previously by Steffensen et al., 1998) and exhibited spontaneous activity of 12.7 ± 2.1 Hz (n = 14). Electrical stimulation of the vBNST produced almost no response in VTA GABA neurons. Only 2 of the 14 VTA GABA neurons that were tested exhibited an excitation, both with long onset latencies (>100 msec for one GABA neuron and >300 msec for the other); the remaining 12 cells were not affected by vBNST stimulation.
Modulation of burst firing in VTA DA neurons by chemical inhibition or stimulation of the vBNST
We studied the effect of chemical inhibition of the vBNST (via GABA injection) on the activity of 24 VTA DA cells in three rats. Microinfusion of GABA (60 nl; 1 m) into the vBNST had no effect on the firing rate of VTA DA neurons but significantly decreased their burst firing (Fig.3A–E). As shown in Figure3A, GABA microinfusion in the vBNST produced an 8.9% decrease of DA neuron burst firing (ANOVA; df = 54;F = 10.78; p < 0.01; n= 24). A typical response of the bursting activity of VTA DA cells before and after microinfusion of GABA in the vBNST is illustrated in Figure 3, B and C. Microinfusion of GABA in the vBNST affected the number of spikes in bursts (Fig. 3D, subtraction histogram), but not the overall firing rate of VTA DA cells (Fig. 3E). These data show that the vBNST exerts a tonic excitatory influence over the firing characteristics of VTA DA neurons.
Fig. 3.

Modulation of burst firing and firing rate in VTA DA neurons by the vBNST. A, E, Infusion of 1m GABA into the vBNST caused a decrease in burst firing without affecting the firing rate of VTA DA neurons. Microinjection of 10 or 50 mm Glu into the vBNST increased both firing rate and bursting activity of VTA DA cells. For burst-firing analysis the scores that are plotted are the percentage of spikes in bursts postdrug minus the percentage of spikes in bursts predrug. For firing rates the scores that are plotted are the change in rates postdrug as a percentage of predrug rates. ANOVAs followed by Student'st tests for pairwise comparisons were performed for bursting activity and firing rate. *p < 0.05 versus control (aCSF). B, C, F, G, Interspike interval histograms (5 msec bins) illustrating the firing pattern of two VTA DA neurons before and after the infusion of 1m GABA (B, C) or before and after the infusion of 50 mm Glu into the vBNST (F, G).Insets show corresponding microelectrode traces before and after microinjections of GABA or Glu. D, H, Subtraction histograms comparing the bursting activity before and after microinjections of GABA or Glu, respectively. D showsC minus B (effect of GABA), whereasH shows G minus F (effect of Glu). Insets show corresponding microelectrode traces before microinjections of GABA or Glu. In B–D andF–H, open bars indicate the number of spikes occurring in bursts (interspike intervals <80 msec), andfilled bars indicate the number of spikes occurring outside of bursts (interspike intervals >80 msec). Note the decrease in bursting activity of VTA DA neurons after microinjection of GABA into the vBNST (B–D, open bars) and the increase in bursting activity of VTA DA neurons after microinjection of Glu into the vBNST (F–H, open bars).
The ability of microinfused GABA to decrease VTA DA neuronal bursting activity was critically dependent on injection placement. GABA microinjection in areas outside the vBNST induced a small increase or no change in the burst firing of VTA DA neurons. For example, microinjection of GABA in the lateral hypothalamus (7 neurons) produced a nonsignificant increase of burst firing (+4.2%). Microinjection of GABA in the substantia innominata had no effect on the bursting activity or firing rate of VTA DA neurons (5 neurons). Administration of vehicle alone (60 nl of aCSF in the vBNST) had no effect on VTA DA bursting activity or firing rate (16 neurons).
We have reported previously that chemical stimulation of the vBNST (via microinfusion of 10 or 50 mm Glu) consistently increased the firing rate of DA cells (Georges and Aston-Jones, 2001). Here we analyzed burst firing in those data. This analysis revealed that microinjection of 10 or 50 mm Glu into the vBNST increased bursting activity of VTA DA cells (10 mm Glu: +4.5%,p = 0.058, n = 6; 50 mm Glu: +9.6%, p < 0.01,n = 14; for each: ANOVA, df = 54,F = 10.78) (Fig. 3A,E). A typical response in the bursting activity of VTA DA cells after microinfusion of Glu 50 mm in the vBNST is illustrated in Figure 3,F and G. The subtraction histogram in Figure3H confirmed the increase of bursting activity. The ability of Glu to alter burst firing of VTA DA neurons was critically dependent on injection placement; Glu microinjection in areas outside the vBNST induced no change in burst firing. For example, microinjection of 10 mm Glu in the lateral preoptic area (5 cells) or in the ventral pallidum (3 cells) produced nonsignificant changes in burst firing of the VTA DA neurons (−0.3 and +0.5%, respectively).
Infusion of EAA antagonists in the VTA-attenuated DA neuron response to vBNST stimulation
As shown in the normalized graphs in Figure4, microinfusion of the broad-spectrum EAA antagonist kynurenic acid (3 mm), the specific NMDA antagonist AP-5 (100 μm), the specific non-NMDA antagonist CNQX (50 μm), or a cocktail of AP-5 (100 μm) plus CNQX (50 μm) into the VTA significantly reduced both the short and long latency excitations evoked in DA neurons by vBNST stimulation (Fig.4A, short latency excitation: ANOVA, df = 69,F = 16.44, p < 0.001; Fig.4C, long latency excitation: ANOVA, df = 75,F = 6.41, p < 0.001). As shown for sample cells in Figures 5 through 8, microinjection of any of the antagonists, including either CNQX (50 μm) or AP-5 (100 μm) alone, prevented or substantially reduced the activation of VTA DA neurons in response to stimulation of the vBNST. Figure 5 reveals that microinjection of kynurenic acid decreased theRmag for the short latency activation by 83.8%, reduced the Rmag for the inhibitory response by 41.3%, and decreased theRmag for the long latency activation by 67.1% (ANOVA, df = 14, F = 12.43 for each;p < 0.05 and 0.001, respectively;n = 15). Similarly, Figure6 shows that microinjection of the AP-5+CNQX cocktail decreased the Rmagfor the short latency activation by 78.9%, reduced theRmag for the inhibitory response by 62.9%, and decreased the Rmag for the long latency activation by 81.1% (ANOVA, df = 10,F = 9.185, p < 0.001 for each;n = 11). As seen in Figure7, microinjection of CNQX alone decreased the Rmag for the short onset latency activation by 78.1%, reduced the Rmagfor the inhibitory response by 40.4%, and decreased theRmag for the long onset latency activation by 60.6% (ANOVA, df = 19, F = 7.072,p < 0.001 for each; n = 20). Finally, Figure 8 shows that microinjection of AP-5 alone decreased the Rmag for the short latency activation by 88.1% and decreased theRmag for the long latency activation by 46.8% (ANOVA, df = 14, F = 8.270;p < 0.05 and 0.001, respectively; n = 15). There was no significant effect of AP-5 alone on the inhibitory response.
Fig. 4.
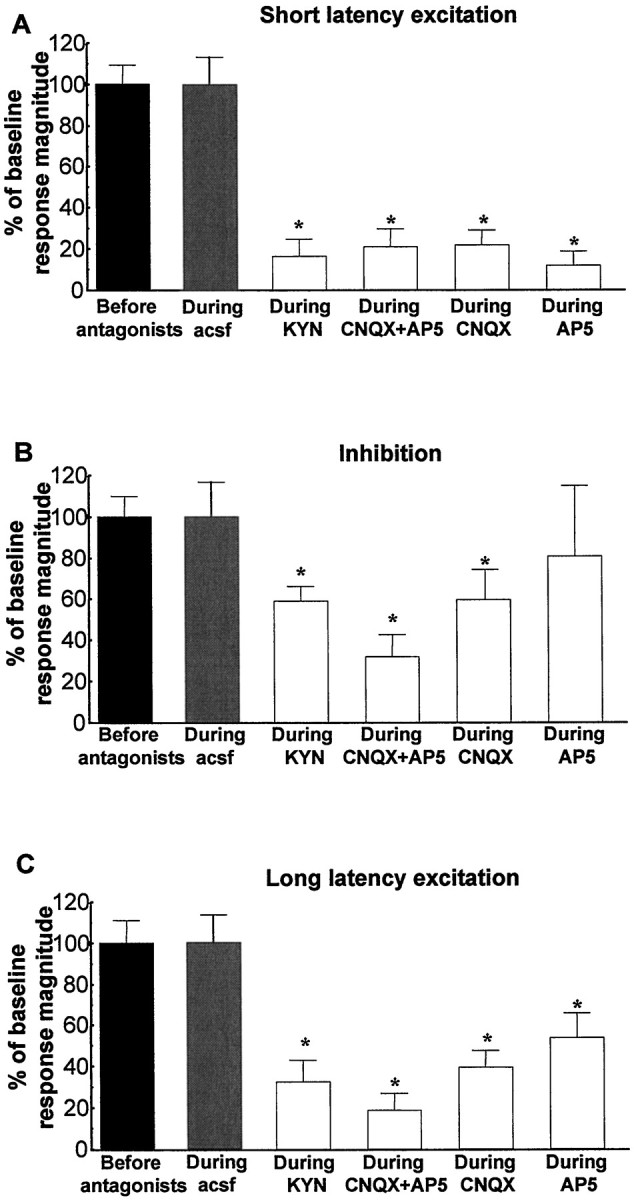
Graphs illustrating the effects of EAA antagonists on the three characteristic responses obtained after stimulation of the vBNST. Scores that are plotted are percentages ofRmags (± SEM) for VTA DA neuronal responses evoked by vBNST electrical stimulation before (black bars) and during microinjection of aCSF (gray bars) or EAA antagonists (white bars). Notably, NMDA or non-NMDA receptor antagonists significantly reduced short and long latency excitation evoked in DA neurons by vBNST stimulation. Only kynurenic acid, CNQX alone, and the mixture of CNQX+AP-5 antagonists significantly reduced the inhibition evoked in DA neurons by vBNST stimulation. For drug concentrations and cell numbers that were tested for each pharmacological agent, refer to Figures 5-8. An ANOVA followed by Student's t test for pairwise comparisons was performed for each characteristic response: short latency activation, inhibition, and long latency activation. *p < 0.05.
Fig. 5.

Effects of kynurenic acid on VTA DA neuronal responses evoked by BNST electrical stimulation.A–D, PSTHs showing VTA-evoked responses before and during kynurenate injection into the VTA for two typical DA neurons. Single-pulse stimuli (5 mA, 0.5 msec, 0.5/sec) were delivered at time 0. E, Mean ± SEM Rmags of VTA DA neuronal responses evoked by vBNST stimulation before (black bars) and during (white bars) microinjection of 3 mm kynurenic acid into the VTA. Microinjection of kynurenic acid prevented the short latency activation of DA VTA neurons evoked by electrical stimulation of the vBNST and decreased the inhibition and long latency excitation. The same cells were used before and after drug application (n = 15). An ANOVA followed by Student's t test for pairwise comparisons was performed for each characteristic response: short latency activation, inhibition, and long latency activation. *p < 0.05.
Fig. 8.

Effects of AP-5 on VTA DA neuronal responses evoked by BNST electrical stimulation. A–D, PSTHs showing VTA-evoked responses before and during AP-5 injection into the VTA for two typical DA neurons. Single-pulse stimuli (5 mA, 0.5 msec, 0.5/sec) were delivered at time 0. E, Mean ± SEM Rmags of VTA DA neuronal responses evoked by vBNST stimulation before (black bars) and during (white bars) microinjection of 100 μm AP-5 into the VTA. Microinjection of AP-5 prevented the short latency activation of DA VTA neurons evoked by vBNST stimulation and decreased the long latency excitation. The same cells were used before and after drug application (n = 15). An ANOVA followed by Student'st test for pairwise comparisons was performed for each characteristic response: short latency activation, inhibition, and long latency activation. *p < 0.05.
Fig. 6.

Effects of AP-5+CNQX on VTA DA neuronal responses evoked by BNST electrical stimulation. A–D, PSTHs showing VTA-evoked responses before and during drug injection into the VTA for two typical DA neurons. Single-pulse stimuli (5 mA, 0.5 msec, 0.5/sec) were delivered at time 0. E, Mean ± SEM Rmags of VTA DA neuronal responses evoked by vBNST stimulation before (black bars) and during (white bars) microinjection of 100 μm AP-5 plus 50 μm CNQX into the VTA. Microinjection of AP-5+CNQX prevented the short latency activation of DA VTA neurons evoked by vBNST stimulation and decreased the inhibition and long latency excitation. The same cells were used before and after drug application (n = 11). An ANOVA followed by Student's t test for pairwise comparisons was performed for each characteristic response: short onset latency activation, inhibition, and long onset latency. *p < 0.05.
Fig. 7.

Effects of CNQX on VTA DA neuronal responses evoked by BNST electrical stimulation. A–D, PSTHs showing VTA-evoked responses before and during CNQX injection into the VTA for two typical DA neurons. Single-pulse stimuli (5 mA, 0.5 msec, 0.5/sec) were delivered at time 0. E, Mean ± SEM Rmags of VTA DA neuronal responses evoked by vBNST stimulation before (black bars) and during (white bars) microinjection of 50 μm CNQX into the VTA. Microinjection of CNQX prevented the short latency activation of DA VTA neurons evoked by vBNST stimulation and decreased the long latency excitation. The same cells were used before and after drug application (n = 20). An ANOVA followed by Student's t test for pairwise comparisons was performed for each characteristic response: short latency activation, inhibition, and long latency activation. *p < 0.05.
It is noteworthy that either AP-5 or CNQX alone virtually eliminated the short-latency excitatory response. Post hoc analyses revealed that there was no significant difference between the degrees of antagonism produced by AP-5 or CNQX alone (ANOVA; df = 69;F = 16.44; p = 0.07). In addition, neither the cocktail of AP-5+CNQX nor kynurenic acid produced a significantly greater antagonism that either AP-5 or CNQX alone (ANOVA; df = 69; F = 16.44; p > 0.2).
Partial or full recovery had occurred for most cells by 15 min after microinjection of EAA antagonists. Baseline firing rates of VTA DA neurons in the absence of stimulation were slightly, but not significantly, affected by microinjection of AP-5+CNQX (3.7 ± 0.6 spikes/sec before injection vs 2.3 ± 0.7 spikes/sec during injection; p = 0.060; n = 11) or AP-5 alone (3.7 ± 0.7 spikes/sec before injection vs 3.0 ± 0.6 spikes/sec during injection; p = 0.231;n = 15). Baseline firing rates of DA VTA neurons in the absence of stimulation were decreased significantly by microinjections of CNQX (3.9 ± 0.7 spikes/sec before injection vs 2.4 ± 0.5 spikes/sec during injection; p = 0.049;n = 20) or kynurenic acid (5.06 ± 0.6 spikes/sec before injection vs 2.7 ± 0.5 spikes/sec during injection;p = 0.001; n = 15).
As shown in Figure 9, microinjection of 50 mm Glu into the vBNST activated VTA DA neurons, as we described recently (Georges and Aston-Jones, 2001), and increased bursting (as described above). In addition, microinfusion of the cocktail of AP-5 (100 μm) plus CNQX (50 μm) into the VTA blocked both the increase in bursting as well as the increase in the firing rate of VTA DA neurons evoked by Glu stimulation of the vBNST (Fig. 9A,B; 4 rats, 8 neurons). As shown for a sample cell in Figure 9C, microinjection of Glu into the vBNST activated a VTA DA neuron, and this activation was blocked by a microinfusion of EAA antagonists into the VTA (Fig. 9C,D). The cocktail of antagonists produced a small (but nonsignificant) decrease in spontaneous firing rate (p < 0.058) and bursting activity (n = 13 neurons).
Fig. 9.

Effects of AP-5 plus CNQX on VTA DA neuronal responses evoked by BNST stimulation by Glu microinjection. A, B, Graphs comparing bursts (A) and firing rate (B) of VTA DA neurons before (control) and during local microinfusion of 50 μm CNQX plus 100 μm AP-5 into the VTA. Note that the EAA antagonists blocked both the increase in bursting as well as the increase in firing rate of VTA DA neurons evoked by chemical stimulation of the vBNST. Data are the results from eight neurons recorded in four rats. Data were analyzed by two-way ANOVA to determine possible interactions between Glu and EAA antagonists. ForA, F = 4.901 andp = 0.035; for B,F = 4.778 and p = 0.037. *p < 0.01 compared with before Glu (paired Student's t tests). C, D, Firing rate histograms showing the blockade of response in a typical VTA DA neuron to Glu stimulation of the vBNST with local microinjection of 50 μm CNQX plus 100 μm AP-5 into the VTA. In C, note the activation of the VTA neuron by Glu stimulation of the vBNST and the lack of such activation when Glu was applied in the vBNST during EAA antagonist microinfusion in the VTA, as marked above. Note also the elevated activity of the VTA neuron after the end of the EAA antagonist microinfusion, with oscillatory activity as typically observed with 50 mm Glu injection into the vBNST (Georges and Aston-Jones, 2001). D shows the period of Glu stimulation of the vBNST during EAA antagonist microinfusion into the VTA in greater temporal detail. Injection of Glu into the dorsocaudal BNST did not activate any of the four VTA DA neurons that were tested.
Antidromic activation of vBNST neurons after VTA stimulation
Antidromicity was determined by collision tests and by response to high-frequency stimulation. A characteristic example of collision and high-frequency activation of a vBNST neuron driven from the VTA is shown in Figure 10. Electrical stimulation of the VTA antidromically activated 11 vBNST neurons and 1 dorsal BNST neuron (n = 4 rats). Nineteen penetrations were made through the vBNST to get the 11 antidromic neurons. The antidromic responses had a mean latency of 12.5 ± 0.4 msec (n = 11). The absolute refractory period was 1.25 ± 0.1 msec (n = 11), as determined with paired pulse stimulation at a frequency of 200 Hz or greater. For each antidromic response the latency for driving was invariant. Collision of spontaneous spikes with antidromically activated spikes occurred when a spontaneous spike fell within a critical period of 12.71 ± 0.4 msec (n = 8 spontaneously active neurons). This critical period is approximately equal to the latency for driving plus the absolute refractory period, as expected for antidromic activation. The onset of the short latency excitation after vBNST stimulation was slightly, but not significantly, longer than the latency of antidromic responses in the vBNST after VTA stimulation (16.4 ± 1.3 vs 12.5 ± 0.4 msec; p = 0.22, Student'st test). This result is consistent with a direct excitatory pathway from the vBNST to the VTA mediating the short latency EAA-dependent response.
Fig. 10.

Characteristic antidromic response of a vBNST neuron after VTA stimulation. A, D, Photomicrographs of coronal sections through the VTA and the BNST showing stimulation and recording sites. A, An electrical stimulation site in the VTA, marked by passing positive current through the stimulation electrode (dark lesioned area, arrow). Tissue was stained for TH (brown) to indicate the DA neuron area. D, Iontophoretic injection of pontamine sky blue (dark spot, arrow; coronal section) marks the recording location for a vBNST neuron (neutral red stain). Scale bars: A, 0.5 mm; D, 1.0 mm. B, C, E, F, Five superimposed traces illustrating high-frequency activation and collision test for a vBNST cell driven from the VTA. B, Driven spikes (black circles) elicited by each of the paired stimuli (vertical lines; 2 msec interpulse interval) indicating the frequency following for this cell at 500 Hz. The second driven spike is smaller than the first, presumably showing an isolated axon spike and failure to invade the soma-dendritic membrane. C, Spikes driven by the second stimulus are occluded when the interpulse interval is decreased to 1 msec.E, Stimulation of VTA (vertical lines) 12 msec after spontaneous spikes (left side of traces) elicit driven spikes (black circle) at 13.1 msec latency. F, Driven spikes are occluded for similar stimuli delivered 11 msec after spontaneous impulses, indicating collision between spontaneous and driven spikes. Theasterisk signifies where driven spikes would have occurred had there been no collision. Calibration in Fapplies to B, C, E, F.
Tract tracing study
We also examined projections from the vBNST to the VTA by using anatomical methods. We made microinjections of CTb, a sensitive retrograde and anterograde tracer (Luppi et al., 1995), into either the VTA or vBNST and analyzed tissue for retrogradely labeled neurons or anterogradely labeled fibers. In each of four animals with CTb injections in the VTA, we observed numerous retrogradely labeled cell bodies throughout the BNST (Fig.11C,D). The intensity of retrograde labeling appeared to be strongest in the vBNST, especially in the dense noradrenergic projection area, compared with other subregions of the BNST. No anterograde labeling was detected in the vBNST.
Fig. 11.
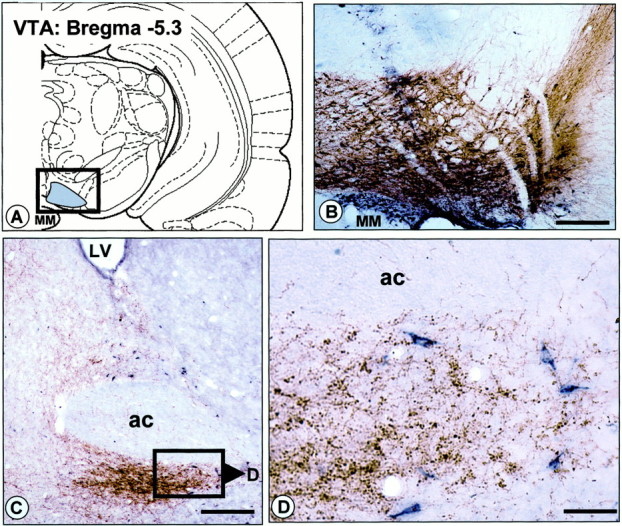
Projection from the vBNST to the VTA revealed by retrograde labeling in the vBNST after injection of CTb into the VTA.A, Schematic diagram illustrating the location of CTb injections in the VTA. B, Photomicrograph illustrating a representative CTb injection site in the VTA (dark bluelabeling). The section has been counterstained with TH immunohistochemistry (in brown), to delineate dopamine neurons and processes in the VTA (boxed area inA). C, D, Bright-field photomicrographs illustrating retrograde labeling in the vBNST after CTb injection into the VTA. The sections have been processed dually for CTb (dark blue) and DBH (brown). Note that numerous CTb+ cell bodies are observed in the dorsal and ventral BNST. Cell bodies retrogradely labeled in the vBNST are shown at higher power in D. ac, Anterior commissure; MM, medial mamillary nucleus. Scale bars:B, C, 1.0 mm; D, 0.1 mm.
We also made injections of CTb into the vBNST of five animals to examine anterograde labeling in the VTA. CTb injections in the vBNST produced a large number of anterogradely labeled fibers in the VTA (Fig. 12D). These fibers were characterized by punctuate labeling and were distributed throughout the entire TH+ area of the VTA. As illustrated in Figure 12, B and C (enlarged in Fig. 12D), there was substantial overlap between CTb+ anterogradely labeled terminals and TH+ neurons and processes in the VTA. After CTb injections in the vBNST only one or two cell bodies per section were labeled retrogradely in the VTA (Fig.12D).
Fig. 12.
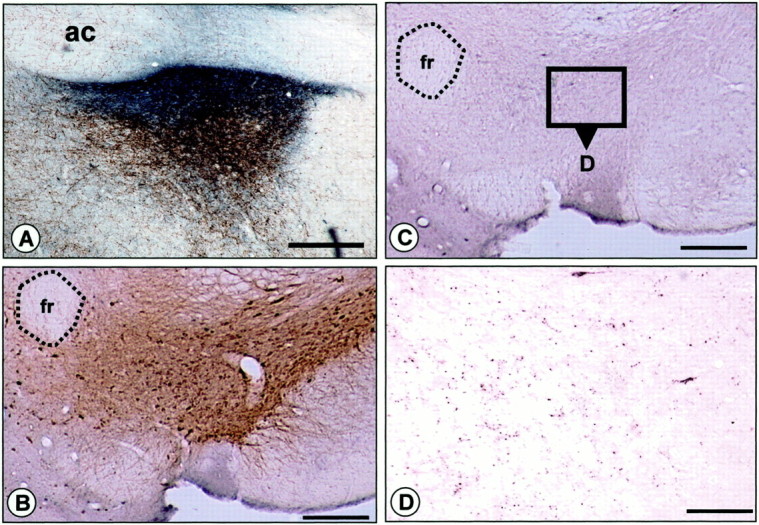
Projection from the vBNST to the VTA revealed by anterograde transport of CTb to the VTA after injection into the vBNST.A, Photomicrograph showing a representative iontophoretic CTb injection site in the vBNST (dark bluelabeling). The section has been counterstained with DBH immunohistochemistry (in brown) to delineate the region of dense noradrenergic innervation in the BNST. B–D, Photomicrographs of the anterograde transport of CTb in the VTA after injection into the vBNST. B, C, Adjacent sections counterstained with CTb immunohistochemistry (C, which is enlarged in D) or TH immunohistochemistry (B) confirmed the overlap between CTb+ terminals (dark blue punctate labeling) and TH+ neurons and processes in the VTA. Note that anterogradely labeled CTb terminals from the vBNST (dark blue dots) are distributed throughout the VTA (D). ac, Anterior commissure;fr, fasciculus retroflexus. Scale bars,A–C, 1.0 mm; D, 0.1 mm.
DISCUSSION
Our study provides anatomical and functional evidence for a novel EAA regulation of VTA DA neurons by the vBNST. In addition, we have found that the vBNST tonically can modulate the burst firing of VTA DA neurons.
Response of VTA DA neurons to electrical stimulation of the vBNST
The short latency excitations for many cells after vBNST stimulation (<25 msec) and the distance between the vBNST and the VTA (∼6 mm) indicate that at least some of these responses may be mediated by a direct input to the VTA from the vBNST. If we consider a straight-line distance between the vBNST and the VTA, the conduction velocity would average 0.4 m/sec. This conduction velocity is within the range of other slow-conducting CNS pathways such as the dopaminergic nigrostriatal pathway (Deniau et al., 1978) or locus ceruleus neurons (Faiers and Mogenson, 1976). Our findings for antidromic activation of vBNST neurons after stimulation of the VTA and retrograde and anterograde labeling between the vBNST and VTA strongly support the hypothesis of a direct projection from the vBNST to the VTA. Our recent results for a similar activation of VTA DA neurons after microinjections of Glu into the vBNST indicate that the effects reported here are mediated by the activation of vBNST neurons and not by fibers of passage (Georges and Aston-Jones, 2001). Although the short latency responses appear to be mediated by a direct vBNST–VTA pathway, it also seems likely that the long latency responses were conveyed indirectly to the VTA via other EAA inputs. This conclusion is consistent with our finding that the conduction latency of vBNST–VTA projections (as determined by antidromic tests) was similar to the short latency synaptic responses but much shorter than the long latency synaptic activation. The BNST projects to the pedunculopontine nucleus and to the amygdala, both of which send EAA inputs the VTA (Cassell et al., 1986; Di Loreto et al., 1992; Semba and Fibiger, 1992). Further experiments are needed to establish which of these (or other) structures act as relays between the BNST and the VTA. Regardless of the pathway involved, our results show that the vBNST is a significant regulator of VTA DA neural activity via EAA inputs.
Influence of the vBNST on burst activity of VTA DA neurons
Microinfusion of the excitatory agent Glu into the vBNST increased burst activity of DA neurons, whereas microinfusion of the inhibitory agent GABA into the vBNST decreased burst activity in DA cells. These results reveal that the vBNST can regulate tonically the number of bursting of VTA DA neurons. Striatal DA efflux has been shown to correlate strongly with DA neuron activity and is highly dependent on the pattern of discharge (Moore et al., 1998). In fact, it has been shown that bursts of action potentials are twice as potent as the same number of regularly spaced spikes to trigger DA release (Suaud-Chagny et al., 1992). Thus the modulation of burst firing by the vBNST could be an important element in its regulation of function of the VTA DA system.
Effect of EAA antagonists on the response of VTA DA neurons to vBNST stimulation
The present results demonstrate that VTA DA activation evoked by vBNST stimulation is mediated primarily by EAA inputs acting at NMDA as well as AMPA/kainate receptors. The concentrations of CNQX and AP-5 that were used were similar to those used previously in in vivo (Chergui et al., 1993; Jodo and Aston-Jones, 1997) orin vitro studies (Seutin et al., 1990; Mereu et al., 1991) and are within the ranges for these compounds to act specifically on non-NMDA or NMDA receptors, respectively. Although such specificity cannot be certain, it appears to be highly likely in our experiments where substantial dilution of the antagonists occurs after microinfusion.
It is noteworthy that either AP-5 or CNQX alone almost completely blocked the short latency activation and markedly attenuated the long latency excitation evoked by vBNST stimulation. For the short latency response there was no greater effect produced by administration of the AP-5+CNQX mixture than for either antagonist alone. In addition, the broad-spectrum EAA antagonist kynurenic acid produced similar effects on both responses, confirming that EAA receptors are prominently involved. This spectrum of results indicates that the short latency excitation of VTA DA neurons by the vBNST requires the activation of both NMDA and non-NMDA receptors simultaneously; blockade of either one alone is sufficient to eliminate the response. The mechanism for this joint receptor activation requirement is unknown but may involve the interaction of Mg2+ with NMDA receptors, as has been well established in other systems (Ascher and Nowak, 1988). One possibility is that the bulk of the response is mediated through the NMDA receptor but that activation of the NMDA receptor requires previous activation of the non-NMDA receptor (e.g., to remove Mg2+ blockade from the NMDA receptor channel). Our results indicate that activation of the non-NMDA receptor alone is not sufficient for evoking impulse activity (as the response is blocked by AP-5). This could occur if, for example, the non-NMDA response rapidly desensitizes (as described for other systems; Geoffroy et al., 1991), limiting the ability to produce driven spikes despite facilitating NMDA receptors. Further experiments are necessary to determine the mechanism underlying this requirement for joint NMDA and non-NMDA receptor activation.
This requirement for simultaneous NMDA and non-NMDA receptor activation has not been described for VTA responses to other EAA inputs. For example, activation of VTA neurons after stimulation of the prefrontal cortex is attenuated strongly by NMDA antagonists, but not by non-NMDA antagonists alone (Tong et al., 1996). In contrast, responses of VTA or nigral DA neurons to stimulation of the pedunculopontine or subthalamic nuclei are blocked by non-NMDA antagonists, but not by NMDA antagonists given alone (Di Loreto et al., 1992; Chergui et al., 1994). Studies of VTA neurons in brain slices have not examined this issue specifically, although they have shown both NMDA and non-NMDA receptor-mediated responses to local field stimulation (Mereu et al., 1991; Bonci and Malenka, 1999). The response profile seen here is also unusual among other brain sites that have been examined with EAA synaptic inputs. However, a similar property is seen for oxytocin release from neurons in the supraoptic nucleus (Parker and Crowley, 1995). The strong NMDA receptor responses that we observed may indicate a role for BNST inputs in synaptic plasticity within the VTA, because NMDA receptor-dependent long-term potentiation has been found for these DA neurons (Bonci and Malenka, 1999).
Although our results indicate a potent EAA-mediated influence of the vBNST on VTA DA neurons, they do not exclude involvement of other neurotransmitters in this pathway. Thus inhibitory responses may be mediated by GABA projections from the vBNST to the VTA. It is also possible that the vBNST sends EAA afferents to GABA terminals that synapse onto DA neurons in the VTA. Indeed, pharmacological studies suggest that NMDA and non-NMDA receptors may act presynaptically to modulate the release of GABA (Young and Bradford, 1993; Matsuyama et al., 1997; Glitsch and Marty, 1999). Thus BNST projections may act on presynaptic ionotropic glutamate receptors and modulate the release of GABA in the VTA to produce the inhibition of DA neurons. It is also possible, however, that the inhibitory responses that have been observed are mediated by DA release onto the recorded neuron from other cells nearby that are activated at an early latency. Additional studies are needed to test these and other possible mechanisms mediating the inhibition produced by vBNST stimulation.
Anatomical and electrophysiological characterization of the connection from the vBNST to the VTA
Earlier retrograde tracing studies demonstrated neuronal projections to the VTA from various brain structures, including the BNST (Phillipson, 1979; Holstege et al., 1985). However, these previous studies used relatively large injections of less selective tract tracers than were used here and did not analyze projections specifically from the ventrolateral subregion of the BNST. Here we made injections of anterograde tracer, or analyzed retrograde labeling, specifically within the vBNST region containing dense NE fibers. Our results extend previous studies by specifying that the ventrolateral subregion of the BNST containing dense NE fibers projects directly to the VTA. We also identified vBNST neurons projecting to the VTA by using antidromic activation. As illustrated in Figure10D for a characteristic recording localization, vBNST neurons driven antidromically from the VTA were localized in the subcommissural level of the vBNST, corresponding to the area of cells retrogradely labeled from the VTA in our anatomic experiments (Fig.11D). These results support the hypothesis that at least some of the responses of DA neurons to vBNST stimulation are mediated by a direct projection.
Functional implications
The above analysis also indicates that neurons in the vBNST projecting to the VTA may be influenced by NE inputs. This would be of some interest, because it has been found recently that NE in this area is involved importantly in the affective response to opiate withdrawal (Delfs et al., 2000). Other studies reveal that withdrawal alters DA levels in the accumbens in an NE-dependent manner (Pothos et al., 1991;Acquas and Di Chiara, 1992) and that the DA in the nucleus accumbens is critically involved in opiate withdrawal (Harris and Aston-Jones, 1994). In view of these findings, our present results suggest that the NE regulation of the vBNST and subsequent effects on VTA DA neurons may be involved in responses to opiate drugs and withdrawal.
EAA afferents to the VTA play an important role in regulating the activity of DA neurons (Johnson et al., 1992; Overton and Clark, 1992;Wang and French, 1993; White, 1996; Bonci and Malenka, 1999) and the subsequent release of dopamine in efferent targets (Karreman et al., 1996; Westerink et al., 1996, 1998). The present results identify a previously unknown and potent EAA influence on VTA DA neuronal function and demonstrate that both NMDA and non-NMDA receptors are involved prominently in the activation of VTA DA neurons by vBNST stimulation. As reported recently, EAA synapses onto VTA DA neurons are involved in a long-term potentiation phenomenon and may contribute to the development of drug addiction (Ungless et al., 2001). Considering that glutamatergic systems are involved in both acute and chronic responses to drugs of abuse (for review, see Wolf, 1998), our results support the view that the BNST is an important component of the brain systems involved in drug addiction. Additional work is required to establish the role of this novel EAA input in the normal behaviors and behavioral changes in response to drugs of abuse that are mediated by the VTA DA neurons.
Footnotes
This work was supported by United States Public Health Service Grant DA06214 and the Foundation Fyssen. We thank Dr. G. Harris for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Gary Aston-Jones, Laboratory for Neuromodulation and Behavior, Department of Psychiatry, Veterans Affairs Medical Center, Mail Box 151, University and Woodland Avenues, Philadelphia, PA 19104. E-mail: gaj@mail.med.upenn.edu.
REFERENCES
- 1.Acquas E, Di Chiara G. Depression of mesolimbic dopamine transmission and sensitization to morphine during opiate abstinence. J Neurochem. 1992;58:1620–1625. doi: 10.1111/j.1471-4159.1992.tb10033.x. [DOI] [PubMed] [Google Scholar]
- 2.Aghajanian GK, Bunney BS. Dopamine “autoreceptors”: pharmacological characterization by microiontophoretic single cell recording studies. Naunyn Schmiedebergs Arch Pharmacol. 1977;297:1–7. doi: 10.1007/BF00508803. [DOI] [PubMed] [Google Scholar]
- 3.Akaoka H, Aston-Jones G. Opiate withdrawal-induced hyperactivity of locus coeruleus neurons is substantially mediated by augmented excitatory amino acid input. J Neurosci. 1991;11:3830–3839. doi: 10.1523/JNEUROSCI.11-12-03830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ascher P, Nowak L. The role of divalent cations in the N-methyl-d-aspartate responses of mouse central neurones in culture. J Physiol (Lond) 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aston-Jones G, Shaver R, Dinan TG. Nucleus basalis neurons exhibit axonal branching with decreased impulse conduction velocity in rat cerebrocortex. Brain Res. 1985;325:271–285. doi: 10.1016/0006-8993(85)90323-3. [DOI] [PubMed] [Google Scholar]
- 6.Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter CJ. Topographical distribution of possible glutamatergic pathways from the frontal cortex to the striatum and substantia nigra in rats. Neuropharmacology. 1982;21:379–383. doi: 10.1016/0028-3908(82)90019-3. [DOI] [PubMed] [Google Scholar]
- 8.Cassell MD, Gray TS, Kiss JZ. Neuronal architecture in the rat central nucleus of the amygdala: a cytological, hodological, and immunocytochemical study. J Comp Neurol. 1986;246:478–499. doi: 10.1002/cne.902460406. [DOI] [PubMed] [Google Scholar]
- 9.Chergui K, Charlety PJ, Akaoka H, Saunier CF, Brunet JL, Buda M, Svensson TH, Chouvet G. Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur J Neurosci. 1993;5:137–144. doi: 10.1111/j.1460-9568.1993.tb00479.x. [DOI] [PubMed] [Google Scholar]
- 10.Chergui K, Akaoka H, Charlety PJ, Saunier CF, Buda M, Chouvet G. Subthalamic nucleus modulates burst firing of nigral dopamine neurones via NMDA receptors. NeuroReport. 1994;5:1185–1188. doi: 10.1097/00001756-199406020-00006. [DOI] [PubMed] [Google Scholar]
- 11.Chiodo LA. Dopamine-containing neurons in the mammalian central nervous system: electrophysiology and pharmacology. Neurosci Biobehav Rev. 1988;12:49–91. doi: 10.1016/s0149-7634(88)80073-3. [DOI] [PubMed] [Google Scholar]
- 12.Danbolt NC. Glutamate uptake [review]. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 13.Delfs JM, Zhu Y, Druhan JP, Aston-Jones GS. Origin of noradrenergic afferents to the shell subregion of the nucleus accumbens: anterograde and retrograde tract-tracing studies in the rat. Brain Res. 1998;806:127–140. doi: 10.1016/s0006-8993(98)00672-6. [DOI] [PubMed] [Google Scholar]
- 14.Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- 15.Deniau JM, Hammond C, Riszk A, Feger J. Electrophysiological properties of identified output neurons of the rat substantia nigra (pars compacta and pars reticulata): evidences for the existence of branched neurons. Exp Brain Res. 1978;32:409–422. doi: 10.1007/BF00238711. [DOI] [PubMed] [Google Scholar]
- 16.Di Loreto S, Florio T, Scarnati E. Evidence that non-NMDA receptors are involved in the excitatory pathway from the pedunculopontine region to nigrostriatal dopaminergic neurons. Exp Brain Res. 1992;89:79–86. doi: 10.1007/BF00229003. [DOI] [PubMed] [Google Scholar]
- 17.Faiers AA, Mogenson GJ. Electrophysiological identification of neurons in locus coeruleus. Exp Neurol. 1976;53:254–266. doi: 10.1016/0014-4886(76)90295-8. [DOI] [PubMed] [Google Scholar]
- 18.Geoffroy M, Lambolez B, Audinat E, Hamon B, Crepel F, Rossier J, Kado RT. Reduction of desensitization of a glutamate ionotropic receptor by antagonists. Mol Pharmacol. 1991;39:587–591. [PubMed] [Google Scholar]
- 19. Georges F, Aston-Jones G. Potent regulation of midbrain dopamine neurons by the bed nucleus of the stria terminalis. J Neurosci 21 2001. RC160:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons. I. Identification and characterization. Neuroscience. 1983;10:301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- 22.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guyenet PG, Aghajanian GK. Antidromic identification of dopaminergic and other output neurons of the rat substantia nigra. Brain Res. 1978;150:69–84. doi: 10.1016/0006-8993(78)90654-6. [DOI] [PubMed] [Google Scholar]
- 24.Harris GC, Aston-Jones G. Involvement of D2 dopamine receptors in the nucleus accumbens in the opiate withdrawal syndrome [see comments]. Nature. 1994;371:155–157. doi: 10.1038/371155a0. [DOI] [PubMed] [Google Scholar]
- 25.Holstege G, Meiners L, Tan K. Projections of the bed nucleus of the stria terminalis to the mesencephalon, pons, and medulla oblongata in the cat. Exp Brain Res. 1985;58:379–391. doi: 10.1007/BF00235319. [DOI] [PubMed] [Google Scholar]
- 26.Jodo E, Aston-Jones G. Activation of locus coeruleus by prefrontal cortex is mediated by excitatory amino acid inputs. Brain Res. 1997;768:327–332. doi: 10.1016/s0006-8993(97)00703-8. [DOI] [PubMed] [Google Scholar]
- 27.Jodo E, Chiang C, Aston-Jones G. Potent excitatory influence of prefrontal cortex activity on noradrenergic locus coeruleus neurons. Neuroscience. 1998;83:63–79. doi: 10.1016/s0306-4522(97)00372-2. [DOI] [PubMed] [Google Scholar]
- 28.Johnson SW, Seutin V, North RA. Burst firing in dopamine neurons induced by N-methyl-d-aspartate: role of electrogenic sodium pump. Science. 1992;258:665–667. doi: 10.1126/science.1329209. [DOI] [PubMed] [Google Scholar]
- 29.Kalivas PW, Alesdatter JE. Involvement of N-methyl-d-aspartate receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1993;267:486–495. [PubMed] [Google Scholar]
- 30.Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- 31.Karreman M, Westerink BH, Moghaddam B. Excitatory amino acid receptors in the ventral tegmental area regulate dopamine release in the ventral striatum. J Neurochem. 1996;67:601–607. doi: 10.1046/j.1471-4159.1996.67020601.x. [DOI] [PubMed] [Google Scholar]
- 32.Kita H, Kitai ST. Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol. 1987;260:435–452. doi: 10.1002/cne.902600309. [DOI] [PubMed] [Google Scholar]
- 33.Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 34.Kornhuber J, Kim JS, Kornhuber ME, Kornhuber HH. The cortico-nigral projection: reduced glutamate content in the substantia nigra following frontal cortex ablation in the rat. Brain Res. 1984;322:124–126. doi: 10.1016/0006-8993(84)91189-2. [DOI] [PubMed] [Google Scholar]
- 35.Luppi PH, Aston-Jones G, Akaoka H, Chouvet G, Jouvet M. Afferent projections to the rat locus coeruleus demonstrated by retrograde and anterograde tracing with cholera toxin B subunit and Phaseolus vulgaris leucoagglutinin. Neuroscience. 1995;65:119–160. doi: 10.1016/0306-4522(94)00481-j. [DOI] [PubMed] [Google Scholar]
- 36.Matsuyama S, Nei K, Tanaka C. Regulation of GABA release via NMDA and 5-HT1A receptors in guinea pig dentate gyrus. Brain Res. 1997;761:105–112. doi: 10.1016/s0006-8993(97)00318-1. [DOI] [PubMed] [Google Scholar]
- 37.Meltzer LT, Christoffersen CL, Serpa KA. Modulation of dopamine neuronal activity by glutamate receptor subtypes. Neurosci Biobehav Rev. 1997;21:511–518. doi: 10.1016/s0149-7634(96)00030-9. [DOI] [PubMed] [Google Scholar]
- 38.Mereu G, Costa E, Armstrong DM, Vicini S. Glutamate receptor subtypes mediate excitatory synaptic currents of dopamine neurons in midbrain slices. J Neurosci. 1991;11:1359–1366. doi: 10.1523/JNEUROSCI.11-05-01359.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore H, Todd CL, Grace AA. Striatal extracellular dopamine levels in rats with haloperidol-induced depolarization block of substantia nigra dopamine neurons. J Neurosci. 1998;18:5068–5077. doi: 10.1523/JNEUROSCI.18-13-05068.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J Neurosci. 1996;16:4135–4145. doi: 10.1523/JNEUROSCI.16-13-04135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Overton P, Clark D. Iontophoretically administered drugs acting at the N-methyl-d-aspartate receptor modulate burst firing in A9 dopamine neurons in the rat. Synapse. 1992;10:131–140. doi: 10.1002/syn.890100208. [DOI] [PubMed] [Google Scholar]
- 42.Parker S, Crowley W. Central stimulation of oxytocin release in the lactating rat by NMDA: requirement for coactivation through non-NMDA glutamate receptors or the glycine coagonist site. Neuroendocrinology. 1995;62:467–478. doi: 10.1159/000127036. [DOI] [PubMed] [Google Scholar]
- 43.Phillipson OT. Afferent projections to the ventral tegmental area of Tsai and interfascicular nucleus: a horseradish peroxidase study in the rat. J Comp Neurol. 1979;187:117–143. doi: 10.1002/cne.901870108. [DOI] [PubMed] [Google Scholar]
- 44.Pothos E, Rada P, Mark GP, Hoebel BG. Dopamine microdialysis in the nucleus accumbens during acute and chronic morphine, naloxone-precipitated withdrawal, and clonidine treatment. Brain Res. 1991;566:348–350. doi: 10.1016/0006-8993(91)91724-f. [DOI] [PubMed] [Google Scholar]
- 45.Rinvik E, Ottersen OP. Terminals of subthalamonigral fibres are enriched with glutamate-like immunoreactivity: an electron microscopic, immunogold analysis in the cat. J Chem Neuroanat. 1993;6:19–30. doi: 10.1016/0891-0618(93)90004-n. [DOI] [PubMed] [Google Scholar]
- 46.Semba K, Fibiger HC. Afferent connections of the laterodorsal and the pedunculopontine tegmental nuclei in the rat: a retro- and anterograde transport and immunohistochemical study. J Comp Neurol. 1992;323:387–410. doi: 10.1002/cne.903230307. [DOI] [PubMed] [Google Scholar]
- 47.Sesack SR, Pickel VM. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol. 1992;320:145–160. doi: 10.1002/cne.903200202. [DOI] [PubMed] [Google Scholar]
- 48.Seutin V, Verbanck P, Massotte L, Dresse A. Evidence for the presence of N-methyl-d-aspartate receptors in the ventral tegmental area of the rat: an electrophysiological in vitro study. Brain Res. 1990;514:147–150. doi: 10.1016/0006-8993(90)90448-k. [DOI] [PubMed] [Google Scholar]
- 49.Shepard P, Bunney B. Effects of apamin on the discharge properties of putative dopamine-containing neurons in vitro. Brain Res. 1988;463:380–384. doi: 10.1016/0006-8993(88)90414-3. [DOI] [PubMed] [Google Scholar]
- 50.Steffensen SC, Svingos AL, Pickel VM, Henriksen SJ. Electrophysiological characterization of GABAergic neurons in the ventral tegmental area. J Neurosci. 1998;18:8003–8015. doi: 10.1523/JNEUROSCI.18-19-08003.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suaud-Chagny MF, Chergui K, Chouvet G, Gonon F. Relationship between dopamine release in the rat nucleus accumbens and the discharge activity of dopaminergic neurons during local in vivo application of amino acids in the ventral tegmental area. Neuroscience. 1992;49:63–72. doi: 10.1016/0306-4522(92)90076-e. [DOI] [PubMed] [Google Scholar]
- 52.Tepper JM, Young SJ, Groves PM. Autoreceptor-mediated changes in dopaminergic terminal excitability: effects of increases in impulse flow. Brain Res. 1984;309:309–316. doi: 10.1016/0006-8993(84)90598-5. [DOI] [PubMed] [Google Scholar]
- 53.Tokuno H, Moriizumi T, Kudo M, Nakamura Y. A morphological evidence for monosynaptic projections from the nucleus tegmenti pedunculopontinus pars compacta (TPC) to nigrostriatal projection neurons. Neurosci Lett. 1988;85:1–4. doi: 10.1016/0304-3940(88)90417-x. [DOI] [PubMed] [Google Scholar]
- 54.Tong ZY, Overton PG, Clark D. Antagonism of NMDA receptors but not AMPA/kainate receptors blocks bursting in dopaminergic neurons induced by electrical stimulation of the prefrontal cortex. J Neural Transm. 1996;103:889–904. doi: 10.1007/BF01291780. [DOI] [PubMed] [Google Scholar]
- 55.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 56.Walker JR, Ahmed SH, Gracy KN, Koob GF. Microinjections of an opiate receptor antagonist into the bed nucleus of the stria terminalis suppress heroin self-administration in dependent rats. Brain Res. 2000;854:85–92. doi: 10.1016/s0006-8993(99)02288-x. [DOI] [PubMed] [Google Scholar]
- 57.Wang T, French ED. l-Glutamate excitation of A10 dopamine neurons is preferentially mediated by activation of NMDA receptors: extra- and intracellular electrophysiological studies in brain slices. Brain Res. 1993;627:299–306. doi: 10.1016/0006-8993(93)90334-j. [DOI] [PubMed] [Google Scholar]
- 58.Westerink BH, Kwint HF, De Vries JB. The pharmacology of mesolimbic dopamine neurons: a dual-probe microdialysis study in the ventral tegmental area and nucleus accumbens of the rat brain. J Neurosci. 1996;16:2605–2611. doi: 10.1523/JNEUROSCI.16-08-02605.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Westerink BH, Enrico P, Feimann J, De Vries JB. The pharmacology of mesocortical dopamine neurons: a dual-probe microdialysis study in the ventral tegmental area and prefrontal cortex of the rat brain. J Pharmacol Exp Ther. 1998;285:143–154. [PubMed] [Google Scholar]
- 60.White FJ. Synaptic regulation of mesocorticolimbic dopamine neurons. Annu Rev Neurosci. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- 61.Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 62.Young AM, Bradford HF. N-methyl-d-aspartate releases γ-aminobutyric acid from rat striatum in vivo: a microdialysis study using a novel preloading method. J Neurochem. 1993;60:487–492. doi: 10.1111/j.1471-4159.1993.tb03176.x. [DOI] [PubMed] [Google Scholar]