

Abstract
Transgenic mice expressing high levels of familial amyotrophic lateral sclerosis (FALS)-associated mutant superoxide dismutase 1 (SOD1) under the control of a human SOD1 minigene (hMg) accumulate mutant protein ubiquitously and develop motoneuron disease. However, restricted expression of SOD1 mutants in neurons apparently does not cause motor impairments in mice. Here, we investigated the possible pathogenic roles of mutant SOD1 accumulation in motoneurons. First, we used a Thy1 expression cassette to drive high constitutive expression of transgene in postnatal mouse neurons, including upper and lower motoneurons. Second, we expressed human (h) SOD1(G93A) and hSOD1(G85R) as transgenes (i.e., two SOD1 mutants with aggressive pathogenic properties in inducing FALS). Third, in addition to clinical signs of disease, we monitored early signs of disease onset and pathogenesis, including muscle innervation, astrogliosis in the spinal cord, and accumulation of ubiquitinated deposits in motoneurons and astrocytes. We report that high-level expression and accumulation of the mutant proteins in neurons failed to produce any detectable sign of pathology or disease in these transgenic mice. Crossing hMg-SOD1(G93A) mice (Gurney et al., 1994) with Thy1-SOD1(G93A) mice produced double-transgenic mice with spinal cord SOD1(G93A) levels that were approximately twofold higher than in the hMg-SOD1(G93A) single transgenics but did not affect the onset or progression of pathology or motoneuron disease. The accumulation of mutant SOD1 in postnatal motoneurons is thus not sufficient and probably also not critical to induce or accelerate motoneuron disease in FALS mice. The pathogenic process in FALS may involve non-neuronal cells, and selective vulnerability of motoneurons to this process may lead to motoneuron pathology and disease.
Keywords: amyotrophic lateral sclerosis, pathophysiological process, motoneuron, neurodegeneration, neuronal pathology, mutant SOD1
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disease involving upper and lower motoneurons. Approximately 15% of the total number of ALS cases are familial (FALS), and of these, ∼20% are attributable to point mutations in cytosolic Cu/Zn superoxide dismutase (SOD1) (Deng et al., 1993; Pardo et al., 1995). Three FALS-associated SOD1 mutants (G93A, G85R, and G37R) were introduced into mice using a human SOD1 minigene (hMg) approach, leading to ubiquitous high-level expression of the human transgene and late-onset ALS-like motoneuron disease in these mice (Gurney et al., 1994; Ripps et al., 1995; Wong et al., 1995;Bruijn et al., 1997; Tu et al., 1997).
Because they consistently develop motoneuron disease with predictable onset times and progression rates, FALS mice have provided powerful model systems to identify early pathophysiological mechanisms associated with ALS. These studies have revealed that mitochondrial pathology (Kong and Xu, 1998), fragmentation of the Golgi apparatus (Mourelatos et al., 1996), and a decrease in the rate of fast axonal transport (Williamson and Cleveland, 1999) are detected in spinal motoneurons long before the onset of detectable clinical signs of disease. In addition, a selective and progressive loss of muscle innervation is an early, sensitive, and reliable indicator of advancing neuromuscular pathology in diseases affecting motoneurons in the mouse (Frey et al., 2000).
Despite the availability of appropriate genetic models, the etiology of FALS has remained difficult to define, and the cellular targets critical to the disease process are still not known (Price et al., 1996; Julien, 2001). Motoneuron disease in FALS mice is attributable to a dose-dependent toxic gain-of-function mechanism specifically associated with the accumulation of mutant SOD1 (Wong et al., 1995). In the hMg-transgenic mice, mutant SOD1 accumulated at high levels in many and possibly all tissues and cell types. However, because pathology and dysfunction affected primarily motoneurons, it has seemed reasonable to assume that accumulation of mutant protein in motoneurons is the main cause of disease. This view was challenged by a recent report that transgenic expression of SOD1(G37R) restricted to mouse neurons failed to produce clinical signs of motoneuron disease. Although transgene expression decreased in the adult in that study, and although the G37R mutant requires particularly high expression levels to produce disease in mice, these results suggested that FALS may not be a cell-autonomous disease of motoneurons.
Here, we investigated the possible pathogenic roles of mutant SOD1 accumulation in motoneurons. We used a mouse Thy1.2 expression cassette (Caroni, 1997) to drive high constitutive expression of human (h) SOD1(G93A) or hSOD1(G85R) in postnatal mouse neurons, including upper and lower motoneurons, and monitored clinical signs of disease as well as early signs of disease onset and pathogenesis. We report that high-level expression and accumulation of the mutant proteins in neurons failed to produce any detectable sign of motoneuron pathology or disease in these transgenic mice. In addition, double-transgenic hMg-SOD1(G93A)/Thy1-SOD1(G93A) mice with spinal cord SOD1(G93A) levels that were approximately twofold higher than in the hMg-SOD1(G93A) single-transgenic mice were undistinguishable from the hMg-SOD1(G93A) single-transgenic mice with respect to the onset and progression of motoneuron pathology and disease. The accumulation of mutant SOD1 in postnatal motoneurons is thus not sufficient and possibly also not critical to cause ALS-type pathology in motoneurons. A pathogenic process involving the accumulation of SOD1 mutants in non-neuronal cells may thus lead to motoneuron pathology and dysfunction attributable to selective vulnerability properties of these long-projection neurons.
MATERIALS AND METHODS
Transgenic mice. Transgenic mice expressing FALS-associated hSOD1 mutants, specifically in adult neurons, were generated using the mouse Thy1.2 expression cassette, as described previously (Caroni, 1997). The G93A or G85R mutations were engineered into wild-type human SOD1 cDNA using a PCR/oligonucleotide primer-directed mutagenesis kit (Stratagene, La Jolla, CA). All cDNAs used in this study were confirmed twice by DNA sequencing. Mutated SOD1 cDNAs were cloned upstream of an internal ribosomal entry site (IRES)–enhanced green fluorescent protein (EGFP) sequence to generate bicistronic transcripts expressing SOD1 mutants and GFP. These constructs were subsequently cloned into the mouse Thy1.2 expression cassette. Transgenic mice were generated and screened according to conventional methods. hMg-SOD1(G93A) mice (Gurney et al., 1994; Frey et al., 2000) were obtained from The Jackson Laboratory (Bar Harbor, ME) (catalog number JR2726). Thy1-SOD1(G93A), Thy1-SOD1(G85R), and hMg-SOD1(G93A) mice were all bred in the C57BL/6 background.
Transgene detection, histology, and immunocytochemistry. To produce total protein extract fractions, neural tissues [brain, lumbar spinal cord, lumbar ventral roots (L3–L5)] were homogenized in 120 mm NaCl, 50 mm Tris, pH 7.5, 1% Triton X-100, 0.1% SDS, and 5 mm EDTA, plus protease inhibitors (aprotinin and PMSF). To determine SOD1 contents, equal amounts of total protein were separated on 15% polyacrylamide gels and transferred onto nitrocellulose filters. Endogenous mouse and mutant human SOD1 were subsequently detected using a polyclonal antibody (Stressgen, Victoria, Canada) that binds to both human (apparent molecular mass, 22 kDa) and mouse (apparent molecular mass, 19 kDa) SOD1. Bound antibody was detected with peroxidase-labeled secondary antibodies, followed by a chemiluminescence reaction (Amersham Biosciences, Arlington Heights, IL) and x-ray film exposure. For quantitative analysis, films were analyzed with NIH Image software. Superoxide dismutase activity was assayed on nondenaturing polyacrylamide gels as described previously (Beauchamp and Fridovich, 1971). Briefly, lumbar spinal cord or brain was homogenized in buffer containing 20 mmTris-Cl, pH 7.2, 1 mm EDTA, and 1% Triton X-100, and then 20 μg of protein supernatant (centrifuged at 10,000 ×g for 5 min) was loaded on each gel slot. As a positive control and to determine the migration position of the human enzyme, human erythrocyte SOD1 was used as a marker.
Transgene transcripts were detected by in situ hybridization using a digoxigenin-labeled cRNA EGFP probe (Baumeister et al., 1997).
For immunocytochemistry, mice were anesthetized with Rompun (Bayer AG, Leverhusen, Germany) and perfused with PBS, followed by 4% paraformaldehyde in PBS. Cryostat sections (12 μm thick) of spinal cord or brain were subsequently processed for immunocytochemistry according to standard procedures. The following antibodies were used: rabbit anti-GFAP (Dako, Carpinteria, CA), rabbit anti-ubiquitin (Dako, Copenhagen, Denmark), rabbit anti-human SOD1 (Stressgen), and monoclonal antibody against phosphorylated neurofilaments (SMI31; Stenberger Monoclonals Inc., Lutherville, MD).
Neuromuscular synapses and their innervation patterns were visualized on fresh-frozen sections using a combined silver esterase reaction, as described previously (Frey et al., 2000). Alternatively, innervation of muscle was analyzed using an affinity-purified antiserum against p75 (kind gift from U. Mueller, Friedrich Miescher Institute, Basel, Switzerland) and RITC-α-bungarotoxin (Molecular Probes, Eugene, OR), as described previously (Frey et al., 2000).
To analyze peripheral nerve regeneration, the right sciatic nerve of 2- and 6-month-old mice was crushed at midthigh level, and nerve regeneration and reinnervation of muscle were analyzed 14 d after the crush, as described previously (Aigner et al., 1995).
Quantitative analysis of data. Mice were tested for hindlimb muscle strength using the loaded grid protocol (Barneoud et al., 1997). Testing was started at 6 weeks of age and repeated weekly. For the quantitative analysis of GFAP immunoreactivity and ubiquitin deposit accumulation, sets of lumbar spinal cords (L3–L5) to be compared were processed for immunocytochemistry, and photographs of the labeled sections were processed in the same way. To quantify areas of GFAP-immunoreactive cells, the outlines of all labeled cells in the ventral spinal cord were traced, and total labeled areas were determined with a computer program. Fifty sections per mouse were analyzed, and the values are given in arbitrary units (average values per section; pooled data from two mice each). To quantify ubiquitin deposit accumulations, all ubiquitin-positive structures in the ventral and dorsal spinal cord were outlined, and their areas were determined. Forty sections per mouse were analyzed. The values are in arbitrary units per section (averages of all data from two mice).
Muscle denervation was quantified by two independent methods. Percentage values of nerve-free, acetylcholine esterase-positive neuromuscular junctions (NMJs) were determined for adjacent segments of medial gastrocnemius (MGC) and lateral gastrocnemius (LGC) synaptic regions, as described previously (Frey et al., 2000). In separate experiments, muscle sections were double-labeled for p75 and postsynaptic acetylcholine receptor clusters, and the percentage of p75-positive NMJs was determined for all NMJs in the medial compartment of the LGC on five sections per mouse. Data from two mice each were pooled.
RESULTS
Transgenic mice expressing high levels of FALS-associated SOD1(G93A) or SOD1(G85R) in postnatal neurons
To generate transgenic mice expressing high levels of FALS-associated SOD1 mutants constitutively and specifically in neurons, we expressed hSOD1(G93A) or hSOD1(G85R) under the control of the mouse Thy1.2 expression cassette (Caroni, 1997). Neuron-selective expression driven by this promoter construct starts during the first postnatal week and is unusually strong (Caroni, 1997). To monitor transgene expression patterns in the absence of cross-reactivity with endogenous mouse SOD1, transgenic transcripts included an IRES followed by the coding sequence for EGFP (see Materials and Methods). Transgenic lines were selected for high-level expression in most spinal cord and motor cortex neurons.
Four Thy1-SOD1(G93A) lines (lines 1, 11, 13, and 16) and two Thy1-SOD1(G85R) lines (lines 5 and 6) were retained. Figure1 shows lumbar spinal cord and motor cortex in situ hybridization data for Thy1-SOD1(G93A) line 13 (Fig. 1A,B) and Thy1-SOD1(G85R) line 6 (Fig.1D,E) mice. Prominent transgenic transcript accumulation was detected in large ventral horn neurons (Fig.1A,D) and in layer 5 motor cortex projection neurons (Fig. 1B,E), starting around postnatal day 5 (P5) (data not shown). The other transgenic lines selected for additional analysis (lines 1, 11, 16, and 5) exhibited comparable expression patterns (data not shown). The transgene hSOD1(G93A) was metabolically active (Fig. 1C). To monitor the accumulation levels of mutant SOD1 protein in the transgenic mice, we performed an immunoblot analysis of lumbar spinal cord (Fig. 1C,F), ventral roots 3–5 (Fig. 1C), and brain homogenates (data not shown). Transgene accumulation levels for the Thy1-SOD1(G93A) lines were compared with those of high-expression hMg-SOD1(G93A) mice (Gurney et al., 1994), which die of motoneuron disease at ∼136 d. As shown in Figure 1C, all Thy1-SOD1(G93A) lines selected for this study expressed transgene levels that were nearly as high as those detected in the reference line hMg-SOD1(G93A). Densitometric analysis of the immunoblots revealed total SOD1(G93A) lumbar spinal cord levels that ranged between 58 and 85% of the values in the reference line. SOD1(G85R) is known to have a low metabolic stability, and transgene accumulation levels in the corresponding Thy1 lines were substantially lower than those in the Thy1-SOD1(G93A) lines (Fig.1F). However, when the transgene signal is compared with that of endogenous mouse SOD1, relative transgene accumulation levels are equal to if not higher than those reported for the highest expressing hMg-SOD1(G85R) lines.
Fig. 1.
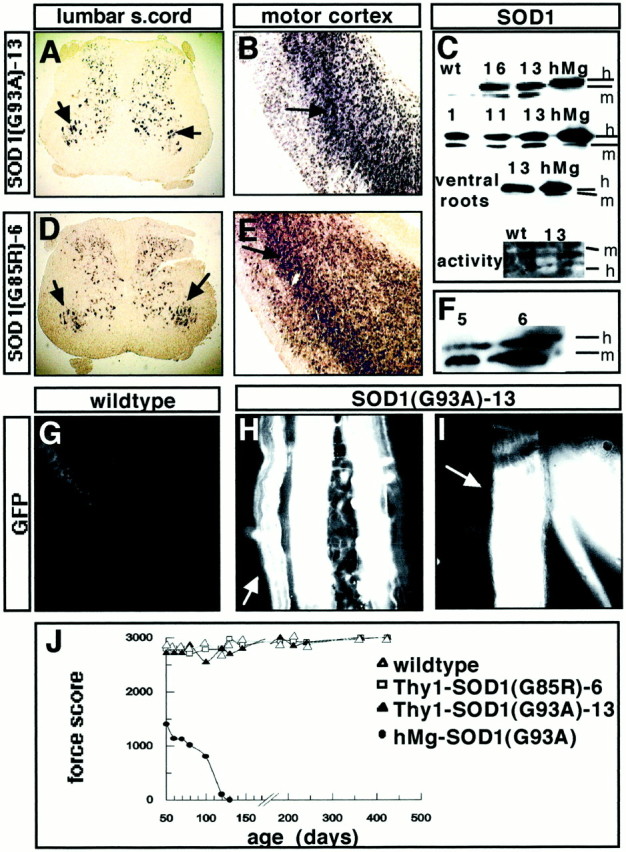
Establishment of transgenic mouse lines expressing high levels of mutant SOD1(G93A) or SOD1(G85R) in postnatal neurons, including spinal and upper motoneurons. A–C, Thy1-SOD1(G93A) transgenic lines. A, Expression of transgene in lumbar spinal cord (s.cord) neurons [Thy1-SOD1(G93A) line 13, 1 month; EGFP mRNA; arrowspoint to transgene-positive large ventral horn neurons]. In control experiments, when the EGFP probe was applied to sections from nontransgenic mice, it yielded no detectable signals at up to fivefold longer color development times (data not shown). B, Expression of transgene in motor cortex neurons [Thy1-SOD1(G93A) line 13, 1 month; EGFP mRNA; arrow points to transgene-positive layer 5 neurons]. C, Top three rows, Immunoblots of human (h) and mouse (m) SOD1 in 1 month lumbar spinal cord homogenates (top two rows) and 1 month L3–L5 ventral roots from wild-type (wt), hMg-SOD1(G93A) high-expression line (hMg) (Gurney et al., 1994), and Thy1-SOD1(G93A) transgenic lines 1, 11, 13, and 16. Bottom row, SOD activity in 1 month spinal cord homogenates from wild-type (left) and Thy1-SOD1(G93A) 13 mice (right, brain homogenate). Note that on this nondenaturing gel, human SOD1 migrated faster than endogenous mouse SOD1. D–F, Thy1-SOD1(G85R) transgenic lines. D, E, Expression of transgene in lumbar spinal cord (D) and motor cortex layer 5 (E) neurons (1 month mouse, details as inA and B). F, Immunoblot of human and mouse SOD1 in lumbar spinal cord homogenates (1 month) from Thy1-SOD1(G85R) transgenic lines 5 and 6. G–I, EGFP fluorescence in peripheral nerves from a wild-type (G) and Thy1-SOD1(G93A) line 13 mouse (H, I; 14 months). G, Level of peroneal nerve (as inI). H, Level of the tibial nerve, where three muscular branches arise that run along the external sural (arrow), internal sural, and posterior tibial vessels, respectively. I, Level of common peroneal nerve, where it divides into superficial (arrow) and deep peroneal branches. J, Hindlimb muscle strength as a function of age in wild-type, Thy1-SOD1(G93A) line 13, Thy1-SOD1(G85R) line 6, and high-expressing hMg-SOD1(G93A) mice. The values are averages from three mice each.
To verify that the transgene was expressed in neurons innervating hindlimb muscles in our mice, we monitored the accumulation of EGFP in ventral roots and in distal sections of peripheral nerves. Figure1H,I shows that in the transgenic lines, peripheral nerves exhibited strong fluorescent GFP signals, whereas no fluorescence was detected in corresponding nontransgenic littermates (Fig. 1G). Closer examination of ventral roots, peripheral nerves, and intramuscular nerves suggested that in these mice, the transgenes were expressed in most and possibly all motoneurons.
Absence of motoneuron disease and FALS-associated pathology in Thy1-SOD1(G93A) and Thy1-SOD1(G85R) mice
The Thy1-transgenic lines were bred and followed longitudinally for survival and signs of motor dysfunction. None of the lines exhibited any obvious behavioral phenotype, and survival curves were undistinguishable from those of wild-type mice. Hindlimb muscle strength in the transgenic mice was assayed with a loaded grid protocol. Mice from the reference line hMg-SOD1(G93A) lost most of their hindlimb muscle strength between P70 and P110 (Fig.1J). In addition, presumably because they were smaller than their nontransgenic littermates, hMg-SOD1(G93A) mice exhibited reduced strength as early as P50. In contrast, all Thy1-SOD1(G93A) and Thy1-SOD1(G85R) mice exhibited hindlimb muscle strength values undistinguishable from those of wild-type littermates at any age tested (Fig. 1J). For the data shown in Figure 1J, muscle strength values were monitored systematically between P50 and P500. In addition, we analyzed a smaller sample of Thy1-SOD1(G93A) line 13 mice between 18 and 20 months of age, and these were again undistinguishable from wild-type littermates (data not shown).
To determine whether Thy1-SOD1(G93A) and Thy1-SOD1(G85R) transgenic mice exhibited any sign of muscle denervation, an early hallmark of disease in FALS mice, we analyzed triceps sural innervation patterns using a combined silver esterase method. This histological method reveals nerves in black (silver reaction precipitate) and NMJs in blue (synaptic acetylcholine esterase reaction product). As shown in Figure 2A, extensive denervation of lateral segments of the LGC was detected as early as P50 in the reference line hMg-SOD1(G93A). In contrast, no denervation was detected in Thy1-SOD1 mutant mice (Fig. 2B shows LGC data for a 14-month-old line 13 mouse). A detailed quantitative analysis revealed that none of the Thy1-SOD1 transgenic lines exhibited any significant denervation of LGC, MGC, or soleus between 1 and 15 months of age (Fig. 2C).
Fig. 2.
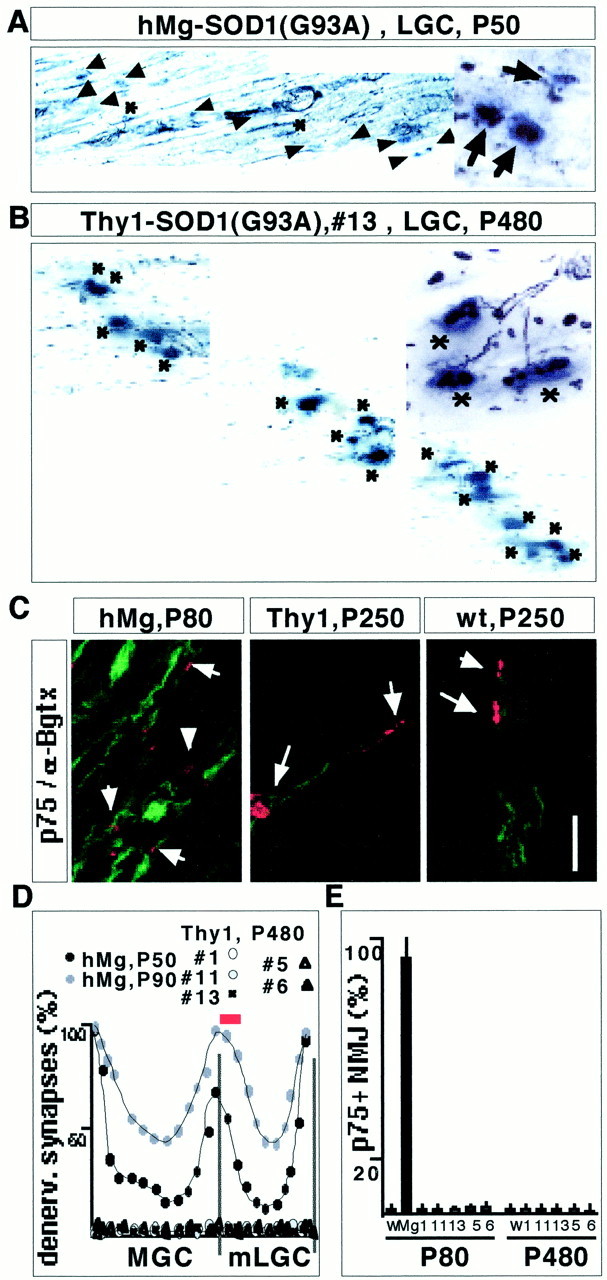
Absence of muscle denervation in Thy1-SOD1(G93A) and Thy1-SOD1(G85R) transgenic mice. A, B, Combined silver esterase reaction of LGC [medial end of medial compartment (mLGC), as indicated by the red bar inD] from an hMg-SOD1(G93A) mouse at P50 (A) and a Thy1-SOD1(G93A) line 13 mouse at P480.Insets on the right show a detail from the same region. Synaptic regions are in blue(acetylcholine esterase reaction product), and nerves are inblack. Some of the nuclei on the section yield ablack background color. Asterisks, Innervated NMJs; arrows, denervated NMJs.C, Double-labeling immunocytochemistry for p75 and α-bungarotoxin in LGC (medial compartment). The Thy1 transgenic mouse was from the Thy1-SOD1(G93A) 13 line. Arrows point to some of the α-bungarotoxin-positive NMJs. The weak p75 signal on the sections from wild-type and Thy1 transgenic mice was associated with sensory nerves, whereas the strong signal in the hMg mice was attributable to Schwann cells that had lost contact with motor nerve. The small size of the acetylcholine receptor cluster signals in the hMg mouse is attributable to chronic denervation of mLGC in this mouse. Scale bars: A, B, 120 μm; insetin A and B, 50 μm; C, 60 μm. D, Quantitative analysis of denervation (denerv.) along the synaptic band of MGC and LGC muscle (see Materials and Methods for details). Each curve represents average values from two mice. E, Quantitative analysis of mLGC denervation (p75-positive NMJs; see Materials and Methods for details). W, Wild type; Mg, minigene, hMg-SOD1(G93A). Average values from two mice each are shown.
Schwann cell p75 immunoreactivity is slightly upregulated at paralyzed NMJs and strongly upregulated at denervation. To further investigate the possibility that a few individual NMJs may exhibit signs of denervation in the Thy1-SOD1 mice, we performed a detailed analysis of p75 immunoreactivity at the NMJ and in peripheral nerves. As expected, prominent upregulation of p75 was detected in the LGC of hMg-SOD1(G93A) mice (Fig. 2D). In contrast, and as in wild-type controls (Fig. 2F,G), Thy1-SOD1(G93A) (Fig.2E,G) and Thy1-SOD1(G85R) mice (Fig. 2G) exhibited no signs of muscle denervation at any of the times analyzed in this study.
To determine whether, despite the absence of FALS-related functional deficits, Thy1-SOD1(G93A) or Thy1-SOD1(G85R) mice may exhibit signs of FALS-associated pathology in the spinal cord, we monitored the distribution and appearance of ubiquitin-rich deposits in these mice. As expected, hMg-SOD1(G93A) reference mice exhibited a profusion of ubiquitin-rich deposits in ventral spinal cord neurons (Fig. 3A; P120 mouse). In contrast, no ubiquitin deposits were detected in Thy1-SOD1(G93A) mice (Fig. 3A; P430 mouse) or in Thy1-SOD1(G85R) mice (data not shown). In reference line hMg-SOD1(G93A) mice, ubiquitin deposit accumulation was evident from P80 on, and the density of the deposits increased with age. In contrast, we failed to detect ubiquitin-rich spinal cord deposits in Thy1-SOD1(G93A) and Thy1-SOD1(G85R) mice between P30 and P520. In additional experiments, we also did not detect abnormal accumulations of SOD1 or phosphorylated neurofilament immunoreactivity in the Thy1-SOD1 mutant mice, whereas these signs of pathology were prominent in the hMg-SOD1(G93A) mice (data not shown).
Fig. 3.
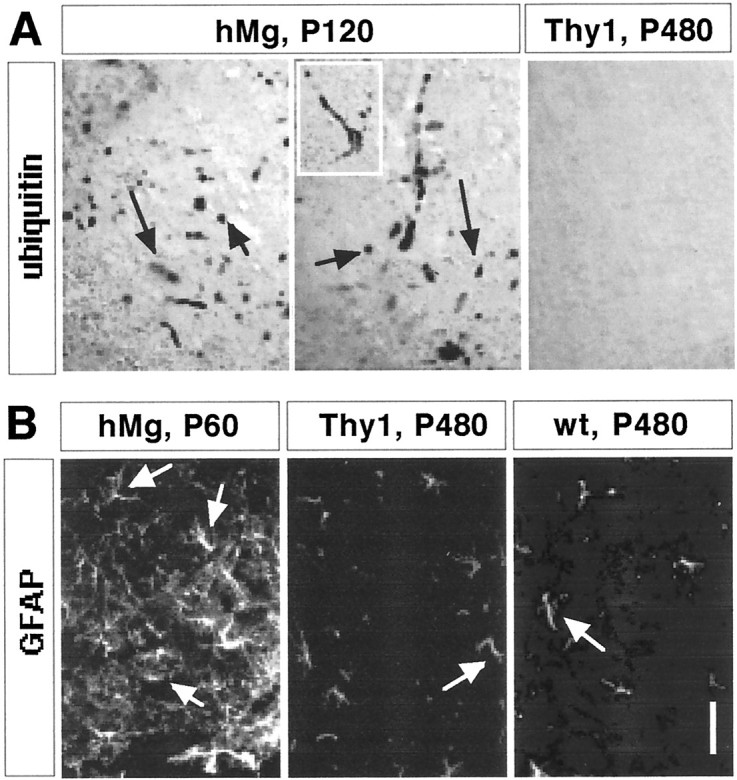
Absence of ubiquitinated deposits and astrocytosis in Thy1-SOD1(G93A) mice. The data are from Thy1-SOD1(G93A) line 13 mice, but similar results were obtained with the other Thy1-SOD1(G93A) lines and with Thy1-SOD1(G85R) lines. A, Ubiquitin immunoreactivity in lumbar spinal cord (ventral horn). Thearrows point to ubiquitin-positive inclusions; theinset shows a large neuron with inclusions.B, GFAP-positive astrocytes (arrows) in lumbar spinal cord (ventral horn). Scale bar: A, 40 μm; B, 100 μm.
Reactive astrocytosis, as revealed by the appearance of hypertrophic astrocytes with high expression levels of GFAP, is a prominent early marker of dysfunction in FALS mice. As expected, reference line hMg-SOD1(G93A) mice exhibited extensive astrocytosis in the spinal cord (Fig. 3B). In contrast, spinal cord GFAP immunoreactivity in Thy1-SOD1(G93A) (Fig. 3B) and Thy1-SOD1(G85R) mice (data not shown) was indistinguishable from that detected in wild-type littermates (Fig. 3B; for a quantitative analysis, see Fig.4).
Fig. 4.
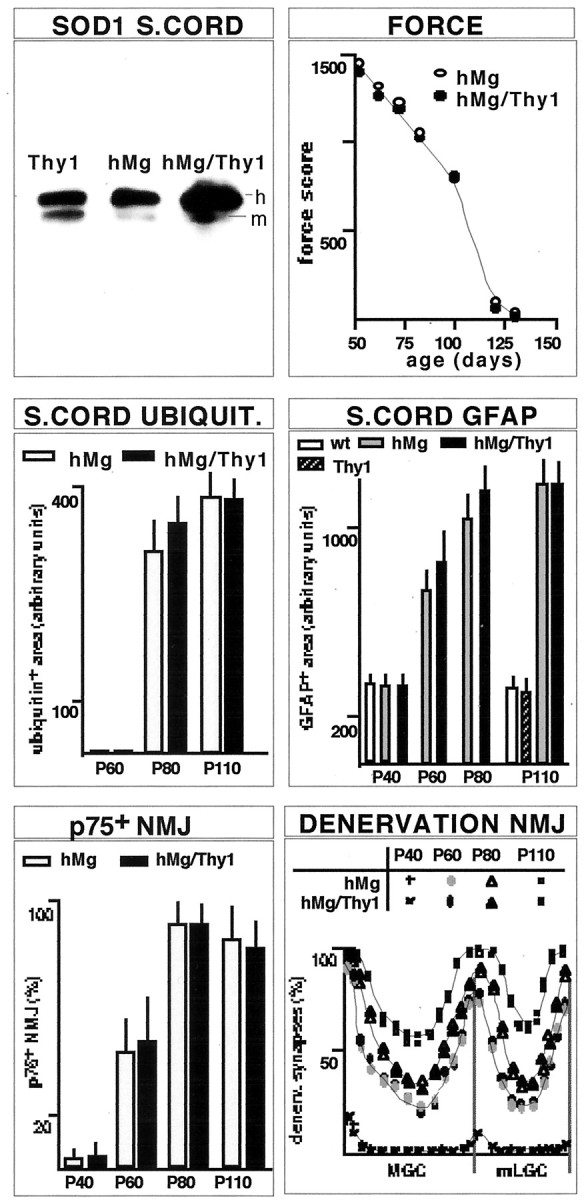
Compared with single-transgenic hMg-SOD1(G93A) mice, double-transgenic hMg-SOD1(G93A)/Thy1-SOD1(G93A) mice express at least twice the amount of mutant SOD1(G93A) in postnatal neurons but exhibit no acceleration of motoneuron pathology or disease. The immunoblot analysis of human (h) and mouse (m) SOD1 in spinal cord (S.CORD) from 1 month Thy1-SOD1(G93A) line 13, hMg-SOD1(G93A), and double-transgenic hMg-SOD1(G93A)/Thy1-SOD1(G93A) mice was as described in Figure 1C. Force was determined as described for Figure 1J, and the values are averages from two mice each. Lumbar spinal cord ubiquitin deposits and areas of GFAP-immunoreactive cells were analyzed as described in Materials and Methods. Values are averages from 40 (ubiquitin) and 50 (GFAP) spinal cord sections; data from two mice each were pooled. The quantitative analysis of muscle denervation (p75: mLGC) was as described for Figure2D,E. All transgenic mice analyzed in the figure expressed the SOD1(G93A) mutant protein.
To determine whether spinal motoneurons in the Thy1-SOD1 mutant mice may be more vulnerable to stress, we monitored peripheral nerve regeneration and muscle reinnervation after sciatic nerve crush in pairs of 2- and 6-month-old Thy1-SOD1(G93A) line 13 mice. These experiments also failed to reveal differences between the transgenic mice and corresponding wild-type littermates (data not shown).
Doubling the levels of mutant SOD1(G93A) in motoneurons of postnatal FALS mice does not affect onset time and progression of motoneuron pathology and disease
Although overexpression of high levels of FALS-associated SOD1 mutants in postnatal neurons was not sufficient to induce motoneuron disease or motoneuron pathology in mice, the levels of SOD1 mutant protein in motoneurons may nevertheless affect the onset time and/or the progression of dysfunction and pathology in FALS mice. To address this possibility, we crossed reference line hMg-SOD1(G93A) mice with Thy1-SOD1(G93A) line 13 mice. As shown in Figure 4, double-transgenic hMg-SOD1(G93A)/Thy1-SOD1(G93A) mice accumulated mutant SOD1(G93A) protein levels in the spinal cord that were approximately twice as high as those detected in single-transgenic hMg-SOD1(G93A) littermates. Despite these elevated accumulation levels in postnatal neurons, single- and double-transgenic mice were undistinguishable with respect to muscle strength values between P50 and P130 (Fig. 4), the time course of denervation in LGC (Fig. 4), and the time at which paralysis prevented locomotion in these mice. In addition, although we did detect minor transient increases during the intermediate phase of spinal cord ubiquitin accumulation and astrogliosis in the transgenic mice, pathology values during the onset time and the advanced phase of the disease were undistinguishable from those in single-transgenic hMg-SOD1(G93A) mice (Fig. 4). Therefore, although onset time and disease progression are highly dependent on transgene expression levels in these ALS models, doubling the levels of mutant SOD1(G93A) in neurons did not affect the kinetic properties of pathology and disease. These results suggest that the accumulation of mutant SOD1(G93A) in neurons is not a critical pathogenic factor in this model of familial ALS.
DISCUSSION
We have provided evidence that overexpression of FALS-associated SOD1 mutants in postnatal neurons of mice fails to produce muscle denervation, motoneuron disease, or motoneuron pathology. In addition, we have shown that duplicating the load of SOD1(G93A) in motoneurons does not affect disease onset, disease progression, and motoneuron pathology in reference hMg-SOD1(G93A) mice. In the following, we discuss the implications of these findings for the role of mutant SOD1 in motoneuron pathology and disease and the pathophysiology of motoneuron disease.
In the absence of motoneuron disease, the accumulation of mutant SOD1 in motoneurons fails to induce motoneuron pathology
Our results provide evidence that overexpression and accumulation of FALS-associated SOD1 mutants in postnatal neurons of transgenic mice is not sufficient to cause motoneuron disease or to induce neuromuscular synapse loss (i.e., an early premonitory sign of motoneuron dysfunction in motoneuron disease). The Thy1-SOD1(G93A) lines described in this study accumulated mutant protein levels in the spinal cord and ventral roots that were 58–85% of those detected in hMg-SOD1(G93A) mice with the highest published expression levels (Gurney et al., 1994). Unlike the Thy1 transgenics, those mice die at 135 d, and hMg-SOD1(G93A) line 20 mice, which accumulate ∼33% of the SOD1(G93A) levels detected in our transgenics, are completely paralyzed at 1 year of age (Dal Canto and Gurney, 1997). hMg transgenic mice with expression levels of SOD1(G85R) lower than those of our Thy1-SOD1(G85R) mice die at 8–14 months, depending on expression levels, whereas none of our mutant mice exhibited any sign of dysfunction at 18 months. These transgene accumulation comparisons do not take into account the fact that transgene expression is not restricted to neurons in hMg mice, whereas it is in the Thy1 transgenics. Consequently, when comparisons with hMg lines are based on immunoblots of spinal cord or ventral root tissue, relative levels of mutant SOD1 accumulation in neurons of Thy1 transgenic lines are underestimated. Therefore, had the accumulation of mutant SOD1 in postnatal motoneurons been sufficient to induce motoneuron dysfunction and disease, we should have detected it in our mice.
A possible explanation for our findings could be that transgene expression may have been restricted to a fraction of the motoneurons in any motor pool, or that transgene expression levels in motoneurons varied substantially, with only a fraction of the motoneurons accumulating sufficiently high and toxic levels of mutant SOD1. However, most if not all axons in distal sections of peripheral nerves to hindlimb muscles exhibited bright green fluorescence, and ventral roots were homogeneously fluorescent, suggesting that most if not all motor neurons also accumulated high levels of mutant SOD1. In addition, if a fraction of the spinal motoneurons had been affected by the transgene in our mice, in situ hybridization patterns with transgene-specific probes should have revealed a selective loss of neurons with high transgene signals as the transgenic mice became older. However, when lumbar spinal cord in situhybridization samples were compared for transgenic mice aged 1 and 8 months, we did not detect alterations in the number of transgene-expressing neurons or in the fraction of large ventral horn cells with the highest apparent expression levels of transgene. In addition, a detailed analysis of peripheral and intramuscular nerves failed to reveal any sign of denervation-related upregulation of p75 immunoreactivity in transgenic mice aged 1–16 months. Therefore, SOD1 mutant accumulation restricted to postnatal neurons is not sufficient to induce loss of peripheral synapses or clinical signs of motoneuron disease in FALS mice.
An important result of our study is that the accumulation of SOD1 mutants in postnatal motoneurons not only failed to cause motoneuron disease but also failed to induce FALS-associated pathology in motoneurons or neighboring cells in the spinal cord. Thus, no SOD1, neurofilament, or ubiquitin-accumulating deposits were detected in spinal cord neurons of the Thy1 transgenic mice at any age. In addition, we did not detect any sign of astrocytosis in the transgenic mice. Together with the finding that loss of vulnerable neuromuscular junctions was never detected in the transgenic mice, these results suggest that the accumulation of high levels of FALS-associated SOD1 mutants did not significantly affect the viability of motoneurons. An important implication of these findings is that the accumulation of deposits, including those containing SOD1, in motoneurons is probably a consequence of disease, not a causative factor in the pathophysiology of FALS (for a model, see Fig. 5).
Fig. 5.
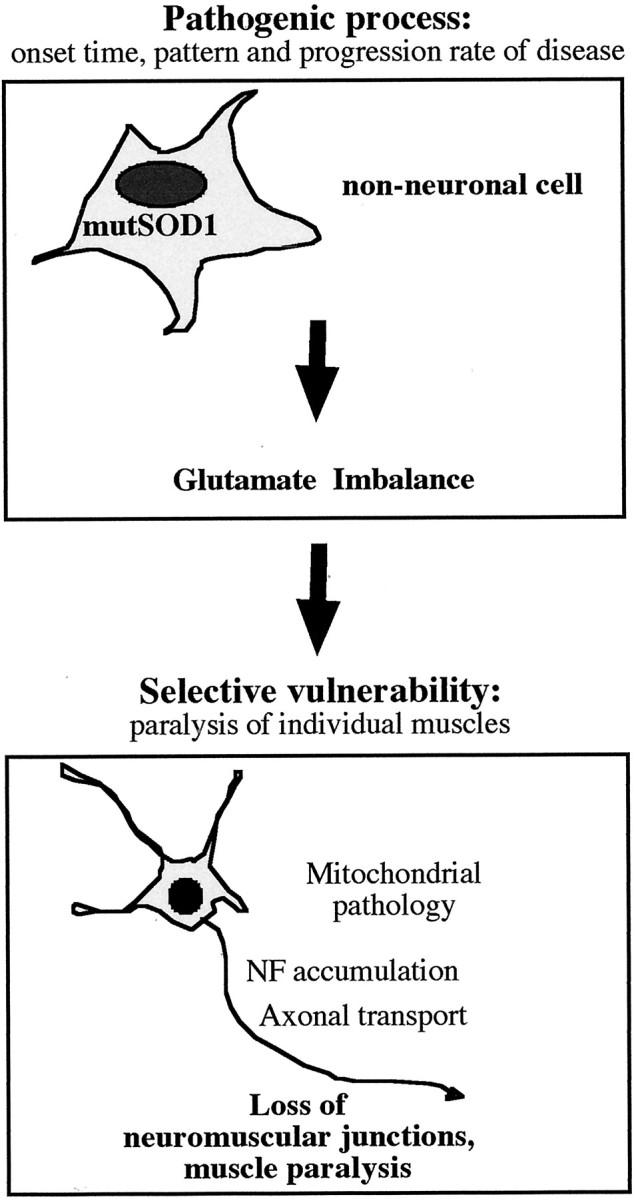
Proposed model of motoneuron disease initiation and progression in mutant SOD1 FALS mice. The model postulates the existence of a pathogenic process driven by the accumulation of mutant SOD1 in non-neuronal cells. This process may lead to a chronic local imbalance in extracellular glutamate handling in the CNS, to which motoneurons are selectively vulnerable. In motoneurons, this would lead to deficits in energy metabolism, axonal transport, and neuromuscular junction maintenance, followed by cell death. According to this model, the timing of motoneuron disease onset and the kinetics of its progression would be determined primarily by the pathogenic process in non-neuronal cells. Although the accumulation of mutant SOD1 in motoneurons may aggravate damage to motoneurons, it may not be necessary for motoneuron disease to be induced.
Accumulation levels of SOD1 mutants in motoneurons are not a critical determinant of motoneuron pathology or disease progression
Double-transgenic hMg-SOD1(G93A)/Thy1-SOD1(G93A) mice accumulated mutant SOD1 levels in the spinal cord and ventral roots that were approximately twice as high as those detected in corresponding single-transgenic hMg mice. Nevertheless, we did not detect differences in the timing of muscle denervation or in the appearance of muscle weakness or paralysis between single hMg-SOD1(G93A) and double-transgenic hMg/Thy1 mice. In addition, the onset time and extent of ubiquitin deposits and astrocytosis in the spinal cord were comparable in single- and double-transgenic mice. There is substantial experimental evidence to support the notion that transgene expression levels in FALS mice determine the timing of disease onset and its progression rate. Because of the expression properties of the Thy1 promoter, the excess load of mutant SOD1 in the double-transgenic mice must have been restricted to neurons, including motoneurons. Therefore, if neurons and motoneurons were the site at which SOD1 mutant accumulation affects disease onset and progression in a dose-dependent manner, the double-transgenic mice would have to exhibit accelerated pathology and disease. Our results therefore suggest that the levels of mutant SOD1 in postnatal neurons are not a critical determinant of motoneuron disease initiation or progression.
Our findings are consistent with the possibility that the accumulation of mutant SOD1 in motoneurons is not necessary to induce motoneuron disease or motoneuron pathology. However, we cannot exclude the possibility that a minimal level of mutant SOD1 in motoneurons is necessary to cause motoneuron disease and/or that the critical concentrations of mutant SOD1 in motoneurons are those that accumulate in embryonic neurons. The latter possibility appears to us unlikely, because it would imply that the extent of an insult to motoneurons induced by mutant SOD1 during embryonic development would be predictive of the onset time and progression rate of disease in the adult. We did detect a small increase in ubiquitin deposits and astrocytosis at intermediate times during disease progression in the double-transgenic mice. These differences between single- and double-transgenic mice may be attributable to some toxic effect of mutant SOD1 overexpression in the motoneurons.
Implications for the mechanisms that lead to motoneuron disease
The results of this study suggest that neurons, and in particular spinal motoneurons, are not the cells in which mutant SOD1 acts primarily to initiate and propagate motoneuron disease. Instead, motoneurons appear to be selectively vulnerable to a pathological process caused by the accumulation of FALS-associated SOD1 in a different type of cell (Fig. 5). That process subsequently leads to mitochondrial pathology, the accumulation of deposits, a reduction in axonal transport, and a loss of peripheral synapses in motoneurons, ultimately causing paralysis and death.
What could be the nature of the pathological process in FALS, and in what cells may SOD1 mutants act to initiate and propagate the disease? There is substantial experimental evidence to support the notion that an excess of extracellular glutamate and glutamate excitotoxicity may be a critical factor in motoneuron disease (Rothstein, 1995; Ludolph et al., 2000). Astrocytes play an important role in removing glutamate from the extracellular space of the neuropil, and a deficit in excitatory amino acid transporter-2, the glutamate transporter on astrocytes, was detected in a mouse model of FALS (Trotti et al., 1999). In addition, early damage to astrocytes was detected in hMg-SOD1(G85R) mice (Bruijn et al., 1997). However, transgenic mice overexpressing SOD1(G85R) in astrocytes exhibited some astrocytosis but did not develop motoneuron disease (Gong et al., 2000). Damage to astrocytes may thus be involved in the pathogenesis of motoneuron disease (Levine et al., 1999), although astrocytes may not be the initial site of action of mutant SOD1. A recent study provided evidence that augmenting CXC chemokine receptor-4 signaling in microglia can lead to excess release of tumor necrosis factor-α, excess extracellular glutamate, and motoneuron pathology (Bezzi et al., 2001). Accordingly, one possibility is that microglia may be the cells in which mutant SOD1 acts to initiate FALS in the transgenic mouse models. However, regardless of whether FALS may be initiated in microglia, the sequence of pathogenic events leading to a chronic local imbalance in glutamate handling most likely involves multiple cellular elements.
If the pathogenic process in FALS does not involve primarily motoneurons, why are these cells selectively vulnerable to the disease? Motoneurons have particularly long axonal projections and appear to be particularly sensitive to excess glutamate (Ludolph et al., 2000). Axonal transport puts a high energy load on neurons, and mitochondrial pathology in motoneurons has been detected early in presymptomatic FALS mice, suggesting that a deficit in energy production may be an early component in the sequence of events leading to motoneuron dysfunction and loss (Kong and Xu, 1999). An energy deficit in axons can lead to the formation of cofilin-containing aggregates because of excess dephosphorylated, active cofilin (Minamide et al., 2000). In addition, the accumulation of neurofilaments and neurofilament aggregates in motoneuron axons has been linked to the pathogenesis of human motor neuron diseases, including ALS (Lee and Cleveland, 1994;Lee et al., 1994; Tu et al., 1997; Couillard-Despres et al., 1998;Williamson et al., 1998). Therefore, as a consequence of glutamate excitotoxicity, deficits in energy production and axonal transport in motoneurons (Zhang et al., 1997; Williamson and Cleveland, 1999) may lead to neuromuscular junction dysfunction, loss of neuromuscular synapses (Frey et al., 2000), and muscle paralysis.
Footnotes
We are grateful to S. Arber for valuable comments on this manuscript. The Friedrich Miescher Institute is a branch of the Novartis Research Foundation.
Correspondence should be addressed to Pico Caroni, Friedrich Miescher Institute, Maulbeerstrasse 66, CH-4058 Basel, Switzerland. E-mail:caroni@fmi.ch.
REFERENCES
- 1.Aigner L, Arber S, Kapfhammer JP, Laux T, Schneider C, Botteri F, Brenner HR, Caroni P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 2.Barneoud P, Lolivier J, Sanger DJ, Scatton B, Moser P. Quantitative motor assessment in FALS mice: a longitudinal study. NeuroReport. 1997;8:2861–2865. doi: 10.1097/00001756-199709080-00012. [DOI] [PubMed] [Google Scholar]
- 3.Baumeister A, Arber S, Caroni P. Accumulation of muscle ankyrin repeat protein transcript reveals local activation of primary myotube end compartments during muscle morphogenesis. J Cell Biol. 1997;139:1231–1242. doi: 10.1083/jcb.139.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beauchamp C, Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 5.Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 6.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 7.Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/s0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- 8.Couillard-Despres S, Zhu Q, Wong PC, Price DL, Cleveland DW, Julien JP. Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc Natl Acad Sci USA. 1998;95:9626–9630. doi: 10.1073/pnas.95.16.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dal Canto MC, Gurney ME. A low expressor line of transgenic mice carrying a mutant human Cu,Zn superoxide dismutase (SOD1) gene develops pathological changes that most closely resemble those in human amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 1997;93:537–550. doi: 10.1007/s004010050650. [DOI] [PubMed] [Google Scholar]
- 10.Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 11.Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 14.Julien JP. Amyotrophic lateral sclerosis: unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- 15.Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kong J, Xu Z. Peripheral axotomy slows motoneuron degeneration in a transgenic mouse line expressing mutant SOD1 G93A. J Comp Neurol. 1999;412:373–380. doi: 10.1002/(sici)1096-9861(19990920)412:2<373::aid-cne13>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 17.Lee MK, Marszalek JR, Cleveland DW. A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron. 1994;13:975–988. doi: 10.1016/0896-6273(94)90263-1. [DOI] [PubMed] [Google Scholar]
- 18.Levine JB, Kong J, Nadler M, Xu Z. Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS). Glia. 1999;28:215–224. [PubMed] [Google Scholar]
- 19.Ludolph AC, Meyer T, Riepe MW. The role of excitotoxicity in ALS: what is the evidence? J Neurol. 2000;247:I7–I16. doi: 10.1007/s004150050552. [DOI] [PubMed] [Google Scholar]
- 20.Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000;2:628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- 21.Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci USA. 1995;92:954–958. doi: 10.1073/pnas.92.4.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price DL, Koliatsos VE, Wong PC, Pardo CA, Borchelt DR, Lee MK, Cleveland DW, Griffin JW, Hoffman PN, Cork LC, Sisodia SS. Motor neuron disease and model systems: aetiologies, mechanisms and therapies. Ciba Found Symp. 1996;196:3–13. doi: 10.1002/9780470514863.ch2. [DOI] [PubMed] [Google Scholar]
- 24.Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothstein JD. Excitotoxic mechanisms in the pathogenesis of amyotrophic lateral sclerosis. In: Serratrice G, Munsat T, editors. Pathogenesis and therapy of ALS. Lippincott-Raven; Philadelphia: 1995. pp. 7–20. [PubMed] [Google Scholar]
- 26.Trotti D, Rolfs A, Danbolt NC, Brown RH, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:848. doi: 10.1038/12227. [DOI] [PubMed] [Google Scholar]
- 27.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tu PH, Gurney ME, Julien JP, Lee VM, Trojanowski JQ. Oxidative stress, mutant SOD1, and neurofilament pathology in transgenic mouse models of human motor neuron disease. Lab Invest. 1997;76:441–456. [PubMed] [Google Scholar]
- 29.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 30.Williamson TL, Bruijn LI, Zhu Q, Anderson KL, Anderson SD, Julien JP, Cleveland DW. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1998;95:9631–9636. doi: 10.1073/pnas.95.16.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 32.Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol. 1997;139:1307–1315. doi: 10.1083/jcb.139.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]