

Abstract
Many ligand/receptor families are known to contribute to axonal growth and targeting. Thus far, there have been no reports implicating Wnts and Frizzleds in this process, despite their large numbers and widespread expression within the CNS. In this study, we show that targeted deletion of the mouse fz3 gene leads to severe defects in several major axon tracts within the forebrain. In particular, fz3(−/−) mice show a complete loss of the thalamocortical, corticothalamic, and nigrostriatal tracts and of the anterior commissure, and they show a variable loss of the corpus callosum. Peripheral nerve fibers and major axon tracts in the more caudal regions of the CNS are mostly or completely unaffected. Cell proliferation in the ventricular zone and cell migration to the developing cortex proceed normally until at least embryonic day 14. Extensive cell death in the fz3(−/−) striatum occurs late in gestation, perhaps secondary to the nearly complete absence of long-range connections. In contrast, there is little cell death, as assayed by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, in the cortex. These data provide the first link between Frizzled signaling and axonal development.
Keywords: axonal growth, fiber tracts, frizzled, mouse brain development, forebrain, Wnt signaling
The founding member of the Frizzled (Fz) family of transmembrane proteins was identified as the product of a Drosophila gene required for the correct orientation of cuticular hairs (trichomes) and bristles (Gubb and Garcia-Bellido, 1982; Vinson et al., 1989). In wild-type flies, these cuticular structures have a definite and highly stereotyped orientation with respect to the body axes, a property referred to as tissue or planar cell polarity. Based on their finding that Fz is an integral membrane protein, Adler and colleagues suggested that it might be the receptor for an extracellular ligand that communicates directional information (Vinson et al., 1989; Park et al., 1994). Consistent with this general hypothesis, experiments in cell culture and in Drosophilaembryos have demonstrated that Fz and a related receptor, Fz2, function as receptors for Wingless, a member of the Wnt family of extracellular signaling proteins (Bhanot et al., 1996, 1999; Bhat, 1998; Kennerdell and Carthew, 1998; Chen and Struhl, 1999). Ligand-receptor relationships between Wnts and Fzs have also been inferred from coinjection experiments in Xenopus embryos (Yang-Snyder et al., 1996; He et al., 1997; Deardorff et al., 2001). However, intracellular signaling in tissue polarity appears to be distinct from Wingless/Fz signaling in the embryo because the former uses a rho GTPase pathway (Strutt et al., 1997; Winter et al., 2001), whereas the latter depends on the modulation of gene expression by β-catenin-lymphocyte enhancer factor (LEF)/T-cell factor (TCF) (van de Wetering et al., 1997).
Current models of Wnt/Fz signaling envision any of three downstream signaling pathways, depending on the biological or experimental context: (1) β-catenin-LEF/TCF mediated transcriptional control, as seen in segment polarity during Drosophila embryogenesis (van de Wetering et al., 1997) and in mammalian cell division/tumorogenesis (Morin et al., 1997), (2) rho- and rac-dependent control of cytoskeletal dynamics as seen in Drosophilatissue polarity (Strutt et al., 1997; Winter et al., 2001) and inXenopus gastrulation (Habas et al., 2001), and (3) G-protein-coupled calcium mobilization, as observed in mammalian cell culture and early zebrafish development (Kuhl et al., 2000). An additional complexity, and one that presents a major challenge in dissecting the role of individual Wnt and Fz family members in vivo, arises from the partial overlap of function within these families. For example, in Drosophila, Fz and Fz2 are almost completely redundant in mediating Wingless action in the embryonic cuticle, gut, CNS, and heart (Bhanot et al., 1999; Chen and Struhl, 1999), and in mice, Wnt1 and Wnt3a have mostly redundant roles in promoting cell division in the developing neural tube and crest (Ikeya et al., 1997). Considering that typical mammalian genomes code for at least nine Fzs and 18 Wnts (http://www.stanford.edu/∼rnusse/wntwindow.html), many of which have partially overlapping patterns of expression, it is likely that at least partial redundancy will be the rule rather than the exception.
Among developmental processes, two of the most complex are the specification of neurons within the CNS and the correct targeting of their axons. The latter process bears some formal resemblance to the establishment of tissue polarity in the Drosophila cuticle because it involves the sensing of directional cues and a local polarized response. A wide variety of ligands, including netrins, ephrins, semaphorins, slits, and neurotrophins, together with their corresponding receptors, are now known to contribute to axonal growth and targeting. However, there have thus far been no reports implicating Wnts and Fzs in this process, despite their large numbers and widespread expression within the CNS (Wang et al., 1996; Grove et al., 1998; Borello, 1999). Instead, Wnts and/or Fzs have been implicated in CNS development in the context of cell fate specification, cell proliferation, cell survival, and synaptogenesis. For example, in chick and Xenopus embryos, Wnt/Fz signaling is involved in early decisions regarding neural crest development and specification (Deardorff et al., 2001; Kiecker and Niehrs 2001; Wilson et al., 2001), and, in the mouse, Wnt1 plays a critical role in the early stages of cerebellar development (McMahon and Bradley, 1990;Thomas and Capecchi, 1990), Wnt3a and LEF/TEF are required for cell proliferation in the developing hippocampus (Dickinson et al., 1994; Galceran et al., 2000), Fz4 is required for cell survival in the cerebellum (Wang et al., 2001), and Wnt7a plays a role in cerebellar synaptogenesis (Lucas and Salinas, 1997; Hall et al., 2000).
In the present study, we report the phenotype of mice carrying a targeted deletion of the fz3 gene. fz3(−/−)mice exhibit massive defects in the development of fiber tracts within the forebrain while leaving other aspects of CNS development mostly unaffected. These data provide the first link between Fz function and axonal development.
MATERIALS AND METHODS
Generation of fz3(−/−) mice. The fz3knock-in construct (see Fig. 1) was electroporated into R1 cells, and colonies were grown in medium containing G418 and gancyclovir. Colonies were picked 8 d after plating and screened by Southern blot hybridization. Positive embryonic stem cell clones were injected into C57BL/6 blastocysts.
Fig. 1.

Targeted replacement of the first coding exon offz3 by lacZ. Top, Partial map of the murine fz3 locus. The first threefz3 coding exons are indicated by filled rectangles. B, BamHI;Bg, BglII; E,EcoRI; H, HindIII;N, NcoI; X,XbaI. Parenthesis indicates elimination of that restriction enzyme site. Bottom, Structure of the fz3 targeting construct and Southern blot hybridization probe. A flanking segment of 8.3 kb located immediately 5′ of the initiator methionine codon of fz3 was joined to the initiator methionine codon of a β-galactosidase expression cassette. The β-galactosidase coding region is followed by an intron and poly(A) site from the mouse protamine-1 gene (lacZ-mp1) (Peschon et al., 1987) and by aphosphoglycerate kinase-neo selectable marker, both with the same orientation. The 5 kb 3′ homology segment encompasses the 5′ 454 bp of the third coding exon and adjacent upstream intron sequences and is followed by a thymidine kinase-negative selectable marker (MC1-TK).Right, Genotyping of fz3(+/+),fz3(+/−), and fz3(−/−) mice byBamHI digestion and Southern blotting with the 5′ flanking BamHI–XbaI probe indicated atleft. The wild-type and gene-targeted alleles generate fragments of 18 and 12 kb, respectively.
Histochemistry. Throughout this study, the first day after overnight mating is counted as embryonic day 0 (E0). Tissues for 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal), NADPH diaphorase, and acetylcholine esterase staining were immersion fixed 2–6 hr at 4°C in PBS, 2.5% formalin, and 0.2% gluteraldehyde; embedded in 3% agarose in PBS; and sectioned at 300 μm on a vibratome. Embryos younger than embryonic day E15 were fixed intact; for later stage embryos, fixation proceeded with isolated heads from which the skin had been removed. Before cutting vibratome sections, later stage heads were incubated at 4°C for 5–7 d in 0.5× PBS, 20 mm Na EDTA to decalcify the skull. For X-gal staining of adult brains, perfusion with 2.5% formalin, 0.2% gluteraldehyde, 2 mm MgCl2, and PBS was followed by a 30 min room temperature incubation in the same fixative before vibratome sectioning. Complete serial sections were collected in the wells of 12-well tissue culture trays. X-gal staining was performed for 24 hr at 37°C in standard X-gal staining solution supplemented with 0.02% Tween 20. Acetylcholine esterase staining was performed at room temperature with minor modifications of the method ofKarnovsky and Root (1964). The reaction mixture contained 0.1m sodium phosphate, pH 6.0, 4 mm acetylcholine iodide, 5 mm sodium citrate, 3 mmcopper sulfate, 0.5 mm potassium ferricyanide, and 100 μm tetraisopropylpyrophosphoramide. NADPH diaphorase staining was performed at 37°C for 1–3 hr in PBS with 0.3% Triton X-100, 0.1 mm NADPH, and 0.25 mg/ml nitroblue-tetrazolium-chloride as described by Bancroft and Stevens (1982).
Immunohistochemistry, bromodeoxyuridine, and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. Immunostaining and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) were performed on brains, whole heads, or various organs that were either (1) fresh frozen, cryosectioned at 16 μm, and then postfixed in 4% paraformaldehyde, PBS, or (2) immersion fixed in cold Carnoy's fixative, dehydrated, embedded in paraffin, and cut at 8 μm. Reagents were obtained from the following sources: anti-neurofilament (anti-neurofilament M; Chemicon, Temecula, CA, or anti-neurofilament 200; Sigma, St. Louis, MO), anti-tyrosine hydroxylase (Chemicon), anti-calretinin (Swant, Bellinzona, Switzerland), anti-dopamine transporter (Chemicon), anti-GFAP and anti-MAP2 (Sigma), anti-acetylated tubulin (TuJ1; a gift from Dr. Anthony Frankfurter, University of Virginia, Charlottesville, VA), anti-bromodeoxyuridine (BrdU) (Harlan Sera-lab, Loughborough, UK), and fluorescein TUNEL reagents (Roche, Indianapolis, IN).
For BrdU labeling, pregnant females were given a single intraperitoneal injection of 400 μl of 10 mm BrdU at the indicated gestational ages. Fetuses were recovered either 1 hr later (for pulse-labeling experiments) or at E18, immersion fixed in cold Carnoy's fixative for 6 hr at 4°C, dehydrated, and embedded in paraffin. Deparaffinized 8 μm sections were incubated in 4N HCl for 20 min, washed in PBS, blocked in 10% normal goat serum in 0.3% Triton/PBS, and incubated in anti-BrdU antibody (1:10 in blocking solution) overnight at 4°C. Sections were washed in PBS, incubated in biotinylated goat anti-rat antibody (Vector Laboratories, Burlingame, CA; 1:200) for 1 hr, followed by horseradish peroxidase-conjugated ExtrAvidin (Sigma, 1:100) for 30 min, and visualized with a standard diaminobenzidine/peroxidase reaction.
1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate tracing. For 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) tracing, intact E12–E15 embryos or embryonic day 18 heads in which the dorsal aspect of the brain was exposed were fixed overnight in PBS with fresh 4% paraformaldehyde. For E12–E15 embryos, a small hole was made through the skull with a 30 gauge needle through which DiI crystals were placed on and in the cortex. For E18 brains, DiI crystals were placed on the exposed surface of the cortex or tectum using a flame-drawn glass needle or on the cut surface of the striatum after the most anterior aspect of the telencephalon had been cut away. Tissues were incubated for 3–6 weeks in 0.5× PBS, 2% paraformaldehyde, and 20 mm Na EDTA (for decalcification), at either room temperature or 37°C, then embedded in 3% agarose in PBS, and sectioned at 300 μm on a vibratome.
Electron microscopy. E18 brains were fixed overnight in PBS, 2% paraformaldehyde, 2% gluteraldehyde, and 1 mm MgCl2, embedded in 3% agarose in PBS, and sectioned at 300 μm on a vibratome. Sections containing cortex and striatum were osmicated, stained with uranyl acetate, embedded in Unicryl resin (British Biotechnology, Inc., Cardiff, UK), and cut with a glass knife.
Dissociated neuronal cultures. Freshly dissected E18 cortices or striata were digested in 0.5 mg/ml papain, 0.5 mm EDTA, and 0.13 mg/mll-cysteine in Earl's Balanced Salt Solution (EBSS) at 37°C for 30 min, and then transferred to 1 mg/ml BSA, 1 mg/ml trypsin inhibitor, and 10 μg/ml DNaseI in EBSS, and gently triturated. Cell suspensions were layered on a shelf of 10 mg/ml BSA and 10 mg/ml trypsin inhibitor in EBSS and pelleted. Cell pellets were washed and resuspended in 10% fetal calf serum, 5% horse serum, 200 μml-cysteine, and 2 mm glutamine in Minimum Essential Medium. Cells were plated on poly-l-ornithine-coated coverslips at 5 × 105 cells per well in 24-well plates, and the plating media was replaced with neurobasal media supplemented with B27 16 hr later. Half of the culture media was replaced with fresh media every other day. Four days after plating, uridine and 5-fluoro-2′-deoxyuridine were added to the culture to inhibit glial cell division.
RESULTS
fz3 expression assayed with alacZ knock-in
To examine the role of Fz3 in vivo, a targeted disruption of the fz3 gene was constructed in which the first 129 codons, present within the first two coding exons, were replaced by a lacZ reporter, followed by aprotamine-1 intron and polyadenylation site (Fig.1). The deleted region codes for the N-terminal signal sequence and the conserved extracellular cysteine-rich domain; the latter has been shown to mediate Wnt binding in a variety of Fz proteins (Bhanot et al., 1996; Hsieh et al., 1999;Dann et al., 2001). The targeted allele is therefore presumed to be a null.
The pattern of lacZ expression in developingfz3(+/−) and fz3(−/−) embryos and in the adultfz3(+/−) brain is in good agreement with existing in situ hybridization and RNase protection data showing transcripts predominantly or exclusively in the nervous system (Fig.2) (Wang et al., 1996; Borello et al., 1999). At midgestation, X-gal staining is seen throughout the developing CNS (Fig. 2A), and at E18, it is seen in the cortex, diencephalon (including the major thalamic nuclei), and brainstem, with the most intense staining in the striatum and trigeminal ganglia (Fig. 2B–D). Figure2B–D shows the pattern of X-gal staining in afz3(−/−) head at E18; a nearly identical pattern is seen in fz3(+/−) heads but with reduced intensity. In the adultfz3(+/−) brain, X-gal staining is most intense in the cortex, the diencephalon, and the rostral brainstem, and little or no staining is seen in the striatum or cerebellum (Fig.2E–H).
Fig. 2.
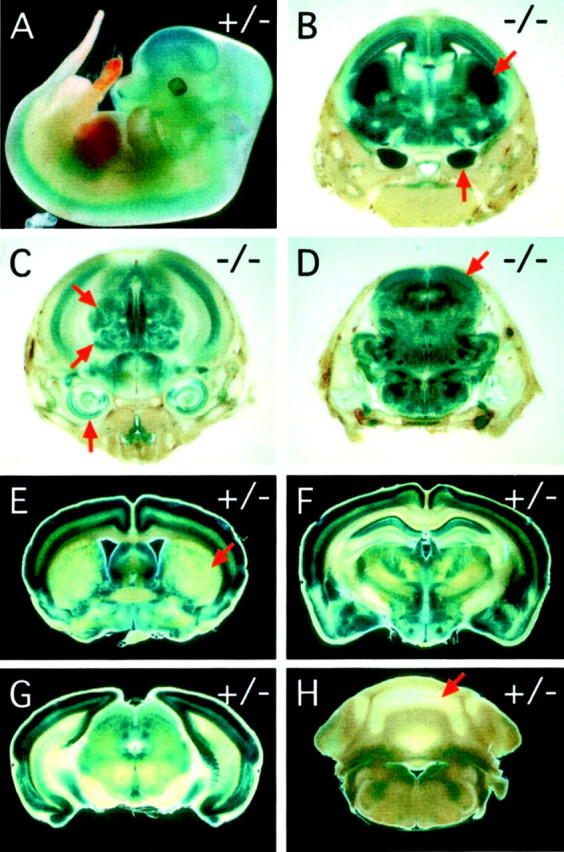
Pattern of fz3 expression as determined by X-gal staining of the knocked-in lacZreporter in fz3(+/−) and fz3(−/−)mice. A, fz3(+/−) embryos at E12 show widespread X-gal staining in the developing CNS. B–D,fz3(−/−) embryos at E18; coronal series from rostral (B) to caudal (D).B, Centered on the striatum; C, centered on the thalamus; D, centered on the tectum. Widespread X-gal staining is seen in the cerebral cortex, diencephalon, and brainstem, with the highest levels in the developing striatum (arrow at 45° angle in B) and the trigeminal ganglia (vertical arrow in B). Staining is also seen in the inner ear (vertical arrowin C), dorsal and ventral thalamic nuclei (45°arrows in C), tectum (arrow in D), and retina (data not shown). fz3(+/−) embryos at E18 show a nearly identical pattern of X-gal staining but at lower intensity. E–H, Adult fz3(+/−) brains show X-gal staining in the cerebral cortex and select midbrain structures with minimal staining in the striatum (arrow in E), cerebellum (arrow in H), and brainstem. Coronal series from rostral (E) to caudal (H). E, Centered on the striatum; F, centered on the thalamus; G, centered on the colliculus; H, centered on the cerebellum.
Functional and anatomic defects infz3(−/−) mice
fz3(+/−) mice are indistinguishable from theirfz3(+/+) littermates in viability, growth, appearance, fertility, and the gross anatomic and histologic appearance of the brain. Therefore, in the remainder of this study, we will present bothfz3(+/+) and fz3(+/−) mice as interchangeable representatives of the wild-type phenotype. fz3(−/−)neonates have a curly tail [a sensitive indicator of neural tube defects (Peeters et al., 1998)] and flexed lower limbs (Fig.3A,B), and they breathe irregularly and typically die within 30 min of birth.fz3(−/−) neonates show a normal withdrawal reflex when pinched, but unlike their fz3(+/−) and fz3(+/+)littermates, they do not vocalize in response to the stimulus.fz3(−/−) mice do not appear to be lost during gestation, because a survey of 122 E18 embryos from 15 consecutive litters derived from fz3(+/−) parents revealedfz3(+/+):fz3(+/−):fz3(−/−) progeny in a ratio of 37:54:31, close to the expected Mendelian ratio of 1:2:1. This analysis also revealed a perfect correlation between thefz3(−/−) genotype and the curled tail phenotype and between the fz3(+/+) and fz3(+/−) genotypes and the straight tail phenotype.
Fig. 3.
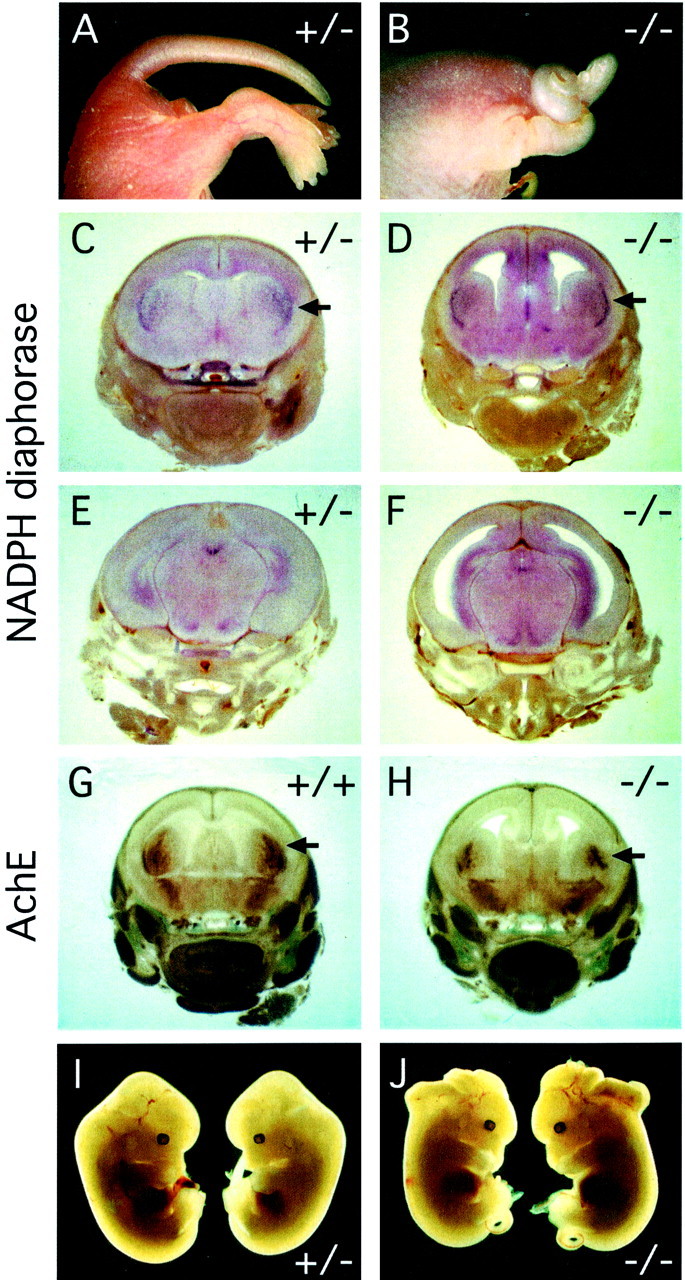
Gross anatomic anomalies infz3(−/−) mice. A, B, Newborn fz3(−/−) mice have a curled tail and flexed hindlimbs. C–H, E18 fz3(−/−) mice have enlarged lateral ventricles, a thinned cerebral cortex, and a smaller striatum but essentially normal patterns of NADPH diaphorase (C–F) and acetylcholine esterase (G, H) activity in the CNS.Arrows in C, D,G, and H point to the higher density of stained cells in the lateral striatum. I,J, fz3(+/−) andfz3(−/−) littermates at E18 from a litter in which 2/2fz3(−/−) fetuses have an open cephalic neural tube.
At present, the pathogenic mechanism of the fz3(−/−)breathing defect is unknown. fz3(−/−) neonates appear cyanotic in room air but become as well oxygenated as their wild-type littermates when placed in 100% oxygen. When the lungs of oxygenatedfz3(−/−) animals are examined several hours after birth, they are typically found to be partially inflated, and in those regions which are inflated, the fz3(−/−) alveoli and bronchi look microscopically normal. Whole-mount acetylcholine esterase staining shows no abnormalities of the intercostal muscles or their innervation. The characteristically irregular breathing together with the finding of gross neuroanatomic defects in the cortex and striatum (described below) suggest that there may be a defect in the brainstem control of respiration (Richter and Spyer, 2001), although we have not observed histologic abnormalities in this region of the CNS.
In the mixed 129/SVJ × C57BL/6 background of thefz3(+/−) line, there is a failure of cephalic neural tube closure in a small percentage of fz3(−/−) embryos but not in fz3(+/−) or fz3(+/+) littermates (Fig.3I,J). This phenotype most likely arises from the effect of genetic modifiers, because it occurs at higher frequency in some crosses, as seen, for example, in the two affected fz3(−/−) littermates in Figure 3J. In one experiment aimed at examining possible interactions betweenfz3 and Wnt-1, 3 of 44 E18 progeny obtained from crossing fz3(+/−);Wnt-1(+/−) double heterozygous parents exhibited a failure of cephalic neural tube closure. Two of these three animals werefz3(−/−);Wnt-1(+/+), and the third wasfz3(−/−);Wnt-1(+/−) [out of a total of 11fz3(−/−) progeny]. This small series supports a model in which the cephalic closure phenotype requires both loss offz3 and one or more genetic modifiers, and it shows no indication that this phenotype is enhanced by loss ofWnt-1.
Defects in major fiber tracts in thefz3(−/−) CNS
At E18, fz3(−/−) mice exhibit a single gross neuroanatomic defect: a marked enlargement of the lateral ventricles secondary to shrinkage of the striatum and thinning of the cortex (Figs. 3C–H). A complete survey of acetylcholine esterase and NADPH diaphorase histochemistry throughout thefz3(−/−) brain at E18 revealed minimal differences from the wild type (Figs. 3C–H). A developmental survey of brain structure using cresyl violet staining revealed little or no difference between the wild-type and fz3(−/−) striatum and cortex at E13 (Fig.4A,B,E,F). By E15, the lateral ventricles of the fz3(−/−) brain are enlarged, but the cortex still appears nearly normal (Fig.4C,D,G,H). However, by E18, the fz3(−/−) cortex is significantly thinner than the wild-type cortex, mostly because of changes within the ventricular, subventricular, and intermediate zones (Fig.4I,J), and thefz3(−/−) striatum is more compact and lacks the internal capsule.
Fig. 4.
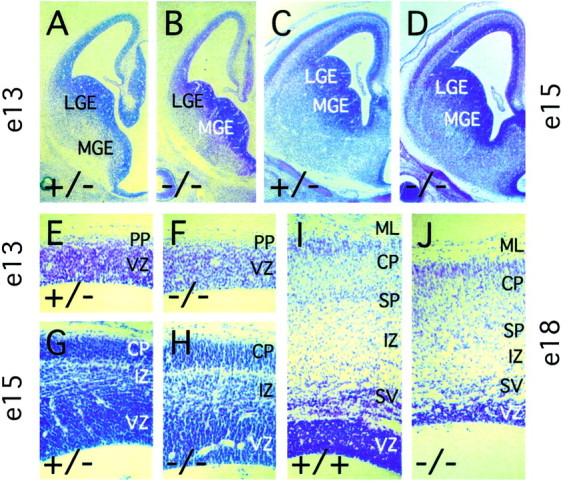
Morphology of the fz3(−/−)cerebral cortex between E13 and E18. Cresyl violet-stained paraffin sections at E13 (A, B, E,F), E15 (C, D,G, H), and E18 (I,J). The fz3(−/−) cortex has a normal or nearly normal thickness and lamination at E13 (E, F) and E15 (G,H); by E18, the intermediate, subventricular, and ventricular zones are reduced in thickness (I,J). The fz3(−/−) lateral ganglionic eminence (LGE) and medial ganglionic eminence (MGE) are of normal size at E13 (A,B) but are reduced in size by E15 (C,D). CP, Cortical plate;ML, marginal layer; PP, preplate;SP, subplate; IZ, intermediate zone; SV, subventricular zone; VZ, ventricular zone.
A survey of major fiber tracts in the E18 fz3(−/−) brain, visualized by anti-neurofilament immunostaining and DiI tracing, revealed a complete absence of the anterior commissure, a nearly complete absence of major corticofugal and thalamocortical fibers, a variable decrease in the size of the corpus callosum, and a marked thinning or in some cases a complete loss of the intermediate zone of the developing cortex (Figs. 5, 6; Table1); none of these defects were observed in fz3(+/+) or fz3(+/−) littermates. Infz3(−/−) brains at E18, the hippocampus appears minimally affected (Fig. 5I,J). DiI placed in the cortex of fz3(+/+) and fz3(+/−)brains at E18 efficiently diffuses along fibers in the intermediate zone of the cortex (Fig. 6I–L) and through the internal capsule toward the thalamus (Fig.6D–H,I–L), but DiI similarly placed in thefz3(−/−) cortex shows mostly local diffusion (Fig.6M–P). A similar result is obtained when DiI crystals are placed in the caudal region of the striatum: in wild-type brains, the internal capsule and adjacent intermediate zone of the cortex are labeled on the ipsilateral side, and the anterior commissure is labeled on the contralateral side (Fig. 6A), whereas in fz3(−/−) brains, DiI labels only local structures (Fig. 6B). An examination of well separated cortical cells filled by application of microcrystals of DiI to the cortical surface revealed a range of approximately normal morphologies among cortical pyramidal cells (Fig. 6C).
Fig. 5.
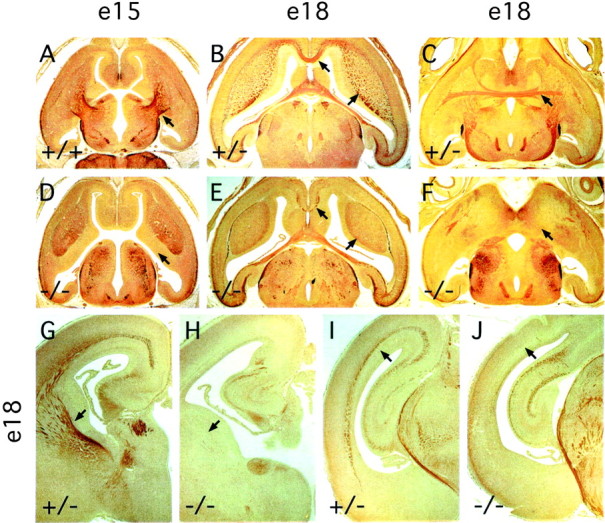
Absence of major fiber tracts in the cortex and striatum in fz3(−/−) brains. Anti-NF200 immunostaining of horizontal sections of the forebrain at E15 (A, D) and at E18 (B,E, dorsal region; C, F, ventral region) and of coronal sections of the forebrain (G, H) and midbrain (I, J) regions. Corticothalamic and thalamocortical fibers passing through the striatum are missing at E15 (arrows in A and D). A complete or nearly complete absence of callosal (leftwardarrows in B andE), corticofugal and thalamocortical (rightward arrows in B and E;arrows in G and H), anterior commissural (arrows in C andF), and cortical (arrows inI and J) fibers is apparent infz3(−/−) embryos at E18. The loss of fibers is most prominent in the rostral cortex and striatum (G,H) and is minimal in the hippocampus (I, J).
Fig. 6.
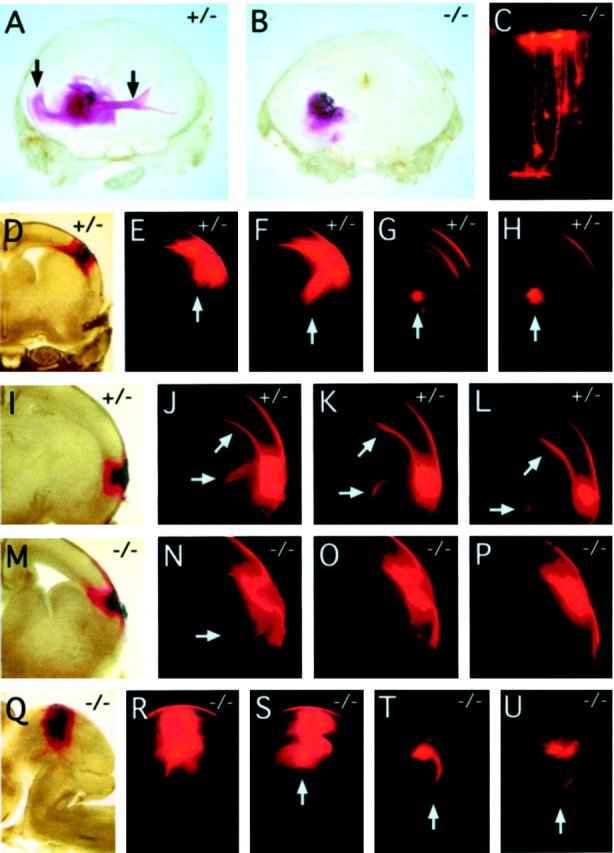
DiI labeling of cortical, commissural, and tectal fiber tracts at E18. A, DiI crystals placed in the caudal region of the striatum of a fz3(+/−) brain label the external capsule and the genu of the corpus callosum in the ipsilateral cortex and the anterior commissure in the contralateral hemisphere (vertical arrows). B, In thefz3(−/−) brain, there is little diffusion of the DiI beyond the site of crystal placement. C, Individualfz3(−/−) cortical neurons labeled with microcrystals of DiI from the cortical surface (top) show normal morphologies. D–L, Serial coronal sections through twofz3(+/−) brains in which DiI crystals were placed in the dorsolateral (D) or lateral (I) cortex. Prominent labeling is seen in cortical fibers traversing the internal capsule (vertical arrows in E–H and horizontal arrows in J–L) and in the genu of the corpus callosum (arrows at 45° angle in J–L).M–P, Serial coronal sections through afz3(−/−) brain in which DiI crystals were placed in the lateral cortex. The DiI has spread locally but has labeled only a small number of fibers traversing the internal capsule (arrow in N). Q–U, Serial sagittal sections through an fz3(−/−) brainstem in which DiI crystals were placed in the lateral tectum.R–U, Efficient labeling of the tectospinal tract (arrows in S–U) with midline crossing in T, a labeling pattern indistinguishable from that seen in fz3(+/+) or fz3(+/−)brains. Sections are 300 μm in thickness.
Table 1.
DiI tracing of fiber tracts in wild-type and fz3 (−/−) brains
| fz3 (+/+) and fz3 (+/−) | fz3 (−/−) | |
|---|---|---|
| E12 cortex | 4 | 4 |
| E13 cortex | 5 | 4 |
| E15 cortex | 4 | 3 |
| E18 cortex | 8 | 6 |
| E18 striatum | 6 | 5 |
| E18 tectum | 6 | 6 |
The table indicates the number of wild-type, i.e., fz3 (+/+), fz3 (+/−), and mutant fz3 (−/−), brains analyzed for each location of the DiI crystals and developmental age. Phenotypes were scored based on whether the tail was curled or straight.
The defects in major fiber tracts appear to be mostly confined to the cortex and diencephalon, because neurofilament immunostaining within the spinal cord and brainstem appears normal in fz3(−/−)mice at E18 (data not shown). DiI tracing of tectospinal fibers at E18 shows the normal decussation of fibers at the isthmus of the brain stem with no apparent differences between fz3(−/−) mice and their fz3(+/+) or fz3(+/−) littermates (Fig.6Q–U). Similarly, a normal pattern of cranial and spinal nerves was observed in whole-mount E11 fz3(−/−)embryos stained for neuron-specific tubulin with monoclonal antibody (mAb) TuJ1 (Lee et al., 1990). At E12, fz3(−/−) and wild-type spinal cords show indistinguishable patterns of immunostaining with antibodies to Nkx2.2, HNF3-β, Lim2, Isl1, and neurofilament. Whether the loss of fz3 produces some subtle alteration in spinal cord development that is not evident from this analysis, as seen for example in the Wnt1/Wnt3a double mutant (Muroyama et al., 2002), remains to be determined. In the enteric nervous system, E18 fz3(−/−) mice showed normal densities and morphologies of enteric neurons, as visualized with NADPH diaphorase and acetylcholine esterase staining.
To examine a single immunochemically defined fiber tract within the affected region of the CNS, we immunostained the striatum for tyrosine hydroxylase (TH) and the dopamine transporter, two well characterized presynaptic markers for the dopaminergic nigrostriatal pathway. At E18, the wild-type striatum shows robust immunostaining for both markers, whereas the fz3(−/−) striatum is devoid of immunoreactivity (Fig. 7A–D). Importantly, dopaminergic neurons appear to be present in normal numbers within the fz3(−/−) substantia nigra as revealed by TH immunostaining (Fig.7E,F).
Fig. 7.
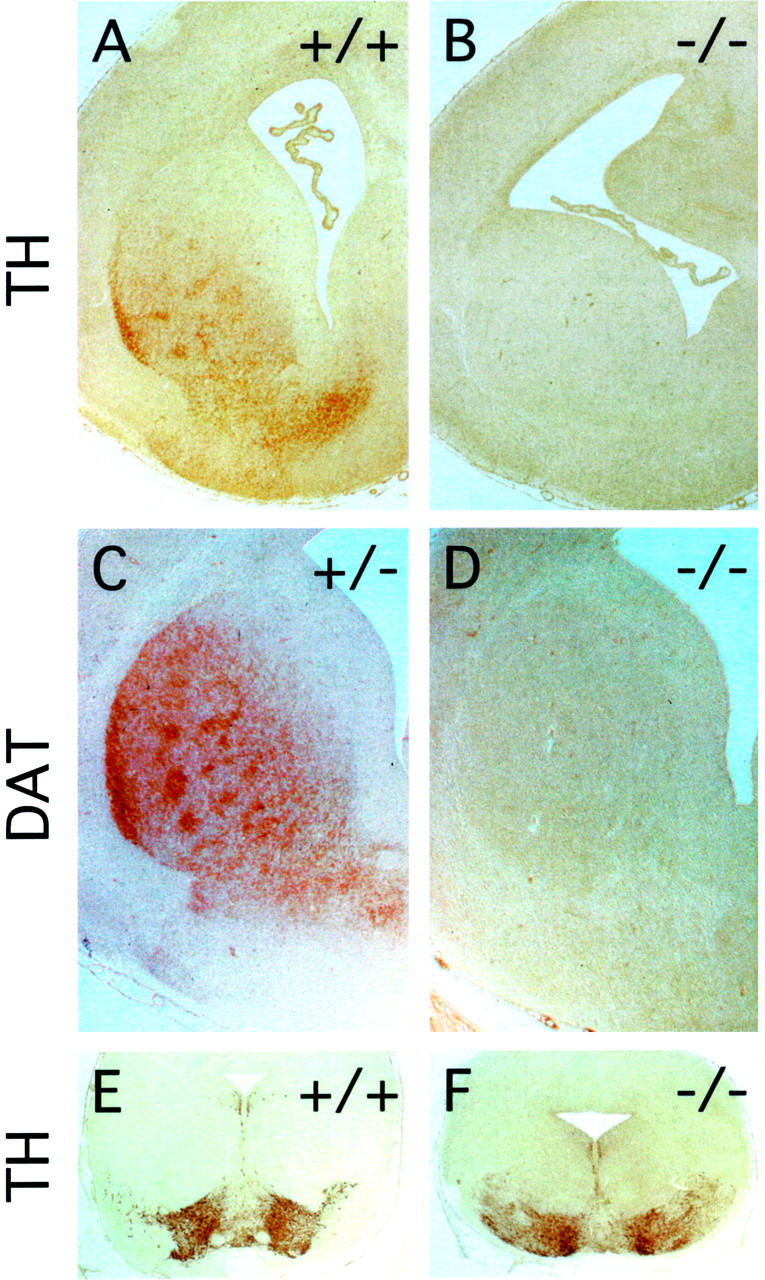
Dopaminergic neurons from the substantia nigra fail to innervate the striatum in fz3(−/−) brains. Tyrosine hydroxylase (A, B) and the dopamine transporter (C, D), markers for presynaptic nigrostriatal processes, are absent from thefz3(−/−) striatum at embryonic day 18.E, F, Tyrosine hydroxylase staining of cell bodies in the substantia nigra demonstrates normal numbers of dopaminergic neurons in the fz3(−/−) brain at embryonic day 18.
To distinguish between an early failure of axonal outgrowth or pathfinding versus a subsequent loss of axons from their targets, corticofugal and thalamocortical axons were visualized by DiI tracing at E12, E13, and E15 (Sheth et al., 1998; Auladell et al., 2000) (Table1), and axon tracts throughout the forebrain were visualized by anti-neurofilament immunostaining at E13, E14, E15, and E16. DiI tracing at E13 and E15 revealed a clear failure of thalamocortical and corticothalamic tract formation in the fz3(−/−) brain (Fig. 8 and data not shown). In contrast, robust and indistinguishable DiI labeling of the developing trigeminal ganglia was observed in fz3(+/+), fz3(+/−), andfz3(−/−) preparations at E12, E13, and E15, presumably via DiI labeling of fibers that innervate the scalp (Fig.8F,L,R,X). Anti-neurofilament staining at these early gestational ages similarly reveals a massive failure in the development of the major forebrain fiber tracts (Fig. 5A,D).
Fig. 8.

DiI tracing from the cortex at E13. Paired bright-field (A–F) and fluorescent (G–L) images of 300-μm-thick serial horizontal sections of an E13 fz3(+/−) head. M–X, The analogous paired series for an E13 fz3(−/−) head. Local spreading of DiI within the cortex and adjacent striatum and labeling of the ipsilateral trigeminal ganglion (arrowsin F, L, R, andX), presumably from DiI in the scalp, are seen in both samples. However, DiI labeling of the thalamus is only seen in thefz3(+/−) brain (arrows inC–E and I–K).
The ultrastructural consequences of the forebrain fiber defects are seen in Figure 9. In the wild-type striatum at E18, densely packed clusters of axons course between groups of striatal neurons, whereas in the fz3(−/−) striatum, few if any axons are seen, and instead, numerous spaces separate the resident cells (Fig. 9A,B). Similarly, in the intermediate zone of the wild-type cortex at E18, large numbers of axons are separated by sparsely distributed cell bodies, whereas in the fz3(−/−) intermediate zone, the intercellular space is mostly devoid of axons or other cellular material (Fig. 9C,D). The presence of lacunae rather than fiber tracts within the fz3(−/−) striatum and cortex suggests either that some axonal outgrowth occurred followed by a subsequent loss or that there are pre-existing extracellular spaces through which axons normally course.
Fig. 9.
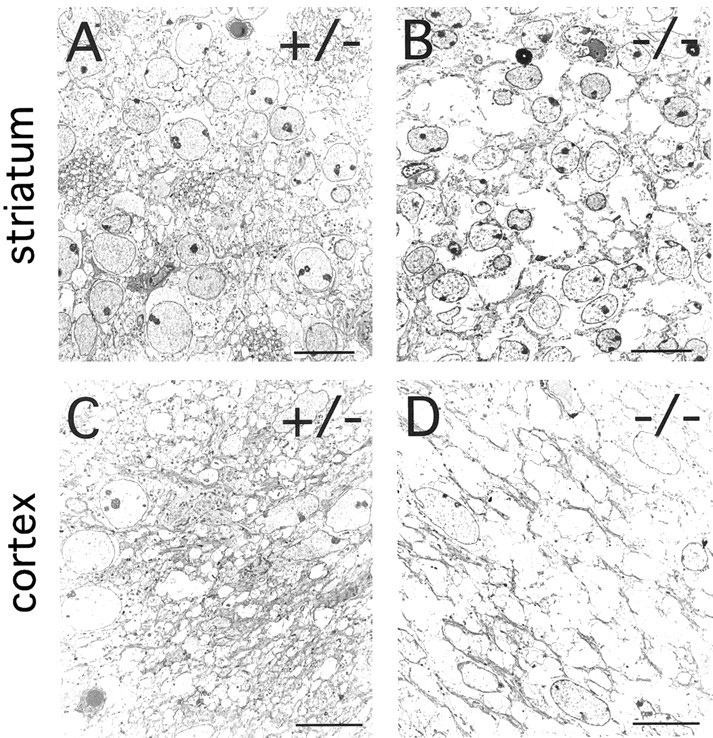
Ultrastructural defects in thefz3(−/−) striatum and cortex at E18. A,B, A high density of synapses, dendritic processes, and axons is seen in the fz3(+/+) striatum. These structures are mostly missing in the fz3(−/−)striatum, which show numerous spaces between neurons. C,D, The subventricular region of thefz3(+/+) cortex shows a dense packing of fibers and cell bodies. In the corresponding region of the fz3(−/−)cortex, fibers are mostly missing, and numerous spaces separate the remaining cell bodies. Scale bars, 10 μm.
Generation, differentiation, and survival of neurons in thefz3(−/−) cortex and striatum
The diminution of striatal volume and thinning of the cortex infz3(−/−) mice at E18 could simply reflect a decrease in tissue mass referable to the missing axons. It could also be caused in part by a decrease in the number of cortical or striatal cells as a result of decreased proliferation and/or increased cell death. To examine these possibilities, we analyzed cell proliferation by BrdU pulse labeling for 1 hr at E12, E14, and E15, during the peak of cortical and striatal neurogenesis (Bayer and Altmann, 1995), and also at E18 (Fig. 10A–D). In 8 μm sections, the number of BrdU-labeled cells in thefz3(−/−) forebrain ventricular zone is larger than that from the corresponding region from wild-type littermates by 0, 6, 12, and 18% at E12, E14, E15, and E18, respectively. These differences are likely to be within the limits of experimental variability of the BrdU method. Consistent with this interpretation, wild-type andfz3(−/−) brains show virtually identical densities of proliferating cells within the ventricular zone at E15 and E18 as determined by immunostaining for proliferating cell nuclear antigen.
Fig. 10.
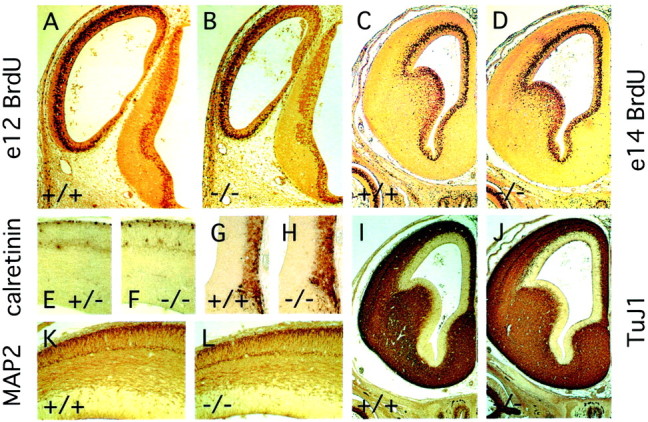
Normal proliferation and differentiation of cortical neurons in fz3(−/−) brains.A–D, One hour BrdU pulse labeling at E12 (A, B) or E14 (C,D) shows nearly identical numbers of proliferating cells in the ventricular zone of wild-type and fz3(−/−)forebrains. E, F, Calretinin immunoreactivity in the dorsal cortex at E15 reveals Cajal-Retzius cells at the marginal zone (top) together with a subset of neurons in the cortical plate. G, H, Calretinin immunoreactivity in the medial forebrain at E14 reveals a subset of neurons in the most superficial cortical layers (the lateral ventricle is at the top left; the midline is at theright). I, J, TuJ1 immunostaining at E14 shows widespread expression of brain-specific tubulin in postmitotic neurons. K, L, MAP2 immunostaining shows the differentiation and lamination of cortical neurons at E14.
Differentiation of cortical neurons was assessed by immunostaining for MAP2, calretinin, and neuron-specific tubulin (with mAb TuJ1). At E13 and E14, anti-MAP2 shows indistinguishable patterns of immunostaining in the wild-type and fz3(−/−) forebrain, revealing the early lamination of the cortex (Fig. 10K–L). At E14, uniform and intense TuJ1 immunostaining is seen in all regions outside of the ventricular zone (Fig.10I,J). In the developing rodent cortex, calretinin immunoreactivity marks Cajal-Retzius cells in the marginal zone and a subset of neurons in the subplate (Fonseca et al., 1995). At E14 and E15, wild-type and fz3(−/−)forebrains show indistinguishable patterns of calretinin immunoreactivity in both of these classes of cells, as well as in a broad band of cortical cells adjacent to the midline (Fig.10E–H). The indistinguishable patterns of calretinin staining in the marginal zone persist through at least E17. At E15 and E17, calretinin staining is also seen in axons in the wild type but not the fz3(−/−) cortex.
Migration and layering of cortical neurons were examined by pulse labeling with BrdU at E13 or E14, followed by histologic analysis at E18. As seen in Figure11A–H, wild-type andfz3(−/−) cortex and striata at E18 exhibit similar numbers and arrangements of BrdU-labeled cells. The principal differences between the two appear to be a relative thinning of the intermediate and subventricular zones of the fz3(−/−) cortex, as noted earlier, and a greater packing density of labeled cells in thefz3(−/−) striatum. In summary, cell proliferation and differentiation in the forebrain are remarkably normal infz3(−/−) mice during a time window when axonogenesis is severely disrupted.
Fig. 11.
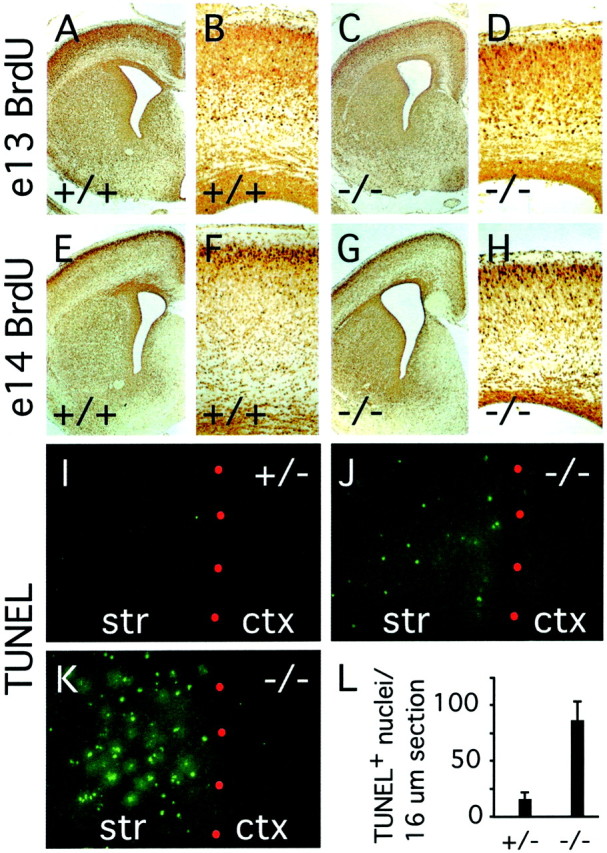
Cortical migration and striatal cell death infz3(−/−) brains at E18. A–H, BrdU labeling at E13 (A–D) or E14 (E–H); animals were killed at E18. The density of striatal neurons and the density and layering pattern of cortical neurons are similar between fz3(+/−) andfz3(−/−) brains. The fz3(−/−) cortex shows a thinning of the genu of the corpus callosum and the subventricular zone. I–L, TUNEL-labeled cells in 16 μm sections of striatum and cortex at E18. Red dots, The border between the striatum (str) and cortex (ctx). I, J, Typical appearance of fz3(−/−) and fz3(+/−)sections; K, a fz3(−/−) section with an unusually high density of TUNEL-positive cells. L, Mean and SDs of TUNEL-positive cells per 16 μm section of striatum at E18, counted from fz3(+/−) (n = 24 sections) or fz3(−/−) (n = 28 sections) brains.
Cell death in the developing brain was analyzed at E16 and E18 by quantitating the number of TUNEL-positive nuclei (Gavrieli et al., 1992). Although minimal labeling was observed in wild-type andfz3(−/−) brains at E16, at E18 fz3(−/−)brains showed a dramatic increase in the number of TUNEL-positive nuclei in the striatum (Fig. 11I–L). In contrast, TUNEL-positive cells were exceedingly rare in other brain regions, including the cortex. It will be of interest to determine whether the absence of striatal afferants or efferants secondarily triggers the death of striatal neurons or whether their death results from some other fz3-dependent process.
If the absence of major fiber tracts in vivo reflects an intrinsic defect in the production, growth, or maintenance of axons, such a defect might be manifest by individual dissociated cells in culture. To test this possibility, we harvested and dissociated cells from either the cortex alone or from the striatum and pyriform cortex at E18 and examined their survival and morphology over the ensuing 25 d in culture. Both wild-type and fz3(−/−) cultures show similar high levels of cell viability and similar ratios of neurons to glia (identified by immunostaining for MAP2 and GFAP, respectively) (Fig.12A–D,K–N). Moreover, no differences between wild-type and fz3(−/−)cultures were observed in glial or neuronal morphologies or in the density of either dendrites or axons, as determined by immunostaining for MAP2 or neurofilaments (Fig.12E,F), and by transfecting green fluorescent protein (GFP) cDNA and subsequent visualization of isolated GFP-expressing cells by immunostaining (data not shown). Synapse formation in vitro also appears to be unaffected by loss of fz3 as assessed by immunostaining for synaptophysin (Fig. 12G,H) or bassoon, a presynaptic cytoplasmic matrix protein found at both excitatory and inhibitory synapses (Fig.12I,J) (Dieck et al., 1998). Thus, the axon tract defects in the fz3(−/−) brain appear to reflect a defect in the interaction between axons and their microenvironment in vivo rather than an intrinsic defect in axonal outgrowth per se.
Fig. 12.
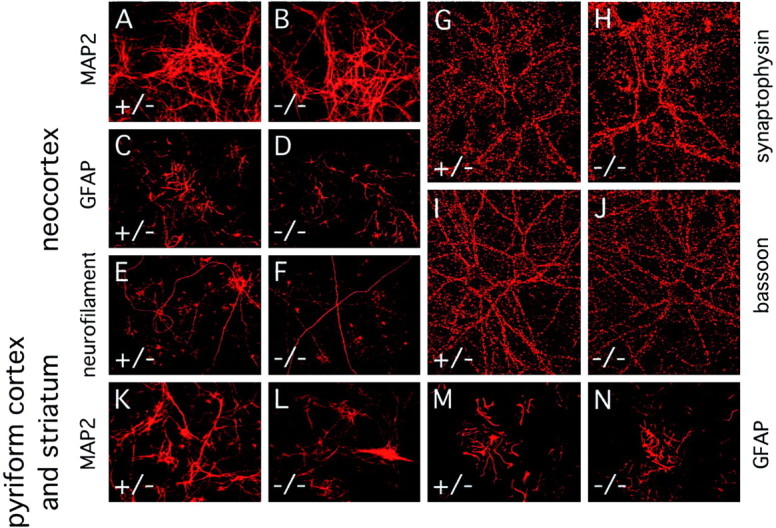
Normal numbers and morphologies of dissociated neurons and glia cultured from E18 neocortex or combined E18 striatum and pyriform cortex. Cultures were prepared from pooledfz3(+/−) and fz3(+/+) brains (designated “+/−”) or from pooled fz3(−/−)brains. A–J, Neocortex; K–N, combined striatum and pyriform cortex. Immunostaining was performed after 3–25 d in culture. A, B, K,L, Dendrites visualized by anti-MAP2 immunostaining.C, D, M, N, Glia visualized by anti-GFAP immunostaining. E,F, Axons visualized with anti-neurofilament immunostaining. G–J, Synapses visualized with anti-synaptophysin (G, H) or anti-bassoon (I, J) antibodies.
DISCUSSION
In this study, we show that targeted deletion of the mousefz3 gene leads to severe defects in several major axon tracts within the forebrain. In particular, fz3(−/−) mice show a complete loss of the thalamocortical, corticothalamic, and nigrostriatal tracts and of the anterior commissure, and they show a variable loss of the corpus callosum. Axonal defects are apparent beginning at E13. In contrast, peripheral nerve fibers and major axon tracts in the more caudal regions of the CNS are mostly or completely unaffected. Cell proliferation in the ventricular zone and cell migration to the developing cortex proceed normally. Extensive cell death in the fz3(−/−) striatum occurs late in gestation, perhaps secondary to the nearly complete absence of long-range connections. Curiously, the cortex exhibits little or no cell death, as determined by histochemical detection of DNA fragmentation, despite a nearly complete absence of connections to subcortical structures. Thefz3(−/−) axonal phenotype is in marked contrast to the phenotypes reported for mutations that affect axon guidance, in which cases axonal outgrowth is readily observed, but the axons follow erroneous trajectories (Bagri et al., 2002). As discussed more fully below, it is also in marked contrast to the phenotypes described thus far for Wnt mutants.
Models for Fz3 function
How might the absence of Fz3 cause defects in axon growth? One clue comes from the observation that fz3(−/−) neurons grown in culture exhibit grossly normal axonal and dendritic growth and synaptogenesis. This observation suggests that the axonal growth defects in the fz3(−/−) brain may involve inhibitory interactions within the developing CNS that are relieved or bypassed when neurons are grown at relatively low density in culture. For example, axonal growth in the forebrain might require Fz3 to overcome inhibitory or adhesive interactions with ECM components. If such a mechanism were to apply generally within the CNS, then we would presume that in more caudal regions, it is mediated by other Fz family members either alone or redundantly with Fz3.
Alternatively, Fz3 may function within developing neurons to coordinate cell and cytoskeletal polarity in a manner analogous to the action ofDrosophila Fz in tissue polarity. In the developing wing epithelium, Fz function is required to determine the location from which each cell's single cuticular hair will emerge (Adler and Lee, 2001). In the wild type, the hair emerges from the tip of the cell that is furthest from the thorax, and it grows away from the thorax (Wong and Adler, 1993). The initial outgrowth of axons within the CNS presents a conceptually similar, albeit vastly more complex, cell biological challenge. If Fz3 functions at this point in neuronal development, then the data presented here would imply that a failure to correctly polarize the budding axon leads to abortive axonal outgrowth.
In the developing Drosophila wing epithelium, Fz appears to localize within each cell to the point from which the single hair emerges (Strutt, 2001). [We note that this observation was made with an overexpressed Fz-GFP fusion protein; the results are presumed to hold for the endogenous Fz protein, the low abundance of which has thus far hindered its immunolocalization (Park et al., 1994).] This localization, together with genetic evidence that rac andrho act downstream of fz in the tissue polarity pathway (Strutt et al., 1997; Winter et al., 2001), suggest that in this developmental context, Fz acts locally to organize the cytoskeleton. Therefore, determining the subcellular localization of Fz3 within developing neurons and, in particular, determining whether it is uniformly distributed over the plasma membrane or clustered within a subdomain, although technically challenging given the low abundance of the Fz3 protein, might provide a significant clue to its action in the CNS.
The best characterized role for Wnts in the CNS, and, by inference, a role for Fzs, is in cell proliferation. Loss of Wnt3a or LEF/TCF function produces a massive failure to generate cells within the developing hippocampus (Galceran et al., 2000; Lee et al., 2000). The partial or complete absence of the cerebellum seen inWnt1(−/−) mice (McMahon and Bradley, 1990; Thomas and Capecchi, 1990) may represent an analogous failure of cell proliferation within the midbrain. In support of this general idea, Wnt1/Wnt3a double mutant embryos show a defect in cell proliferation among dorsal neural crest precursors (Ikeya et al., 1997), and ectopic expression of Wnt-1 in the developing spinal cord causes a local increase in cell proliferation (Dickinson et al., 1994). In contrast, the normal or nearly normal pattern of cell proliferation seen in the fz3(−/−) CNS argues against mechanisms common to those implicated for the Lef-1, Wnt-1, andWnt-3a mutants. Similarly, the late demise of postmitotic cerebellar granule and Purkinje cells reported in fz4(−/−)mice does not fit into the paradigm of a cell proliferation defect (Wang et al., 2001).
As the above discussion indicates, it is unclear which one or more of the three characterized Fz signaling pathways is used by Fz3. Indeed, this remains an open question for all of the mammalian Fz proteins. As noted in the introductory remarks, the precedent from studies ofDrosophila Fz action in the embryo and wing would suggest that the answer is likely to be specific to the biological context.
Diversity and redundancy in Fz3 function
Studies in Xenopus using expression of wild-type and dominant negative Xfz3 mutants suggest that, in theXenopus embryo, XFz3 functions in the specification of the developing eye fields and, in conjunction with XWnt1, in the specification of neural crest (Shi et al., 1998; Deardorff et al., 2001; Rasmussen et al., 2001). The absence of effects on either of these processes in fz3(−/−) mice suggests that the phenotypes we report here may represent only a small fraction of the functions normally mediated by Fz3, the others being covered in the mouse by the redundant action of one or more of the remaining eight Fz proteins encoded in the genome. This level of redundancy among mammalian Fzs is certainly plausible given the nearly complete redundancy shown by Drosophila Fz and Fz2 in mediating a variety of Wingless actions in embryonic development (Bhat, 1998;Kennerdell and Carthew, 1998; Bhanot et al., 1999; Chen and Struhl, 1999). Thus, one challenge for the future will be to dissect the multiple layers of redundancy to fully define the role of each Fz in CNS development.
Footnotes
This work was supported by the Howard Hughes Medical Institute (Y.W., P.M.S., J.N.) and the Medical Scientist Training Program (N.T.). We thank the following: Drs. Pradeep Bhide, Anirvan Ghosh, Lori Redmond, Hengye Man, and Lin Ding for advice; the Johns Hopkins Transgenic Core for blastocyst injections; Carol Cooke for assistance with electron microscopy; and Drs. Anirvan Ghosh, Alex Kolodkin, Tudor Badea, and Hui Sun for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Jeremy Nathans, 805 PCTB, 725 North Wolfe Street, Johns Hopkins University School of Medicine, Baltimore, MD 21205. E-mail: jnathans@jhmi.edu.
REFERENCES
- 1.Adler PN, Lee H. Frizzled signaling and cell-cell interactions in planar polarity. Curr Opin Cell Biol. 2001;13:635–640. doi: 10.1016/s0955-0674(00)00263-5. [DOI] [PubMed] [Google Scholar]
- 2.Auladell C, Perez-Sust P, Super H, Soriano E. The development of thalamocortical and corticothalamic projections in the mouse. Anat Embryol. 2000;201:169–179. doi: 10.1007/pl00008238. [DOI] [PubMed] [Google Scholar]
- 3.Bagri A, Marin O, Plump AS, Mak J, Pleasure SJ, Rubenstein JL, Tessier-Lavigne M. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 2002;33:233–248. doi: 10.1016/s0896-6273(02)00561-5. [DOI] [PubMed] [Google Scholar]
- 4.Bancroft JD, Stevens A. Theory and practice of histological techniques, pp 396–399. Churchill Livingstone; New York: 1982. [Google Scholar]
- 5.Bayer SA, Altmann J. Neurogenesis and neuronal migration. In: Paxinos G, editor. The rat nervous system. Academic; New York: 1995. pp. 1041–1078. [Google Scholar]
- 6.Bhanot P, Brink M, Harryman Samos C, Hsieh J-C, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382:225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- 7.Bhanot P, Fish M, Jemison J, Nusse R, Nathans J, Cadigan KM. Frizzled and frizzled-2 function as redundant receptors for Wingless during Drosophila embryonic development. Development. 1999;126:4175–4186. doi: 10.1242/dev.126.18.4175. [DOI] [PubMed] [Google Scholar]
- 8.Bhat KM. Frizzled and frizzled2 play a partially redundant role in wingless signaling and have similar requirements to wingless in neurogenesis. Cell. 1998;95:1027–1036. doi: 10.1016/s0092-8674(00)81726-2. [DOI] [PubMed] [Google Scholar]
- 9.Borello U, Buffa V, Sonnino C, Melchionna R, Vivarelli E, Cossu G. Differential expression of the Wnt putative receptors Frizzled during mouse somitogenesis. Mech Dev. 1999;89:173–177. doi: 10.1016/s0925-4773(99)00205-1. [DOI] [PubMed] [Google Scholar]
- 10.Chen C, Struhl G. Wingless transduction by the Frizzled and Frizzled2 proteins of Drosophila. Development. 1999;126:5441–5452. doi: 10.1242/dev.126.23.5441. [DOI] [PubMed] [Google Scholar]
- 11.Dann CE, Hsieh JC, Rattner A, Sharma D, Nathans J, Leahy DJ. Insights into Wnt binding and signaling from the structures of two Frizzled cysteine-rich domains. Nature. 2001;412:86–90. doi: 10.1038/35083601. [DOI] [PubMed] [Google Scholar]
- 12.Deardorff MA, Tan C, Saint-Jeannet JP, Klein PS. A role for frizzled 3 in neural crest development. Development. 2001;128:3655–3663. doi: 10.1242/dev.128.19.3655. [DOI] [PubMed] [Google Scholar]
- 13.Dickinson ME, Krumlauf R, McMahon AP. Evidence for a mitogenic effect of Wnt-1 in the developing central nervous system. Development. 1994;120:1453–1471. doi: 10.1242/dev.120.6.1453. [DOI] [PubMed] [Google Scholar]
- 14.Dieck S, Sanmarti-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kampf U, Franzer JT, Stumm M, Garner CC, Gundelfinger ED. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol. 1998;142:499–509. doi: 10.1083/jcb.142.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fonseca M, Del Rio JA, Martinez A, Gomez S, Soriano E. Development of calretinin immunoreactivity in the neocortex of the rat. J Comp Neurol. 1995;361:177–192. doi: 10.1002/cne.903610114. [DOI] [PubMed] [Google Scholar]
- 16.Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R. Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development. 2000;3:469–482. doi: 10.1242/dev.127.3.469. [DOI] [PubMed] [Google Scholar]
- 17.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grove EA, Tole S, Limon J, Yip Y, Ragsdale CW. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development. 1998;125:2315–2325. doi: 10.1242/dev.125.12.2315. [DOI] [PubMed] [Google Scholar]
- 19.Gubb D, Garcia-Bellido A. A genetic analysis of the determination of cuticular polarity during development in Drosophila melanogaster. J Embryol Exp Morphol. 1982;68:37–57. [PubMed] [Google Scholar]
- 20.Habas R, Kato Y, He X. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell. 2001;107:843–854. doi: 10.1016/s0092-8674(01)00614-6. [DOI] [PubMed] [Google Scholar]
- 21.Hall AC, Lucas FR, Salinas PC. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell. 2000;100:525–535. doi: 10.1016/s0092-8674(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 22.He X, Saint-Jeannet J-P, Wang Y, Nathans J, Dawid I, Varmus R. A member of the Frizzled protein family mediating axis induction by Wnt-5A. Science. 1997;275:1652–1654. doi: 10.1126/science.275.5306.1652. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh JC, Rattner A, Smallwood PM, Nathans J. Biochemical characterization of Wnt-frizzled interactions using a soluble, biologically active vertebrate Wnt protein. Proc Natl Acad Sci USA. 1999;96:3546–3551. doi: 10.1073/pnas.96.7.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeya M, Lee SMK, Johnson JE, McMahon AP, Takada S. Wnt signaling required for expansion of neural crest and CNS progenitors. Nature. 1997;389:966–970. doi: 10.1038/40146. [DOI] [PubMed] [Google Scholar]
- 25.Karnovsky MJ, Root L. A “direct coloring” thiocholine method for choline esterases. J Histochem Cytochem. 1964;12:219–221. doi: 10.1177/12.3.219. [DOI] [PubMed] [Google Scholar]
- 26.Kennerdell JR, Carthew RW. Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell. 1998;95:1017–1026. doi: 10.1016/s0092-8674(00)81725-0. [DOI] [PubMed] [Google Scholar]
- 27.Kiecker C, Niehrs C (2001) A morphogen gradient of Wnt/beta-catenin signalling regulates anteroposterior neural patterning in Xenopus Development. 128:4189–4201. [DOI] [PubMed]
- 28.Kuhl M, Sheldahl LC, Park M, Miller JR, Moon RT. The Wnt/Ca2+ pathway: a new vertebrate Wnt signaling pathway takes shape. Trends Genet. 2000;16:279–283. doi: 10.1016/s0168-9525(00)02028-x. [DOI] [PubMed] [Google Scholar]
- 29.Lee MK, Tuttle JB, Rebhun LI, Cleveland DW, Frankfurter A. The expression and posttranslational modification of a neuron-specific beta-tubulin isotype during chick embryogenesis. Cell Motil Cytoskelet. 1990;17:118–132. doi: 10.1002/cm.970170207. [DOI] [PubMed] [Google Scholar]
- 30.Lee SMK, Tole S, Grove E, McMahon AP. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development. 2000;127:457–467. doi: 10.1242/dev.127.3.457. [DOI] [PubMed] [Google Scholar]
- 31.Lucas FR, Salinas PC. WNT-7a induces axonal remodeling and increases synapsin I levels in cerebellar neurons. Dev Biol. 1997;192:31–44. doi: 10.1006/dbio.1997.8734. [DOI] [PubMed] [Google Scholar]
- 32.McMahon AP, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- 33.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 34.Muroyama Y, Fujihara M, Ikeya M, Kondoh H, Takada S. Wnt signaling plays an essential role in neuronal specification of the dorsal spinal cord. Genes Dev. 2002;16:548–553. doi: 10.1101/gad.937102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park WJ, Liu J, Adler PN. The frizzled gene of Drosophila encodes a membrane protein with an odd number of transmembrane domains. Mech Dev. 1994;45:127–137. doi: 10.1016/0925-4773(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 36.Peeters MC, Schutte B, Lenders MH, Hekking JW, Drukker J, Van Straaten HW. Role of differential cell proliferation in the tail bud in aberrant mouse neurulation. Dev Dyn. 1998;211:382–389. doi: 10.1002/(SICI)1097-0177(199804)211:4<382::AID-AJA9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 37.Peschon JJ, Behringer RR, Brinster RL, Palmiter RD. Spermatid-specific expression of protamine 1 in transgenic mice. Proc Natl Acad Sci USA. 1987;84:5316–5319. doi: 10.1073/pnas.84.15.5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasmussen JT, Deardorff MA, Tan C, Rao MS, Klein PS, Vetter ML. Regulation of eye development by frizzled signaling in Xenopus. Proc Natl Acad Sci USA. 2001;98:3861–3866. doi: 10.1073/pnas.071586298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter DW, Spyer KM. Studying rhythmogenesis of breathing: comparison of in vivo and in vitro models. Trends Neurosci. 2001;24:464–472. doi: 10.1016/s0166-2236(00)01867-1. [DOI] [PubMed] [Google Scholar]
- 40.Sheth AN, McKee ML, Bhide PG. The sequence of formation and development of corticostriate connections in mice. Dev Neurosci. 1998;20:98–112. doi: 10.1159/000017306. [DOI] [PubMed] [Google Scholar]
- 41.Shi DL, Goisset C, Boucaut JC. Expression of Xfz3, a Xenopus frizzled family member, is restricted to the early nervous system. Mech Dev. 1998;70:35–47. doi: 10.1016/s0925-4773(97)00166-4. [DOI] [PubMed] [Google Scholar]
- 42.Strutt DI. Asymmetric localization of frizzled and the establishment of cell polarity in the Drosophila wing. Mol Cell. 2001;7:367–375. doi: 10.1016/s1097-2765(01)00184-8. [DOI] [PubMed] [Google Scholar]
- 43.Strutt DI, Weber U, Mlodzik M. The role of RhoA in tissue polarity and Frizzled signaling. Nature. 1997;387:292–295. doi: 10.1038/387292a0. [DOI] [PubMed] [Google Scholar]
- 44.Thomas KR, Capecchi MR. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature. 1990;346:847–850. doi: 10.1038/346847a0. [DOI] [PubMed] [Google Scholar]
- 45.van de Wetering M, Cavallo R, Dooijes D, van Beest M, van Es J, Loureiro J, Ypma A, Hursh D, Jones T, Bejsovec A, Peifer M, Mortin M, Clevers H. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 1997;88:789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]
- 46.Vinson CR, Conover S, Adler PN. A Drosophila tissue polarity locus encodes a protein containing seven potential transmembrane domains. Nature. 1989;338:263–264. doi: 10.1038/338263a0. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Macke JP, Abella BS, Andreasson K, Worley P, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. A large family of putative transmembrane receptors homologous to the product of the Drosophila tissue polarity gene frizzled. J Biol Chem. 1996;271:4468–4476. doi: 10.1074/jbc.271.8.4468. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Huso D, Cahill H, Ryugo D, Nathans J. Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J Neurosci. 2001;21:4761–4771. doi: 10.1523/JNEUROSCI.21-13-04761.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilson SI, Rydstrom A, Trimborn T, Willert K, Nusse R, Jessell TM, Edlund T. The status of Wnt signaling regulates neural and epidermal fates in the chick embryo. Nature. 2001;411:325–330. doi: 10.1038/35077115. [DOI] [PubMed] [Google Scholar]
- 50.Winter CG, Wang B, Ballew A, Royou A, Karess R, Axelrod JD, Luo L. Drosophila Rho-associated kinase (Drok) links Frizzled-mediated planar cell polarity signaling to the actin cytoskeleton. Cell. 2001;105:81–91. doi: 10.1016/s0092-8674(01)00298-7. [DOI] [PubMed] [Google Scholar]
- 51.Wong LL, Adler PN. Tissue polarity genes of Drosophila regulate the subcellular location for prehair initiation in pupal wing cells. J Cell Biol. 1993;123:209–221. doi: 10.1083/jcb.123.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang-Snyder J, Miller JR, Brown JD, Lai CJ, Moon RT. A frizzled homolog functions in a vertebrate Wnt signaling pathway. Curr Biol. 1996;6:1302–1306. doi: 10.1016/s0960-9822(02)70716-1. [DOI] [PubMed] [Google Scholar]