

Abstract
Synaptotagmin 1, a Ca2+ sensor for fast synaptic vesicle exocytosis, contains two C2 domains that form Ca2+-dependent complexes with phospholipids. To examine the functional importance of Ca2+ binding to the C2A domain of synaptotagmin 1, we studied two C2A domain mutations, D232N and D238N, using recombinant proteins and knock-in mice. Both mutations severely decreased intrinsic Ca2+ binding and Ca2+-dependent phospholipid binding by the isolated C2A domain. Both mutations, however, did not alter the apparent Ca2+affinity of the double C2 domain fragment, although both decreased the tightness of the Ca2+/phospholipid/double C2 domain complex. When introduced into the endogenous synaptotagmin 1 gene in mice, the D232N and D238N mutations had no apparent effect on morbidity and mortality and caused no detectable alteration in the Ca2+-dependent properties of synaptotagmin 1. Electrophysiological recordings of cultured hippocampal neurons from knock-in mice revealed that neither mutation induced major changes in synaptic transmission. The D232N mutation, however, caused increased synaptic depression during repetitive stimulation, whereas the D238N mutation did not exhibit this phenotype. Our data indicate that Ca2+ binding to the C2A domain of synaptotagmin 1 may be important but not essential, consistent with the finding that the two C2 domains cooperate and may be partially redundant in Ca2+-dependent phospholipid binding. Moreover, although the apparent Ca2+ affinity of the synaptotagmin 1/phospholipid complex is critical, the tightness of the Ca2+/phospholipid complex is not. Our data also demonstrate that subtle changes in the biochemical properties of synaptotagmin 1 can result in significant alterations in synaptic responses.
Keywords: synaptotagmin, neurotransmitter release, exocytosis, C2 domain, Ca2+-binding site, synaptic plasticity
When an action potential invades a nerve terminal, voltage-gated Ca2+channels open, and Ca2+ triggers fast neurotransmitter release with an overall probability of 0.05–1.00 (Hessler et al., 1993; Rosenmund et al., 1993; Xu-Friedman et al., 2001). Because of the steep Ca2+sensitivity of release (Bollmann et al., 2000; Schneggenburger and Neher, 2000), the final step in triggering release requires cooperative interaction of multiple Ca2+ ions with its receptor(s). Although several Ca2+ sensors are likely involved in neurotransmitter release, the synaptic vesicle protein synaptotagmin 1 probably is the primary sensor for synaptic vesicle exocytosis. This conclusion is based on the fact that synaptotagmin 1 binds multiple Ca2+ ions (Ubach et al., 1998; Fernández et al., 2001) and is essential for fast, Ca2+-triggered exocytosis in central synapses (Geppert et al., 1994) in a manner whereby its apparent Ca2+ affinity dictates the Ca2+ responsiveness of release (Fernández-Chacón et al., 2001).
Synaptotagmin 1 is the prototype of a large family of proteins, many of which are probably Ca2+ sensors for exocytosis (for review, see Südhof, 2002). All synaptotagmins are composed of an N-terminal transmembrane region, a central linker sequence, and two C-terminal C2 domains. The two C2 domains, C2A and C2B, bind multiple Ca2+ ions and form Ca2+-dependent phospholipid complexes (Davletov and Südhof, 1993; Chapman and Jahn, 1994; Ubach et al., 1998; Fernández et al., 2001). In addition, the synaptotagmin C2 domains bind to several proteins in vitro as a function of Ca2+, most prominently the soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) syntaxin 1 (Chapman et al., 1995; Li et al., 1995; Kee and Scheller, 1996). Because the C2 domains account for the majority of the synaptotagmin 1 sequence (Perin et al., 1990), synaptotagmin 1 can be considered a membrane-tethered Ca2+-binding machine.
C2 domains are composed of stable eight-stranded β-sandwiches with flexible loops emerging from the top and bottom (Sutton et al., 1995). Ca2+ binds exclusively to the top loops at sites that are formed by residues that are widely separated in the primary sequence and often coordinate multiple Ca2+ ions. As a result, C2 domains bind Ca2+ions in a cluster with a low intrinsic Ca2+ affinity (Ubach et al., 1998;Fernández et al., 2001). This affinity increases dramatically in the presence of phospholipid membranes that potentiate the apparent Ca2+ affinity of C2domains up to 1000-fold (Zhang et al., 1998). Mutations in the top loops of C2 domains differentially alter their intrinsic and apparent Ca2+ affinities. For example, a point mutation in the C2A domain of synaptotagmin 1, R233Q, increases its intrinsic Ca2+ affinity but decreases its apparent Ca2+ affinity (Fernández-Chacón et al., 2001). When introduced into the endogenous mouse synaptotagmin 1 gene by homologous recombination, this mutation decreased by approximately twofold the apparent Ca2+ affinity of the double C2 domain fragment of native synaptotagmin 1 for phospholipids but not for SNARE complexes. Because the R233Q mutation lowered the Ca2+ sensitivity of neurotransmitter release twofold in the mutant mice, these results suggested that the Ca2+-dependent synaptotagmin/phospholipid complex is the driving force behind the Ca2+ triggering of neurotransmitter release (Fernández-Chacón et al., 2001). However, it is unclear why synaptotagmins contain two independently folded C2-domains with similar Ca2+-dependent properties. In the present study, we have tested the effects of mutations in the Ca2+-binding sites of the C2A domain of synaptotagmin 1 using a combination of biophysical, biochemical, genetic, and electrophysiological approaches.
MATERIALS AND METHODS
Nuclear magnetic resonance spectroscopy measurements of intrinsic Ca2+ binding by wild-type and mutant C2A domains.1H-15N heteronuclear single quantum coherence (HSQC) spectra were acquired with 1 hr total acquisition time at 25°C in a Varian INOVA500 nuclear magnetic resonance (NMR) spectrometer (spectral widths = 7600 and 1163 Hz in the 1H and 15N dimensions, respectively) using uniformly 15N-labeled C2A domains (Zhang et al., 1994). The Ca2+ titrations were recorded at 150–160 μm protein concentrations with successive additions of 0, 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.375, 0.45, 0.525, 0.6, 0.675, 0.75, 1, 1.2, 2, 5, 10, 20, 30, and 40 mmCa2+. The Ca2+ affinities of the different sites were calculated by curve-fitting the Ca2+dependence of selected chemical shifts to standard protein-ligand equilibrium equations using Sigma Plot (Fernández-Chacón et al., 2001).
Ca2+-dependent phospholipid binding studies of wild-type (WT) and mutant recombinant C2A domains were performed with two assays. (1) Glutathione S-transferase (GST)-pulldown assays using 3H-labeled liposomes containing 30% phosphatidyl serine(PS)/70% phosphatidyl choline (PC) and immobilized GST-fusion proteins were performed essentially as described (Fernández-Chacón et al., 2001;Gerber et al., 2001). Data obtained with identical amounts of recombinant proteins, liposomes, and glutathione beads were evaluated in terms of both Ca2+ affinity and amount of binding (see Fig. 2A,B). Results from multiple experiments are summarized in Table 2. (2) Liposome centrifugation assays using soluble C2 domain GST-fusion proteins eluted from glutathione columns after purification and heavy liposomes were performed essentially as described by Sugita et al. (2002). Briefly, dried lipids (PS/PC = 25:75, w/w) were suspended in HEPES buffer (50 mm HEPES, 100 mm NaCl, 4 mm EGTA, pH 6.8) containing 0.5 m sucrose, vortexed, and sonicated. Heavy liposomes were isolated by centrifugation (100,000 × g for 30 min) after adding 4 vol of HEPES buffer without sucrose. The heavy liposomes were then washed with HEPES buffer, precipitated by centrifugation (13,000 rpm for 10 min), and resuspended in HEPES buffer. Recombinant GST-synaptotagmin 1 C2 domains (6 μg protein) were incubated with 100 μg of liposomes with various concentrations of free Ca2+ clamped with Ca2+/EGTA buffers and centrifuged (13,000 rpm for 10 min). The pellets were washed with the corresponding Ca2+/EGTA buffer, treated with chloroform/methanol (1:2, v/v), and centrifuged again (13,000 rpm for 15 min). The protein precipitate was then analyzed by SDS-PAGE and Coomassie Blue staining.
Fig. 2.

Apparent Ca2+-binding affinities of the wild-type (WT) and mutant C2A domains of synaptotagmin 1 measured by Ca2+-dependent GST pulldowns of radiolabeled liposomes. A, Ca2+ titrations of phospholipid binding with equal amounts of recombinant C2A domains (25 μg protein) and radiolabeled liposomes. Note that both mutations (D232N and D238N) inactivate >80% of Ca2+-dependent phospholipid binding, but that the remaining binding is still Ca2+ dependent.B, Same data as in B but normalized for maximal binding to illustrate the shift in apparent Ca2+ affinities of the D232N mutant. Note that because of the low signal-to-noise, in particular for the D238N mutant C2A domain, which exhibits almost no Ca2+-dependent phospholipid binding, the curve fits are rather inaccurate in single experiments as shown here but can be averaged from multiple experiments as summarized in Table 2.
Table 2.
Apparent Ca2+-binding affinities of wild-type and mutant C2A domains measured as Ca2+/phospholipid complexes
| C2A domain | EC50 | Hillcoeff | Absolute binding | n |
|---|---|---|---|---|
| WT | 14 ± 0.2 μm | 4.0 ± 0.2 | 33863 ± 227 dpm | 3 |
| D232N | 5 ± 1.0 μm | 1.5 ± 0.1 | 5461 ± 147 dpm | 3 |
| D238N | 17 ± 1.3 μm | 1.6 ± 0.2 | 3411 ± 115 dpm | 3 |
The apparent Ca2+ affinities of the indicated purified C2A domains were measured as described in Figure 2using pulldowns of radiolabeled phospholipid vesicles composed of 30% phosphatidylserine/70% phosphatidylcholine with immobilized GST-fusion proteins containing the C2A domains. Data shown are mean ± SEMs from the number of experiments indicated (n) performed in triplicate as described in Materials and Methods. Each reaction contained ∼25 μg of protein and 4.5 μg of phospholipids. Binding constants and Hill coefficients were calculated using GraphPad software and are not accurate for the D232N and D238N mutants because of the decrease in absolute binding. “Absolute binding” gives the amount of phospholipid vesicles, expressed as counts of radiolabeled phospholipids, that are pulled down at saturating Ca2+ concentrations.
Ca2+-dependent phospholipid and syntaxin binding by native synaptotagmins was examined essentially as described (Fernández-Chacón et al., 2001), and bound proteins were analyzed by SDS-PAGE and immunoblotting.
Generation of knock-in mutant mice by homologous recombination. Mice carrying the D232N or the D238N point mutations in the endogenous synaptotagmin 1 gene were generated essentially as described previously for the R233Q and the K236Q knock-in mutants (Fernández-Chacón et al., 2001). Mice were genotyped by PCR as described (Fernández-Chacón et al., 2001), and PCR fragments were digested with specific restriction enzymes to detect diagnostic restriction sites that were inserted in conjunction with the point mutations (BstBI for D232N andEcoRI for D238N). As in our previous study on the R233Q and K236Q mutants, all analyses were performed on the offspring of matings between compound heterozygotes carrying one allele of the D232N or D238N knock-in, and a second allele of the control knock-in that includes the same neomycin gene in the intron but lacks the mutation in an exon. This procedure ensured that all analysis was performed on precisely matched controls.
Cell culture and electrophysiology. Microislands of astrocyte feeder cells were prepared 4–5 d before hippocampal neurons were plated. Briefly, islands of substrate (0.1 gm/l poly-d-lysine; 1 gm/l rat tail collagen in 1 mm acetic acid) were applied to round glass coverslips using a stamp containing regularly spaced squares (200 × 200 μm). Type 1 astrocytes (5000/cm2) were preplated in DMEM containing 10% fetal calf serum. Once astrocytes reached confluency, 5-fluoro-2′deoxyuridine (10 μm) was added to inhibit further proliferation. Newborn animals were decapitated according to the rules of the state and animal welfare committee. Brains were cleaned of meninges and vascular tissue, and then the hippocampi were removed in physiological salt solution. The tissue was enzymatically dissociated in papain (2 U/ml) in DMEM for 60 min at 37°C. Before plating the dissociated hippocampal neurons at a density of 500/cm2, the medium of the astrocyte feeder cells was replaced with serum-free medium (Neurobasal medium A supplemented with B27, Glutamax, and Pen/strep; Invitrogen). Neurons were allowed to mature for 14–21 d before they were used for experiments, and only islands containing single neurons were examined. Patch pipette solutions contained (in mm): 135 K-Gluconate, 10 HEPES, 1 EGTA, 4.6 MgCl2, 4 Na-ATP, 15 creatine phosphate, 50 U/ml phosphocreatine kinase (300 mOsm, pH 7.3). The extracellular saline solution contained (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 CaCl2, and 4 MgCl2 unless noted otherwise (305 mOsm, pH 7.3). All chemicals, except for MK-801 (Tocris, Bristol, UK), were purchased from Sigma. All solutions were applied using a fast flow system at room temperature (Pyott and Rosenmund, 2002).
Electrophysiology and statistics. Cells were whole-cell voltage clamped at −70 mV with either an Axopatch 200B amplifier (Axon Instruments) under control of the Clampex 8.0 (Axon Instruments) program or an EPC9 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany). Currents were low-pass filtered at 1 or 5 kHz and stored at either 10 or 20 kHz. The series resistance was compensated to 70–90%. Only cells with series resistances below 10 MΩ were analyzed. Statistical significance was tested using unpaired, nonparametric Wilcoxon tests. All values are presented as the mean ± SEM.
RESULTS
D232N and D238N mutations severely impair Ca2+binding to the C2A domain of synaptotagmin 1
The three Ca2+-binding sites of the synaptotagmin 1 C2A domain are formed by six amino acid side chains, five aspartate residues and one serine residue, located on two separate loops that extend from the top of the domain (Ubach et al., 1998). Intrinsic Ca2+binding by the C2A domain is of low affinity (>20 mm for the third Ca2+ion) and lacks cooperativity (Table 1). To perturb Ca2+ binding to the C2A domain without affecting its structure, we introduced point mutations into the C2A domain Ca2+ binding sites. These mutations, D232N and D238N, target key aspartate residues that coordinate multiple Ca2+ ions (Ubach et al., 1998) but are nevertheless conservative, and they preserve the overall structure of the domain (Shao et al., 1998; Contreras et al., 1999). We then analyzed purified recombinant wild-type and mutant C2A domains by NMR spectroscopy, using HSQC spectra in the presence of increasing Ca2+concentrations to monitor Ca2+ binding to the C2A domains. Our data revealed that the mutations severely altered the Ca2+-binding properties of the C2A domain (Fig. 1) (data not shown). Both mutations caused a large decrease in the intrinsic Ca2+ affinities of the second and third Ca2+-binding sites, and the D238N but not the D232N mutation also reduced the intrinsic affinity of the first Ca2+-binding site (Table 1).
Table 1.
Intrinsic Ca2+-binding affinities of wild-type and mutant C2A domains
| Ca2+-binding site | WT | D232N | D238N |
|---|---|---|---|
| Ca11-a | 54 | 53 | 136 |
| Ca21-b | 530 | >40,0001-c | >20,0001-c |
| Ca3 | >20,0001-c | >100,0001-d | >100,0001-d |
Ca2+ binding to the isolated wild-type (WT) C2A domain of synaptotagmin 1 and the D232N and D238N point mutants of the C2A domain was monitored by1H–15N HSQC acquired with 150–165 μm of purified recombinant C2A domains. Dissociation constants were calculated from the Ca2+titrations as described (Fernández-Chacón et al., 2001) and are expressed in micromolar Ca2+.
Calculated from the Ca2+ dependence of the 15N chemical shift of the K200 NH resonance between 0 and 600 μm total Ca2+.
Calculated from the Ca2+ dependence of the 1H chemical shift of the S235 NH resonance.
Only a lower limit can be estimated because the site was not saturated at the highest Ca2+ concentration used (40 mm).
Estimated based on the fact that no cross-peak movement corresponding to this site is observed up to 40 mm Ca2+.
Fig. 1.

Intrinsic Ca2+ binding to the wild-type (WT) and the D232N and D238N mutant C2A domains of synaptotagmin 1 monitored by NMR spectroscopy. Ca2+ titrations were examined by1H-15N HSQC spectra acquired with 150–165 μm of purified recombinant C2A domains.Panels illustrate the Ca2+-dependent shifts of selected cross-peaks corresponding to the D172 (top panels) and D230 (bottom panels) NH groups in all three C2A domains. The cross-peaks from these NH groups are shown in red, and other cross-peaks are shown in black. Numbers next to the resonances indicate the Ca2+ concentration (in millimolar) for that particular position of the cross-peak. Note the typical triphasic movement of the cross-peaks in the wild-type C2A domain, with the corresponding Ca2+-binding sites (Ca1,Ca2, and Ca3) indicated next to each phase. In the mutants, only biphasic movements with a different Ca2+ dependence are detectable. For quantitation of the various Ca2+-binding parameters, see Table1.
To determine the effect of the mutations on Ca2+-dependent phospholipid binding by the isolated C2A domain, we used two methods: pulldowns of radiolabeled liposomes with immobilized GST-fusion proteins of the C2A domains (Fig.2) and a liposome centrifugation assay (Fig. 3). The two assays gave fundamentally the same results. In the isolated C2A domain, both mutations severely depressed Ca2+-dependent phospholipid binding. Consistent with the decrease in intrinsic Ca2+ binding by the mutant domains (Table1), phospholipid binding was more impaired by the D238N than the D232N mutation (Table 2). Curiously, the small amount of remaining Ca2+-dependent phospholipid binding in the D232N mutant exhibited a higher apparent Ca2+ affinity than that of the wild-type C2A domain or of the D238N mutant C2A domain (Fig. 2, Table 2). Viewed together, these data demonstrate that in the isolated C2A domain, the D232N and D238N mutations severely impair Ca2+ binding and Ca2+-dependent phospholipid binding.
Fig. 3.
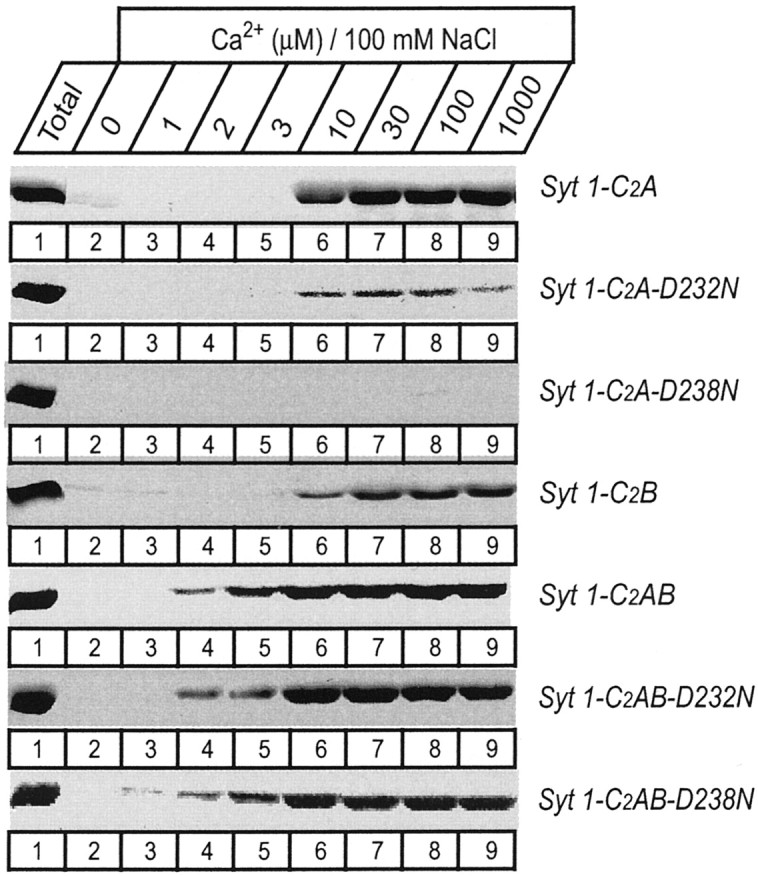
Apparent Ca2+ affinities of wild-type and mutant single and double C2 domain fragments from synaptotagmin 1 measured by Ca2+-dependent binding to liposomes. The isolated wild-type and D232N and D238N mutant C2A domain, the wild-type C2B domain, and the wild-type and mutant double C2 domain fragments were analyzed. Liposomes composed of 25% PS/75% PC were incubated with the indicated C2 domains (present as soluble purified GST-fusion proteins) at the Ca2+ concentrations shown on top (clamped with Ca2+/EGTA buffers) and centrifuged, and bound proteins were analyzed by SDS-PAGE and Coomassie blue staining. Data shown are from a single representative experiment repeated multiple times.
Generation of knock-in mice carrying D232N or D238N mutations
To test the functional consequences of the D232N and D238N mutations in vivo, we generated mice that carried these mutations separately (Fig.4A). These mice were obtained by the same approach used to make knock-in mice with the R233Q and K236Q mutations (Fernández-Chacón et al., 2001) and analyzed similarly by breeding to control mice. These control mice had gone through the same genetic manipulations as the mutant mice and also contained a neomycin resistance gene in an intron but lacked a mutation. The D232N and D238N mutant mice exhibited no outward abnormality. They bred normally and displayed no noticeable morbidity or premature mortality and no obvious behavioral defects. In both types of mutant mice, the apparent levels of synaptotagmin 1 and other proteins (vasolin-containing protein, Syntaxin 1A/B, Synapsins 1A, 1B, 2A, and 2B, SNAP-25, synaptobrevin 2, Munc18–1, synaptophysin 1, Rab3A, rabphilin, and synaptotagmin 7) were evaluated by immunoblotting with detection by enhanced chemiluminescence (Fig.4B) (data not shown). No major changes in protein levels were detected.
Fig. 4.

Strategy for generating knock-in mice with D232N and D238N mutations in the synaptotagmin 1 C2A domain.A, Design of knock-in vectors for homologous recombination. Similar to previous experiments (Fernández-Chacón et al., 2001), a genomic clone containing a single exon from the murine synaptotagmin 1 gene (top) was used to generate targeting vectors in which D232N or D238N mutations were introduced into the exon (middle), with two copies of the thymidine kinase (TK) gene for negative selection and a neomycin resistance cassette (neo) for positive selection. The mutant exons were then introduced into the endogenous synaptotagmin 1 gene by homologous recombination, which also introduces the neomycin resistance gene cassette into the intron (bottom). Numbered arrows identify oligonucleotides (1872 and1873) used for genotyping. The position of the outside probe for detection of homologous recombination by Southern blotting is indicated on the right. The location of selected restriction sites are shown (H, HindIII;B, BglII; E,EcoRI; N, NheI;C, ClaI; P,PstI), and the scale is given on theright. The mutant mice produced by homologous recombination were then crossed with control mice obtained in a previous study (Fernández-Chacón et al., 2001) in which the neomycin gene was introduced into the intron without any mutations in the coding region, and all analyses were performed on littermate offspring from matings between double heterozygous mice carrying one mutant allele (either D232N or D238N) and one control allele with the neomycin cassette but without a mutation. B, Immunoblots of total brain homogenates (30 μg of protein) from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) littermates stained with monoclonal antibodies specific for synaptotagmin 1 and rab3a.
Enhanced short-term depression in synaptotagmin 1 D232N mutant mice
To test the effects of the D232N and D238N mutations on synaptotagmin 1 function, we cultured hippocampal neurons from newborn knock-in mice and performed electrophysiological recordings in neurons that had formed autapses. In all experiments, neurons from mutant and wild-type littermates were cultured and analyzed simultaneously to control for culture-dependent variabilities in synaptic responses. EPSCs in response to action potentials induced at low frequency by brief somatic depolarizations (0 mV, 1–2 msec) appeared similarly robust for all mice tested (data not shown), consistent with a fundamentally normal release apparatus in the mutant mice. We next measured the short-term plasticity properties of wild-type and synaptotagmin 1 mutants by evoking trains of action potentials at 10 Hz (50 stimuli) and 50 Hz (5 stimuli). Although the D238N mutation had no major effect on the time course of EPSCs at both frequencies, the D232N mutation produced significantly faster depression of EPSC amplitudes at both frequencies (Fig. 5). This effect is opposite to that caused by the neighboring R233Q mutation that resulted in strong facilitation under the same conditions (Fernández-Chacón et al., 2001).
Fig. 5.

Effect of the D232N and D238N mutations on short-term synaptic plasticity analyzed in cultured hippocampal neurons. A, Average synaptic currents (EPSCs) recorded from “microisland” cultures in response to 10 Hz stimulation. EPSCs are normalized to the first response; top traces show a comparison of D232N mutant neurons with wild-type (WT) neurons, and bottom traces show a comparison of D238N mutant neurons with wild-type neurons. Stimulus artifacts areblanked. B, Plot of the average EPSC amplitudes normalized to the first response recorded during 10 Hz stimulation. The comparison of D232N mutant with wild-type responses is shown on the left, and the comparison of D238N mutant with wild-type responses is shown on the right.C and D are same as A andB but with 50 Hz stimulation.
In the R233Q mutant mice, the change in short-term plasticity was paralleled by a reduction in the basal Ca2+-triggered response, a right shift in the apparent Ca2+ sensitivity of evoked responses, and a decrease in the vesicular and synaptic release probabilities (Fernández-Chacón et al., 2001). We therefore tested whether the D232N mutation caused an increased release probability, because accelerated EPSC depression during trains of action potentials is indicative for this change in release efficacy. Release efficacy can be calculated by quantifying evoked responses and the size of the readily releasable pool (RRP) in the same cell. The average absolute EPSC amplitudes recorded from neurons from mutant and wild-type littermates were similar in all comparisons (D232N 3.6 ± 0.5 nA, n = 48 vs WT 3.7 ± 0.7 nA,n = 39; D238N 3.6 ± 0.6 nA, n = 41 vs WT 3.4 ± 0.8 nA, n = 31) (Fig.6). The size of the readily releasable vesicle pool can be quantified by integration of the transient inward current component of the response induced by a 4 sec application of 500 mOsm hypertonic sucrose solution applied to the entire cell (Rosenmund and Stevens, 1996). Similar to the absolute EPSC amplitudes, no significant changes were observed between mutants and wild-type controls in the readily releasable pool (D232N 598 ± 86 pC,n = 44 vs WT 551 ± 105 pC, n = 36) and D238N (D238N 655 ± 102 pC, n = 41 vs WT 857 ± 181 pC, n = 30).
Fig. 6.

Analysis of the vesicle pools, release probabilities, and Ca2+ sensitivities in D232N and D238N mutant neurons in comparison with wild-type (WT) neurons. A, Quantitative analysis of evoked responses and the sizes of the readily releasable vesicle pools of excitatory neurons of D232N and D238N excitatory neurons. Mean amplitudes and pool sizes were normalized to the mean values from the littermate wild-type neurons. B, The vesicular release probability was then calculated as the percentage of action potential evoked vesicles compared with the readily releasable pool. Data shown are means ± SEMs from the number of cells shown in parentheses in the bars. C, Evaluation of synaptic release probability from D232N and wild-type neurons. The rate of use-dependent block of NMDA-EPSCs in the presence of the irreversible open channel blocker MK-801 (5 μm) was not different between D232N and wild-type neurons, indicating that release probability was unchanged by the mutation. D, Ca2+ dependence of evoked release. EPSCs were evoked in 12 mm external Ca2+ and 1 mm Mg2+. Presynaptic Ca2+ influx was varied by adding various concentrations of Cd2+ (3–100 μm) to the external medium (n = 7–35 per concentration).Solid lines are best fits to a logistic hill function to determine the IC50 value for Cd2+.
Accordingly, because neither evoked response nor RRP was different between mutants and wild type, we expect that the probability of an individual fusion competent vesicle to exocytose after arrival of an action potential, the vesicular release probability (Pvr), is also unchanged. This probability can be reliably determined when both the charge of the synaptic responses and the charge of the RRP are recorded in the same neuron and can be computed by simply dividing EPSC charge by readily releasable pool charge. Indeed, we found that the averagePr was unchanged and was ∼6% for all groups tested (Fig. 6) and similar to other previously reported values for wild-type cells (Rosenmund and Stevens, 1996;Fernández-Chacón et al., 2001).
In the R233Q mutant mice, the reduced vesicular release probability was accompanied by a reduced synaptic release probability and a reduced apparent Ca2+ sensitivity of triggered release. A detailed analysis of the synaptic release probability and apparent Ca2+ sensitivity in the D232N mutant mice did not reveal any significant changes compared with wild types (Fig. 6C,D). Synaptic release probability can be reliably determined by the degree of NMDA–EPSC depression during synaptic stimulation in the presence of the use-dependent NMDA channel blocker MK-801 (Rosenmund et al., 1993). The stimulus-dependent decrease of the NMDA–EPSC amplitude was not significantly changed in the D232N mutant neurons compared with wild-type cells (Fig.6C). The apparent sensitivity of Ca2+-triggered release was tested by systematically blocking Ca2+ channel with Cd2+. We added different concentrations of Cd2+ (3–100 μm) to the external solution with constant concentrations of other external divalent cations (12 mmCa2+, 1 mmMg2+). In this routine, only the amount of Ca2+ influx is altered without changing axonal excitability. The inhibition of EPSC amplitude as a function of [Cd2+]0 was very similar in D232N mutants and wild types, with IC50 values for Cd2+of 25 and 27 μm, respectively (Fig.6D).
Effects of the D232N and D238N mutations on the double C2 domain fragment
The relatively mild phenotype caused by the D232N mutation and the absence of a phenotype in the D238N mice were initially surprising considering the severe effects of these mutations on intrinsic Ca2+ binding and on Ca2+-dependent phospholipid binding to the isolated C2A domain. To test whether the native synaptotagmin 1 proteins expressed in the knock-in mice also exhibit a severe impairment on phospholipid binding, we obtained native soluble C2A/C2B domain fragments by limited trypsin digestion of brain membranes from the mice and analyzed their ability to bind to phospholipids (Fig.7A). Because synaptotagmin 1 also binds to the SNARE protein syntaxin 1 in a Ca2+-dependent manner, we also tested the solubilized C2A/C2B domain fragments for syntaxin 1 binding (Fig. 7B). No changes in either phospholipid or syntaxin 1 binding were detected, as far as was discernable with the relatively limited assays available. Thus, in contrast to the isolated C2A domain, the D232N and D238N mutations do not cause a major impairment of the Ca2+-binding properties of the double C2 domain fragment as opposed to the R233Q mutation, which affected both the isolated C2A domain and the double C2 domain fragment (Fernández-Chacón et al., 2001).
Fig. 7.

The D232N and D238N mutations do not cause a significant change in Ca2+-dependent phosphopholipid and syntaxin binding by native synaptotagmin 1. A, Soluble C2A/C2B domain fragments of native synaptotagmin 1 were obtained from wild-type and mutant littermate mice by partial trypsin digestion of brain membranes followed by centrifugation. The soluble C2A/C2B domain fragment released into the supernatant was used for binding experiments with liposomes at the indicated free Ca2+ concentrations. Input and bound proteins were then analyzed by immunoblotting using a monoclonal synaptotagmin 1 antibody. The two mutations were analyzed in independent experiments with their littermate wild-type controls, resulting in separate wild-type controls for each mutant. Numbers on theleft indicate positions of size markers.B, GST-pulldown experiments of the soluble C2A/C2B domain fragment of synaptotagmin 1 isolated as described above. Proteins were bound to GST and GST-syntaxin (residues 180–264) at the indicated concentrations of free Ca2+ as described in A, and bound proteins were visualized by immunoblotting.
The different effects of the D232N and D238N mutations on the Ca2+-dependent phospholipid binding properties of the isolated C2A domain and the native C2A/C2B domain fragments obtained from the knock-in mice suggest that there is functional cooperation between the two C2 domains of synaptotagmin 1. Although initially this explanation seemed unlikely because the C2B domain was believed to be unable to bind phospholipids in a Ca2+-dependent manner (Schiavo et al., 1996; Bai et al., 2000), during the course of this study we found that the isolated C2B domain indeed exhibits Ca2+-dependent phospholipid binding when properly purified (Fernández et al., 2001). In addition, partial redundancy between the C2A and C2B domain in phospholipid binding to the double C2 domain region has been described recently (Earles et al., 2001), although these results should be interpreted with caution because the isolated C2B domain did not bind phospholipids in the assay used in this study. To further understand the interplay between the synaptotagmin 1 C2 domains in phospholipid binding, we used recombinant proteins spanning the double C2 domain region. Similar to the C2B domain, the double C2domain fragment has a tendency to bind bacterial contaminants that are difficult to remove on GST-affinity resins (Ubach et al., 2001). Therefore we purified the recombinant proteins in solution and used a liposome centrifugation assay rather than GST pulldowns. The wild-type double C2 domain fragment displayed a significantly higher apparent Ca2+affinity in the presence of phospholipids (∼3 μmCa2+) than each isolated C2 domain (∼10 μmCa2+) (Fig. 3), confirming that the two C2 domains cooperate in lipid binding. In addition, the D232N and D238N mutations had no significant effect on the apparent Ca2+ affinity or the amount of phospholipid binding to the recombinant double C2 domain fragment at physiological ionic strength (Fig. 3), as observed for the native proteins isolated from the knock-in mice (Fig. 7). To explore whether the mutations caused a change in the strength of phospholipid binding, we performed centrifugation assays at different salt concentrations. NaCl had a marked effect on Ca2+-dependent phospholipid binding to the wild-type double C2domain fragment (Fig. 8), showing that the mutations have an effect on the tightness of binding.
Fig. 8.

Salt sensitivity of the Ca2+-dependent complex of the wild-type and mutant double C2 domain fragment from synaptotagmin 1. The double C2 domain fragments were bound to liposomes in the presence of 50 μm Ca2+ and the indicated concentrations of NaCl. Double C2 domain proteins attached to the liposomes after centrifugation were analyzed by SDS-PAGE and Coomassie staining. Note especially that the D238N mutation destabilized the Ca2+-dependent phospholipid complex.
DISCUSSION
In the present study we have probed the functional importance of Ca2+ binding to the C2A domain of synaptotagmin 1. Two point mutations in the Ca2+ ligands of the C2A domain, D232N and D238N, severely affected intrinsic Ca2+ binding and Ca2+-dependent phospholipid binding to the isolated C2A domain. Interestingly, these mutations had little effect on the Ca2+-dependent phospholipid binding properties of the double C2 domain region of synaptotagmin 1 and did not cause major changes in synaptic transmission when introduced into knock-in mice by homologous recombination. D238N mutant mice exhibited no synaptic phenotype, whereas D232N mutant mice displayed only a small but significant increase in synaptic depression. These data show that C2 domain mutations like the D232N or D238N substitutions, which alter only the Ca2+-dependent properties of an isolated C2A domain but not of the double C2 domain fragment, do not have a major effect on the overall function of synaptotagmin 1. In contrast, C2 domain mutations like the R233Q substitution, which decreases the apparent Ca2+ affinity of both the isolated C2A domain and the double C2 domain fragment, result in a major corresponding change in the function of synaptotagmin 1 (Fernández-Chacón et al., 2001). Together these results suggest that the two C2 domains of synaptotagmin 1 function as a single cooperative unit whose overall Ca2+-binding properties, and not the Ca2+-binding properties of its isolated constituents, determine Ca2+-triggered neurotransmitter release.
Together with previous studies, our data lead to a model of how the synaptotagmin 1 C2 domains form Ca2+-dependent phospholipid complexes that emphasizes both the interplay between different types of forces and the cooperation between the two C2 domains (Fig.9). This model is based on three fundamental observations. First, both C2 domains individually engage in ternary complexes with phospholipids and Ca2+ (Davletov and Südhof, 1993;Chapman and Jahn, 1994; Fernández et al., 2001). Second, intrinsic Ca2+ binding to the C2 domains is noncooperative and exhibits a low Ca2+ affinity but becomes cooperative and assumes a high Ca2+ affinity when C2 domain/phospholipid complexes are formed (Zhang et al., 1998; Fernández-Chacón et al., 2001). Third, the C2domain/phospholipid/Ca2+ complex involves direct interactions of positively charged and hydrophobic residues of the C2 domain with the phospholipid bilayer that can be as critical for the complex as the bound Ca2+ ions (Chapman and Davis, 1998; Zhang et al., 1998; Fernández-Chacón et al., 2001; Gerber et al., 2002). Thus at least three forces attach each C2domain to the phospholipid bilayer: the positive charges of the Ca2+ ions, positively charged amino acids (in particular R233 and K366), and hydrophobic residues that insert into the bilayer (in particular M173, F234, V304, and I367).
Fig. 9.

Model of the Ca2+-dependent binding of synaptotagmin 1 C2 domains to phospholipid membranes. The two C2 domains of synaptotagmin 1 (which account for two-thirds of the total sequence) engage in similar but parallel interactions with phospholipid membranes that are fueled by three forces: positive charges supplied by bound Ca2+ ions that are sandwiched between negatively charged phospholipid head groups and C2 domain aspartate residues; positive charges supplied by arginine and lysine residues such as R233 and K366 at the top of the domain; and hydrophobic residues that insert at least partially into the bilayer, such as M173, F234, V305, and I367. In the absence of Ca2+, repulsion by the negatively charged aspartate residues on top of the C2 domains and the negatively charged phospholipid head groups prevents the positively charged and hydrophobic residues at the top of the domain to engage in interactions. Thus in addition to forming a bridge between the phospholipid head groups and the top loops of the C2 domains, Ca2+ ions also neutralize repulsive negative charges. The double C2domains exhibit an approximately threefold higher apparent Ca2+ affinity than the individual isolated C2 domains when the two independent C2 domains become linked physically.
In agreement with this model, our data show that the Ca2+-dependent complex of the double C2 domain fragment with phospholipids is sensitive to increases in ionic strength (Fig. 8), exhibits an increased apparent Ca2+ affinity compared with the individual C2 domains (Fig. 3), and is less susceptible than the individual C2 domains to mutations in Ca2+-binding sites (Figs.3, 4). Thus, limited Ca2+ binding is sufficient to “switch on” the double C2domain fragment and to trigger complete membrane translocation of the entire fragment. Although the disruption of some C2A domain Ca2+-binding sites by the D232N and D238N mutations yields a Ca2+/phospholipid complex with the double C2 domain fragment that is less tight and more sensitive to ionic strength (Fig. 8), the complex is still sufficiently strong to persist at physiological salt concentrations. We have observed previously that the R233Q mutation decreases significantly the apparent Ca2+affinity of both the single C2A domain and the double C2 domain region (Fernández-Chacón et al., 2001), whereas our current data show that the D232N and D238N mutations lead to a decrease only in the C2A domain but not in the double C2 domain. The differential impact of these mutations can be attributed to differences in the energetic contributions to phospholipid binding by the R233 side chain versus the second and third Ca2+-binding sites of the C2A domain. Such differential contributions to overall binding energies are typical in protein–protein interactions where only a subset of the contacts in the interface, called “hot spots,” usually accounts for most of the energy of binding (Cunningham and Wells, 1993). A possible alternative explanation for the synergistic effect revealed by the D232N and D238N mutations on the double C2 domain fragment is that new Ca2+-binding sites are created in the double C2 domain fragment that are not present in the individual C2 domains and are formed by direct interactions between the C2 domains (Garcia et al., 2000). This explanation was evoked previously to explain unexpected properties of the double C2domain fragment compared with the single C2domains, leading to the suggestion of “cryptic” Ca2+-binding sites in the C2B domain (Bai et al., 2002). However, there is nothing cryptic about the Ca2+-binding sites of the C2B domain because the isolated C2B domain is a fully functional Ca2+-binding and Ca2+-dependent phospholipid binding domain (Fernández et al., 2001). Furthermore, direct NMR studies of the double C2 domain fragment indicate that the two C2 domains do not interact with each other at Ca2+ concentrations between 0 and 20 mm in the absence of ternary components (J. Rizo, unpublished observation). Viewed together, it thus seems likely that the properties of the double C2 domain fragment can be explained entirely by the model shown in Figure 9.
Our previous observation on the R233Q knock-in mice showed that a decrease in the apparent Ca2+ affinity of the double C2 domain leads to a decrease in release probability. Now we have observed that the D232N and D238N mutations do not alter the phospholipid binding of the double C2 domain and that these mutations do not have a major effect on neurotransmitter release in vivo. The observation that the D232N and D238N mutations in the knock-in mice did not cause a large change in synaptotagmin function shows that the biochemical cooperation of the synaptotagmin C2domains in phospholipid binding translates into functional cooperationin vivo. We did observe, however, a significant enhancement in synaptic depression after repetitive stimulation in the D232N knock-in mice. Because analysis of cultured hippocampal neurons is sensitive to artifacts caused by differences in the genetic background of mice and drifts in culture conditions, we analyzed the synaptic properties of the mutant neurons under tightly controlled conditions. All analyses were performed simultaneously for mutant and control neurons obtained from littermates, with the control consisting of mice derived by homologous recombination with a neomycin gene cassette in an intron, but without a mutation in the synaptotagmin 1 gene. The best internal but somewhat involuntary control for the specificity of the D232N mutant phenotype, however, was provided by the D238N mutant mice, which did not exhibit such phenotype. In contrast to our previous study examining the impact of the R233Q mutation, we were unable to link the significant change in short-term plasticity for the D232N mutant (Fig.5) to any significant changes in vesicular or synaptic release probability or to a shift in the apparent Ca2+ sensitivity of neurotransmitter release. Either the change is too small to be detectable by these methods, or perhaps more likely, the synaptic depression is not caused directly by a change in release probability or Ca2+ sensitivity of release.
We were also unable to find a defined correlation between the phenotype of the D232N mice and the Ca2+-dependent phospholipid or syntaxin 1-binding properties of the double C2 domain region of synaptotagmin 1. The slight increase in the apparent Ca2+ affinity of the residual phospholipid binding to the isolated D232N C2A domain mutant (Fig. 2) could potentially underlie this phenotype. However, we could not detect such an increase in the context of the double C2 domain fragment within the limits of the centrifugation assay used. In any case, neurotransmitter release is an exquisitely regulated process, and it is thus not surprising if subtle biochemical changes in the Ca2+ sensor, which may be difficult to detect with assays performed in vitro with limited components, translate into more significant changes in the properties of neurotransmitter release. Moreover, the lack of a correlation of short-term plasticity with release probabilities or Ca2+ sensitivity of release, and the differential effects of the D232N mutation and D238N mutations in phospholipid binding and synaptic physiology, support the notion that the function of the C2A domain in triggering Ca2+-dependent neurotransmitter release is complex. Therefore, additional yet unidentified molecular targets may be important for regulating Ca2+-triggered release, although the Ca2+-dependent interaction of synaptotagmin 1 with syntaxin 1 appeared unchanged. Future experiments will have to address the relative merits of these possibilities.
Note added in proof. Similar results were recently obtained in Drosophila with a single DN mutant, although the biochemical consequences of the mutation were not studied with assays that detect phospholipid binding to the C2B domain (Robinson et al., 2002).
Footnotes
This study was supported by Grant NS40944 from the National Institutes of Health to J.R., Grant Ro1296/5-1 from the Deutsche Forschungsgemeinschaft to C.R., and postdoctoral fellowships from the Spanish Ministry of Education and the Fulbright Commission to R.F.C. and the Deutsche Forschungsgemeinschaft to S.H.G, and a fellowship from the Boehringer Ingelheim Fonds to A.C.M. We thank Ina Herfort, Izabella Leznicki, and Andrea Roth for excellent technical assistance, Nicole Hamlin (University of Texas Southwestern Medical Center, Dallas, TX) and Dr. Hermann Riedesel, Jürgen Krause, and Stefan Röcklin (Max-Planck-Institut für experimentelle Medizin, Göttingen) for outstanding help with mouse husbandry, and Dr. E. Neher and Dr. R. Jahn for advice.
Correspondence should be addressed to Dr. Christian Rosenmund, Department Membranbiophysik, Max-Planck-Institut für biophysikalische Chemie, Am Fassberg, D-37070 Göttingen, Germany. E-mail: crosenm@gwdg.de.
R. Fernández-Chacón's present address: Departamento Fisiología Médica y Biofísica, Universidad de Sevilla, Avenida Sánchez-Pizjuán 4, 41009 Sevilla, Spain.
S. Gerber's present address: Universitaet Heidelberg, 69115 Heidelberg, Germany.
REFERENCES
- 1.Bai J, Earles CA, Lewis JL, Chapman ER. Membrane-embedded synaptotagmin penetrates cis or trans target membranes and clusters via a novel mechanism. J Biol Chem. 2000;275:25427–25435. doi: 10.1074/jbc.M906729199. [DOI] [PubMed] [Google Scholar]
- 2.Bai J, Wang P, Chapman ER. C2A activates a cryptic Ca2+-triggered membrane penetration activity within the C2B domain of synaptotagmin 1. Proc Natl Acad Sci USA. 2002;99:1665–1670. doi: 10.1073/pnas.032541099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bollmann JH, Sakmann B, Borst JG. Calcium sensitivity of glutamate release in a calyx-type terminal. Science. 2000;289:953–957. doi: 10.1126/science.289.5481.953. [DOI] [PubMed] [Google Scholar]
- 4.Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem. 1998;273:13995–14001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 5.Chapman ER, Jahn R. Calcium-dependent interaction of the cytoplasmic region of synaptotagmin with membranes. Autonomous function of a single C2-homologous domain. J Biol Chem. 1994;269:5735–5741. [PubMed] [Google Scholar]
- 6.Chapman ER, Hanson PI, An S, Jahn R. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J Biol Chem. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- 7.Contreras MA, Ubach J, Millet O, Rizo J, Pons M. A Lanthanide induced orientation of a calcium binding protein. J Am Chem Soc. 1999;121:8947–8948. [Google Scholar]
- 8.Cunningham BC, Wells JA. Comparison of a structural and functional epitope. J Mol Biol. 1993;237:554–563. doi: 10.1006/jmbi.1993.1611. [DOI] [PubMed] [Google Scholar]
- 9.Davletov BA, Südhof TC. A single C2 domain from synaptotagmin 1 is sufficient for high affinity Ca2+/phospholipid binding. J Biol Chem. 1993;268:26386–26390. [PubMed] [Google Scholar]
- 10.Earles CA, Bai J, Wang P, Chapman ER. The tandem C2 domains of synaptotagmin contain redundant Ca2+ binding sites that cooperate to engage t-SNAREs and trigger exocytosis. J Cell Biol. 2001;154:1117–1123. doi: 10.1083/jcb.200105020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernández I, Arac D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RG, Südhof TC, Rizo J. Three-dimensional structure of the synaptotagmin 1 C2B-domain. Synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32:1057–1069. doi: 10.1016/s0896-6273(01)00548-7. [DOI] [PubMed] [Google Scholar]
- 12.Fernández-Chacón R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC. Synaptotagmin 1 functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- 13.Garcia RA, Forde CE, Godwin HA. Calcium triggers an intramolecular association of the C2 domains in synaptotagmin. Proc Natl Acad Sci USA. 2000;97:5883–5888. doi: 10.1073/pnas.100127197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin 1: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 15.Gerber SH, Rizo J, Südhof TC. The top loops of the C2 domains from synaptotagmin and phospholipase A2 control functional specificity. J Biol Chem. 2001;276:32288–32292. doi: 10.1074/jbc.C100108200. [DOI] [PubMed] [Google Scholar]
- 16.Gerber SH, Rizo J, Südhof TC. Role of electrostatic and hydrophobic interactions in Ca2+-dependent phospholipid binding by the C2A-domain from synaptotagmin 1. Diabetes. 2002;51[Suppl 1]:S12–18. doi: 10.2337/diabetes.51.2007.s12. [DOI] [PubMed] [Google Scholar]
- 17.Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- 18.Kee Y, Scheller RH. Localization of synaptotagmin-binding domains on syntaxin. J Neurosci. 1996;16:1975–1981. doi: 10.1523/JNEUROSCI.16-06-01975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Südhof TC. Ca2+-dependent and -independent activities of neural and non-neural synaptotagmins. Nature. 1995;375:594–599. doi: 10.1038/375594a0. [DOI] [PubMed] [Google Scholar]
- 20.Perin MS, Fried VA, Mignery GA, Jahn R, Südhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–263. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- 21.Pyott S, Rosenmund C. The effects of temperature on vesicular supply and release in autaptic cultures of hippocampal neurons. J Physiol (Lond) 2002;539:523–535. doi: 10.1113/jphysiol.2001.013277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson IM, Ranjan R, Schwarz TL. Synaptotagmins I and IV promote transmitter release independently of a Ca2+ binding in the C2A domain. Nature. 2002;418:336–340. doi: 10.1038/nature00915. [DOI] [PubMed] [Google Scholar]
- 23.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 24.Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- 25.Schiavo G, Gu QM, Prestwich GD, Söllner TH, Rothman JE. Ca2+-dependent switching of the specificity of phosphoinositide binding to synaptotagmin. Proc Natl Acad Sci USA. 1996;93:13327–13332. doi: 10.1073/pnas.93.23.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- 27.Shao X, Fernández I, Südhof TC, Rizo J. Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin 1: does Ca2+ induce a conformational change? Biochemistry. 1998;37:16106–16115. doi: 10.1021/bi981789h. [DOI] [PubMed] [Google Scholar]
- 28.Südhof TC. Synaptotagmins: why so many? J Biol Chem. 2002;277:7629–7632. doi: 10.1074/jbc.R100052200. [DOI] [PubMed] [Google Scholar]
- 29.Sugita S, Shin OH, Han W, Lao Y, Südhof TC. Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutton RB, Davletov BA, Berghuis AM, Südhof TC, Sprang SR. Structure of the first C2 domain of synaptotagmin 1: a novel Ca2+/phospholipid-binding fold. Cell. 1995;80:929–938. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- 31.Ubach J, Zhang X, Shao X, Südhof TC, Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J. 1998;17:3921–3930. doi: 10.1093/emboj/17.14.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ubach J, Lao Y, Fernandez I, Arac D, Südhof TC, Rizo J. The C2B domain of synaptotagmin 1 is a Ca2+-binding module. Biochemistry. 2001;40:5854–5860. doi: 10.1021/bi010340c. [DOI] [PubMed] [Google Scholar]
- 33.Xu-Friedman MA, Harris KM, Regehr WG. Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J Neurosci. 2001;21:6666–6672. doi: 10.1523/JNEUROSCI.21-17-06666.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang O, Kay LE, Olivier JP, Forman-Kay JD. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Rizo J, Südhof TC. Mechanism of phospholipid binding by the C2A-domain of synaptotagmin 1. Biochemistry. 1998;37:12395–12403. doi: 10.1021/bi9807512. [DOI] [PubMed] [Google Scholar]