

Abstract
The function of nuclear factor (NF)-κB within the developing and mature CNS is controversial. We have generated transgenic mice to reveal NF-κB transcriptional activity in vivo. As expected, constitutive NF-κB activity was observed within immune organs, and tumor necrosis factor-inducible NF-κB activity was present in mesenchymal cells. Intriguingly, NF-κB activity was also prominent in the CNS throughout development, especially within neocortex, olfactory bulbs, amygdala, and hippocampus. NF-κB in the CNS was restricted to neurons and blocked by overexpression of dominant-negative NF-κB-inducible kinase or the IκBαM super repressor. Blocking endogenous neuronal NF-κB activity in cortical neurons using recombinant adenovirus induced neuronal death, whereas induction of NF-κB activity increased levels of anti-apoptotic proteins and was strongly neuroprotective. Together, these data demonstrate a physiological role for NF-κB in maintaining survival of central neurons.
Keywords: transgenic mouse, adenovirus, NF-κB, NIK, RelA, TRAF, apoptosis
Nuclear factor (NF)-κB transcription factors are required for regulating cell survival and differentiation and for inflammatory and immune responses. The five mammalian NF-κB subunits [RelA (p65), NFkB2 (p52/p100), NFkB1 (p50/p105), RelB, and c-Rel] each contain a Rel homology domain that allows these factors to dimerize and bind DNA (for review, see Gilmore, 1999; Perkins, 2000). In lymphocytes and other activated immune cells, NF-κB is retained in the nucleus and is constitutively active. In most cells, however, NF-κB dimers are normally rendered inactive in the cytosol by virtue of their interaction with one of the inhibitory IκB proteins (IκBα, IκBβ, IκBε, IκBγ, and Bcl-3). Translocation to the nucleus occurs only after stimuli-induced IκB protein degradation. This process requires the activation of kinase cascades that converge on IκB kinase (IKK)1 and IKK2, related catalytic kinase subunits that, together with NF-κB essential modulator/IKKγ, form a complex that phosphorylates IκB family members and subsequently targets them for ubiquitination and proteosomal degradation (for review, see Karin and Ben-Neriah, 2000).
In non-neuronal cells, three major functions have been ascribed to the NF-κB family. Inducible NF-κB activity is crucial for activating genes mediating the proinflammatory response, a key component of the host defense system (Hatada et al., 2000). NF-κB activation also induces the transcription of anti-apoptotic genes and thereby promotes survival (Van Antwerp et al., 1998; Wang et al., 1998; Barkett and Gilmore, 1999). Finally, NF-κB plays a crucial role in maturation of the skin and skeletal systems (Q. Li et al., 1999). The precise role(s) of NF-κB within the nervous system is less clear. Several studies have found that NF-κB activity facilitates neuronal survival (Barger et al., 1995; Guo et al., 1998; Lezoualc'h et al., 1998; Maggirwar et al., 1998; Hamanoue et al., 1999; Kaltschmidt et al., 1999), yet others report that NF-κB activation is required for neuronal death (Grilli et al., 1996; Post et al., 1998; Schneider et al., 1999). The use of genetically altered mice will no doubt resolve this issue, but so far, the results have been equivocal. For example, mice lacking the p50/p105 NF-κB subunit show increased hippocampal damage in response to kainate-induced excitotoxicity (Yu et al., 1999), yet p50/p105 nulls also show reduced neuronal damage after focal cerebral ischemia (Schneider et al., 1999)
To clarify the role of NF-κB in the CNS, the pattern of transcriptionally active neuronal NF-κB needs to be established. To address this, we generated transgenic mice that provide a sensitive readout of NF-κB activity, particularly within the nervous system. Primary fibroblasts derived from these mice show appropriate reporter gene activation in response to known NF-κB inducers, and this response is blocked by overexpression of IκBαM, a specific NF-κB repressor. Transgenic mice show constitutive NF-κB activation in peripheral lymphoid tissues and display high levels of NF-κB activity in developing epidermal appendages. Intriguingly, the reporter mice also reveal high levels of NF-κB activity within developing and mature neurons of the CNS. Reducing neuronal NF-κB activity through overexpression of an IκB super repressor or dominant inhibitory NF-κB-inducing kinase (NIK) induces cortical neuron death. Conversely, adenovirus-mediated overexpression of p65/RelA in primary neurons induces accumulation of Bcl-XL, inhibitor of apoptosis protein (IAP)1, and IAP2 and confers strong protection against neuronal apoptosis induced by etoposide or camptothecin. Together, these studies demonstrate that active NF-κB activity is present throughout the developing and adult nervous system and indicate that NF-κB plays an important role in survival of CNS neurons.
MATERIALS AND METHODS
DNA construct and production of transgenic mice. To create a high-fidelity NF-κB reporter minigene, an NF-κB tandem repeat derived from the long terminal repeat of human immunodeficiency virus (HIV-LTR) was placed just upstream of a minimal SV40 promoter. Overlapping oligonucleotides were used to make additional identical NF-κB tandem repeats, which when combined, produced three tandem NF-κB repeats upstream of the SV40 minimal promoter. The enhancer/promoter fragment was cloned upstream of an Escherichia coli β-galactosidase gene modified to contain a mammalian Kozak consensus, an SV40 T-antigen-derived nuclear localization signal, and a polyA tract and splicing signal derived from the protamine I gene (Mercer et al., 1991). The minigene cassette was isolated from parental vector and injected into pronuclei to produce a total of 10 transgenic founder mice in a C3HxBALB/c background. Genotyping of transgenic mice was performed by PCR analysis from tail biopsies (Laird et al., 1991) using primers directed to β-galactosidase (5′-CTGCAGATAACTGCCGTCACTCC-3′, 5′-CTTAATCGCCTTGCAGCACAT-3′).
Cell culture and reagents. Primary mouse embryonic fibroblasts (MEF) were derived from the dorsal skin of embryonic day (E) 15–16 transgenic embryos and maintained in DMEM containing 10% bovine calf serum, 2 mml-glutamine, and 100 μg/ml penicillin/streptomycin. Human embryonic kidney (HEK) 293A cells were maintained in DMEM containing 10% bovine calf serum, 2 mml-glutamine, and 100 μg/ml penicillin/streptomycin. Cortical cultures were prepared from E15–16 transgenic mouse telencephalon and maintained 8–10 d in vitro (DIV) in Neurobasal media (Invitrogen), supplemented with a final concentration of 0.5 × B27 supplement (Invitrogen), 0.5 × N2 supplement (Invitrogen), 2 mml-glutamine, and 100 μg/ml penicillin/streptomycin. IκBα, p100, c-IAP1, and Bcl-XL antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA); p65/RelA monoclonal antibody was from Transduction Laboratories; GFP polyclonal antibody was from Clontech (Cambridge, UK); β-galactosidase polyclonal antibody was from ICN Biochemicals; and the anti-hemagglutinin (HA) monoclonal antibody 12CA5 was from Boehringer Mannheim. Secondary donkey anti-rabbit or donkey anti-mouse horseradish conjugates were from Jackson ImmunoResearch(West Grove, PA).
Generation of recombinant adenovirus. The IκBαM cDNA (Van Antwerp et al., 1998) was provided by Inder Verma and subcloned into the cytomegalovirus (CMV) 5′ transfer vector, and recombinant adenovirus was generated in 293A cells using standard techniques (Hitt et al., 1997). Human p65/RelA cDNA was cloned into the pAdTrack-CMV shuttle vector, and recombinant adenovirus was generated as described previously (He et al., 1998). The HA-tagged dominant-negative NIK (dnNIK) consists of a truncated protein where the kinase domain and tumor necrosis factor (TNF) receptor-associated factor (TRAF)-interacting domain were deleted (amino acid 1–623 deletion) (Natoli et al., 1997). The adenoviral dnNIK was constructed using the Cre-lox recombination method as described previously (Russo et al., 2002). IκBαM, dnNIK, and control green fluorescence protein (GFP) or β-galactosidase adenovirus were amplified in 293 cells and purified over sucrose gradients, and stock titer values were obtained using the tissue culture infectious dose 50 method. For primary cell infections, appropriate titers of virus were diluted into 10% of the culture volume and added directly to MEFs or cortical neurons at the time of plating or on cells plated 2 or 5 d earlier.
β-Galactosidase assays and immunofluorescence. Embryos, organs, or cultured cells were fixed for 20 min at 4°C in 4% paraformaldehyde (PFA) in PBS and then assayed for β-galactosidase activity by incubation in 37°C in 80 mm dibasic sodium phosphate, 20 mm monobasic sodium phosphate, 2 mm MgCl2, 0.2% Nonidet P-40, 1 mg/ml sodium deoxycholate, 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, and 800 μg/ml 4-chloro-5-bromo-3-indolyl-β-galactoside (X-gal; Sigma, St. Louis, MO) for 4–16 hr. Samples were then washed in PBS and postfixed in 4% paraformaldehyde in PBS. Immunostaining was performed on parallel PFA-fixed cultures using antibodies directed against β-galactosidase (polyclonal; ICN Biomedicals, Cleveland, OH), MAP2 (monoclonal, clone AP-20 from Sigma), β-III-tubulin (monoclonal, Sigma clone SDL.3D10), and glial fibrillary acidic protein (GFAP) (monoclonal; Boehringer Mannheim), and using donkey anti-mouse-conjugated fluorescein isothiocyanate and donkey anti-rabbit CY3 (The Jackson Laboratory, Bar Harbor, ME) as fluorescent secondary antibodies.
Transcriptional assays. For HEK293 cells, cells on six-well plates were transfected with CaPO4 precipitates containing the reporter plasmid on day 0, induced with cytokines beginning on day 1, and harvested in lysis buffer 16 hr later. β-galactosidase activity was assessed by O-nitrophenyl β-d-galactopyranoside conversion (Promega, Madison, WI). To quantify β-galactosidase activity in primary transgenic MEFs, cells were placed into 96-well plates, left uninfected or infected with recombinant adenovirus for 24 hr, induced with TNFα for 16 hr, and then harvested in radioimmunoprecipitation assay buffer. Lysates were assayed for β-galactosidase activity using Galactostar, a chemiluminescent substrate (Tropix). The same chemiluminescent technique was performed on transgenic cortical cultures that were plated at 20,000 cells/well on a 96-well plate, infected with recombinant adenovirus on day 5 in vitro, and harvested 4 d later.
Survival and apoptotic assays. Dissociated cortical neurons were plated as above and infected with recombinant adenovirus. After the periods indicated in the figure legends, neurons were assayed for viability using 3(4,5-dimethylthio-zol-2-yl)2,5-diphenyltetrazolium bromide (MTT; Sigma), which was added at a final concentration of 1 mg/ml for 4 hr. The reaction was ended by the addition of 1 vol of solubilization buffer (20% SDS, 10% dimethylformamide, and 20% acetic acid). After overnight solubilization, specific and nonspecific absorbance were read at 570 and 630 nm, respectively. Each condition was tested in four to six wells, experiments were performed in triplicate, and results were analyzed for statistical significance by multiple ANOVA. For terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assays, infected neurons were treated 24 hr later with 20 μmcamptothecin or etoposide (Calbiochem, La Jolla, CA) for 16 hr, fixed, and permeabilized in 4% PFA and 1:1 acetone/methanol, incubated with Biotin-dUTP (Boehringer Mannheim) and TdT as per the manufacturer's protocol (Promega), and followed with streptavidin-CY3 (Boehringer Mannheim) for visualization.
RESULTS
Previous studies have demonstrated that κB elements within the HIV-LTR promoter are sensitive to neuronal NF-κB activity in vitro (Rattner et al., 1993) and in vivo (Corboy et al., 1992; Buzy et al., 1995). To produce a reporter construct that would reflect endogenous NF-κB activity in neurons, we generated an NF-κB responsive minigene containing three tandem HIV-derived κB binding element repeats placed proximal to a minimal promoter derived from SV40. This construct drives expression of β-galactosidase that was modified to include an SV40 T-antigen-derived nuclear localization sequence (Fig. 1A). When transfected into HEK293 cells, the reporter construct exhibited low basal activity and could be readily induced to express β-galactosidase after treatment with TNFα, a well established NF-κB activator (Fig. 1B). The NF-κB responsive minigene cassette was injected into pronuclei, and a total of 10 transgenic founder mice were produced in a C3HxBALB/c background. Of these, five (17812, 17813, 17816, 17817, and 17820) showed no developmental β-galactosidase expression, suggesting that the transgene was incorporated into areas of inactive chromatin. Of the remainder, three founders (17814, 17815, and 17819) showed identical β-galactosidase expression patterns, which are described in detail below. The two remaining founder lines (17818 and 17821) showed differing subsets of the expression patterns observed in the 17814, 17815, and 17819 lines. The variable expression in the 17818 and 17821 lines likely represents local enhancer effects on transgene expression, and these lines were not studied further.
Fig. 1.
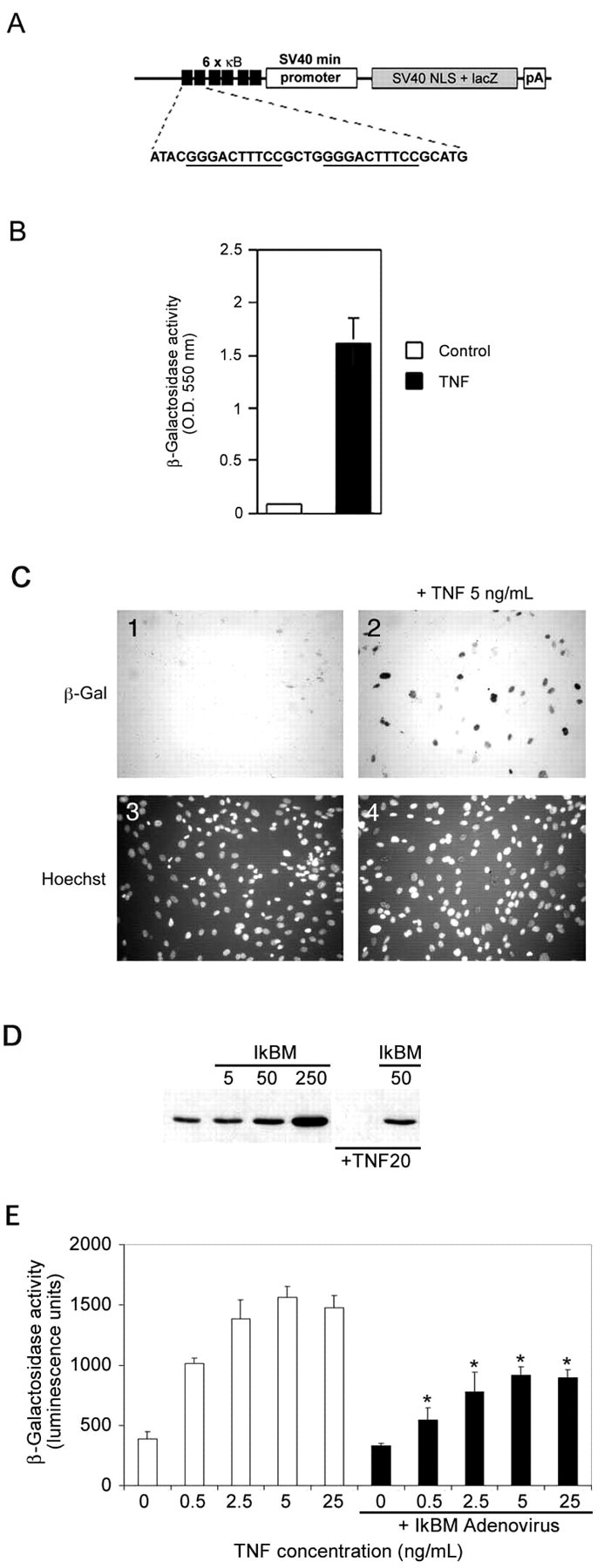
Transgene design and in vitrovalidation of the κB-dependent β-galactosidase construct.A, The NF-κB reporter minigene contains three tandem HIV-LTR repeats upstream of the SV40 minimal promoter, an E. coli β-galactosidase cDNA modified to contain a mammalian Kozak consensus, an SV40 T-antigen-derived nuclear localization signal, and a polyA tract derived from the protamine I gene. B, HEK293 cells were transiently transfected with the minigene and induced with DMEM + TNFα (5 ng/ml) or with DMEM alone (indicated as control) for 16 hr and analyzed for β-galactosidase activity.C, Primary MEFs derived from transgenic mice were incubated with (panels 2,4) or without (panels 1,3) TNFα (5 ng/ml) for 16 hr and then assessed for β-galactosidase activity. Cultures were counterstained with Hoechst 33342 (panels 3, 4) to show cell nuclei. D, MEFs were infected with 0, 5, 50, or 250 MOI of recombinant IκBαM adenovirus for 24 hr, and total cell lysates were prepared and analyzed by immunoblotting for IκBα. Wells that were mock infected or infected with 50 MOI of recombinant IκBαM adenovirus were exposed to TNFα (20 ng/ml) for 10 min. Endogenous IκBα is completely degraded by this treatment, but IκBαM is unaffected. E, Transgenic MEFs were incubated with 0, 0.5, 2.5, 5, and 25 ng/ml TNFα for 16 hr in the absence (whitebars) or presence (blackbars) of IκBαM adenovirus (∼50% infection efficiency). β-galactosidase activity was quantified using a chemiluminescent assay (Galacto-Star; Tropix). Each data point represents the average of six wells of a 24-well plate, and error bars indicate SD. Results were analyzed for statistical significance by ANOVA [Tukey honestly significant difference (HSD) multiple comparison], and statistically significant differences ofp < 0.001 are indicated by anasterisk.
To confirm appropriate in vivo activity of the incorporated minigene, primary fibroblast cells (MEFs) were derived from transgenic mouse embryos and analyzed for TNFα-induced β-galactosidase activity. Figure 1C shows that β-galactosidase activity was not detected in cultured transgenic MEFs under normal growth conditions but was present after exposure to a low concentration of TNFα. The MEF cultures used in these experiments represent a mixture of cells derived from transgenic and nontransgenic embryos and therefore likely under-represents the responsiveness of a pure transgenic population. To confirm that the TNFα-mediated induction of β-galactosidase in transgenic MEFs reflects bona fide NF-κB transcriptional activity, we created a recombinant adenovirus encoding IκBαM, a modified form of IκBα that is resistant to proteolytic degradation and represses NF-κB signaling by constitutively retaining NF-κB subunits in the cytosol in latent form. Figure1D shows that endogenous IκBα is rapidly degraded in cells exposed to TNFα (compare first and fourth lanes), but IκBαM remains intact under these conditions and is therefore available to bind and retain cytosolic NF-κB subunits in the cytoplasm. Transgenic MEFs that were left uninfected or were infected with recombinant adenovirus encoding IκBαM were exposed to increasing concentrations of TNFα and then analyzed for β-galactosidase activity. Figure 1E shows that the TNFα-mediated increase in β-galactosidase is attenuated in cells coexpressing IκBαM and demonstrates that β-galactosidase activity induced by TNFα in these primary cultures occurs through the NF-κB signaling pathway.
To examine NF-κB transcriptional activity during development, transgenic litters from lines 17814, 17815, and 17819 were fixed and whole-mount stained for β-galactosidase activity at different stages after implantation. Results for these three lines are identical, and only those from the 17814 line are shown in Figure2A–K. At E13, NF-κB activity is observed in prominent tactile and sinus hair follicles and in vibrissae primordia (Fig. 2A). The telencephalon is prominently stained, and NF-κB activity is present at the roof plate of the midbrain. Staining is also observed at the midbrain–hindbrain junction (Fig. 2B), within mammary gland primordia in the thoracic region, and in the gonadal area (Fig. 2C). Comparative NF-κB activity in transgenic versus nontransgenic negative control littermates is shown in Figure2D.
Fig. 2.

β-galactosidase expression pattern in discrete locations in embryonic and adult transgenic reporter mice.A, Whole-mount X-gal staining of an E13 transgenic mouse shows high basal NF-κB activity in the telencephalon and along the roof plate of the midbrain. Facial staining is visible within the primordia of the vibrissae (5 parallel rows) and in the prominent tactile hair follicles. B, Dorsal view of E13 transgenic embryos shows staining at the roof plate of the midbrain and at the midbrain–hindbrain junction. C, Close up of thoracic region. NF-κB activity is present in mammary gland primordia and in the gonadal area. D, β-galactosidase staining in transgenic (right) and in control littermate (left). E, E16 transgenic embryo showing prominent staining in vibrissae of the snout, in the olfactory lobes, and in the developing eyelid. F, NF-κB activity in the pads of the plantar surface of the E16 hindpaw (left) and of the palmer surface of the E16 forepaw (right).G, NF-κB activity within nuclei, likely multinucleated muscle fibers, beneath superficial layers of P1 skin. H, Robust NF-κB activity in P1 cortex, olfactory lobes, and roof plate of the midbrain. I–K, Lymphoid organs from postnatal day (P) 60 transgenic mice were analyzed for β-galactosidase activity as described in Materials and Methods. Constitutive NF-κB activity was detected along the trachea and bronchial tubes (I), in the thoracic lymph nodes (J and indicated by arrows inI), and in the thymus (K).
At E16, increased numbers of vibrissae are stained, and NF-κB activity is prominent over the presumptive eyelid crease (Fig.2E). Prominent NF-κB activity is also visible in epidermis on plantar and palmer surfaces of forepaws and hindpaws (Fig.2F). In neonates, high levels of NF-κB activity were observed in the CNS within the cortex, olfactory lobes, roof plate of the midbrain (Fig. 2H), and the midbrain–hindbrain junction (data not shown). The dermal surface of skin also had NF-κB activity, with a beaded appearance that likely represents staining within multinucleated muscle fibers (Fig.2G).
NF-κB is retained in the cytoplasm in an inactive form in most cells, but constitutive NF-κB activity occurs in various immune cells and immunocompetent organs (Lernbecher et al., 1993; Carrasco et al., 1994;Weih et al., 1994). Consistent with this, high β-galactosidase activity was observed in trachea and bronchial tubes, areas of primary immune defense that have high numbers of lymphocytes (Fig.2I), and in lymphoid organs of transgenic-positive animals (Fig. 2J,K). Together, these results show that these transgenic mice provide accurate reporting of endogenous NF-κB activity and indicate that NF-κB activity is present within the developing brain during murine development.
Figure 3 shows that NF-κB activity remains elevated in the CNS into adulthood, particularly in the forebrain. Serial brain sections reveal NF-κB activity in the olfactory bulbs (granule cell layer of the main olfactory bulb and the anterior olfactory nucleus) (Fig. 3A), the olfactory tubercle (islands of Calleja) (Fig. 3A–C), in all layers of the neocortex (Fig. 3A–H), in amygdala and claustrum, and within the dentate gyrus and hippocampus (Fig.3D–G). Lower levels of NF-κB activity are present in the piriform and entorhinal cortices (Fig. 3I) and within the hypothalamus (Fig. 3J). Cingulate and parietal cortex contain abundant blue nuclei in all cortical layers, with staining most prominent in layers 2, 4, and 5 (Fig.3K). In the hippocampus, β-galactosidase-positive nuclei are visible throughout the deep and superficial pyramidal layers of Ammon's horn, and robust activity is present in the CA1 and CA2 regions. Neurons within the CA3 region of the hippocampus show markedly reduced NF-κB activity compared with those in CA1 and CA2 (Fig.3E,K). NF-κB-positive nuclei can also be found throughout the stratum oriens and radiatum (Fig. 3K) and within a layer of interneurons along the stratum lacunosum moleculare (Fig.3F,G,K).
Fig. 3.
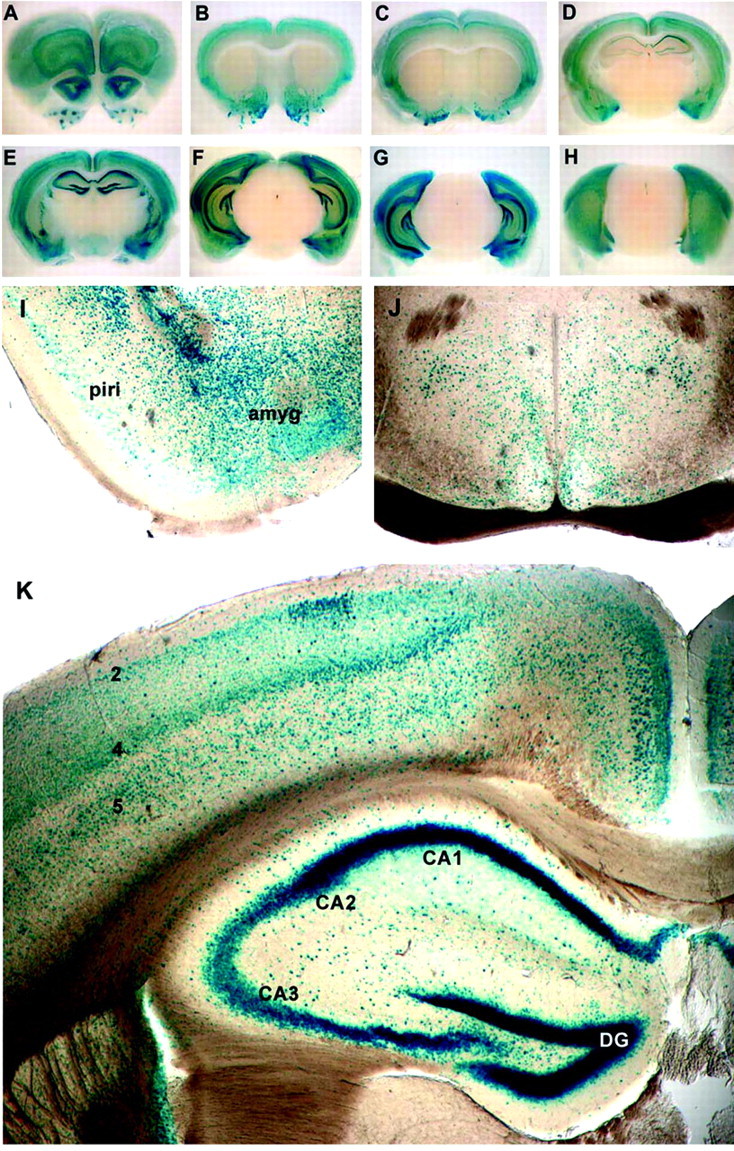
NF-κB activity in the adult brain.A–H, Serial sections (3 mm) of P180 transgenic brain were stained for β-galactosidase activity. Robust activity is visible in cortical layers 2, 4, and 5 (A–G), in the outer layers of the olfactory lobes (A), and in the islands of Calleja (olfactory tubercle) (A–C). Lower levels of β-galactosidase activity are present in the entorhinal and piriform cortices (D–F) and in the amygdala (D,E), claustrum (C–F), dentate gyrus, and hippocampus (D–G). I, NF-κB activity is present in the piriform–entorhinal cortex (piri) and is prominent in the amygdala (amyg). J, NF-κB activity is present in cells throughout the hypothalamus. K, NF-κB activity is prominent in the dentate gyrus (DG) and in CA1 and CA2 regions of the hippocampus. NF-κB activity is present in the CA3 region but lower than in CA1 and CA2 (also see D,E). Positive nuclei are also found in the stratum oriens, radiatum, and lacunosum moleculare of Ammon's horn. Within the cingulate and parietal cortex, positive nuclei are found in all cortical layers, but layers 2, 4, and 5 are stained most prominently.
To confirm that the β-galactosidase activity observed within the embryonic telencephalon reflects neuronal NF-κB activity, cortical cultures were prepared from E16 transgenic embryos, maintainedin vitro for 8–10 d, and then analyzed for β-galactosidase accumulation by immunohistochemistry. Figure4A shows that cells exhibiting typical pyramidal neuronal morphology contain β-galactosidase activity, and Figure 4B shows that the β-galactosidase-positive cells in these cultures coexpress the neuronal markers βIII-tubulin and MAP2. GFAP-positive glial cells consistently lacked β-galactosidase activity (data not shown).
Fig. 4.

NF-κB transcriptional activity is abundant in cultured primary cortical neurons. A, E16 primary cortical neurons derived from a heterozygote litter were grown for 10 DIV and then fixed and assessed for β-galactosidase activity.Arrows, Transgenic nuclei. B, E16 primary cortical neurons were immunostained for β-galactosidase and β-III-tubulin. β-galactosidase immunoreactivity is shown inred, β-III-tubulin is green, and nuclei stained with Hoescht 33342 are blue. C, Transgenic E16 cortical neurons derived from a heterozygote litter were infected with indicated MOIs of recombinant adenovirus expressing GFP, GFP and p65/RelA, or IκBαM for 48 hr, lysed, normalized for protein content, and analyzed for β-galactosidase content by immunoblot. Levels of β-III-tubulin assessed in parallel blots confirmed equivalent protein loading between lanes. D, Primary cortical neurons were infected with adenovirus encoding β-galactosidase (whitebars) or IκBαM (black bars), and survival was measured by MTT conversion 48 hr later. Error bars indicate SD. Results were analyzed for statistical significance by ANOVA (Tukey HSD multiple comparison). Statistically significant differences of p < 0.001 are indicated by an asterisk. A–D, Each experiment was performed at least three times.
To confirm that β-galactosidase levels within transgenic neurons are regulated by NF-κB transcriptional activity, NF-κB activity was disrupted in transgenic neurons using recombinant adenovirus encoding elements of the NF-κB signaling cascade. Figure 4C shows that expression of the IκBαM super repressor, which retains NF-κB subunits in the cytosol, reduced levels of β-galactosidase in transgenic neurons, whereas β-galactosidase levels were elevated in transgenic neurons infected with an adenovirus encoding the p65/RelA NF-κB subunit, which increases NF-κB transcriptional activity. Together, these results indicate that the constitutive β-galactosidase activity observed within transgenic neurons is regulated by NF-κB activity.
In many cells, activation of NF-κB induces transcription of anti-apoptotic genes, and the presence of NF-κB transcriptional activity in central neurons therefore raised the possibility that constitutive neuronal NF-κB activity may play a role in the maintenance of central neuron survival. To address this, NF-κB activity was reduced in cortical neurons using adenovirus encoding the IκBαM super repressor and, 48 hr later, assessed for survival using MTT assays. Figure 4D shows that infection with a control β-galactosidase adenovirus had no significant effect on neuronal survival, whereas infection with the IκBαM super repressor significantly reduced survival, at each multiplicity of infection (MOI) tested. To address whether this neuronal loss occurs through activation of apoptotic cascades, cortical neurons were infected with IκBαM and treated with zVAD, a broad-spectrum caspase inhibitor. Under these conditions, cortical neuron survival was significantly enhanced (p < 0.03; data not shown).
These results indicated that constitutive NF-κB activation is necessary for the survival of primary cortical neurons. To confirm this and begin to address the signaling events that might contribute to this effect, NF-κB signaling was disrupted in primary cortical neurons using a recombinant adenovirus encoding dnNIK, a MAP3K implicated in NF-κB activation in non-neuronal cells (Malinin et al., 1997; Regnier et al., 1997). To first confirm that dominant-negative NIK reduced NF-κB activity in primary neurons, transgenic cortical neurons were infected with dominant-negative NIK adenovirus and assessed for reductions in β-galactosidase activity. Figure5A shows that infection with a control adenovirus encoding GFP had no effect, whereas infection with equivalent titers of dominant-negative NIK adenovirus virus resulted in a significant reduction in β-galactosidase activity. At adenovirus titers >5 (MOI), NIK expression appeared to cause neuronal cell death. To investigate this effect, primary cortical neurons were infected with increasing titers of adenovirus encoding either dominant-negative NIK or GFP and assessed for survival using MTT assays. Figure 5B shows that the adenovirus encoding GFP had no significant effect on neuronal survival, whereas infection with adenovirus encoding dominant-negative NIK resulted in substantial neuronal death. These results indicate that constitutive NF-κB signaling plays an important role in the maintenance of primary cortical neuron survival.
Fig. 5.
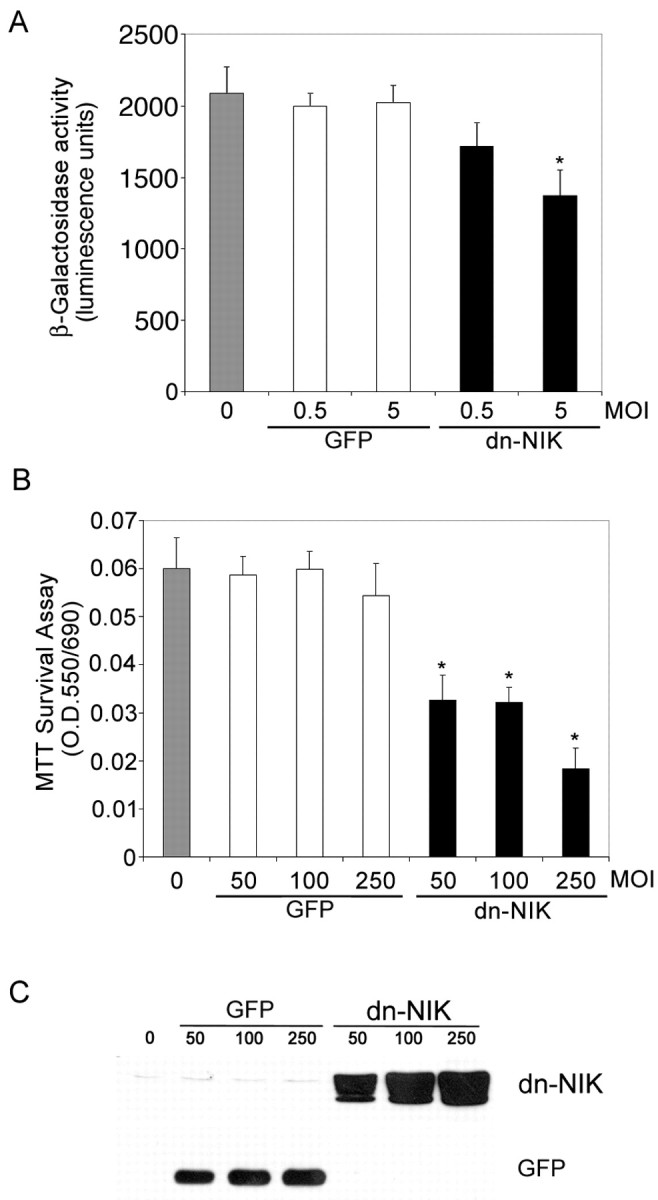
NIK signaling is required for NF-κB transcriptional activity and for neuronal viability in primary cortical neurons. A, E16 primary cortical neurons derived from a heterozygote litter were mock infected (gray bar) or infected with control GFP adenovirus (white bars) or dnNIK adenovirus (blackbars) at 0.5 or 5 MOI and harvested 4 d later. Lysates were analyzed for β-galactosidase activity using a chemoluminescence assay (Tropix). β-galactosidase activity was significantly reduced in cells infected with 5 MOI of dnNIK (*p < 0.03). B,C, Cortical neurons were infected with 0 (gray bar), 50, 100, or 250 MOI of recombinant adenovirus encoding GFP (white bars) or dnNIK (black bars) for 72 hr and then analyzed for viability by MTT dye conversion (B) and for GFP and dnNIK expression by immunoblotting (C). β-galactosidase overexpression had no significant effect on neuronal survival, but overexpression of dnNIK reduced survival at each MOI tested (*p < 0.001). A,B, Six wells were analyzed per condition, and results were analyzed for statistical significance by ANOVA (Tukey HSD multiple comparison). A–C, Each experiment was repeated at least three times.
The profound reduction in survival induced by the IκBαM super repressor and by dominant-negative NIK suggests that NF-κB activity may normally regulate a prosurvival response in neurons. Therefore, we tested whether activation of NF-κB confers a survival advantage in cortical neurons. Adenovirus encoding GFP alone or encoding both GFP and p65/RelA was used to infect primary cortical neurons before treatment with camptothecin and etoposide, inhibitors of topoisomerase I and II that are highly neurotoxic and cause apoptotic cell death of 50–80% of primary cortical neurons (Fig.6I). After 18 hr of drug treatment, infected cells were assessed for apoptotic death using TUNEL assays and by examination of nuclear morphology. Figure6A–D shows that essentially all neurons that were infected with control adenovirus encoding GFP alone displayed pyknotic nuclei and were TUNEL positive, indicating widespread apoptotic cell death. In contrast, Figure 6E–H shows neurons infected with adenovirus encoding p65/RelA and GFP that were uniformly TUNEL negative and contained healthy nuclei (whereas uninfected neurons in the same field were apoptotic). Quantification of these studies (Fig. 6I) revealed that >90% of neurons infected with p65/RelA were protected from apoptosis normally induced by etoposide or camptothecin. Together, these data indicate that NF-κB activation confers a profound survival advantage to cortical neurons.
Fig. 6.

p65/RelA protects cortical neurons from apoptotic death. E15–16 cortical neurons were infected with 75 MOI of recombinant adenovirus encoding GFP alone (A–D) or with recombinant virus encoding both p65/RelA and GFP (E–H) for 24 hr. Cells were then exposed to etoposide (20 μm) for an additional 18 hr and then fixed and analyzed for GFP fluorescence (B, F,green), for apoptosis using Hoescht 33342 nuclear staining (A, E, blue), and TUNEL labeling (C, G,red). D, H, Merged images of A–C and E–G, respectively. Cells infected with GFP alone (and uninfected cells) rapidly underwent apoptosis when exposed to etoposide, whereas neurons infected with p65/RelA were robustly viable under these conditions. I, Cells were infected with 75 MOI of adenovirus expressing either GFP or expressing GFP together with p65/RelA for 48 hr and then treated with camptothecin (20 μm) or etoposide (20 μm) for 18 hr. GFP-positive cells were scored for TUNEL-positive nuclei. Expression of p65/RelA conferred robust protection from apoptosis because of campthothecin (*p < 0.001) or etoposide (*p < 0.0001). At least 300 cells were assessed for each condition, and results were analyzed for statistical significance by Student's t test. J, E16 cortical neurons were either left uninfected or were infected with 75 MOI of recombinant adenovirus expressing GFP or expressing both GFP and p65/RelA for 48 hr. Neurons were then lysed and analyzed by immunoblot. Levels of endogenous IκBα, NFkB1, IAP1, IAP2, and Bcl-XL were specifically increased by p65/RelA overexpression.
To determine whether protein products of anti-apoptotic genes were induced by NF-κB in cortical neurons, cells were infected with adenovirus encoding GFP alone or encoding both GFP and p65/RelA for 48 hr, lysed, and analyzed by immunoblot. Genes encoding NF-κB signaling elements are themselves very sensitive to NF-κB activation and therefore provide useful internal controls to demonstrate NF-κB activation. Levels of endogenous IκBα and NFkB1 protein were unaffected in neurons infected with GFP alone, but both were strongly induced in cells expressing p65/RelA and GFP (Fig.6J), indicating that NF-κB activity was induced in cortical neurons by p65/RelA overexpression. Anti-apoptotic genes of the Bcl-2 family and IAPs represent two main classes of anti-apoptotic genes that are regulated by NF-κB in non-neuronal cells, and we therefore examined Bcl-XL, IAP1, and IAP2 as representative members of these two families. Figure6J shows that levels of each of these genes were increased in cells overexpressing p65/RelA and GFP but not in cells overexpressing GFP alone. Therefore, activation of NF-κB in primary cortical neurons appears to increase levels of anti-apoptotic proteins and thereby elevate the survival threshold of primary cortical neurons.
DISCUSSION
In this study, we have produced transgenic reporter mice that provide a reliable means for assessing NF-κB transcriptional activityin vivo. Mouse embryonic fibroblasts derived from these animals display an inducible NF-κB transcriptional readout that is inhibited by IκBαM, a repressor of NF-κB signaling, whereas transgenic lymphoid cells display constitutive NF-κB activity. Intriguingly, the transgenic reporter mouse reveals prominent constitutive NF-κB activity within neurons of the developing and mature CNS. Blockade of NF-κB activity results in neuronal death, whereas p65/RelA overexpression confers protection against insults and induces expression of anti-apoptotic gene products that include Bcl-XL, IAP1, and IAP2, indicating an important role for NF-κB activity in the regulation of neuronal survival.
NF-κB refers to transcriptional activity that is mediated by the Rel family of gene products through NF-κB cis-elements (for review, see Karin and Ben-Neriah, 2000). There is considerable diversity in the DNA binding properties of the different NF-κB proteins, and the NF-κB consensus sequence has >60 variants with different binding properties. The DNA binding potential of Rel family members is regulated by IκB binding and IκB-independent post-translation mechanisms. Transcription regulated through NF-κBcis-elements will therefore reflect the presence and affinities of subsets of Rel family members that are present in various tissues during specific developmental windows. The NF-κB enhancer element that we used to generate the transgenic mice reported here was derived from a fragment of the HIV-LTR that is particularly sensitive to neuronal NF-κB activity (Corboy et al., 1992; Buzy et al., 1995). p65/RelA and p50/p105 bind this NF-κB element in cultured neurons (Rattner et al., 1993), but the precise complement of NF-κB proteins that mediate the activation of this element in neurons remains unknown. Our identification of constitutive NF-κB activity within CNS neurons expands on previous studies that have used electrophoretic mobility shift assay and immunological methods to identify constitutive NF-κB in developing cortex and in CA1 and CA3 regions of the hippocampus, with much lower activity in the cerebellum (Bakalkin et al., 1993; Kaltschmidt et al., 1993, 1994; Rattner et al., 1993).
Other groups have used distinct NF-κB cis-elements to generate reporter mice that identify endogenous NF-κB activity. Using the κB motif from the immunoglobin κB enhancer, Lernbecher et al. (1993) identified NF-κB activity only in lymphoid tissues, whereas the use of the p105 Rel enhancer or regions of the immunoglobin κB enhancer revealed β-galactosidase activity in lymphoid tissues, as well as in developing rhombencephalon, spinal medulla, and blood vessels (Schmidt-Ullrich et al., 1996). It is likely that any single NF-κB element will provide a readout of only a subset of endogenous NF-κB activities, and it is therefore not surprising to see differences between animals generated using distinct NF-κBcis-elements. Indeed, a generation of mice null for various members of the Rel family has revealed that the physiological sites of NF-κB action extend well beyond those revealed in a single transcriptional reporter mouse line (Lernbecher et al., 1993; Beg et al., 1995; Weih et al., 1995; Beg and Baltimore, 1996; Schmidt-Ullrich et al., 1996; Franzoso et al., 1997; Iotsova et al., 1997).
A crucial step in validating the transgenic reporter mice is to ablate NF-κB signaling and demonstrate concomitant reductions in β-galactosidase reporter gene activity. Primary fibroblasts derived from the transgenic reporter mouse revealed TNFα-induced β-galactosidase activity, which was strongly attenuated in cells expressing the IκBαM repressor, a mutated form of IκBα that cannot be phosphorylated by IKK proteins and therefore retains NF-κB dimers in the cytosol. Several recent studies have shown that TNFα-dependent NF-κB induction in MEFs occurs through an IKK2-dependent signaling mechanism, and our results suggest that peripheral cells derived from these transgenic animals provide a sensitive transcriptional readout of this pathway.
Primary cortical neurons showed constitutive β-galactosidase activity, which was reduced by infection with adenovirus encoding the IκBαM repressor or using a dominant inhibitory form of NIK, a MAP3K that binds both TRAF and IKK1 proteins and normally regulates activation of NF-κB in response to some, but not all, cytokines (Yin et al., 2001). IκBαM repressor should retain Rel family members in the cytosol, whereas the dominant-negative NIK variant used in our studies is a deletion mutant that lacks the kinase domain but contains the TRAF and IKK binding domains that function by blocking the recruitment of endogenous NIK and titrating upstream and downstream effectors (Malinin et al., 1997; Natoli et al., 1997; Ling et al., 1998; Van Antwerp et al., 1998; Delhase et al., 1999; Foehr et al., 2000). Together, these distinct approaches show that the β-galactosidase activity present in primary cortical neurons is indeed regulated by NF-κB activity. The precise signaling elements that contribute to NF-κB-dependent activation of β-galactosidase in these animals are not certain and likely to be complex. It will be particularly interesting to examine the roles of the IKK proteins in this regard; mice rendered null for IKK1 or IKK2 display no apparent neuronal phenotype (Q. Li et al., 1999; Z. W. Li et al., 1999), yet mice lacking both genes show enhanced apoptosis in the neuroepithelium and a defect in neurulation (Li et al., 2000), consistent with the hypothesis that together these genes regulate NF-κB-dependent survival pathways in developing neurons.
We have shown that expression of the IκBαM repressor or dominant-negative NIK results in a profound reduction in cortical neuron viability, consistent with the hypothesis that NF-κB normally plays an important role in promoting central neuron survival. These results are consistent with several studies that indicate that NF-κB promotes the survival of peripheral neurons. In sympathetic neurons, overexpression of a mutated derivative of c-Rel lacking the transactivation domain blocks neurotrophin-dependent survival, whereas c-Rel overexpression facilitates survival (Maggirwar et al., 1998), in part by inducing gene products that block cytochrome crelease (Sarmiere and Freeman, 2001). Similarly, in developing dorsal root sensory neurons, members of the neurotrophin and ciliary neurotrophic factor families activate NF-κB-dependent survival pathways that require p65/RelA (Hamanoue et al., 1999; Middleton et al., 2000).
The impact of NF-κB on the survival of CNS neurons is more controversial, with some studies suggesting a role for NF-κB in the promotion of survival, whereas others indicate that NF-κB may facilitate apoptosis (for review, see Mattson and Camandola, 2001). For example, NF-κB activation appears to protect central neurons against amyloid β-peptide toxicity (Barger et al., 1995) and excitotoxic or oxidative stress (Goodman and Mattson, 1996; Mattson et al., 1997), yet NF-κB exerts a pro-apoptotic effect that facilitates glutamate-induced toxicity (Grilli and Memo, 1999). Furthermore, mice rendered null for p50/p105 show increased death in response to kainate-induced excitotoxicity but are resistant to damage induced by ischemia (Schneider et al., 1999; Yu et al., 1999). Our data showed that induction of NF-κB mediated by p65/RelA overexpression effectively protected primary cortical neurons from death induced by etoposide or camptothecin, likely through upregulation of IAPs and anti-apoptotic BclII family members. This finding is in agreement with recent results showing that Jak2-dependent activation of NF-κB resulted in accumulation of X-linked inhibitor of apoptosis protein and IAP2 proteins and conferred neuroprotection to toxic concentrations of S-nitrosocystein, a nitric oxide donor (Digicaylioglu and Lipton, 2001). Together, these results suggest that physiological stimuli that increase NF-κB activation in neurons will confer neuroprotection, and these data are consistent with recent findings that show that preconditioning stimuli that confer neuroprotection on central neurons in vivo result in increased neuronal NF-κB activation (Blondeau et al., 2001; Ravati et al., 2001). Thus, our results support the hypothesis that constitutive NF-κB is necessary for neuronal survival and that further increases in NF-κB activation are neuroprotective. However, the complexity of NF-κB signaling pathways that results in specific gene activation events under physiological situations should not be underestimated, and we cannot rule out the possibility that there may be pathological conditions that activate sets of NF-κB-dependent genes that may induce distinct effects that include facilitating apoptosis.
The precise stimuli that contribute to constitutive NF-κB activation within neurons are unclear. NF-κB is activated by numerous stimuli, and it is possible that constitutive paracrine or autocrine activation loops act to increase NF-κB activity. It is also possible that NF-κB is a retrograde signal that links synaptic events to transcription (Kaltschmidt et al., 1993; Meberg et al., 1996). Glutamate, kainic acid, and nitric oxide all activate neuronal NF-κB (Guerrini et al., 1995; Simpson and Morris, 1999), and recent studies have demonstrated activity-dependent translocation of p65/RelA from neurites to the nucleus of living neurons stimulated with glutamate, kainate, or potassium chloride (Wellmann et al., 2001). These studies therefore raise the intriguing possibility that NF-κB activity may link neuronal activity to cell survival pathways.
In summary, we have established that NF-κB transcriptional activity is prominent in the developing and adult nervous system. We have shown that NF-κB activity is necessary for neuronal survival and found that overexpression of NF-κB in primary neurons confers a high degree of neuroprotection through production of anti-apoptotic genes. Together, these studies demonstrate an important role for NF-κB in the development and maintenance of the nervous system.
Footnotes
This work was supported by a Studentship from the Canadian Institutes of Health Research (A.L.B.) and a Killam Foundation Scholarship (P.A.B.). We thank Jean-Pierre Julien and the Canadian Neuroscience Network Transgenic Mouse Facility for pronuclei injections and implantations, Inder Verma for the IκBαM construct, Tim Kennedy for assistance with imaging, and Angel Alonso for useful discussions.
Correspondence should be addressed to Philip A. Barker, Montreal Neurological Institute, McGill University, 3801 University Street, Montreal, Quebec, Canada H3A 2B4. E-mail: phil.barker@mcgill.ca.
REFERENCES
- 1.Bakalkin G, Yakovleva T, Terenius L. NF-kappa B-like factors in the murine brain. Developmentally-regulated and tissue-specific expression. Brain Res Mol Brain Res. 1993;20:137–146. doi: 10.1016/0169-328x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- 2.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- 4.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 5.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 6.Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci. 2001;21:4668–4677. doi: 10.1523/JNEUROSCI.21-13-04668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buzy JM, Lindstrom LM, Zink MC, Clements JE. HIV-1 in the developing CNS: developmental differences in gene expression. Virology. 1995;210:361–371. doi: 10.1006/viro.1995.1352. [DOI] [PubMed] [Google Scholar]
- 8.Carrasco D, Weih F, Bravo R. Developmental expression of the mouse c-rel proto-oncogene in hematopoietic organs. Development. 1994;120:2991–3004. doi: 10.1242/dev.120.10.2991. [DOI] [PubMed] [Google Scholar]
- 9.Corboy JR, Buzy JM, Zink MC, Clements JE. Expression directed from HIV long terminal repeats in the central nervous system of transgenic mice. Science. 1992;258:1804–1808. doi: 10.1126/science.1465618. [DOI] [PubMed] [Google Scholar]
- 10.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 11.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 12.Foehr ED, Lin X, O'Mahony A, Geleziunas R, Bradshaw RA, Greene WC. NF-κ B signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J Neurosci. 2000;20:7556–7563. doi: 10.1523/JNEUROSCI.20-20-07556.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilmore TD. The Rel/NF-kappaB signal transduction pathway. Oncogene. 1999;18:6842–6844. doi: 10.1038/sj.onc.1203237. [DOI] [PubMed] [Google Scholar]
- 15.Goodman Y, Mattson MP. Ceramide protects hippocampal neurons against excitotoxic and oxidative insults, and amyloid beta-peptide toxicity. J Neurochem. 1996;66:869–872. doi: 10.1046/j.1471-4159.1996.66020869.x. [DOI] [PubMed] [Google Scholar]
- 16.Grilli M, Memo M. Possible role of NF-kappaB and p53 in the glutamate-induced pro-apoptotic neuronal pathway. Cell Death Differ. 1999;6:22–27. doi: 10.1038/sj.cdd.4400463. [DOI] [PubMed] [Google Scholar]
- 17.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 18.Guerrini L, Blasi F, Denis-Donini S. Synaptic activation of NF-kappa B by glutamate in cerebellar granule neurons in vitro. Proc Natl Acad Sci USA. 1995;92:9077–9081. doi: 10.1073/pnas.92.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Q, Robinson N, Mattson MP. Secreted beta-amyloid precursor protein counteracts the proapoptotic action of mutant presenilin-1 by activation of NF-kappaB and stabilization of calcium homeostasis. J Biol Chem. 1998;273:12341–12351. doi: 10.1074/jbc.273.20.12341. [DOI] [PubMed] [Google Scholar]
- 20.Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM. p75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci. 1999;14:28–40. doi: 10.1006/mcne.1999.0770. [DOI] [PubMed] [Google Scholar]
- 21.Hatada EN, Krappmann D, Scheidereit C. NF-kappaB and the innate immune response. Curr Opin Immunol. 2000;12:52–58. doi: 10.1016/s0952-7915(99)00050-3. [DOI] [PubMed] [Google Scholar]
- 22.He TC, Zhou S, Da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hitt MM, Addison CL, Graham FL. Human adenovirus vectors for gene transfer into mammalian cells. Adv Pharmacol. 1997;40:137–206. doi: 10.1016/s1054-3589(08)60140-4. [DOI] [PubMed] [Google Scholar]
- 24.Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285–1289. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 25.Kaltschmidt B, Baeuerle PA, Kaltschmidt C. Potential involvement of the transcription factor NF-kappa B in neurological disorders. Mol Aspects Med. 1993;14:171–190. doi: 10.1016/0098-2997(93)90004-w. [DOI] [PubMed] [Google Scholar]
- 26.Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:9409–9414. doi: 10.1073/pnas.96.16.9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-kappa B activity in neurons. Mol Cell Biol. 1994;14:3981–3992. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 29.Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lernbecher T, Muller U, Wirth T. Distinct NF-kappa B/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature. 1993;365:767–770. doi: 10.1038/365767a0. [DOI] [PubMed] [Google Scholar]
- 31.Lezoualc'h F, Sagara Y, Holsboer F, Behl C. High constitutive NF-κB activity mediates resistance to oxidative stress in neuronal cells. J Neurosci. 1998;18:3224–3232. doi: 10.1523/JNEUROSCI.18-09-03224.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Z. W.Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, Izpisua-Belmonte JC, Verma IM. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q, Estepa G, Memet S, Israel A, Verma IM. Complete lack of NF-kappaB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev. 2000;14:1729–1733. [PMC free article] [PubMed] [Google Scholar]
- 34.Li ZW, Chu W, Hu Y, Delhase Hu, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ling L, Cao Z, Goeddel DV. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc Natl Acad Sci USA. 1998;95:3792–3797. doi: 10.1073/pnas.95.7.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-kappaB contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–10365. doi: 10.1523/JNEUROSCI.18-24-10356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 38.Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 40.Meberg PJ, Kinney WR, Valcourt EG, Routtenberg A. Gene expression of the transcription factor NF-kappa B in hippocampus: regulation by synaptic activity. Brain Res Mol Brain Res. 1996;38:179–190. doi: 10.1016/0169-328x(95)00229-l. [DOI] [PubMed] [Google Scholar]
- 41.Mercer EH, Hoyle GW, Kapur RP, Brinster RL, Palmiter RD. The dopamine beta-hydroxylase gene promoter directs expression of E. coli lacZ to sympathetic and other neurons in adult transgenic mice. Neuron. 1991;7:703–716. doi: 10.1016/0896-6273(91)90274-4. [DOI] [PubMed] [Google Scholar]
- 42.Middleton G, Hamanoue M, Enokido Y, Wyatt S, Pennica D, Jaffray E, Hay RT, Davies AM. Cytokine-induced nuclear factor kappa B activation promotes the survival of developing neurons. J Cell Biol. 2000;148:325–332. doi: 10.1083/jcb.148.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Natoli G, Costanzo A, Moretti F, Fulco M, Balsano C, Levrero M. Tumor necrosis factor (TNF) receptor 1 signaling downstream of TNF receptor-associated factor 2. Nuclear factor kappaB (NFkappaB)-inducing kinase requirement for activation of activating protein 1 and NFkappaB but not of c-Jun N-terminal kinase/stress-activated protein kinase. J Biol Chem. 1997;272:26079–26082. doi: 10.1074/jbc.272.42.26079. [DOI] [PubMed] [Google Scholar]
- 44.Perkins ND. The Rel/NF-kappa B family: friend and foe. Trends Biochem Sci. 2000;25:434–440. doi: 10.1016/s0968-0004(00)01617-0. [DOI] [PubMed] [Google Scholar]
- 45.Post A, Holsboer F, Behl C. Induction of NF-κB activity during haloperidol-induced oxidative toxicity in clonal hippocampal cells: suppression of NF-κB and neuroprotection by antioxidants. J Neurosci. 1998;18:8236–8246. doi: 10.1523/JNEUROSCI.18-20-08236.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rattner A, Korner M, Walker MD, Citri Y. NF-kappa B activates the HIV promoter in neurons. EMBO J. 1993;12:4261–4267. doi: 10.1002/j.1460-2075.1993.tb06110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ravati A, Ahlemeyer B, Becker A, Klumpp S, Krieglstein J. Preconditioning-induced neuroprotection is mediated by reactive oxygen species and activation of the transcription factor nuclear factor-kappaB. J Neurochem. 2001;78:909–919. doi: 10.1046/j.1471-4159.2001.00463.x. [DOI] [PubMed] [Google Scholar]
- 48.Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 49.Russo MP, Bennett BL, Manning AM, Brenner DA, Jobin C. Differential requirement for NF-kB-inducing kinase (NIK) in the induction of NF-kB by IL-1β, TNFa and Fas. Am J Physiol. 2002;283:C347–C357. doi: 10.1152/ajpcell.00166.2001. [DOI] [PubMed] [Google Scholar]
- 50.Sarmiere PD, Freeman RS. Analysis of the NF-kappa B and PI 3-kinase/Akt survival pathways in nerve growth factor-dependent neurons. Mol Cell Neurosci. 2001;18:320–331. doi: 10.1006/mcne.2001.1021. [DOI] [PubMed] [Google Scholar]
- 51.Schmidt-Ullrich R, Memet S, Lilienbaum A, Feuillard J, Raphael M, Israel A. NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development. 1996;122:2117–2128. doi: 10.1242/dev.122.7.2117. [DOI] [PubMed] [Google Scholar]
- 52.Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- 53.Simpson CS, Morris BJ. Activation of nuclear factor kappaB by nitric oxide in rat striatal neurones: differential inhibition of the p50 and p65 subunits by dexamethasone. J Neurochem. 1999;73:353–361. doi: 10.1046/j.1471-4159.1999.0730353.x. [DOI] [PubMed] [Google Scholar]
- 54.Van Antwerp DJ, Martin SJ, Verma IM, Green DR. Inhibition of TNF-induced apoptosis by NF-kappa B. Trends Cell Biol. 1998;8:107–111. doi: 10.1016/s0962-8924(97)01215-4. [DOI] [PubMed] [Google Scholar]
- 55.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 56.Weih F, Carrasco D, Bravo R. Constitutive and inducible Rel/NF-kappa B activities in mouse thymus and spleen. Oncogene. 1994;9:3289–3297. [PubMed] [Google Scholar]
- 57.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 58.Wellmann H, Kaltschmidt B, Kaltschmidt C. Retrograde transport of transcription factor NF-kappa B in living neurons. J Biol Chem. 2001;276:11821–11829. doi: 10.1074/jbc.M009253200. [DOI] [PubMed] [Google Scholar]
- 59.Yin L, Wu L, Wesche H, Arthur CD, White JM, Goeddel DV, Schreiber RD. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- 60.Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]