

Abstract
The tetrodotoxin-resistant sodium channel α subunit Nav1.8 is expressed exclusively in primary sensory neurons and is proposed to play an important role in sensitization of nociceptors. Here we compared visceral pain and referred hyperalgesia in Nav1.8-null mice and their wild-type littermates in five tests that differ in the degree to which behavior depends on spontaneous, ongoing firing in sensitized nociceptors. Nav1.8-null mice showed normal nociceptive behavior provoked by acute noxious stimulation of abdominal viscera (intracolonic saline or intraperitoneal acetylcholine). However, Nav1.8-null mutants showed weak pain and no referred hyperalgesia to intracolonic capsaicin, a model in which behavior is sustained by ongoing activity in nociceptors sensitized by the initial application. Nav1.8-null mice also showed blunted pain and hyperalgesia to intracolonic mustard oil, which sensitizes nociceptors but also provokes tissue damage. To distinguish between a possible role for Nav1.8 in ongoing activity per se and ongoing activity after sensitization in the absence of additional stimuli, we tried a visceral model of tonic noxious chemical stimulation, cyclophosphamide cystitis. Cyclophosphamide produces cystitis by gradual accumulation of toxic metabolites in the bladder. In this model, Nav1.8-null mice showed normal responses. There were no differences between null mutants and their normal littermates in tissue damage and inflammation evoked by any of the stimuli tested, suggesting that the behavioral differences are not secondary to impairment of inflammatory responses. We conclude that there is an essential role for Nav1.8 in mediating spontaneous activity in sensitized nociceptors.
Keywords: viscera, referred hyperalgesia, tetrodotoxin resistant sodium channels, colitis, cystitis, cyclophosphamide, inflammation, knock-out mice
Tissue damage or inflammation provokes an increase in sensitivity and spontaneous firing of action potentials in primary afferent nociceptors. This process, known as sensitization, gives rise to primary hyperalgesia (hypersensitivity at the site of injury) and ongoing pain. The ongoing nociceptor firing is also essential to establish and maintain secondary hyperalgesia (touch-evoked pain in undamaged tissue). The molecular mechanisms underlying nociceptor sensitization are not fully understood, but there is evidence for the involvement of voltage-gated sodium currents, particularly those that are resistant to tetrodotoxin (TTX). When primary afferent cell bodies in culture are exposed to inflammatory mediators known to produce sensitization of nociceptor terminals, TTX-resistant (TTX-r) sodium currents are enhanced (England et al., 1996; Gold et al., 1996). TTX-r currents are expressed in small- and medium-sized sensory neurons, which are primarily nociceptive (for review, see Waxman et al., 1999; Baker and Wood, 2001).
Two sodium channel α subunits that mediate TTX-r currents in primary afferent neurons have been cloned, Nav1.8 and Nav1.9 (for review, see Waxman et al., 1999). Studies of dorsal root ganglion (DRG) neurons from mice carrying a null mutation of the Nav1.8 gene indicate that Nav1.8 accounts for practically all TTX-r current during action potentials in these neurons (Akopian et al., 1999). Behavioral studies in Nav1.8-null mice have revealed a complete absence of responses to a tonic noxious mechanical stimulus and attenuated primary (thermal) hyperalgesia evoked by intraplantar injection of nerve growth factor (Akopian et al., 1999; Kerr et al., 2001). Inhibiting the expression of Nav1.8 protein using antisense oligonucleotides also reduces primary hyperalgesia produced by intraplantar prostaglandin E2 or Freund's adjuvant in rats (Khasar et al., 1998; Porreca et al., 1999).
Sensitization of primary afferent nociceptors plays a key role in many forms of visceral pain. TTX-r currents are present in the majority of visceral afferents (Yoshimura et al., 1996; Su et al., 1999), and treatment with Nav1.8 antisense reduces spinal Fos expression evoked by bladder irritation with acetic acid (Yoshimura et al., 2001). Both sets of data suggest a role for Nav1.8 in visceral nociceptive pathways, and therefore, we have assessed visceral pain behavior in Nav1.8-null mice. Previous studies have concentrated on the role of Nav1.8 in primary hyperalgesia (Khasar et al., 1998; Akopian et al., 1999; Porreca et al., 1999; Kerr et al., 2001). Here we have chosen instead to focus on spontaneous pain behavior and secondary hyperalgesia. We have used five different tests that differ in the degree to which pain and hyperalgesia depend on spontaneous, ongoing firing in sensitized nociceptors.
MATERIALS AND METHODS
Adult male mice were used. Mice homozygous for the disrupted allele (−/− or null) were compared with littermate wild-type (+/+) mice (Akopian et al., 1999). All experiments were performed blind to the genetic status of the animals in a room in which the temperature and humidity were controlled (21°C ± 1, 45% ± 5, respectively). The animals were habituated to the test environment for ≥30 min before the test started. After testing, the mice were humanely killed. European Union and State legislation regulating animal experiments were followed. Statistical analyses were performed using ANOVA with post hoc tests or Mann–Whitney U or Student's t tests, as appropriate. The level of statistical significance was set at p < 0.05.
Behavioral responses to intraperitoneal injections of acetylcholine. The mice were placed individually in plastic boxes (20 × 27 × 15 cm). Behaviors counted were: (1) licking of the abdomen, (2) stretching, and (3) squashing of the abdomen against the floor. A volume of 0.3 ml of acetylcholine (0.3 mg/ml) was given intraperitoneally, and the behavior of the mice was observed for 10 min (n = 6–7/group).
Behavioral responses to stimulation of the colon. The methods used were as we have described previously (Laird et al., 2001). Briefly, 0.05 ml of the test substance was administered into the colon. In separate experiments, the effect of isotonic saline, 0.1% capsaicin (in 10% ethanol, 10% Tween 80, and 80% saline; Tocris Cookson, Ballwin, MO), and 1% mustard oil (in 70% ethanol; Aldrich, Milwaukee, WI) were tested. The animal was placed on a raised wire mesh. The spontaneous behavior was observed for 20 min. Behaviors counted were: (1) licking of the abdomen, (2) stretching the abdomen, (3) squashing of the lower abdomen against the floor, and (4) abdominal retractions. The latency of the first such behavior was noted, as were the number and type of behaviors displayed. Referred hyperalgesia was measured by testing the frequency of withdrawal responses to the application of von Frey hairs to the abdomen and hindpaws before intracolonic administration (baseline) and 20 min after.
Cyclophosphamide cystitis. Behavioral experiments examining the effect of cyclophosphamide over 4 hr postinjection were performed in +/+ and −/− mice (n = 8–10/group) according to the methods we have described previously (Olivar and Laird, 1999; Laird et al., 2000). The mice were placed individually in plastic cages. After intraperitoneal injection of 300 mg/kg cyclophosphamide (Sigma, Madrid, Spain) dissolved in saline, the mice were observed for 2 min every 30 min for 4 hr, and their behavior was coded according to the following scale: 0, normal; 1, piloerection; 2, strong piloerection; 3, labored breathing; 4, licking of the abdomen; and 5, stretching and contractions of the abdomen. If more than one of these behaviors was noted in one observation period, the sum of the corresponding points was assigned. The day before cyclophosphamide treatment and again at the end of the 4 hr observation period, the responses to mechanical stimulation of the abdomen were tested using von Frey hairs as described above.
After behavioral testing, the animals were killed, and the bladder was removed, opened, and pinned out. Digital photographs of the mucosal side of the entire bladder were taken at 10× magnification. The degree of damage to the mucosa was assessed from the stored digital images.
Plasma extravasation in the viscera evoked by chemical stimulation. Evan's Blue (50 mg/kg, i.v.) was administered 18 hr before behavioral testing to allow subsequent quantification of plasma extravasation in the bladder or colon. The affected viscus was removed postmortem for determination of Evan's Blue content. The tissue was dried and weighed, the dye was extracted in formamide (60°C for 24 hr), and the absorbance was measured with a spectrophotometer (λ = 620 nm).
RESULTS
Nav1.8-null mice showed no overt differences from wild-type littermates in general behavior or locomotor skills, as reported previously (Akopian et al., 1999). We examined the gross morphology of the abdominal viscera and found no differences between genotypes. We also compared the wet weight at postmortem of the stomach, small intestine, large intestine, and urinary bladder in +/+ and −/− mice (n = 6/genotype) and found no significant differences (data not shown).
Nav1.8-null mice show normal responses to acute noxious visceral stimuli
The response to intracolonic instillation of 0.1 ml of isotonic saline was tested in +/+ and −/− mice. This procedure evoked a mild, brief episode of abdominal licking in both genotypes, as shown in Figure 1A. There were no differences in the number, latency, or type of behaviors between genotypes, indicating that Nav1.8 is not required for detection of acute mechanical stimuli. We also tried another acute stimulus, intraperitoneal acetylcholine. Acetylcholine excites nociceptors without sensitizing them (Steen and Reeh, 1993) and also provokes intense smooth-muscle contractions sufficient to excite visceral receptors (Cervero and Sharkey, 1988); therefore, it is an acute mixed noxious stimulus. In +/+ mice (n = 6), intraperitoneal injection of acetylcholine evoked a brief but intense episode of abdominal contractions (mean number of behaviors, 57 ± 4) lasting <10 min. Nav1.8-null mice (n = 6) showed no significant differences from +/+ mice in the number of behaviors evoked by intraperitoneal acetylcholine (mean number, 51 ± 6), nor was there any difference in the type of behaviors or latency to the first behavior, suggesting that Nav1.8 is not required for detection of acute noxious stimuli in viscera.
Fig. 1.
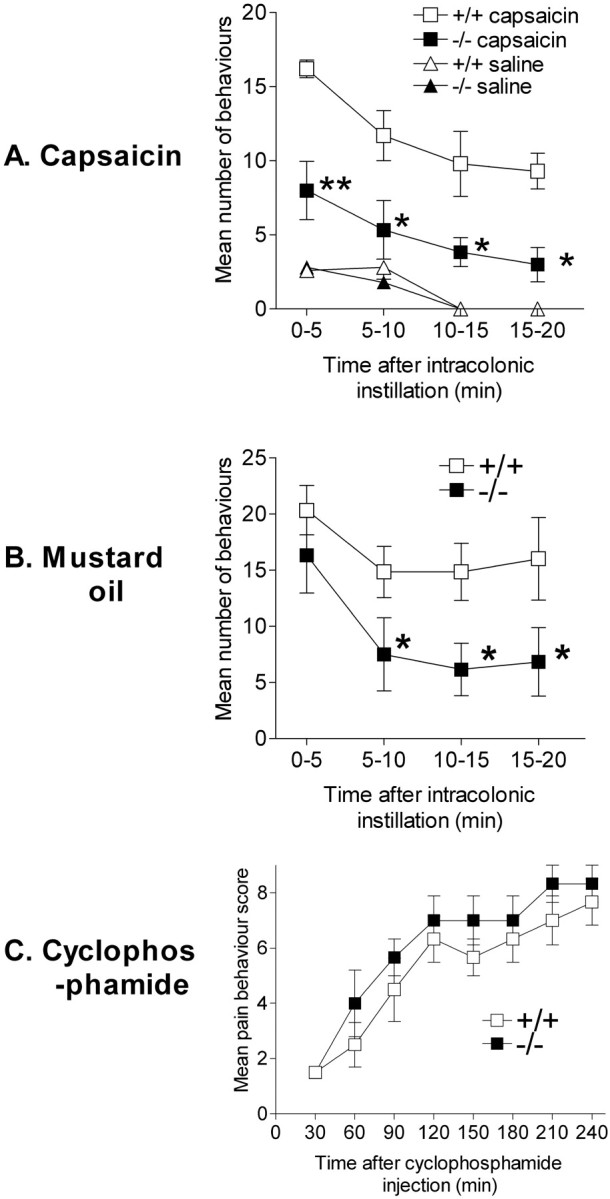
Spontaneous visceral pain-related behavior in wild-type (+/+) and Nav1.8-null (−/−) mice. A, Mean ± SEM number of behaviors per 5 min observation period in the first 20 min after intracolonic instillation of 0.1% capsaicin (n = 6 per group) or isotonic saline (n = 10 per group). B, Mean ± SEM number of behaviors per 5 min observation period in the first 20 min after intracolonic instillation of 1% mustard oil (n = 6 per genotype). C, Time course of mean ± SEM behavioral scores assigned at 30 min intervals over the 4 hr observation period after intraperitoneal injection of 300 mg/kg cyclophosphamide (n = 6 per genotype). For details of scoring system, see Materials and Methods. ∗p < 0.05 and ∗∗p < 0.01 where a significant difference between +/+ and −/− mice was observed, respectively.
Nav1.8-null mice show blunted responses to sensitizing visceral stimuli
Capsaicin produces activation of nociceptors followed by prolonged ongoing activity (because of sensitization) in the absence of additional stimulation (for discussion, see Laird et al., 2001). Intracolonic capsaicin in +/+ mice (n = 6) evoked a strong reaction with a mean latency to the first pain behavior of 17 ± 2 sec. The reaction consisted of abdominal licking and contractions and lasted for ≥20 min with a gradually decreasing intensity over time (Fig. 1A). Null mutants (n = 6) showed a weak pain reaction with significantly fewer behaviors observed (Fig. 1A) after application of intracolonic capsaicin. Furthermore, the latency to the first behavior was significantly longer (mean, 28 ± 4 sec). Although the response of −/− mice to capsaicin was significantly less than that evoked in +/+ mice, it was still greater than that evoked by intracolonic isotonic saline in either genotype.
We subsequently tried the effect of intracolonic 1% mustard oil, because this stimulus activates and sensitizes a mixed population of afferents and also produces tissue damage, in contrast to intracolonic capsaicin, which activates only afferents expressing transient receptor potential V1 (TrpV1 or V1) and produces a pure neurogenic inflammation (Laird et al., 2000, 2001). In +/+ mice (n= 6), intracolonic mustard oil provoked a vigorous pain reaction similar to that seen after intracolonic capsaicin (Fig.1B). The response to intracolonic mustard oil in −/− mice (n = 6) was initially similar. The latency to the first behavior was not significantly different between genotypes, nor was the number of behaviors observed during the first 5 min. However, thereafter, the pain reaction to mustard oil was substantially weaker in −/− mice compared with +/+ mice. These differences in the intensity of the reaction were statistically significant overall (ANOVA; p < 0.05), and post hoc tests revealed significant differences from 5 min onward.
Nav1.8-null mutants show no referred hyperalgesia after intracolonic capsaicin and weak hyperalgesia after intracolonic mustard oil
Tests of mechanical sensitivity of the abdomen and hindpaw 20 min after intracolonic capsaicin in +/+ mice revealed a marked shift of the stimulus response function to the left compared with baseline, evidence of referred hyperalgesia. Significant referred hyperalgesia was seen in both the abdomen (data not shown) and the hindpaws (Fig.2A). In contrast, −/− mice did not show a significant increase in mechanical sensitivity after intracolonic capsaicin either in the hindpaws (Fig.2A) or the abdomen and thus failed to develop signs of referred hyperalgesia. The intracolonic application of mustard oil resulted in significant referred hyperalgesia in both +/+ and −/− mice (Fig. 2B). However, the hyperalgesia observed in −/− mice was significantly weaker than that shown by +/+ mice.
Fig. 2.
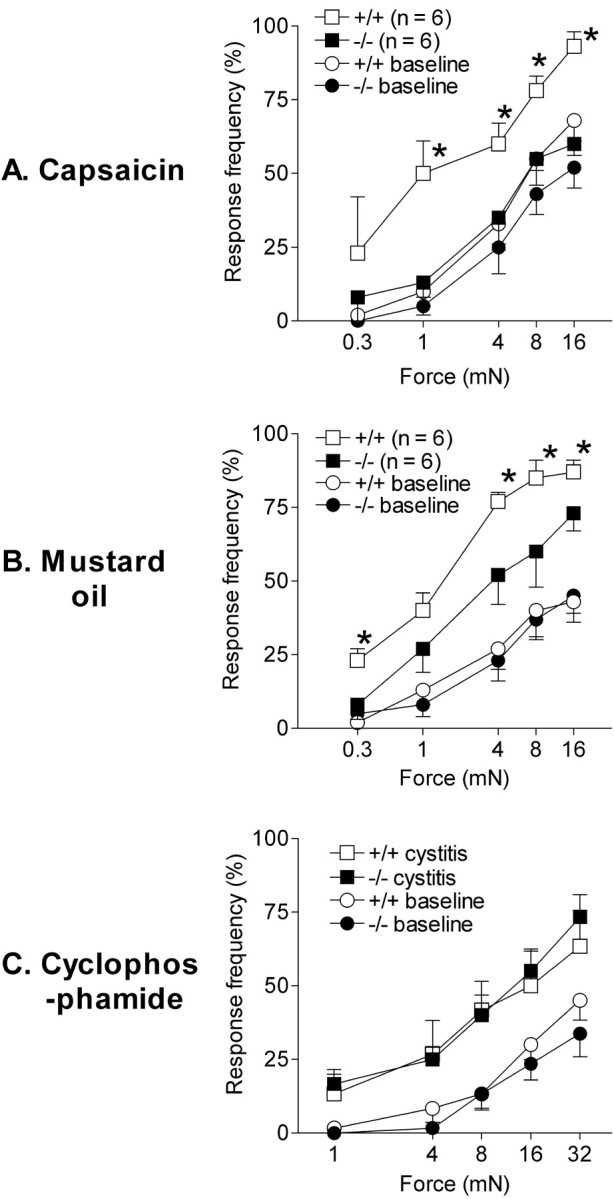
Referred visceral hyperalgesia in wild-type (+/+) and Nav1.8-null (−/−) mice. Referred visceral hyperalgesia was measured as responses to mechanical stimulation of the hindpaws with von Frey hairs of five intensities in wild-type and Nav1.8-null mice. Data are shown as mean percentage response frequency ± SEM before (baseline) and 20 min after intracolonic instillation of 0.1% capsaicin (A), 20 min after intracolonic instillation of 1% mustard oil (B), and 4 hr after 300 mg/kg intraperitoneal cyclophosphamide (C). ∗p < 0.05, indicates forces where a significant difference between +/+ and −/− mice was observed. ANOVA revealed a significant difference overall between genotypes 20 min after intracolonic capsaicin (p = 0.03) and mustard oil (p = 0.02) but not after cyclophosphamide.
Nav1.8-null mice showed blunted pain responses to intracolonic capsaicin or mustard oil. In these models, pain and hyperalgesia, once initiated, are likely maintained by ongoing activity in sensitized nociceptors in the absence of additional stimuli (Laird et al., 2000;Schmelz et al., 2000). These observations could be explained by an involvement of Nav1.8 in sustained spontaneous activity in nociceptors as a result of sensitization, as has been proposed previously (McCleskey and Gold, 1999; Schmelz et al., 2000). However, the present data could also be explained by a role of Nav1.8 in the maintenance of prolonged firing evoked by visceral stimuli. To distinguish between these two possibilities, we tested the responses of Nav1.8-null mice in a model of cyclophosphamide cystitis, where the noxious chemical stimulus tonically increases throughout the test period; thus, the behavior does not depend on sustained spontaneous activity in sensitized nociceptors in the absence of additional stimulation.
Nav1.8-null mice show normal pain responses to cyclophosphamide-induced cystitis
The systemic administration of cyclophosphamide in +/+ mice (n = 6) produced a gradually increasing display of visceral nociceptive behaviors that were scored every 30 min. The behavioral response was clearly established 90–120 min after cyclophosphamide treatment and continued increasing for the remainder of the 4 hr observation period. Nav1.8-null mice (n = 6) showed a very similar behavioral response to systemic cyclophosphamide, such that the number and time course of behaviors were not significantly different (Fig. 1C). Both +/+ and −/− mice also showed marked referred hyperalgesia when tested at 4 hr after cyclophosphamide (Fig. 2C). This referred hyperalgesia was not significantly different between genotypes.
Nav1.8-null mice show normal inflammatory responses to noxious visceral stimuli
At the end of the behavioral experiment, the affected viscus (descending colon or urinary bladder) was removed postmortem, and plasma extravasation was assessed by measuring Evan's Blue content. There were no significant differences between +/+ and −/− mice in the degree of plasma extravasation induced by the different stimuli (Table1). However, there were clear differences in the plasma extravasation between the different types of stimulus. For example, intracolonic mustard oil produced approximately fourfold more plasma extravasation than intracolonic capsaicin in both genotypes.
Table 1.
Plasma extravasation evoked by inflammation of the colon or bladder
| Treatment | +/+ Mice | −/− Mice |
|---|---|---|
| Mustard oil (1%) in colon | 0.51 ± 0.10 | 0.58 ± 0.04 |
| Capsaicin (0.1%) in colon | 0.12 ± 0.01 | 0.12 ± 0.01 |
| Intraperitoneal cyclophosphamide (bladder) | 0.44 ± 0.03 | 0.49 ± 0.03 |
Plasma extravasation was measured as mean Evan's Blue content of the tissue (micrograms of Evan's Blue per milligrams of dry tissue).
In normal animals, cyclophosphamide treatment produces mucosal erosion and hemorrhage of the bladder in addition to edema (Fraiser et al., 1991). An observer who was unaware of the genotype scored bladder mucosal damage from digital images of the tissue obtained at postmortem. No significant differences between genotypes were found (data not shown).
DISCUSSION
In the present study, we found that Nav1.8-null mice showed blunted responses and no referred hyperalgesia in response to intracolonic capsaicin. After application of an acute tissue-damaging inflammatory stimulus, intracolonic mustard oil, Nav1.8-null mice also showed attenuated visceral pain and hyperalgesia. However, Nav1.8-null mice showed no deficits in pain behavior provoked by acute noxious mechanical or mixed mechanical/chemical stimulation of the abdominal viscera or in pain and hyperalgesia evoked by cyclophosphamide cystitis. In all cases, tissue damage and inflammation were no different in Nav1.8-null mice compared with their wild-type littermates.
Nav1.8 is not required for visceral neurogenic or non-neurogenic inflammation
In the present study, Nav1.8-null mice showed no differences from wild type in inflammatory responses to intracolonic capsaicin or to cyclophosphamide, which provokes visceral inflammation wholly or partly via neurogenic mechanisms (Maggi et al., 1992; Laird et al., 2000). Similarly, there were no differences between genotypes in inflammatory responses to intracolonic mustard oil, which is capable of producing inflammation in denervated tissue and thus does not depend on neurogenic mechanisms (Laird et al., 2000). Akopian et al. (1999) have shown previously that Nav1.8 is not required for the development of soft-tissue inflammation after intraplantar carrageenan. Therefore, the differences in pain behavior between Nav1.8 +/+ and −/− mice appear not to be caused by differences in the degree of inflammation evoked by the test stimuli.
Nav1.8 is not required for responses to acute, noxious visceral stimuli
Intracolonic isotonic saline, which produces a brief distension, and intraperitoneal acetylcholine are acute noxious stimuli that do not provoke sensitization of nociceptors or evidence of referred hyperalgesia. Nav1.8-null mice responded normally to these stimuli. We also found that responses to phasic mechanical stimuli applied to the skin were intact in Nav1.8-null mice when testing responses to von Frey hairs applied to the abdomen and paws, as described previously byAkopian et al. (1999). Therefore, Nav1.8 is not essential for normal behavioral responses to acute noxious visceral stimuli.
Nav1.8 is required for the expression of normal visceral pain and hyperalgesia to intracolonic capsaicin or mustard oil
Nav1.8-null mice showed markedly reduced pain responses and no referred hyperalgesia in response to intracolonic capsaicin. After intracolonic mustard oil, Nav1.8-null mice showed attenuated visceral pain and hyperalgesia, but the reduction was less pronounced than after capsaicin. The difference between the responses to these stimuli may be ascribed to the different populations of afferents excited. Capsaicin selectively activates afferent endings expressing VR1 receptors, probably all of which are nociceptors (Caterina and Julius, 2001), and evokes a pure neurogenic inflammation in the colon (Laird et al., 2000). Experiments in isolated DRG neurons labeled from the rat colon have shown that all colon afferents responding to capsaicin expressed large TTX-r sodium currents (Su et al., 1999). This coexpression may explain why the behavioral response to intracolonic capsaicin was so markedly reduced in Nav1.8-null mice in the present study.
Mustard oil excites all classes of unmyelinated afferents, because it is a nonspecific pore-forming substance, and also sensitizes nociceptors (Kress and Reeh, 1996). Tissue damage produced by pore formation in other cell types likely also contributes to a more prolonged activation of nociceptors. The majority of afferents expressing Nav1.8 has unmyelinated axons (Amaya et al., 2000), and most identified nociceptors examined express Nav1.8 (Fang et al., 2001). However, afferents that do not express Nav1.8 but are excited by mustard oil may account for the less pronounced deficit in mustard oil-evoked behavior in Nav1.8-null mice.
Role for Nav1.8 in responses that depend on ongoing activity in sensitized visceral nociceptors
The blunted pain and hyperalgesia to chemical stimuli (intracolonic capsaicin or mustard oil) in Nav1.8-null mice could be attributable to participation of Nav1.8 in sustained spontaneous activity in sensitized nociceptors in the absence of additional stimuli or to an involvement of Nav1.8 in sustained activity per se. The sustained pain and hyperalgesia after capsaicin rely on ongoing activity in nociceptors sensitized by the initial application. As well as sensitizing nociceptors, mustard oil provokes tissue damage, thus providing an additional source of ongoing input that may account for the less pronounced deficit in mustard oil-evoked behavior in Nav1.8-null mice.
A role for Nav1.8 in sustained firing and in control of the firing threshold has been proposed on the basis of the kinetic properties of the channel (McCleskey and Gold, 1999; Baker and Wood, 2001), and a specific role of Nav1.8 in prolonged capsaicin-evoked firing in sensitized nociceptors has also been proposed (Schmelz et al., 2000). An involvement in spontaneous firing in sensitized visceral nociceptors is also supported by the observations of Yoshimura et al. (2001) using a stimulus that acutely sensitizes bladder afferents, infusion of dilute acetic acid into the bladder. They found that spinal Fos expression was inhibited in rats given Nav1.8 antisense compared with rats given mismatch oligonucleotides.
To distinguish between the two possibilities discussed above, we tested the responses of Nav1.8-null mice to intraperitoneal cyclophosphamide. Cyclophosphamide administration produces cystitis as a result of the accumulation in the bladder of a toxic metabolite, acrolein, excreted in urine (Cox, 1979; Fraiser et al., 1991). Acrolein bladder toxicity produces neurogenic edema (Olivar and Laird, 1999; Laird et al., 2000). The toxin accumulates during the course of the 4 hr experiment, and the urothelial barrier gradually breaks down. This slow accumulation is accompanied by a slow increase in pain behaviors likely caused by the increasing stimulus intensity and the progressive recruitment of new populations of bladder nociceptors. Nav1.8-null mice showed no differences from wild types in the pain or referred hyperalgesia induced by cyclophosphamide cystitis, although the majority of bladder afferent neurons express TTX-r currents (Yoshimura et al., 1996). These data suggest that Nav1.8 is not essential for the behavioral response to tonic noxious chemical stimuli. However, the present data do not preclude the possibility that Nav1.8 plays a partial role in the pain behavior produced by this type of stimulus but that compensatory overexpression of other sodium channel subtypes in the −/− mice masks the full Nav1.8-null phenotype. This type of phenotype masking occludes a deficit in thermal hyperalgesia caused by intraplantar carrageenan in these mice (Akopian et al., 1999).
We conclude that the complete lack of referred hyperalgesia and a greatly attenuated pain response after capsaicin observed in Nav1.8-null mice support the idea that Nav1.8 has an essential role in mediating sustained spontaneous activity in sensitized nociceptors. The attenuated response to intracolonic mustard oil in Nav1.8 −/− mice provides additional support for this hypothesis. Our data also suggest that Nav1.8 is not required for normal responses to tonic noxious chemical stimuli or acute noxious stimuli.
Footnotes
This work was supported by the Madrid Regional Government (Contrato Programa), the Ministry of Science and Technology, Spain (SAF-2000-0199), the Medical Research Council, and the Wellcome Trust, UK. J.M.A.L. was a Ramón y Cajal Investigator (Ministry of Science and Technology, Spain). We thank María-JoséGarcía for expert technical assistance and Fleur Geoghegan for help with genotyping.
Correspondence should be addressed to Dr. J. M. A. Laird, Department of Bioscience, AstraZeneca R&D Montreal, 7171 Frederick-Banting, St. Laurent, Quebec H4S 1Z9, Canada. E-mail:jennifer.laird@astrazeneca.com.
REFERENCES
- 1.Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill RG, Stanfa L, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- 2.Amaya F, Decosterd I, Samad T, Plumpton C, Tate S, Mannion RJ, Costigan M, Woolf CJ. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- 3.Baker MD, Wood JN. Involvement of Na+ channels in pain pathways. Trends Pharmacol. 2001;22:27–31. doi: 10.1016/s0165-6147(00)01585-6. [DOI] [PubMed] [Google Scholar]
- 4.Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- 5.Cervero F, Sharkey KA. An electrophysiological and anatomical study of intestinal afferent fibres in the rat. J Physiol (Lond) 1988;401:381–397. doi: 10.1113/jphysiol.1988.sp017168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox PJ. Cyclophosphamide cystitis: identification of acrolein as the causative agent. Biochem Pharmacol. 1979;28:2045–2049. doi: 10.1016/0006-2952(79)90222-3. [DOI] [PubMed] [Google Scholar]
- 7.England JD, Bevan S, Docherty R. PGE2 modulates the tetrodotoxin-sensitive sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol (Lond) 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang X, Djouhri L, Okuse K, Wood JN, Lawson SJ. Sensory and electrophysiological properties of DRG neurones with SNS-like immunoreactivity (SNS-Li) in rats. Soc Neurosci Abstr. 2001;27:819.5. [Google Scholar]
- 9.Fraiser LH, Kanekal S, Kehrer JP. Cyclophosphamide toxicity. Characterising and avoiding the problem. Drugs. 1991;42:781–795. doi: 10.2165/00003495-199142050-00005. [DOI] [PubMed] [Google Scholar]
- 10.Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kerr BJ, Souslova V, McMahon SB, Wood JN. A role for the TTX-resistant sodium channel Nav1.8 in NGF-induced hyperalgesia but not neuropathic pain. NeuroReport. 2001;12:3077–3080. doi: 10.1097/00001756-200110080-00019. [DOI] [PubMed] [Google Scholar]
- 12.Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/s0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- 13.Kress M, Reeh PW. Chemical excitation and sensitization in nociceptors. In: Belmonte C, Cervero F, editors. Neurobiology of nociceptors. Oxford UP; Oxford: 1996. pp. 258–297. [Google Scholar]
- 14.Laird JMA, Olivar T, Roza C, De Felipe C, Hunt SP, Cervero F. Deficits in visceral pain and hyperalgesia of mice with a disruption of the tachykinin NK1 receptor gene. Neuroscience. 2000;98:345–352. doi: 10.1016/s0306-4522(00)00148-2. [DOI] [PubMed] [Google Scholar]
- 15.Laird JMA, Martinez-Caro L, Garcia-Nicas E, Cervero F. A new model of visceral pain and referred hyperalgesia in the mouse. Pain. 2001;92:335–342. doi: 10.1016/S0304-3959(01)00275-5. [DOI] [PubMed] [Google Scholar]
- 16.Maggi CA, Lecci A, Santicioli P, Del Bianco E, Giuliani S. Cyclophosphamide cystitis in rats: involvement of capsaicin-sensitive primary afferents. J Auton Nerv Syst. 1992;38:201–208. doi: 10.1016/0165-1838(92)90031-b. [DOI] [PubMed] [Google Scholar]
- 17.McCleskey EW, Gold MS. Ion channels of nociception. Annu Rev Physiol. 1999;61:835–856. doi: 10.1146/annurev.physiol.61.1.835. [DOI] [PubMed] [Google Scholar]
- 18.Olivar T, Laird JMA. Cyclophosphamide cystitis in mice: behavioural characterisation and correlation with bladder inflammation. Eur J Pain. 1999;3:141–149. doi: 10.1053/eujp.1998.0105. [DOI] [PubMed] [Google Scholar]
- 19.Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels PN3/SNS and NaN/SNS2 in rat models of chronic pain. Proc Natl Acad Sci USA. 1999;96:7640–7644. doi: 10.1073/pnas.96.14.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmelz M, Schmidt R, Handwerker HO, Torebjörk HE. Encoding of burning pain from capsaicin-treated human skin in two categories of unmyelinated nerve fibres. Brain. 2000;123:560–571. doi: 10.1093/brain/123.3.560. [DOI] [PubMed] [Google Scholar]
- 21.Steen KH, Reeh PW. Actions of cholinergic agonists and antagonists on sensory nerve endings in rat skin, in vitro. J Neurophysiol. 1993;70:397–405. doi: 10.1152/jn.1993.70.1.397. [DOI] [PubMed] [Google Scholar]
- 22.Su X, Wachtel R, Gebhart GF. Capsaicin sensitivity and voltage-gated sodium currents in colon sensory neurons from rat dorsal root ganglia. Am J Physiol. 1999;277:G1180–G1188. doi: 10.1152/ajpgi.1999.277.6.G1180. [DOI] [PubMed] [Google Scholar]
- 23.Waxman SG, Cummins TR, Dib-Hajj SD, Fjell J, Black JA. Sodium channels, excitability of primary sensory neurons and the molecular basis of pain. Muscle Nerve. 1999;22:1177–1187. doi: 10.1002/(sici)1097-4598(199909)22:9<1177::aid-mus3>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 24.Yoshimura N, White G, Weight F, De Groat WC. Different types of Na+ and K+ currents in rat dorsal root ganglion neurones innervating the urinary bladder. J Physiol (Lond) 1996;494:1–16. doi: 10.1113/jphysiol.1996.sp021471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshimura N, Seki S, Novakovic SD, Tzoumaka E, Erickson VL, Erickson KA, Chancellor MB, De Groat WC. The involvement of the tetrodotoxin-resistant sodium channel Nav1.8 (PN3/SNS) in a rat model of visceral pain. J Neurosci. 2001;21:8690–8696. doi: 10.1523/JNEUROSCI.21-21-08690.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]