

Abstract
During natural slow-wave sleep (SWS) in nonanesthetized cats, silent (down) states alternate with active (up) states; the down states are absent during rapid-eye-movement sleep and waking. Oscillations (<1 Hz) in SWS and transformation to an activated awake state were investigated with intracellular recordings in vivo and with computational models of the corticothalamic system. Occasional summation of the miniature EPSPs during the hyperpolarized (silent) phase of SWS oscillation activated the persistent sodium current and depolarized the membrane of cortical pyramidal (PY) cells sufficiently for spike generation. In the model, this triggered the active phase, which was maintained by lateral PY–PY excitation and persistent sodium current. Progressive depression of the excitatory interconnections and activation of Ca2+-dependent K+ current led to termination of the 20–25 Hz activity after 500–1000 msec. Including thalamocortical (TC) and thalamic reticular neurons in the model increased the duration of the active epochs up to 1–1.5 sec and introduced waning spindle sequences. An increase in acetylcholine activity, which is associated with activated states, was modeled by the reduction in the K+ leak current in PY and TC cells and by a decrease in intracortical PY–PY synaptic conductances. These changes eliminated the hyperpolarizing phases of network activity and transformed cortical neurons to tonic firing at 15–20 Hz. During the transition from SWS to the activated state, the input resistance of cortical neurons gradually increased and, in a fully activated state, reached the same or even higher values as during silent phases of SWS oscillations. The model describes many essential features of SWS and activated states in the thalamocortical system as well as the transition between them.
Keywords: slow-wave sleep, waking, thalamus, cortex, sensory input, input resistance, network model
When the brain falls asleep, the spatiotemporal patterns in the waking corticothalamic system (Steriade et al., 2001; Timofeev et al., 2001b) are replaced by low-frequency synchronous rhythms that are relatively insensitive to incoming signals (Steriade et al., 1993b). Intracellular and local field potential recordings in vivo and in vitro have revealed that spindles (7–14 Hz) are a result of interactions between reticular (RE) and thalamocortical (TC) neurons in the thalamus (Steriade et al., 1985, 1993b; von Krosigk et al., 1993). Spindle sequences are initiated in the thalamic RE nucleus (Steriade et al., 1987; Bazhenov et al., 1999, 2000) and are terminated through a combination of intrinsic (Bal and McCormick, 1996; Budde et al., 1997; Lüthi et al., 1998) and network (Timofeev et al., 2001a) mechanisms. Thalamic delta (1–4 Hz) oscillations can be generated in a single TC cell by the interplay of intrinsic currents (McCormick and Pape, 1990; Soltesz et al., 1991;Lytton et al., 1996). Delta and spindle oscillations have been studied intensively with computational models (Destexhe et al., 1994a, 1996;Golomb et al., 1994, 1996; Contreras et al., 1996b; Lytton et al., 1996; Bazhenov et al., 2000).
Less is known about mechanisms underlying the slow (<1 Hz) cortical oscillation that occurs during SWS in animals and humans (Steriade et al., 1993b,c; Achermann and Borbely, 1997; Amzica and Steriade, 1997). The depth-positive phase of slow oscillation observed in the depth electroencephalogram (EEG) is associated with significant hyperpolarization of neocortical neurons, whereas the depth-negative component of the slow oscillation in the depth EEG is related to relative depolarization and firing of cortical neurons (Contreras and Steriade, 1995). The survival of slow oscillations after extensive thalamic lesions (Steriade et al., 1993d) and the absence of slow oscillations in the thalamus of decorticated cats (Timofeev and Steriade, 1996) point to an intracortical origin for this rhythm. Intracellular studies on anesthetized and nonanesthetized cats have shown that the hyperpolarizing phase of the slow oscillation is associated with disfacilitation, a temporal absence of synaptic activity (Contreras et al., 1996a; Timofeev et al., 1996, 2001b;Steriade et al., 2001). The long-lasting hyperpolarizations of cortical neurons are absent when brain cholinergic structures are set into action (Metherate and Ashe, 1993; Steriade et al., 1993a) or during REM sleep and waking (Steriade et al., 2001; Timofeev et al., 2001b).
Recent intracellular and local field potential recordings from isolated cortical slabs in vivo revealed patterns of spontaneous activity lasting 0.5–3 sec and appearing every 20–600 sec (Timofeev et al., 2000a). Intracellular recordings of membrane potentials during the silent phase of oscillations revealed the presence of small amplitude depolarizing potentials, but action potentials were never observed between bursts. The frequency of these miniature events was reduced immediately after the last burst and increased a few seconds later. It has been suggested that the random summation of the miniature EPSPs (minis) during the silent state of the network can depolarize the membrane potential in any single pyramidal (PY) cell sufficiently to activate a persistent sodium current, which further depolarizes the neuron leading to the Na+spike (Timofeev et al., 2000a). The firing of one or a few cortical neurons could initiate activity in the whole cortical network. Experiments with varying the slab size and scaling analysis of a cortical model indicated that in a cortical network significantly larger than a small slab, the probability of active state initiation increases and can drive oscillations in the frequency range of SWS activity (Timofeev et al., 2000a). In the present paper we providein vivo data and a model of a corticothalamic system that closely matches activities found during SWS and activated states. Our modeling study suggests that cortically generated slow oscillations can interfere with signal processing in thalamocortical systems.
MATERIALS AND METHODS
In vivo recordings
We performed two groups of experiments. In the first, we used conventional intracellular recordings from neocortical, thalamocortical, and reticular thalamic neurons in cats anesthetized with either ketamine–xylazine (10–15 mg/kg and 2–3 mg/kg, i.m.) or somnotol (35 mg/kg, i.p). The details of these experiments have been described previously (Timofeev et al., 1996; Timofeev and Steriade, 1997; Steriade et al., 1998). The second group of experiments was conducted on nonanesthetized, nonparalyzed cats in different states of vigilance. Details on experimental procedures can be found in Steriade et al. (2001) and Timofeev et al. (2001b). At the end of experiments, cats were injected intravenously with a lethal dose (50 mg/kg) of sodium thiopental.
Computational model
Intrinsic currents: thalamus. We examined single-compartment models of TC and RE cells that included voltage- and calcium-dependent currents described by Hodgkin–Huxley kinetics:
| Equation 1 |
where Cm = 1 μF/cm2 is the membrane capacitance,gL is the leakage conductance (gL = 0.01 mS/cm2 for TC cell andgL = 0.05 mS/cm2 for RE cell), andEL is the reversal potential (EL = −70 mV for TC cell andEL = −77 mV for RE cell).Iint is a sum of active intrinsic currents, andIsyn is a sum of synaptic currents. The area of an RE cell wasSRE = 1.43 · 10−4 cm2, and the area of a TC cell was STC = 2.9 · 10−4cm2.
For both RE and TC cells we considered a fast sodium current,INa, a fast potassium current,IK (Traub and Miles, 1991), a low-threshold Ca2+ current,IT [see Huguenard and Prince (1992)for RE cells and Huguenard and McCormick (1992) for TC cells]), and a potassium leak current, IKL =gKL(V −EKL),EKL = −95mV. A hyperpolarization-activated cation current,Ih(McCormick and Pape, 1990; Destexhe et al., 1996), was also included in TC cells. The expressions for voltage- and Ca2+-dependent transition rates for all currents are given in Bazhenov et al. (1998). The maximal conductances were gK = 10 mS/cm2,gNa = 90 mS/cm2,gT = 2.2 mS/cm2,gh = 0.017 mS/cm2,gKL = 0−0.03 mS/cm2 for TC cells, andgK = 10 mS/cm2,gNa = 100 mS/cm2,gT = 2.3 mS/cm2, andgKL = 0.005 mS/cm2 for RE cells.
Intrinsic currents: cortex. The cortical PY cells and interneurons (INs) were two-compartment models with channels that were modeled by Hodgkin–Huxley kinetics (Mainen and Sejnowski, 1996):
| Equation 2 |
where Cm,gL are the membrane capacitance and the leakage conductance of the dendritic compartment,EL is the reversal potential,VD andVS are the membrane potentials of dendritic and axosomatic compartments,IDint andISint are the sums of active intrinsic currents in axosomatic and dendritic compartments, Isyn is a sum of synaptic currents, and g is the conductance between axosomatic and dendritic compartments. In this model, the axosomatic compartment has no capacitance to speed up simulations. The soma and axon initial segment are characterized by high-conductance densities. When capacitance was included in this compartment, smaller integration steps were needed to ensure stability of calculation, but model firing patterns were unaffected (Mainen and Sejnowski, 1996).
The model included the fast Na+ channels,INa, of a high density in axosomatic compartment and of a low density in the dendritic compartment. A fast delayed rectifier potassium K+ current,IK, was present in the axosomatic compartment. Persistent sodium current,INa(p), was included in the axosomatic and dendritic compartments (Alzheimer et al., 1993; Kay et al., 1998). A slow voltage-dependent noninactivating K+ current,IKm, a slow Ca2+-dependent K+ current,IKCa, a high-threshold Ca2+ current,IHVA, and a potassium leak current,IKL =gKL(V −EKL), were included in the dendritic compartment. The expressions for the voltage- and Ca2+-dependent transition rates for all currents are given in Timofeev et al. (2000a). The maximal conductances and passive properties were Ssoma = 1.0 · 10−6cm2, gNa= 3000 mS/cm2,gK = 200 mS/cm2,gNa(p) = 0.07 mS/cm2 for axosomatic compartment, andCm = 0.75 μF/cm2,gL = 0.033 mS/cm2,gKL = 0−0.0025 mS/cm2,Sdend =Ssomar,gHVA = 0.01 mS/cm2,gNa = 1.5 mS/cm2,gKCa = 0.3 mS/cm2,gKm = 0.01 mS/cm2, andgNa(p) = 0.07 mS/cm2 for dendritic compartment;EL = −68 mV andEKL = −95 mV. NoINa(p) was modeled for IN cells. The resistance between compartments was r = 10 MΩ.
The firing properties of the model in Equation 2 depend on the coupling conductance between compartments (g = 1/R) and the ratio of dendritic area to axosomatic arear (Mainen and Sejnowski, 1996). We used a model of a regular-spiking neuron for PY cells (r = 165) and a model of a fast spiking neuron for IN cells (r = 50).
Synaptic currents
All synaptic currents were calculated according to:
| Equation 3 |
where gsyn is the maximal conductivity, [O](t) is the fraction of open channels, Esyn is the reversal potential.EsynAMPA= 0 mV for AMPA and NMDA receptors,EsynGABAA= −70 mV for GABAA receptors in RE and PY cells,EsynGABAA= −80 mV for GABAA receptors in TC cells (Ulrich and Huguenard, 1997), andEsynGABAB= −95 mV for GABAB receptors. A simple phenomenological model was used to describe short-term depression of intracortical excitatory connections (Abbott et al., 1997; Tsodyks and Markram, 1997; Galarreta and Hestrin, 1998; Timofeev et al., 2000a). According to this, a maximal synaptic conductance was multiplied to depression variable, D ≤ 1, representing the amount of available “synaptic resources.” D = 1 − (1 − Di(1 −U))exp(−(t −ti)/τ), where U = 0.07 is the fraction of resources used per action potential, τ = 700 msec the time constant of recovery of the synaptic resources,Di is the value of Dimmediately before the ith event, and (t − ti) is the time after ith event.
GABAA, NMDA, and AMPA synaptic currents were modeled by first-order activation schemes (Destexhe et al., 1994b). Dependence on postsynaptic voltage for NMDA receptors was 1/(1 +exp(−(Vpost −Vth)/ς)), whereVth = −25 mV, ς = 12.5 mV (Traub et al., 1991; Destexhe et al., 1994b; Golomb and Amitai, 1997). GABAB receptors were modeled by a higher-order reaction scheme that took into account activation of K+ channels by G-proteins (Dutar and Nicoll, 1988; Destexhe et al., 1994b; Destexhe et al., 1996). The equations for all synaptic currents are given in Bazhenov et al. (1998)and Timofeev et al. (2000a). The maximal conductances (for each synapse) weregAMPA(PY−PY)= 0.08–0.15 μS,gNMDA(PY−PY)= 0.01 μS,gAMPA(PY−TC)= 0.08–0.025 μS,gAMPA(PY−RE)= 0.05 μS,gAMPA(TC−PY)= 0.1 μS,gAMPA(PY−IN)= 0.05 μS,gNMDA(PY−IN)= 0.008 μS,gGABAA(IN−PY)= 0.05 μS,gAMPA(TC−IN)= 0.1 μS,gGABAA(RE−RE)= 0.2 μS,gGABAA(RE−TC)= 0.2 μS,gGABAB(RE−TC)= 0.04 μS, andgAMPA(TC−RE)= 0.4 μS.
Spontaneous miniature EPSPs and IPSPs followed the same equations as the regular PSPs, and their arrival times were modeled by Poisson processes (Stevens, 1993), with time-dependent mean rate μ1(t) = (2/(1 +exp(−(t −t0)/400)) − 1)/100 or μ2(t) =log((t −t0 + 50)/50)/400 (see Fig.4B), where t0 is a time instant of the last presynaptic spike (Timofeev et al., 2000a). The mini amplitude was ∼0.75 mV.
Fig. 4.

Patterns of spontaneous activity in the model network of PY and IN cells. A, On theleft, network size increase from 20 PY–5 IN cells to 100 PY–25 IN cells led to higher frequency of spontaneous bursting and increased its regularity. On the right, an increase of the miniature EPSP amplitudes in the network of 20 PY–5 IN cells up to 170% of that on the left had a similar effect as an increase of network size. B, The shape of the function describing the mini average rate increase (logarithmic vs exponential) had little effect on the network dynamics.
Network geometry and stimulation
The cortical model consisted of a one-dimensional two-layer array of N PY and M = N/4 IN cells. The thalamocortical model consisted of a one-dimensional four-layer array of N PY, M = N/4 IN, L = N/2 RE and L =N/2 TC cells (Fig. 1).N was varied between 20 and 200. In most of simulations the connection fan-out was ±5 cells for AMPA- and NMDA-mediated PY–PY synapses; ±1 cell for AMPA- and NMDA-mediated PY–IN synapses; ±5 cells for GABAA-mediated IN–PY synapses; ±10 cells for AMPA-mediated TC–PY synapses; ±2 cells for AMPA-mediated TC–IN synapses; ±5 cells for AMPA-mediated PY–TC and PY–RE synapses; and ±5 cells for AMPA-, GABAA-, and GABAB-mediated synapses between RE and TC cells. Other radii of synaptic interconnections and N/M/L ratios were tested to ensure robustness of the results. We also tested the network including GABAergic synaptic connections between INs. IN–IN coupling did not have any significant effect on the spatiotemporal patterns of network activity, and this type of connection was excluded from most of simulations. Some of the intrinsic parameters of the neurons in the network were initialized with random variability (Gaussian distribution with ς = 5–10%) to ensure the robustness of the results (Bazhenov et al., 1998).
Fig. 1.

Network geometry. Network model included four layers of neurons with N PY, M IN, L RE, and L TC cells. In most simulations we used N = 100, M = 25, and L = 50.
In simulations with external input, 25% of the TC cells were stimulated by Poisson-distributed spike trains that were homogeneous across TC cells and the firing rates of which were calculated asr = ω + (0.9 ω)sin(2πΩt), where the average rate was ω = 25 Hz and Ω = 0.4, 1, and 2.5 Hz.
Computational methods
All simulations described in this paper were performed using the fourth-order Runge-Kutta [RK(4)] method and in some cases the embedded Runge-Kutta [RK6(5)] method (Enright et al., 1995) with a time step of 0.02 msec. Source C++ code was compiled on a Dell Workstation 420 (1 GHz) using a GCC compiler. A network of 225 cells took ∼20 min of computer time to simulate 1 sec of real time.
RESULTS
Intracellular activities of neocortical neurons during three major states of vigilance
We recorded intracellular activities from >700 cortical neurons during SWS, REM sleep, and waking in chronically implanted cats. The major states of vigilance were routinely characterized by recordings of EEG, electromyogram, and electro-oculogram (Steriade and McCarley, 1990). We distinguished SWS by low-frequency, high-amplitude slow waves in the EEG, the presence of muscle tone, and the absence of eye movements. REM sleep was distinguished by activated EEG pattern, REMs, and the absence of muscle tone. The waking state was identified by activated EEG, muscle tone accompanied by episodic contractions, and sporadic eye movements. Intracellular recordings during natural slow-wave sleep revealed that the distribution of the membrane potential of neocortical neurons was bimodal (Fig.2). The depolarizing state of neurons during SWS corresponded to EEG depth-negativity and periods that immediately follow the EEG depth-negative wave. Most of the neurons fired action potentials during the depolarizing state. The hyperpolarizing state of neurons during SWS corresponded to EEG depth-positive waves. During REM sleep and the waking state, neurons displayed a unimodal distribution of the membrane potential centered on −62 mV. In association and sensory cortical areas, regular spiking neurons stopped firing during ocular saccades in REM sleep (Fig. 2) because of increased firing of INs (Timofeev et al., 2001b). The waking state was characterized by tonic firing of neurons and a unimodal distribution of the membrane potential, similar to that in REM sleep.
Fig. 2.

Intracellular activity of neocortical neuron during three major states of vigilance in a chronically implanted, nonanesthetized cat. High-amplitude and low-frequency field potentials, intracellular cyclic hyperpolarizing potentials, and stable muscle tone are distinctive features of SWS. Low-amplitude and high-frequency field potential oscillations, tonic neuronal firing with small fluctuations in the membrane potential, rapid eye movements, and muscle atonia are cardinal features of REM sleep. There is a slight hyperpolarization during REM-related ocular saccades. Low-amplitude and high-frequency field potential oscillations, tonic firing with smaller fluctuations in the membrane potential, and muscle tone with periodic contractions are characteristics of the waking state. The parts indicated byarrows are expanded below. At bottom are histograms of membrane potential distribution during three states of vigilance. Histograms were constructed by sampling of neuronal activity at 10 kHz and counting the number of samples with bins of 0.5 mV.
Intracellular activities of cortical and thalamocortical neurons during slow sleep oscillation in anesthetized cats
Intracellular membrane potentials of neocortical and thalamic neurons were studied in cats anesthetized with ketamine–xylazine by simultaneous EEG and dual intracellular recordings. Injecting ketamine and xylazine to mimic this type of anesthesia induced more stereotyped activity compared with electrophysiological patterns that were found in naturally sleeping cats (Grenier et al., 2001, their Fig. 13). We recorded activities of cortical neurons from area 4 and of TC neurons from the ventrolateral nucleus or the rostrolateral sector of the RE nucleus. As in previous studies, cortical, thalamocortical, and reticular thalamic neurons were hyperpolarized during depth-positive wave of EEG and displayed waning spindles during depth-negative EEG wave (Contreras and Steriade, 1995; Timofeev and Steriade, 1996). We emphasize here that although waning spindles were clearly distinguished in the majority of thalamic neurons during ketamine–xylazine anesthesia, clear-cut spindles were not as obvious in many neocortical neurons. Figure 3 provides an example of dual intracellular recordings from a cortical and a TC neuron during an EEG depth-negative wave, in which the TC neuron revealed spindle-related IPSPs sometimes associated with rebound spike bursts, whereas the firing and subthreshold fluctuations of the membrane potential of the cortical neuron did not show modulation at spindle frequency. Intracellular recordings from cortical neurons in naturally sleeping cats revealed that the vast majority of neurons did not show intracellular signs of spindles (Timofeev et al., 2000b, 2001b;Steriade et al., 2001). Indeed, the intrinsic properties of neocortical neurons in a sleeping (anesthetized) state modify input signals that might result in desynchronization of the corticothalamic network (Timofeev et al., 2001a).
Fig. 3.
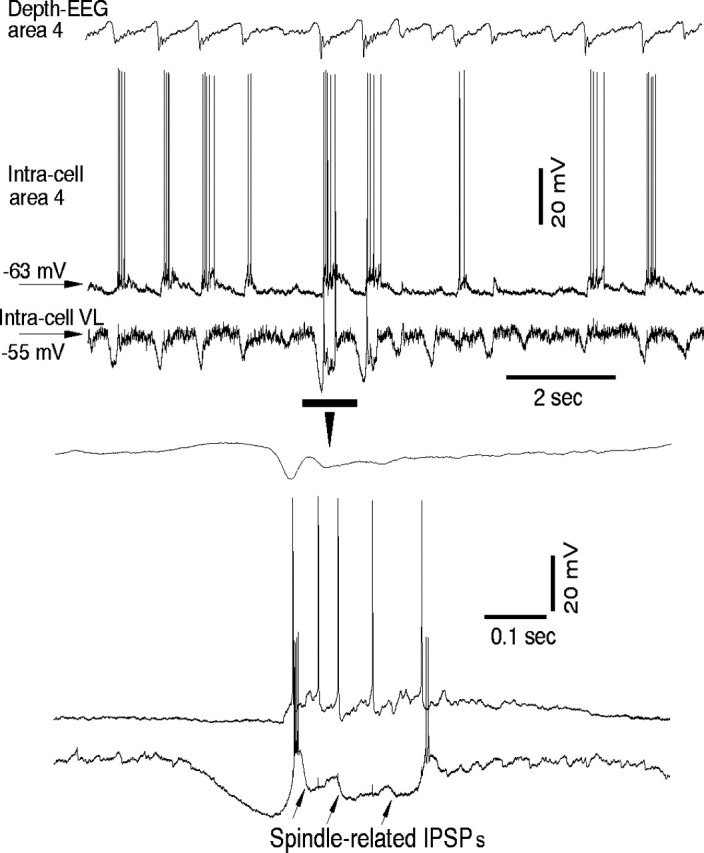
Sleep spindles in cortical and thalamocortical neurons during the slow oscillation under ketamine–xylazine anesthesia. Three traces show depth-EEG from area 4, intracellular activity from an area 4 cortical neuron, and intracellular activity of a thalamocortical neuron from the ventrolateral nucleus of the thalamus. The part indicated by the horizontal bar is expanded at the bottom. Note that the thalamocortical neuron does not fire during all cycles of the slow oscillation, and the cortical neuron does not follow all spike bursts of the thalamocortical neuron.
On the basis of these experimental data, we developed a computational model of the thalamocortical system that closely matched the activities of cortical and thalamic neurons during slow-wave sleep and its transition to activated states that correspond to wakefulness or REM sleep.
Spontaneous activity in the isolated cortical network
Poisson processes with time-dependent mean rate were used to model the arrival times for spontaneous miniature EPSPs at AMPA-mediated synapses between cortical cells (see Materials and Methods) (Timofeev et al., 2000a). Starting from the last Na+spike during a preceding active period, the rate at each synapse increased rapidly during first few seconds and then grew more slowly as observed experimentally (Timofeev et al., 2000a). Examples of different rate functions used in this study are shown in Figure4B. A random summation of miniature EPSPs in one of the PY cells in the network could be sufficient to depolarize this cell to the level of membrane potential where the persistent Na+ current,INa(p), is activated.INa(p) activation was followed then by a Na+ spike, initiating spread of activity over the whole network. A single spike in a PY cell was able to initiate spikes in other neurons that were relatively depolarized by miniature synaptic events. Network activity was maintained byINa(p) activation and PY–PY AMPA- and NMDA-mediated excitatory interconnections. Because a minimal threshold of synaptic excitation was required to maintain firing, depression of PY–PY interconnections supplemented by slow activation of the Ca2+-dependent K+ current eventually terminated firing, and the network switched back to the down (silent) state. A sufficiently high level of IN-mediated inhibition was also required to ensure transitions to the down state (see below).
An example of spontaneous activity in the network with 20 PY–4 IN cells is shown in Figure 4A (left panel,top). In such a small network the probability that one of the PY neurons could reach a level of depolarization sufficient forINa(p) activation was relatively small, producing a low rate of bursting (25–50 sec intervals between bursts) and a high variability of interburst intervals. Because the miniature EPSPs were independent at different synapses and different cells, as the network size increased the probability that a Na+ spike would be generated in one of the neurons increased. Figure 4A (left panel,middle and bottom) shows examples of networks with 60 PY–15 IN cells and 100 PY–25 IN cells. In the latter case the network fired more regularly and at a higher frequency of ∼0.05 Hz. An increase of the miniature EPSP amplitude had a similar effect on the network activity. Figure 4A (right panel) shows a network with 20 PY–4 IN cells, which fired quite regularly when the mini amplitude was increased by ∼50%.
In a previous analytical model based on experimental data for amplitude and rate of miniature events, the mini-dependent mechanism drove periodic network oscillations at frequencies of 0.2–0.5 Hz when the network size exceeded ∼108 neurons (Timofeev et al., 2000a). This is in the frequency range of SWS oscillations (Steriade et al., 1993c,d). Computer simulations involving this many conductance-based model neurons is not yet feasible, so we increased the amplitude of miniature events by ∼50% in our simulations to obtain SWS-like oscillations in relatively small networks.
Figure 4B shows examples of the membrane potential for one PY cell from the same network shown in Figure4A (right panel, bottom) using different functions for the mean rate of Poisson processes (see Materials and Methods). The overall patterns of network activation were virtually identical in both cases, suggesting that the properties of SWS oscillations are independent of the shape of the rate versus time curve when the network size or miniature event amplitudes are large enough.
Spatiotemporal properties of SWS oscillations
In the model, each pattern of activity started from a spike in one of the PY cells and then spread over the network. Figure5A shows two-dimensional (2-D) plots of PY activity in the network with 100 PY–25 IN cells. The spatiotemporal patterns depended on the coupling strengths between PY cells and between PY and IN cells. Generally, an increase of the maximal conductance between PY neurons increased the duration of active phases. An increase of maximal conductances between PY and IN cells or IN and PY cells had more complex effects. The example in Figure5A shows that an increase of the AMPA-mediated conductance from PY to IN cells increased the regularity of SWS oscillations. This occurred because strengthening PY to IN connections enhanced inhibitory feedback and made possible earlier and more reliable termination of the active states. This effect was similar to a direct increase of the GABAA-mediated conductance from IN to PY cells (data not shown). However, an excessive increase of GABAergic coupling between IN and PY cells also significantly reduced the duration of the active states. When GABAA-mediated conductances from IN to PY cells were blocked, the active phase lasted continuously. Introducing GABAergic inhibitory connections between INs had little effect and was generally similar to reducing GABAA-mediated conductance from IN to PY cells (data not shown). This effect of IN–IN coupling was further reduced when the reversal potential for GABAA synapses in INs was depolarized relative to the IN resting potential (Paré et al., 2000). IN cells generally showed patterns of activity that were similar to those in PY cells (see Fig. 7). Thus, silent (down) states were not maintained by GABAA IPSPs but reflected disfacilitation in the cortical network.
Fig. 5.
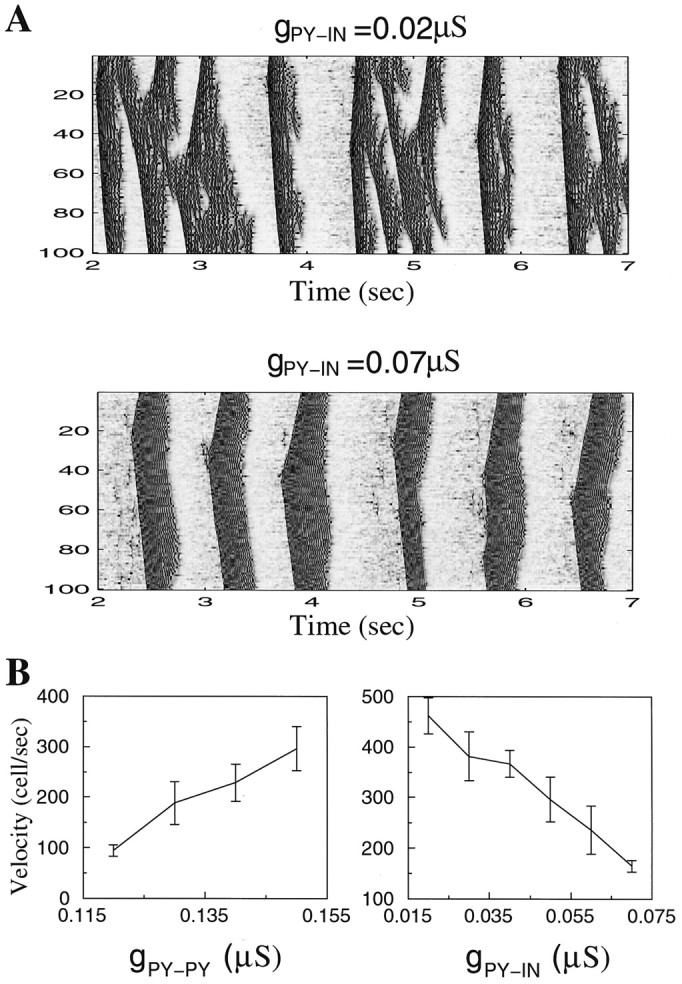
Effect of PY–PY and PY–IN conductances on spontaneous waves propagation. A, Reduction of the maximal conductances for PY–IN synapses decreased the regularity of spontaneous patterns. B, Velocity of propagation (in cells per second) versus maximal conductances for PY–PY and PY–IN synapses.
Fig. 7.

Membrane potential traces of individual PY, IN, RE, and TC cells from the network in Figure6. Spontaneous firing in the PY–IN network initiated waning spindles in the RE–TC network. The spindles in TC cells usually started with two to three cycles of subthreshold (no spikes) oscillations mediated by inhibitory input from RE neurons.
Spiking patterns propagated through the network with a velocity that depended on the maximal conductances for synaptic interconnections, as illustrated in Figure 5B. An increase of the excitatory conductance between PY cells in the network with fixed radii of synaptic interconnections increased the propagation velocity; changes of maximal conductance between PY and IN cells had the opposite effect. This phenomenon has been reported previously in the theoretical study of the network of excitatory and inhibitory cortical neurons (Golomb and Ermentrout, 2001, 2002).
SWS oscillations in the thalamocortical network
To explore the properties of SWS oscillation in a thalamocortical network, we simulated networks with 100 PY–25 IN–50 RE–50 TC cells (Fig. 1). Although thalamic RE and TC cells were not necessary to maintain SWS oscillations in the model, their presence changed spatiotemporal patterns of SWS activity. Figure6 shows 2-D spatiotemporal plots of PY and TC networks during SWS oscillations. Each active phase was initiated in one of the cortical PY cells and then spread over the thalamocortical network. Figure 6 shows that isolated spikes occurred in many PY cells during silent phases of SWS oscillations. These spikes were not able to induce postsynaptic response unless the postsynaptic cell itself was sufficiently depolarized by random mini summation. PY cells fired at a higher frequency during the initial phase of the depolarized state and at a reduced firing rate after 200–300 msec (Fig. 7). The IN cell shown in Figure 7 (bottom) fired before the PY cell because it received direct excitatory input from one of the PY neurons that fired earlier in the cycle as indicated by the local field potential trace. In TC cells, activity patterns always started from hyperpolarization because the immediate effect of PY spiking was a burst of spikes in RE cells (Fig. 7). This hyperpolarization was followed by deinactivation of low-threshold Ca2+ current,IT, and rebound low-threshold spike. Only a few TC cells fired Na+ spikes in the second cycle; in many cases TC cells showed a few cycles of subthreshold ∼10 Hz oscillations followed by a few cycles with low-threshold spikes crowned by action potentials. This pattern is a signature of waning spindle oscillations usually observed during active phases of SWS (Timofeev and Steriade, 1996). The main factor contributing to spindle termination in this model was powerful and nonsynchronous AMPA-mediated PY → RE input depolarizing RE cells that inactivated IT and terminated rebound oscillations in the RE–TC circuit (Fig. 7) [see also Timofeev et al. (2001a)].
Fig. 6.
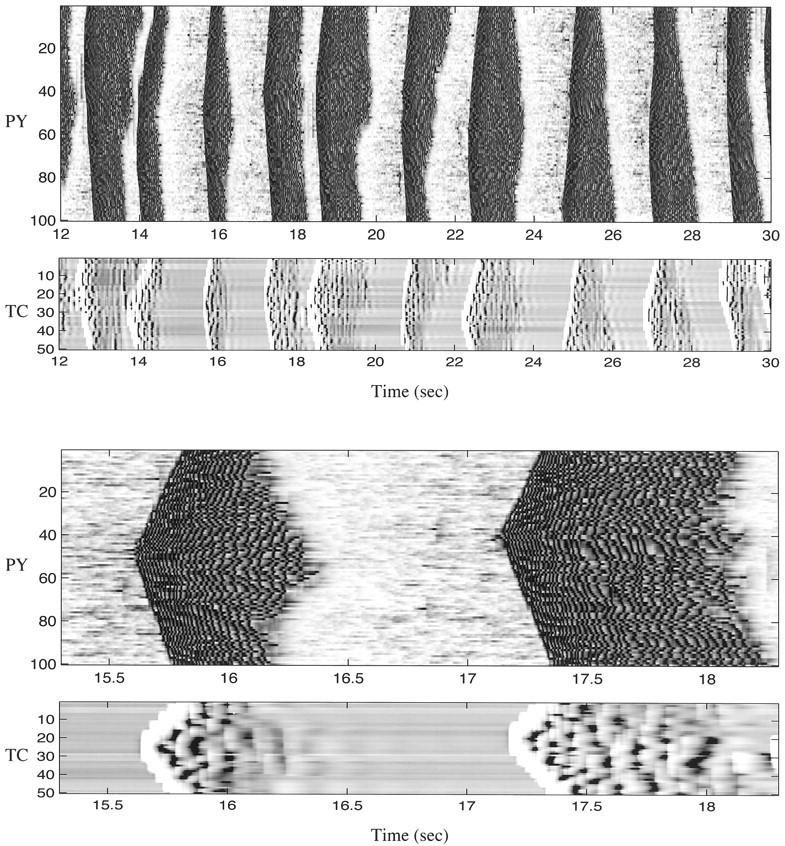
Two-dimensional plots of spontaneous activity in the thalamocortical network. One hundred PY–25 IN–50 RE–50 TC cells were simulated. Two active periods are expanded below. It shows that each pattern of activity was randomly initiated at different foci of the network.
AMPA-mediated input from TC to PY cells induced additional depolarization, increasing the duration of cortical active phases up to 1–1.5 sec (compare Figs. 5A and 6). In some cases, thalamic spindles outlasted cortical activity, which also reduced the duration of corresponding down states in the cortical network; TC-mediated EPSPs arriving at the top of mini-induced depolarization in PY cells could initiate new active patterns after ∼0.5 sec or less. Thus, compared with the isolated cortical network, SWS sleep oscillations in the full thalamocortical model were characterized by prolonged active (up) states and reduced silent (down) states. This prediction is consistent with recordings from cortical slice preparations in which SWS oscillations are characterized by prolonged down and relatively short up states (Sanchez-Vives and McCormick, 2000).
Transition from SWS oscillations to activated state
Activation of neuromodulatory systems controls the transition from sleep to activated states (Steriade and McCarley, 1990). An increase in the level of acetylcholine release associated with activation of nicotinic and muscarinic receptors depolarizes cortical pyramidal cells and thalamic relay cells both in awake states and during REM sleep. This effect is achieved mainly by blocking resting potassium conductances in these cells (McCormick, 1992). We modeled the effect of cholinergic excitation by blocking potassium leak current in PY and TC cells. Figure 8A1 shows examples of PY, RE, and TC cells from the network 100 PY–25 IN–50 RE–50 TC cells in which IKleakcurrent was completely blocked in PY and TC cells. Cortical depolarization eliminated the silent phases of SWS oscillation, and PY cells fired continuously in the frequency range of 30–40 Hz. RE and TC cells received tonic excitation from cortex and were silent.
Fig. 8.

Effects of synaptic conductances and potassium leak current on network activity. Decrease of potassium leak current inPY and TC cells transformed SWS oscillations into persistent firing at frequency depending on PY–PY synaptic conductance. A, In the presence of the strongRE–TC–RE coupling the RE–TC network generated spontaneous spindles even during continuous firing in the PY–IN network.A1,gPY−PY = 0.15 μS, gRE−TC = 0.2 μS,gTC−RE = 0.4 μS. A2,gPY−PY = 0.09 μS, gRE−TC = 0.2 μS,gTC−RE = 0.4 μS. A3,gPY−PY = 0.09 μS, gRE−TC = 0.1 μS,gTC−RE = 0.2 μS. B, Input resistance of PY cells and frequency of spontaneous firing versus maximal conductances for AMPA-mediated PY–PY synapses (gKL = 0,gRE−TC = 0.1 μS, gTC−RE = 0.2 μS) and potassium leak channels (gPY−PY= 0.08 μS,gRE−TC = 0.1 μS, andgTC−RE = 0.2 μS).
The application of the cholinergic agonist muscarine depresses intracortical EPSPs by ∼35–40%; GABABreceptor agonist application has a similar effect (Gil et al., 1997). This suggests that in states of arousal when forebrain activity is high, intracortical synapses are relatively depressed via presynaptic GABAB and muscarinic receptors (Gil et al., 1997). To model this, AMPA-mediated synaptic conductances between PY cells were reduced (Fig. 8A2). One effect was a reduction of the cortical firing rate to ∼20–25 Hz. Reduction of depolarizing input from cortex in turn affected the firing patterns of RE and TC cells. The membrane potential of RE cells became hyperpolarized when the corticothalamic drive was reduced, which partially deinactivated RE ITchannels. This led to persistent activity in frequency range of 10–15 Hz in the RE–TC circuit through the mechanisms similar to those responsible for spindles (Fig. 8A2). Because this activity depended on the synaptic coupling between RE and TC cells, we reduced GABAA- and AMPA-mediated conductances between thalamic neurons. This eliminated persistent firing of RE and TC cells; the cortical firing rate was further reduced to 15–20 Hz (Fig. 8A3). Note that now RE cells occasionally fired spikes that were not able, however, to initiate sustained activity in the RE–TC circuit. The frequency of cortical firing as a function of maximal conductance for AMPA-mediated PY–PY synapses is plotted in Figure 8B (top). For low values of PY–PY coupling, cortical PY cells fired at frequencies of 15–20 Hz in the frequency range of spontaneous cortical activity recorded in vivo (Steriade et al., 2001).
Input resistance of PY cells
Changes of the maximal conductances for the K+ leak current and the PY–PY synaptic current can change the input resistance of PY neurons. We systematically varied gKleak andgPY−PYin PY cells starting from the network state shown in Figure8A3. As AMPA-mediatedgPY−PYincreased, the input resistance quickly decreased from ∼200 MΩ forgPY−PY= 0.08 μS to ∼150 MΩ forgPY−PY= 0.16 μS (Fig. 8B). The frequency of spontaneous firing increased from ∼14 to ∼40 Hz. An increase of the maximal conductance for the K+ leak current had a similar effect: the input resistance was reduced to ∼170 MΩ whengKleak increased from 0 to 3 μS/cm2 (Fig. 8B,bottom). These results suggest that modifications of intrinsic and synaptic properties of cortical cells during the sleep-wake transition can affect their input resistance.
To model the transition from SWS oscillations to an activated state in the thalamocortical network, both the K+leak conductances in PY and TC cells and the PY–PY synaptic current were changed simultaneously. Figure9A shows examples of activity in one PY cell at different stages of transition. The transition to an activated state was characterized by an increased duration of the epochs when neurons fired continuously; the silent (down) states appeared less frequently and finally disappeared. To show changes in the frequency domain, we plotted integrated (in different frequency bands) values of the power spectrum (Fig. 9B) at different transitional stages. There was a dramatic decrease in power in the frequency band below 2 Hz indicating the disappearance of low-frequency periodic switches between up and down states. At higher frequencies the changes were not as significant, except for an increase in power in the 10–20 Hz band, which reflected a reduction of the firing rate in the activated state compared with active (up) states of SWS oscillations.
Fig. 9.

Transition from SWS oscillation to activated state in the thalamocortical network model. A, Decrease of the maximal conductances for potassium leak current in PY and TC cells and synaptic conductance between PY neurons eliminated silent phases in the cortical network activity. The firing became persistent and its frequency stabilized at ∼17 Hz. SWS state:gPY−PY = 0.15 μS, gRE−TC = 0.2 μS,gTC−RE = 0.4 μS, gKL = 0.3 μS/cm2; activated state:gPY−PY = 0.08 μS, gRE−TC = 0.1 μS,gTC−RE = 0.2 μS, gKL = 0. B, Integrated power in different frequency bands (as indicated in this figure) during transition to activated state. C, The input resistance of PY cells increased from ∼110 MΩ (during up phases of SWS oscillations) to ∼190 MΩ (during activated state), thus reaching the level of the input resistance during silent (Down) phases of SWS.
Figure 9C shows the input resistance of PY cells at different stages of transition to an activated state. There was a significant difference in the input resistances between silent and active states of SWS oscillations (Fig. 9C,left). As parameters changed, not only did the down states disappear but also the input resistance in the activated states increased. In a fully activated network the input resistance reached (or even exceeded) the level measured during silent states of SWS oscillations (Fig. 9C, right). Note the jump in the input resistance value after the silent (down) states became irregular (Fig. 9C, middle). During SWS oscillations the firing rate of PY cells was higher right after onset of an active state and was reduced at the end, which affected the input resistance. Input resistance values plotted in Figure 9Cwere averaged over 30 individual measurements similar to our previous study (Steriade et al., 2001). When down states became irregular and rare, there were fewer measurements near the onset of the active states where input resistance was reduced because of the higher firing rate.
External stimulation
It is generally assumed that during SWS the thalamus partially blocks sensory input to the cerebral cortex (Steriade et al., 1993b;Timofeev et al., 1996). We used our model to explore the ability of a thalamocortical network to transmit prethalamic input up to the level of PY cells during SWS oscillations and in an activated state. To model prethalamic stimulation, 25% of TC cells in the model were stimulated by external AMPA synapses with Poisson-distributed spike trains with a mean rate 25 Hz modulated by sinusoidal function at different frequencies: 0.4, 1, and 2.5 Hz (see Materials and Methods). Figure10 shows running spike histograms (RSHs) for input spike trains and spike trains in TC and PY cells. RSHs for TC and PY cells were averaged over 25% of the population; TC cells receiving external stimulation and their target/postsynaptic PY neurons were used to calculate averages. In the activated state, the prethalamic stimulus appeared at a cortical level as modulation of background activity. Responses of cortical PY cells correlated well with the input signal at all tested frequencies (Fig. 10A). This is also illustrated at Figure11A, in which normalized power spectra of the RSHs are plotted. At all frequencies the peaks in the power spectra for input signals and thalamic and cortical activities coincided. When the same input was applied to the network in the SWS state, the cortical response was a superposition of the input signal and an intrinsic oscillation (Fig. 10B). The power spectra for PY neurons innervated by fibers from those TC cells that received input signals were generally shifted to the frequency range 1.5–2.5 Hz, (Fig. 11B), compared with PY cells located remotely from direct stimulation. The durations of the silent (down) phases were reduced in PY neurons located postsynaptically to TC cells receiving external input, which changed the overall frequency of SWS oscillations in these neurons. At 2.5 Hz (Fig. 11B) and higher (data not shown), the power spectra of cortical oscillations contained peaks corresponding to the frequency of stimulation. The maximum of the cross-correlation function between input RSH and PY responses also increased. Thus, during SWS, external inputs at higher frequencies were transferred more accurately, because the local subpopulation of PY cells fired almost continuously (Fig.12).
Fig. 10.

Cortical response during thalamic stimulation. Twenty-five percent of the TC cells were stimulated by Poisson-distributed spike trains; those rates were modulated at 0.4, 1, and 2.5 Hz. A, Activated state. B, SWS state. For each graph, the bottom panelshows an RSH of the input spike trains; the middle panelshows an RSH averaged over all TC cells receiving input (25% of TC population); the top panel shows an RSH averaged over PY cells receiving afferents from those TC neurons that were stimulated (25% of the total PY population). The ability of the thalamocortical network to transmit sensory input was reduced in the SWS state.
Fig. 11.

Precision of PY responses during thalamic stimulation. Power spectra of RSHs from Figure 10 and cross-correlations between input RSHs and PY responses are shown.A, Activated state. B, SWS state. During SWS oscillations the low-frequency input was masked by intrinsic network oscillations more severely than input at higher frequencies.
Fig. 12.

Cortical activity at different distances from the stimulation site. Two PY neurons from the PY–IN–RE–TC network are shown. The cell receiving excitatory drive from the thalamus shows almost persistent firing (bottom panel), while a remotely located cell displays “normal” SWS activity (top panel).
DISCUSSION
Cortical SWS oscillations are low-frequency (0.3–1 Hz) rhythms dominating cortical activity during natural sleep and under some types of anesthesia (Steriade et al., 1993b,c,d). During SWS the entire thalamocortical network switches periodically between up and down states. The down state is characterized by disfacilitation, a state-specific withdrawal of synaptic activity leading to synaptic silence, whereas the up state is characterized by spiking in the frequency range of 20–30 Hz. SWS activity exhibits remarkable synchrony between different cortical areas (Amzica and Steriade, 1995a,b) and between cortex and thalamus (Contreras and Steriade, 1995). A recent study of SWS oscillation during natural sleep indicates that silent (down) states are not associated with IPSPs but rather are the result of activating intrinsic (presumably potassium) currents in cortical neurons and global disfacilitation (Timofeev et al., 2001b). Cortical inhibitory interneurons follow the same pattern of activity as excitatory cells; they are silent during down states of SWS oscillations (Steriade et al., 2001; Timofeev et al., 2001b). Thus, the whole thalamocortical network is silent (inactive) during down states of SWS oscillations, which raises a question about the mechanisms leading to reinitiation of activity at the onset of each active phase.
One possible mechanism for recovery of active states during SWS could be the dynamics of some intrinsic currents in cortical neurons such as the hyperpolarization-activated cation current,Ih, similar to the way that dynamics of the low-threshold Ca2+ current andIh in thalamic relay cells can organize their oscillations in the delta frequency range (McCormick and Pape, 1990; Soltesz et al., 1991; Lytton et al., 1996). Recently, it was shown using a cortical slice preparation that in a relatively high concentration (3.5 mm) of extracellular K+, cortical slices can oscillate in the frequency range of slow sleep oscillations (Sanchez-Vives and McCormick, 2000). This activity was usually initiated in layer 5 and propagated over the whole slice. Although these results do not exclude the possibility of intracellular mechanisms for SWS oscillations, it is not clear how the specific conditions in those slice preparations affected the excitability of cortical neurons and the temporal patterns of their activity.
The approach taken here was to critically test whether SWS oscillations can be maintained by intrinsic mechanisms in an isolated part of the cortical network in vivo. These experiments used a cortical slab preparation: a 10 × 6 mm slab of cortex that was synaptically isolated from the rest of the cortical network but kept with a natural blood supply (Timofeev et al., 2000a). Although neurons outside the slab demonstrated “normal” SWS oscillations, activity in the small slab consisted of small depolarizing potentials, presumably spike-independent minis (Fatt and Katz, 1952), interrupted by bursts of high-amplitude depolarizing events. These bursts lasted 0.5–3 sec, appeared at very low frequencies (2–6 min−1), and were composed of EPSPs and IPSPs (Timofeev et al., 2000b). Miniature EPSPs observed during silent periods had a mean amplitude of 0.54 mV; their incidence decreased significantly immediately after a burst and recovered during the following 3–4 sec (Timofeev et al., 2000a). When a whole gyrus was isolated, its activity was similar to normal SWS activity. This suggests that the relatively low frequency of bursting in the isolated slab could be caused by the relatively small number of neurons in the slab compared with a gyrus or the intact cortex, thus pointing to network mechanisms for SWS oscillations. To test this hypothesis, the mean and SD of interburst intervals were estimated analytically as a function of number of neurons in a network (Timofeev et al., 2000a). For a slab the estimated mean was ∼24 ± 21 sec; the mean decreased with the size of the network and reached 4.9 ± 2.3 sec for a network the size of a gyrus. Thus, cortical SWS oscillations could arise from the same mechanisms as spontaneous slab activity in the limit of a very large neuronal population.
In computer simulations of Hodgkin–Huxley-type model neurons, an increase in the mini rate or amplitude had a similar effect on the frequency of spontaneous bursting as an increase of the network size. In this study, we used a relatively small thalamocortical network comprising a few hundred neurons and a higher mini rate and amplitude to explore properties of SWS activity. On each cycle of oscillation, an active phase was started in one or a few PY neurons driven by the spontaneously occurring coincidence of miniature ESPSs. Mini-evoked depolarization led to INa(p)activation and a burst of spikes. Once started, the activity propagated with a velocity that depended on the maximal conductances of the inhibitory and excitatory synapses between PY and IN cells. In a network model including only cortical PY and IN cells, each pattern of activity lasted 0.5–1 sec and was terminated by synaptic depression and activation of IK(Ca). A sufficiently high level of inhibitory feedback was required to maintain transitions to the down state. Duration of active phases depended on the presence of NMDA receptors between PY cells. When NMDA currents were blocked, only short bursts lasting <0.5 sec were obtained.
Thalamic relay cells have intrinsic properties that generate slow (<1 Hz) oscillations (Hughes et al., 2002). Intrinsic slow oscillations in TC neurons can be synchronized by corticothalamic input (Timofeev and Steriade, 1996) and contribute to enhancing and shaping the slow sleep rhythm. In the full thalamocortical model, the active phases of SWS oscillations usually lasted longer (1–1.5 sec), partially maintained by the depolarizing drive from TC neurons.
Transition to activated states
The transition from slow-wave sleep to REM sleep or waking is controlled by activation of neuromodulatory systems (Steriade and McCarley, 1990). Acetylcholine increases during the transition to both REM sleep and waking, whereas noradrenaline and serotonin are increased during waking only (for review, see Steriade et al., 1997). To model the activation of these neuromodulatory systems, K+ leak currents were blocked in PY and TC cells in the model (McCormick, 1992). This depolarized the neurons and eliminated the silent (down) states of SWS oscillations, and the neurons fired continuously. To ensure realistically low frequencies of spontaneous activity in an activated state, maximal conductances for AMPA-mediated synapses between PY neurons in the model had to be reduced by ∼40%. This is consistent with experimental findings that application of muscarine or a low concentration of acetylcholine depressed intracortical EPSPs both in vitro (Gil et al., 1997) and in vivo (Oldford et al., 2000). We then tested the ability of the thalamocortical network model to process prethalamic sensory input. In an activated state the network precisely transformed the input (rate-modulated Poisson spike train) into responses from cortical PY cells. During SWS oscillations the cortical response was masked by intrinsic activity, especially at low frequencies of stimulation. Surprisingly, at higher frequencies signal transmission was improved because the local groups of PY neurons fired almost continuously during stimulation. This result can explain the blockage of slow EEG rhythms and the appearance of fast oscillations (20–40 Hz) restricted to the motor cortex in experiments with high-frequency stimulation of interpositus and dentate cerebellar nuclei in vivo (Steriade, 1995).
The transition to an activated state is associated with an increase in the input resistance of cortical cells in vivo(Steriade et al., 2001). In the network model there was also a significant difference between the input resistances of PY cells in the down (silent) and up (active) states during SWS oscillations. This difference was explained primary by a high level of synaptic activity during active phases of SWS. The input resistance gradually increased during the transition from SWS to an activated state, eventually reaching the same or a higher level as during silent phases of the SWS oscillations. Both reduction of K+ leak and AMPA-mediated synaptic conductances in cortical neurons contributed to this shift in the passive properties of neurons during waking.
Conclusion and model predictions
The present model provides, for the first time, a complete and realistic mechanism for generating cortical slow-wave sleep rhythms. In the proposed scenario of slow-wave sleep, the “re-excitation” of the cortical network on each cycle of oscillation is driven by the spontaneously occurring coincidence of miniature EPSPs. This mechanism was previously proposed to explain spontaneous active states in the isolated cortical slab (Timofeev et al., 2000a) and was used here to explain periodic transitions between silent and active states of slow sleep oscillations. In large neocortical networks there may be many independent foci where activity could be initiated almost simultaneously. The model requires more than a single spike in a PY cell to induce the active (up) state, and many PY cells fired isolated spikes during silent phases of SWS oscillations (Fig. 6). These spikes were unable to induce postsynaptic response unless the postsynaptic cell was itself sufficiently depolarized by random mini summation. Thus, in a large cortical network, several events (a spike in one PY cell and a sufficiently high depolarization in a few other cells) must occur simultaneously to initiate an active state. The model predicts that the persistent Na+ current is important in initiating transitions from silent (down) to active (up) states and that the Ca2+-dependent K+ current as well as synaptic depression and IN-mediated inhibition are important in terminating active phases.
The model explains the transition from SWS to activated states (such as REM sleep or waking). It predicts that this transition is accomplished by changing both the passive properties of cortical neurons and the nonlinear map of prethalamic signals to cortical responses in a frequency-dependent matter. Some of these predictions were confirmed recently in in vivo studies with naturally awake and sleeping cats (Steriade et al., 2001; Timofeev et al., 2001b).
Recent studies have reported that slow-wave sleep may be essential for memory consolidation and memory formation (Gais et al., 2000; Stickgold et al., 2000). The cellular mechanisms underlying sleep-related memory consolidation including synaptic plasticity and cellular conditioning (Timofeev et al., 2002) can be further integrated into the model.
Footnotes
This research was supported by National Institutes of Health Grant NS40522, the Human Frontier Science Program, and the Canadian Institutes of Health Research (Grant MOP-37862). I.T. is a scholar of Fonds de la Recherche en Santé du Québec.
Correspondence should be addressed to Dr. Maxim Bazhenov, The Salk Institute, 10010 North Torrey Pines Road, La Jolla, CA 92037. E-mail:bazhenov@salk.edu.
REFERENCES
- 1.Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- 2.Achermann P, Borbely AA. Low-frequency (<1 Hz) oscillations in the human sleep electroencephalogram. Neuroscience. 1997;81:213–222. doi: 10.1016/s0306-4522(97)00186-3. [DOI] [PubMed] [Google Scholar]
- 3.Alzheimer C, Schwindt PC, Crill WE. Modal gating of Na+ channels as a mechanism of persistent Na+ current in pyramidal neurons from rat and cat sensorimotor cortex. J Neurosci. 1993;13:660–673. doi: 10.1523/JNEUROSCI.13-02-00660.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amzica F, Steriade M. Disconnection of intracortical synaptic linkages disrupts synchronization of a slow oscillation. J Neurosci. 1995a;15:4658–4677. doi: 10.1523/JNEUROSCI.15-06-04658.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amzica F, Steriade M. Short- and long-range neuronal synchronization of the slow (<1 Hz) cortical oscillation. J Neurophysiol. 1995b;73:20–38. doi: 10.1152/jn.1995.73.1.20. [DOI] [PubMed] [Google Scholar]
- 6.Amzica F, Steriade M. The K-complex: its slow (<1-Hz) rhythmicity and relation to delta waves. Neurology. 1997;49:952–959. doi: 10.1212/wnl.49.4.952. [DOI] [PubMed] [Google Scholar]
- 7.Bal T, McCormick DA. What stops synchronized thalamocortical oscillations? Neuron. 1996;17:297–308. doi: 10.1016/s0896-6273(00)80161-0. [DOI] [PubMed] [Google Scholar]
- 8.Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Cellular and network models for intrathalamic augmenting responses during 10 Hz stimulation. J Neurophysiol. 1998;79:2730–2748. doi: 10.1152/jn.1998.79.5.2730. [DOI] [PubMed] [Google Scholar]
- 9.Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Self-sustained rhythmic activity in the thalamic reticular nucleus mediated by depolarizing GABAA receptor potentials. Nat Neurosci. 1999;2:168–174. doi: 10.1038/5729. [DOI] [PubMed] [Google Scholar]
- 10.Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Spiking-bursting activity in the thalamic reticular nucleus initiates sequences of spindle oscillations in thalamic networks. J Neurophysiol. 2000;84:1076–1087. doi: 10.1152/jn.2000.84.2.1076. [DOI] [PubMed] [Google Scholar]
- 11.Budde T, Biella G, Munsch T, Pape HC. Lack of regulation by intracellular Ca2+ of the hyperpolarization-activated cation current in rat thalamic neurons. J Physiol (Lond) 1997;503:79–85. doi: 10.1111/j.1469-7793.1997.079bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. 1995;15:604–622. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Contreras D, Timofeev I, Steriade M. Mechanisms of long-lasting hyperpolarizations underlying slow sleep oscillations in cat corticothalamic networks. J Physiol (Lond) 1996a;494:251–264. doi: 10.1113/jphysiol.1996.sp021488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Contreras D, Destexhe A, Sejnowski TJ, Steriade M. Control of spatiotemporal coherence of a thalamic oscillation by corticothalamic feedback. Science. 1996b;274:771–774. doi: 10.1126/science.274.5288.771. [DOI] [PubMed] [Google Scholar]
- 15.Destexhe A, Contreras D, Sejnowski TJ, Steriade M. A model of spindle rhythmicity in the isolated thalamic reticular nucleus. J Neurophysiol. 1994a;72:803–818. doi: 10.1152/jn.1994.72.2.803. [DOI] [PubMed] [Google Scholar]
- 16.Destexhe A, Mainen ZF, Sejnowski TJ. Synthesis of models for excitable membranes, synaptic transmission and neuromodulation using a common kinetic formalism. J Comp Neurosci. 1994b;1:195–230. doi: 10.1007/BF00961734. [DOI] [PubMed] [Google Scholar]
- 17.Destexhe A, Bal T, McCormick DA, Sejnowski TJ. Ionic mechanisms underlying synchronized oscillations and propagating waves in a model of ferret thalamic slices. J Neurophysiol. 1996;76:2049–2070. doi: 10.1152/jn.1996.76.3.2049. [DOI] [PubMed] [Google Scholar]
- 18.Dutar P, Nicoll RA. A physiological role for GABAB receptors in the central nervous system. Nature. 1988;332:156–158. doi: 10.1038/332156a0. [DOI] [PubMed] [Google Scholar]
- 19.Enright WH, Higham DJ, Owren B, Sharp PW (1995) A survey of the explicit Runge-Kutta method. Available fromftp://ftp.cs.toronto.edu/ pub/reports/na/cs-94–291.ps.Z.
- 20.Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. J Physiol (Lond) 1952;117:109–128. [PMC free article] [PubMed] [Google Scholar]
- 21.Gais S, Plihal W, Wagner U, Born J. Early sleep triggers memory for early visual discrimination skills. Nat Neurosci. 2000;3:1335–1339. doi: 10.1038/81881. [DOI] [PubMed] [Google Scholar]
- 22.Galarreta M, Hestrin S. Frequency-dependent synaptic depression and the balance of excitation and inhibition in the neocortex. Nat Neurosci. 1998;1:587–594. doi: 10.1038/2822. [DOI] [PubMed] [Google Scholar]
- 23.Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 24.Golomb D, Amitai Y. Propagating neuronal discharges in neocortical slices: computational and experimental study. J Neurophysiol. 1997;78:1199–1211. doi: 10.1152/jn.1997.78.3.1199. [DOI] [PubMed] [Google Scholar]
- 25.Golomb D, Ermentrout GB. Bistability in pulse propagation in networks of excitatory and inhibitory populations. Phys Rev Lett. 2001;86:4179–4182. doi: 10.1103/PhysRevLett.86.4179. [DOI] [PubMed] [Google Scholar]
- 26.Golomb D, Ermentrout GB. Slow excitation supports propagation of slow pulses in networks of excitatory and inhibitory populations. Phys Rev E. 2002;65:061911. doi: 10.1103/PhysRevE.65.061911. [DOI] [PubMed] [Google Scholar]
- 27.Golomb D, Wang XJ, Rinzel J. Synchronization properties of spindle oscillations in a thalamic reticular nucleus model. J Neurophysiol. 1994;72:1109–1126. doi: 10.1152/jn.1994.72.3.1109. [DOI] [PubMed] [Google Scholar]
- 28.Golomb D, Wang XJ, Rinzel J. Propagation of spindle waves in a thalamic slice model. J Neurophysiol. 1996;75:750–769. doi: 10.1152/jn.1996.75.2.750. [DOI] [PubMed] [Google Scholar]
- 29.Grenier F, Timofeev I, Steriade M. Focal synchronization of ripples (80–200 Hz) in neocortex and their neuronal correlates. J Neurophysiol. 2001;86:1884–1898. doi: 10.1152/jn.2001.86.4.1884. [DOI] [PubMed] [Google Scholar]
- 30.Hughes SW, Cope DW, Blethyn KL, Crunelli V. Cellular mechanisms of the slow (<1 Hz) oscillation in thalamocortical neurons in vitro. Neuron. 2002;33:947–958. doi: 10.1016/s0896-6273(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 31.Huguenard JR, McCormick DA. Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol. 1992;68:1373–1383. doi: 10.1152/jn.1992.68.4.1373. [DOI] [PubMed] [Google Scholar]
- 32.Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca2+-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804–3817. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kay AR, Sugimori M, Llinás R. Kinetic and stochastic properties of a persistent sodium current in mature guinea pig cerebellar Purkinje cells. J Neurophysiol. 1998;80:1167–1179. doi: 10.1152/jn.1998.80.3.1167. [DOI] [PubMed] [Google Scholar]
- 34.Lüthi A, Bal B, McCormick DA. Periodicity of thalamic spindle waves is abolished by ZD7288, a blocker of Ih. J Neurophysiol. 1998;79:3284–3289. doi: 10.1152/jn.1998.79.6.3284. [DOI] [PubMed] [Google Scholar]
- 35.Lytton WW, Destexhe A, Sejnowski TJ. Control of slow oscillations in the thalamocortical neuron: a computer model. Neuroscience. 1996;70:673–684. doi: 10.1016/s0306-4522(96)83006-5. [DOI] [PubMed] [Google Scholar]
- 36.Mainen ZF, Sejnowski TJ. Influence of dendritic structure on firing pattern in model neocortical neurons. Nature. 1996;382:363–366. doi: 10.1038/382363a0. [DOI] [PubMed] [Google Scholar]
- 37.McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol. 1992;39:337–388. doi: 10.1016/0301-0082(92)90012-4. [DOI] [PubMed] [Google Scholar]
- 38.McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurons. J Physiol (Lond) 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metherate R, Ashe JH. Ionic flux contributions to neocortical slow waves and nucleus basalis-mediated activation: whole-cell recordings in vivo. J Neurosci. 1993;13:5312–5323. doi: 10.1523/JNEUROSCI.13-12-05312.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oldford E, Jones-Gotman M, Castro-Alamancos MA. Input-specific effects of acetylcholine on sensory and intracortical evoked responses in the barrel cortex in vivo. Soc Neurosci Abstr. 2000;26:133. doi: 10.1016/s0306-4522(02)00663-2. [DOI] [PubMed] [Google Scholar]
- 41.Paré D, Royer S, Martina M. Contrasting synaptic responsiveness of various physiologically identified types of perirhinal neurons. Soc Neurosci Abstr. 2000;26:470. [Google Scholar]
- 42.Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 2000;3:1027–1034. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- 43.Soltesz I, Lightowler S, Leresche N, Jassik-Gerschenfeld D, Pollard CE, Crunelli V. Two inward currents and the transformation of low-frequency oscillations of rat and cat thalamocortical cells. J Physiol (Lond) 1991;441:175–197. doi: 10.1113/jphysiol.1991.sp018745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steriade M. Two channels in the cerebellothalamocortical system. J Comp Neurol. 1995;354:57–70. doi: 10.1002/cne.903540106. [DOI] [PubMed] [Google Scholar]
- 45.Steriade M, McCarley RW. Brainstem control of wakefulness and sleep. Plenum; New York: 1990. [Google Scholar]
- 46.Steriade M, Deschênes M, Domich L, Mulle C. Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J Neurophysiol. 1985;54:1473–1497. doi: 10.1152/jn.1985.54.6.1473. [DOI] [PubMed] [Google Scholar]
- 47.Steriade M, Domich L, Oakson G, Deschênes M. The deafferented reticular thalamic nucleus generates spindle rhythmicity. J Neurophysiol. 1987;57:260–273. doi: 10.1152/jn.1987.57.1.260. [DOI] [PubMed] [Google Scholar]
- 48.Steriade M, Amzica F, Nuñez A. Cholinergic and noradrenergic modulation of the slow (approximately 0.3 Hz) oscillation in neocortical cells. J Neurophysiol. 1993a;70:1385–1400. doi: 10.1152/jn.1993.70.4.1385. [DOI] [PubMed] [Google Scholar]
- 49.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993b;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 50.Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993c;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steriade M, Nuñez A, Amzica F. Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci. 1993d;13:3266–3283. doi: 10.1523/JNEUROSCI.13-08-03266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steriade M, Jones EG, McCormick DA. Thalamus: organization and function. Elsevier Science; Oxford, UK: 1997. [Google Scholar]
- 53.Steriade M, Timofeev I, Dürmüller N, Grenier F. Dynamic properties of corticothalamic neurons and local cortical interneurons generating fast rhythmic (30–40 Hz) spike bursts. J Neurophysiol. 1998;79:483–490. doi: 10.1152/jn.1998.79.1.483. [DOI] [PubMed] [Google Scholar]
- 54.Steriade M, Timofeev I, Grenier F. Natural waking and sleep states: a view from inside neocortical neurons. J Neurophysiol. 2001;85:1969–1985. doi: 10.1152/jn.2001.85.5.1969. [DOI] [PubMed] [Google Scholar]
- 55.Stevens CF. Quantal release of neurotransmitter and long-term potentiation. Cell. 1993;72S:55–63. doi: 10.1016/s0092-8674(05)80028-5. [DOI] [PubMed] [Google Scholar]
- 56.Stickgold R, James L, Hobson JA. Visual discrimination learning requires sleep after training. Nat Neurosci. 2000;3:1237–1238. doi: 10.1038/81756. [DOI] [PubMed] [Google Scholar]
- 57.Timofeev I, Steriade M. Low-frequency rhythms in the thalamus of intact-cortex and decorticated cats. J Neurophysiol. 1996;76:4152–4168. doi: 10.1152/jn.1996.76.6.4152. [DOI] [PubMed] [Google Scholar]
- 58.Timofeev I, Steriade M. Fast (mainly 30–100 Hz) oscillations in the cat cerebellothalamic pathway and their synchronization with cortical potentials. J Physiol (Lond) 1997;504:153–168. doi: 10.1111/j.1469-7793.1997.153bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Timofeev I, Contreras D, Steriade M. Synaptic responsiveness of cortical and thalamic neurones during various phases of slow sleep oscillation in cat. J Physiol (Lond) 1996;494:265–278. doi: 10.1113/jphysiol.1996.sp021489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M. Origin of slow cortical oscillations in deafferented cortical slabs. Cereb Cortex. 2000a;10:1185–1199. doi: 10.1093/cercor/10.12.1185. [DOI] [PubMed] [Google Scholar]
- 61.Timofeev I, Grenier F, Steriade M. Impact of intrinsic properties and synaptic factors on the activity of neocortical networks in vivo. J Physiol (Paris) 2000b;94:343–355. doi: 10.1016/s0928-4257(00)01097-4. [DOI] [PubMed] [Google Scholar]
- 62.Timofeev I, Bazhenov M, Sejnowski TJ, Steriade M. Contribution of intrinsic and synaptic factors in the desynchronization of thalamic oscillatory activity. Thal Relat Syst. 2001a;1:53–69. [Google Scholar]
- 63.Timofeev I, Grenier F, Steriade M. Disfacilitation and active inhibition in the neocortex during the natural sleep-wake cycle: an intracellular study. Proc Natl Acad Sci USA. 2001b;98:1924–1929. doi: 10.1073/pnas.041430398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Timofeev I, Grenier F, Bazhenov M, Houweling AR, Sejnowski TJ, Steriade M. Short- and medium-term plasticity associated with augmenting responses in cortical slabs and spindles in intact cortex of cats in vivo. J Physiol (Lond) 2002;542:583–598. doi: 10.1113/jphysiol.2001.013479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Traub RD, Miles R. Neuronal networks of the hippocampus. Cambridge UP; Cambridge, UK: 1991. [Google Scholar]
- 66.Traub RD, Wong RK, Miles R, Michelson H. A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J Neurophysiol. 1991;66:635–650. doi: 10.1152/jn.1991.66.2.635. [DOI] [PubMed] [Google Scholar]
- 67.Tsodyks MV, Markram H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci USA. 1997;94:719–723. doi: 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ulrich D, Huguenard JR. Nucleus-specific chloride homeostasis in rat thalamus. J Neurosci. 1997;17:2348–2354. doi: 10.1523/JNEUROSCI.17-07-02348.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]