

Abstract
Basolateral amygdala (BLA) activation by emotional arousal modulates memory-related processes in the hippocampus. We have shown (Akirav and Richter-Levin, 1999b) that activating the BLA before perforant path (PP) tetanization has a biphasic effect on hippocampal plasticity; priming the BLA immediately before PP tetanization results in the enhancement of dentate gyrus (DG) long-term potentiation (LTP) (an “emotional tag”), whereas stimulation in a spaced interval results in the suppression of DG-LTP. Here, we aimed to elucidate the mechanisms underlying BLA modulation of DG-LTP and specifically to examine whether the stress hormones norepinephrine (NE) and corticosterone (CORT) are main mediators of the BLA biphasic effects. We found that the BLA affects hippocampal plasticity in a complex manner; BLA priming enhanced DG-LTP, and both NE and CORT mediated this effect. Furthermore, we found that ipsilateral BLA spaced activation (2 hr before PP tetanization) suppressed DG-LTP and that this suppressive effect was also mediated by NE and CORT. Priming the contralateral BLA enhanced DG-LTP similarly to the ipsilateral enhancement, but neither NE nor CORT mediated this effect. The spaced activation of the contralateral BLA did not suppress DG-LTP. Taken together, these results suggest that differential mechanisms underlie the ipsilateral and contralateral BLA effects on hippocampal plasticity.
Hence, the BLA modulates hippocampal memory processes, presumably via the mediation of the stress hormones NE and CORT, to establish a diverse memory of the experience. Possibly, at the onset of an emotional event the stress hormones permissively mediate plasticity. However, their prolonged presence in the system may suppress the cognitive response to stress.
Keywords: long-term potentiation, plasticity, basolateral amygdala, central amygdala, hippocampus, dentate gyrus, norepinephrine, glucocorticoids, corticosterone, serotonin, ipsilateral, contralateral
Several lines of evidence suggest that emotional arousal activates the amygdala and that this activation, specifically that of the basolateral amygdala (BLA), results in modulation of memory-related processes in the hippocampus (for review, see Cahill and McGaugh, 1998; McGaugh, 2000; Richter-Levin and Akirav, 2000; Roozendaal, 2000; Abe, 2001; Packard and Cahill, 2001).
Although emotional experiences can either enhance or impair hippocampal memory and plasticity (Diamond et al., 2000), BLA activation was reported to enhance hippocampal long-term potentiation (LTP) (Ikegaya et al., 1995; Akirav and Richter-Levin, 1999a).
In agreement with the twofold influence of emotional experiences on hippocampal-dependent memory, we have shown (Akirav and Richter-Levin, 1999b) that activating the amygdala before perforant path (PP) stimulation has a biphasic effect on hippocampal plasticity; priming the BLA immediately before PP tetanization results in the enhancement of dentate gyrus (DG)-LTP, whereas spaced BLA stimulation results in the suppression of DG-LTP. We suggested that the fast excitatory phase may serve as a marker that generates strong memories for emotionally charged experiences (an “emotional tag”) and that the slower inhibitory phase may be beneficial in reducing masking effects of subsequent, less-significant events during the initial stages of consolidation.
The stress hormones [norepinephrine (NE) and corticosterone (CORT)], released by emotional arousal, are potent modulators of both learning and brain plasticity, and these effects are presumably mediated by influences involving the amygdala (Liang et al., 1990; Roozendaal and McGaugh, 1996; Cahill and McGaugh, 1998; McGaugh, 2000).
NE has been shown repeatedly to be involved in memory reinforcement of different behavioral tasks (McGaugh, 1989; Cahill et al., 1994) and in the reinforcement of hippocampal LTP (Izquierdo and Medina, 1995;Seidenbecher et al., 1997). Specifically, it has been suggested that noradrenergic activation of the BLA may serve to modulate memory storage and plasticity in the hippocampus (Ikegaya et al., 1997; Ferry et al., 1999; Frey et al., 2001).
Although the BLA contains a moderate density of glucocorticoid receptors (GRs) (Morimoto et al., 1996), the hippocampus contains substantial concentrations of GRs (McEwen and Sapolsky, 1995). CORT has dose-dependent inverted U-shaped effects on hippocampal LTP and primed burst potentiation (PBP) (Diamond et al., 1989, 1992, 1994; Pavlides et al., 1993, 1995; Kerr et al., 1994; Rey et al., 1994). In addition, amygdala electrical stimulation has been shown to increase plasma levels of CORT (Feldman et al., 1982), and it has been suggested that a functioning BLA is required for adrenal steroids to exert their influence on hippocampal memory storage (Roozendaal et al., 1996, 1999;Roozendaal and McGaugh, 1997).
The response to stress involves a biphasic secretion of the stress hormones in which NE represents the first phase and glucocorticoids represent the second phase. Here, we aimed to elucidate the mechanisms underlying BLA modulation of DG-LTP and specifically to examine whether NE is a main mediator of the BLA enhancing effect (the first phase) and CORT is a main mediator of the BLA suppressing effect (the second phase). If so, BLA activation in NE- or CORT-depleted rats should not lead to DG-LTP enhancement or suppression, respectively.
MATERIALS AND METHODS
Animals
Adult male Wistar rats, weighing 280–320 gm (Harlan, Jerusalem, Israel), were maintained five per cage on a 12 hr light/dark cycle with water and laboratory rodent chow ad libitum.
Drug treatment
N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride (DSP-4) (50 mg/kg dissolved in saline, intraperitoneal injection; Sigma) (Richter-Levin et al., 1991) was used to deplete forebrain NE. DSP-4 was injected 1 week before the electrophysiological experiment. This dose causes depletion of >80% of NE in the cortex and hippocampus (Jaim-Etcheverry and Zieher, 1980).
Metyrapone (Met) (50 mg/kg dissolved in a vehicle containing 40% polyethylene glycol and 60% saline, intraperitoneal injection; Aldrich) reduces the synthesis of CORT by inhibiting the 11β-hydroxylation reaction in the adrenal glands. Metyrapone also inhibits the synthesis of other adrenocortical hormones such as aldosterone (de Kloet et al., 1998), and yet with regard to hippocampal LTP, its effect on CORT levels is considered to be a dominant one. Metyrapone was injected 50 min before the stimulation of the BLA.
dl-p-chlorophenylalanine (PCPA) (200 mg/kg dissolved in saline, intraperitoneal injection, once a day for 3 consecutive days; Sigma) depletes serotonin (5-HT) by inhibiting tryptophan hydroxylase (Richter-Levin and Segal, 1989).
Approximately half of the animals in the nondrug groups were injected with either saline or vehicle in the same protocol as the drug groups to control for possible effects of the injection or the vehicle. Because no significant difference was found between these control groups, they were grouped together.
Electrophysiology
Surgical procedure. Rats were anesthetized (40% urethane and 5% chloral hydrate in saline, 0.5 ml/100 gm, i.p.) and mounted in a Stoelting (Wood Dale, IL) stereotaxic frame. The scalp was incised and retracted, and head position was adjusted to place bregma and lambda in the same horizontal plane. Small burr holes (2 mm diameter) were drilled unilaterally in the skull for the placement of recording and stimulating electrodes.
A recording microelectrode (glass; tip diameter, 2–5 μm; filled with 2 m NaCl; resistance, 1–4 MΩ) was placed in the dorsal DG (coordinates: 4 mm posterior, 2.5 mm lateral to bregma; depth adjusted to yield largest EPSP response to stimulation of the PP).
A bipolar 125 μm stimulating electrode was implanted in the ipsilateral medial PP (coordinates: 8 mm posterior, 4 mm lateral to bregma; depth adjusted to yield maximal response of the DG).
In the BLA groups, a second stimulating electrode was implanted in the ipsilateral or the contralateral BLA as indicated (coordinates: 3 mm posterior, 5.3 mm lateral to bregma; depth 6.7 mm). To control for a possible lateralization effect of the BLA, in the contralateral and ipsilateral groups, the DG and PP electrodes were placed in the right or left hemispheres, alternately.
In the central amygdala (CeA) groups, a second stimulating electrode was implanted in the ipsilateral CeA (coordinates: 2.5 mm posterior, 4.5 mm lateral to bregma; depth 7.1 mm).
Baseline stimuli to the PP (monopolar pulses, 100 μsec duration, intensity adjusted to yield an EPSP slope of ∼4 mV/msec.) were delivered at 0.1 Hz. There was no significant difference in stimulus intensities between the groups. After the electrodes were positioned, the rat was left for 60 min before the experiment commenced.
Evoked responses were digitized (10 kHz) and analyzed using theCambridge Electronic Design (Cambridge, UK) 1401+ interface and its Spike2 software. Off-line measurements were made of the slope of the EPSP using averages of five successive responses to a given stimulation intensity applied at 0.1 Hz.
LTP was measured as an increase in EPSP slope. The EPSP slope was measured as a percentage of baseline value immediately before the tetanus.
During the course of the experiment, body temperature was monitored and maintained at 37 ± 0.5°C by a feedback-regulated heating pad.
LTP induction and BLA stimulation. LTP was induced by a theta-like high-frequency stimulation (HFS) to the PP (three sets of 10 trains; each train consisted of 10 pulses at 100 Hz; intertrain interval, 200 msec; interset interval, 1 min). During HFS stimulus, intensity was increased to 2 mV.
The BLA and CeA groups received stimulation (10 trains of five pulses at 100 Hz; intertrain interval, 200 msec) to the BLA/CeA (50 μsec, 1 V), either 30 sec (phase 1) or 2 hr (phase 2) before HFS was applied to the PP.
Histology
Histological verification of the stimulating electrode location was performed on all the rats that were implanted with a stimulating electrode in the BLA or the CeA.
After electrophysiological testing, marking lesions were made by passing anodal currents (10 mA for 15 sec) to the metal bipolar stimulating electrode. Brains were removed, postfixed for one night in formaldehyde (10%), and sectioned (120 μm) on a sledge microtome. The sections were mounted on gelatin-coated slides, stained in cresyl violet, dehydrated, and coverslipped. The electrode tract and lesion locations were then identifiable under a light microscope (Akirav and Richter-Levin, 1999a,b).
Experimental groups
Phase 1: BLA priming. Twelve groups were tested: (1) control LTP (n = 9): HFS to the PP; (2) ipsi BLA priming (n = 12): a priming stimulation to the ipsilateral BLA applied 30 sec before HFS to the PP; (3) CeA priming (n = 5): a priming stimulation to the ipsilateral CeA applied 30 sec before HFS to the PP; (4) DSP-4 LTP (n = 7): rats were injected with DSP-4 1 week before receiving HFS to the PP; (5) DSP-4 ipsi priming (n = 12): rats injected with DSP-4 received a priming stimulation to the ipsilateral BLA 30 sec before HFS to the PP; (6) Met LTP (n = 7): rats were injected with metyrapone 50 min before receiving HFS to the PP; (7) Met ipsi priming (n = 7): rats injected with metyrapone received a priming stimulation to the ipsilateral BLA 30 sec before HFS to the PP; (8) PCPA LTP (n = 5): rats were injected with PCPA once a day for 3 consecutive days before receiving HFS to the PP; (9) PCPA ipsi priming (n = 7): rats injected with PCPA received a priming stimulation to the ipsilateral BLA 30 sec before HFS to the PP; (10) contra priming (n = 10): a priming stimulation to the contralateral BLA was applied 30 sec before HFS to the PP; (11) DSP-4 contra priming (n = 10): rats injected with DSP-4 received a priming stimulation to the contralateral BLA 30 sec before HFS to the PP; (12) Met contra priming (n = 8): rats injected with metyrapone received a priming stimulation to the contralateral BLA 30 sec before HFS to the PP.
Phase 2: spaced activation. Seven groups were tested: (1) control spaced LTP (n = 8): HFS to the PP; (2) ipsi BLA spaced (n = 10): stimulation to the ipsilateral BLA 2 hr before HFS to the PP; (3) CeA spaced (n = 5): stimulation to the ipsilateral CeA 2 hr before HFS to the PP; (4) DSP-4 spaced (n = 8): rats injected with DSP-4 received stimulation to the ipsilateral BLA 2 hr before HFS to the PP; (5) Met spaced (n = 8): rats injected with metyrapone received stimulation to the ipsilateral BLA 2 hr before HFS to the PP; (6) PCPA spaced (n = 5): rats injected with PCPA received stimulation to the ipsilateral BLA 2 hr before HFS to the PP; (7) contra BLA spaced (n = 8): stimulation to the contralateral BLA 2 hr before HFS to the PP.
Statistical analysis
The results are expressed as means ± SEM. For statistical analysis, overall mixed ANOVA, one-way ANOVA, and t test were used as indicated. All post hoc comparisons were made using the least significant difference multiple-comparison test (LSD).
RESULTS
Phase 1: BLA priming
Similar stimulus intensities were applied (F(11,87) < 1; NS), and overall mixed ANOVA [groups × time (12 × 2)] for comparison between the groups before HFS did not reveal a significant difference in EPSP slope at either −30 min or −1 min, indicating a similar baseline in all groups (F(11,87) < 1; NS). Using overall mixed ANOVA [groups × time (12 × 3)] for post-HFS comparison, we found a significant difference in EPSP slope levels between the groups (F(11,87) = 3.811;p = 0.0001) that was further analyzed.
Ipsilateral BLA priming enhances DG-LTP, whereas CeA priming does not
Potentiation levels in the LTP group after HFS to the PP was significantly different from 100% at all times post-HFS [ttest for difference from baseline (100%): +1 min,t(8) = 7.6458, p < 0.0001; +30 min, t(8) = 5.728,p < 0.001; +60 min,t(8) = 6.002; p < 0.001].
One-way ANOVA revealed a significant difference between the groups at +30 and +60 min post-HFS (Fig.1A) (+30 min:F(2,23) = 10.155, p < 0.001; +60 min: F(2,23) = 14.826,p < 0.0001).
Fig. 1.
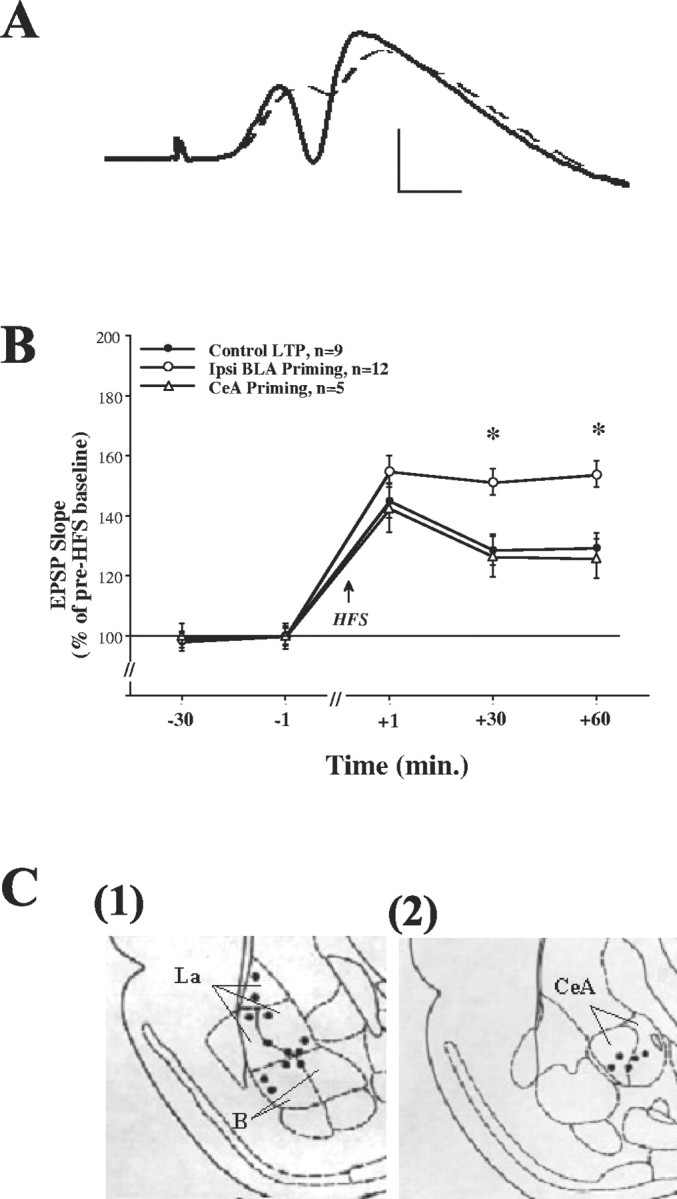
A, Representative evoked potentials recorded from the DG before (dashed line) and after HFS to the PP. Calibration: vertical, 5 mV; horizontal, 1 msec.B, Ipsilateral BLA priming enhances DG-LTP, whereas CeA priming does not. The increase in EPSP slope (LTP) was measured as a percentage of baseline value immediately before HFS to the PP. The levels of DG-LTP in the Control LTP group (n = 9) after HFS were significantly different from 100% at all times post-HFS (see Results). Ipsilateral priming stimulation of the BLA (Ipsi BLA Priming, n=12) induced 30 sec before HFS was applied to the PP significantly increased DG-LTP levels compared with the Control LTP group at +30 min (*p < 0.001) and at +60 min post-HFS (*p < 0.0001), confirming our previous reports of BLA priming enhancing effect on DG-LTP (Akirav and Richter-Levin, 1999a,b). Priming the CeA (CeA Priming, n=5) did not enhance DG-LTP levels compared with the Control LTP group, and the CeA Priming group was significantly different from the Ipsi BLA Priming group at both +30 min (*p < 0.01) and +60 min post-HFS (*p < 0.0001). This suggests that the CeA does not modulate DG plasticity.C, Schematic drawings of BLA and CeA electrode placements. Shown is a coronal view at positions 3.14 and 2.56 mm posterior to bregma for the BLA and the CeA, respectively. Solid black circles indicate the locations: (1) Ipsi BLA Priming group and (2) CeA Priming group (B, basal amygdala; La, lateral amygdala; together they form the BLA).
Post hoc comparisons showed a significant difference at both +30 and +60 min post-HFS between the ipsi BLA priming group and the control LTP group (+30 min: p < 0.001; +60 min:p < 0.0001) and between the ipsi BLA priming group and the CeA priming group (+30 min: p < 0.01; +60 min:p < 0.0001). There was no significant difference between the control LTP group and the CeA priming group at any time point. This confirms our previous results of a BLA priming enhancing effect on DG-LTP (Akirav and Richter-Levin, 1999a,b). Moreover, it suggests that under the conditions applied, the CeA does not modulate DG-LTP because priming the CeA has no enhancing effect.
The placements of the electrode tips located in the ipsi BLA priming and the CeA priming groups are shown in Figure 1C,(1) and (2), respectively.
Ipsilateral BLA priming effects are mediated by NE and CORT, but not by 5-HT
Potentiation levels in animals with NE depletion (using DSP-4), CORT depletion (using metyrapone), or 5-HT depletion (using PCPA) that received HFS to the PP were not significantly different from a vehicle control LTP group (data not shown). Thus, there was no significant effect of these drugs on DG-LTP. This result is in agreement with other reports showing that NE depletion using DSP-4 did not affect LTP (Dunwiddie et al., 1982; Robinson and Racine, 1985), although using other drugs to deplete NE reduced LTP (Bliss et al., 1983; Stanton and Sarvey, 1985). We then tested the effects of these drugs on BLA priming of DG-LTP.
One-way ANOVA revealed a significant difference between the groups at +30 and +60 min post-HFS (Fig.2A) (+30 min:F(3,34) = 7.718, p < 0.0001; +60 min: F(3,34) = 14.075,p < 0.0001), but not at any other time point.
Fig. 2.
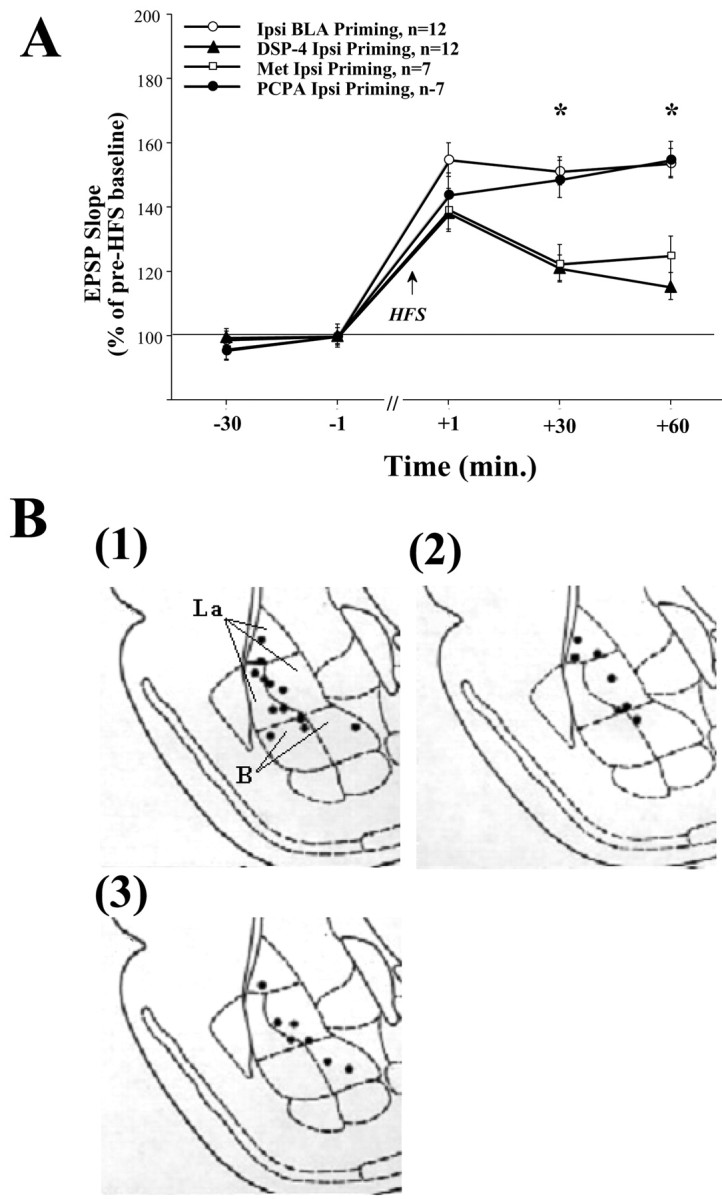
A, Ipsilateral BLA priming is mediated by NE and CORT but not by 5-HT. Priming the BLA in NE-depleted (DSP-4 Ipsi Priming, n=12) or CORT-depleted (Met Ipsi Priming, n=7) rats did not enhance DG-LTP as seen in the Ipsi BLA Priming group (+30 min: DSP-4 Ipsi Priming, *p < 0.0001; Met Ipsi Priming, p < 0.01; +60 min: DSP-4 Ipsi Priming, *p < 0.0001; Met Ipsi Priming, p < 0.001). This suggests that both NE and CORT may mediate the BLA priming enhancing effect on hippocampal LTP. In contrast, priming the BLA in 5-HT-depleted rats (PCPA Ipsi Priming, n=7) enhanced DG-LTP as in the Ipsi BLA Priming group, and this group was significantly different from the other depleted groups at both +30 min (DSP-4 Ipsi Priming, *p < 0.01; Met Ipsi Priming, p < 0.01) and +60 min post-HFS (DSP-4 Ipsi Priming, *p < 0.0001;Met Ipsi Priming, p < 0.01). This suggests that the BLA priming enhancing effect on DG-LTP is not dependent on serotonergic activation. B, Schematic drawings of BLA electrode placements. Solid black circles indicate the locations: (1) DSP-4 Ipsi Priming group, (2) Met Ipsi Priming group, and (3) PCPA Ipsi Priming group.
Post hoc comparisons showed a significant difference between the ipsi BLA priming group and the DSP-4 ipsi priming and the Met ipsi priming groups at both +30 min (DSP-4 ipsi priming: p< 0.0001; Met ipsi priming: p < 0.01) and +60 min post-HFS (DSP-4 ipsi priming: p < 0.0001; Met ipsi priming: p < 0.001). There was no significant difference between the DSP-4 ipsi priming group and the Met ipsi priming group at any time point. This suggests that both NE and CORT may mediate the BLA priming enhancing effect on DG-LTP.
As a control group for the stress modulators depletion, another group of animals was depleted of 5-HT using PCPA. Animals with 5-HT depletion that received BLA priming stimulation showed priming similar to control and were significantly different from the other depleted groups (same ANOVA as above).
Post hoc comparisons showed a significant difference between the PCPA ipsi priming group and the DSP-4 ipsi priming and the Met ipsi Priming groups at both +30 min (DSP-4 ipsi priming: p< 0.01; Met ipsi priming: p < 0.01) and +60 min post-HFS (DSP-4 ipsi priming: p < 0.0001; Met ipsi priming: p < 0.01). There was no significant difference between the PCPA ipsi priming group and the ipsi BLA priming group at any time point. This suggests that the BLA priming enhancing effect on DG-LTP is probably not dependent on serotonergic activation.
The placements of the electrode tips located in the DSP-4 ipsi priming, the Met ipsi priming, and the PCPA ipsi priming groups are shown in Figure 2B, (1),(2), and(3), respectively.
Contralateral priming enhances DG-LTP, but is not mediated by NE or CORT
One-way ANOVA revealed a significant difference between the groups at +30 and +60 min post-HFS (Fig.3A) (+30 min:F(3,33) = 3.88, p < 0.05; +60 min: F(3,33) = 5.508,p < 0.01), but not at any other time point.
Fig. 3.
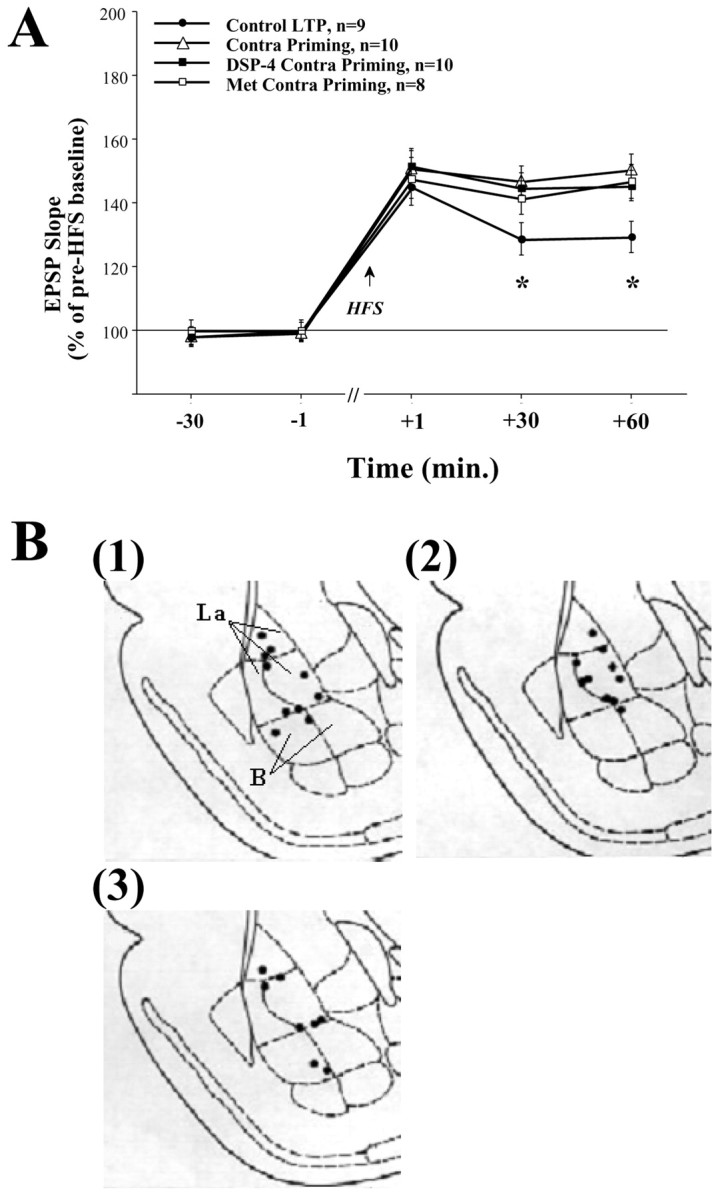
A, Contralateral priming enhances DG-LTP but is not NE or CORT dependent. Priming the contralateral BLA (Contra Priming, n=10) significantly enhanced DG-LTP levels compared with the control LTP group at both +30 min (*p < 0.01) and +60 min post-HFS (*p < 0.001). In addition, there was no significant difference between the Contra Priming group and the Ipsi BLA Priming group shown in Figure 1A. This suggests that priming the contralateral BLA enhances DG-LTP in a way similar to the enhancement seen by ipsilateral priming. Priming the contralateral BLA of rats depleted of NE (DSP-4 Contra Priming, n=10) or CORT (Met Contra Priming, n=8) also resulted in enhanced DG-LTP levels. They were significantly different from the control LTP group at both +30 min (Control LTP, *p < 0.01; DSP-4 Contra Priming,p < 0.05), and +60 min post-HFS (Control LTP, *p < 0.01; DSP-4 Contra Priming, p < 0.01) but were not different from the Contra Priming group. This suggests that, although contralateral BLA priming enhances DG-LTP, this effect is NE and CORT independent. B, Schematic drawings of BLA electrode placements. Solid black circles indicate the locations: (1) Contra Priming group, (2) DSP-4 Contra Priming group, and (3) Met Contra Priming group.
Post hoc comparisons showed a significant difference at both +30 and +60 min post-HFS between the control LTP group and the contra priming group (+30 min: p < 0.01; +60 min:p < 0.001). In addition, there was no significant difference between the contra priming group and the ipsi BLA priming group shown in Figure 1A. This suggests that similar to the enhancement seen by ipsilateral priming, priming of the contralateral BLA enhances DG-LTP.
Animals with depletion of NE or CORT that received priming stimulation to the contralateral BLA were also significantly different from the control LTP group (same ANOVA as above). Post hoccomparisons showed a significant difference at +30 min post-HFS between the control LTP group and the DSP-4 contra priming and the Met contra priming groups (DSP-4 contra priming: p < 0.01; Met contra priming: p < 0.05) and at +60 min post-HFS (DSP-4 contra priming: p < 0.01; Met contra priming:p < 0.01). Moreover, there was no significant difference between the contra priming group and DSP-4 contra priming and the Met contra priming groups at any time point.
This suggests that although contralateral BLA priming enhances DG-LTP, this effect is not dependent on noradrenergic or corticosteroid activation. Thus, differential mechanisms probably underlie the ipsilateral and contralateral BLA enhancing effects on DG-LTP.
The placements of the electrode tips located in the contra priming, DSP-4 contra priming, and Met contra priming groups are shown in Figure3B, (1), (2), and (3), respectively. To control for a possible lateralization effect of the BLA, electrodes were placed in the right and left hemispheres alternately, but are shown only in the right hemisphere for purpose of clarity.
Phase 2: spaced activation
Similar stimulus intensities were applied (F(6,45) < 1; NS) and using overall mixed ANOVA [groups × time (7 × 2)] for comparison between the groups before tetanization did not reveal a significant difference in EPSP slope at either −30 or −1 min before activating the BLA, indicating a similar baseline in all groups (Figs.4A,5A, and6A) (F(6,45) < 1; NS).
Fig. 4.
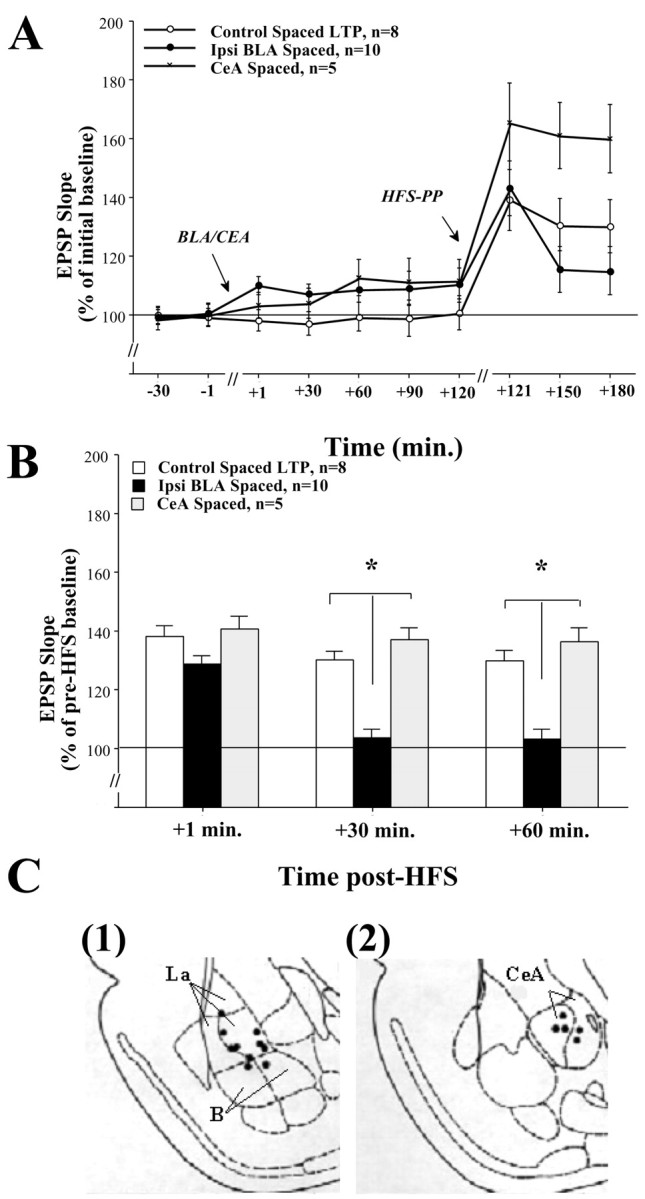
A, DG-EPSP slope after BLA and CeA stimulation. Shown is the DG-EPSP slope (in response to PP stimulation) for the duration of the experiment. B, Ipsilateral BLA spaced activation suppresses DG-LTP, whereas CeA spaced activation does not. The levels of DG-LTP in the Control Spaced LTP group (n = 8) after HFS to the PP were significantly different from 100% at all the times post-HFS to the PP (+1, +30, and + 60 min) (see Results). Spaced stimulation of the BLA (Ipsi BLA Spaced, n=10) significantly reduced DG-LTP levels compared with the Control Spaced LTP group at +30 min (*p < 0.0001) and +60 min post-HFS (*p < 0.0001). This confirms our previous reports showing BLA spaced activation suppressing DG-LTP levels (Akirav and Richter-Levin, 1999b). Spaced activation of the CeA (CeA Spaced, n=5) did not reduce DG-LTP levels compared with the Control Spaced LTP group and was significantly different from the Ipsi BLA Spaced group at both +30 min (*p < 0.0001) and +60 post-HFS (*p < 0.0001). This suggests that under these conditions CeA spaced activation does not modulate DG-LTP. Note that, although BLA and CeA spaced activation induced a shift in baseline EPSP (A), the levels of LTP in the CeA group were similar to that in the control group. This indicates that the lack of potentiation in the BLA spaced group was not caused by saturation of plasticity as a result of the shift in baseline EPSP before HFS.C, Schematic drawings of BLA and CeA electrode placements. Solid black circles indicate the locations: (1) Ipsi BLA Spaced group and (2) CeA Spaced group.
Fig. 5.
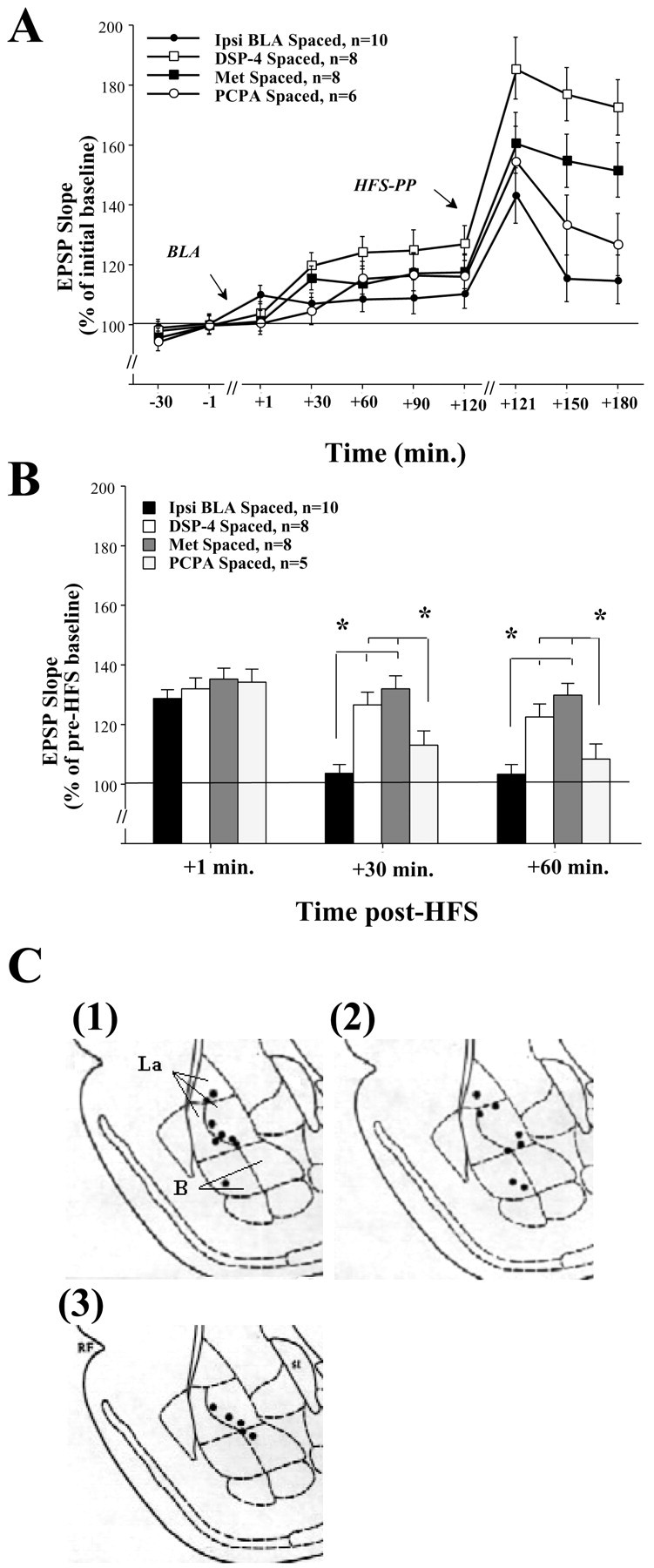
A, DG-EPSP slope after BLA stimulation in NE-, CORT-, and 5-HT-depleted animals. Shown is the DG-EPSP slope (in response to PP stimulation) for the duration of the experiment. B, Ipsilateral BLA spaced activation is mediated by NE and CORT but not by 5-HT. BLA spaced activation in NE-depleted (DSP-4 Spaced, n=8) or CORT-depleted (Met Spaced, n=8) rats did not reduce DG-LTP levels as seen in the Ipsi BLA Spaced rats at both +30 min (DSP-4 Spaced, *p < 0.0001; Met Spaced, *p < 0.0001) and +60 min post-HFS (DSP-4 Spaced, *p < 0.0001;Met Spaced, p < 0.01). Moreover, the NE-and CORT-depleted groups were not significantly different from the Control Spaced LTP group shown in Figure 4B. This suggests that both NE and CORT may mediate the BLA spaced suppressing effect on DG-LTP. BLA spaced activation in 5-HT-depleted rats (PCPA Spaced, n=6) significantly reduced DG-LTP compared with the NE- and CORT-depleted rats at both +30 min (DSP-4 Spaced, *p < 0.05;Met Spaced, p < 0.01) and +60 min post-HFS (DSP-4 Spaced, *p < 0.05;Met Spaced, p < 0.01). Moreover, there was no significant difference between the PCPA Spaced group and the Ipsi BLA Spaced group at any time point. This suggests that the BLA spaced suppressing effect on DG-LTP is not dependent on serotonergic activation. C, Schematic drawings of BLA electrode placements. Solid black circles indicate the locations: (1) DSP-4 Spaced group, (2) Met Spaced group, and (3) PCPA Spaced group.
Fig. 6.
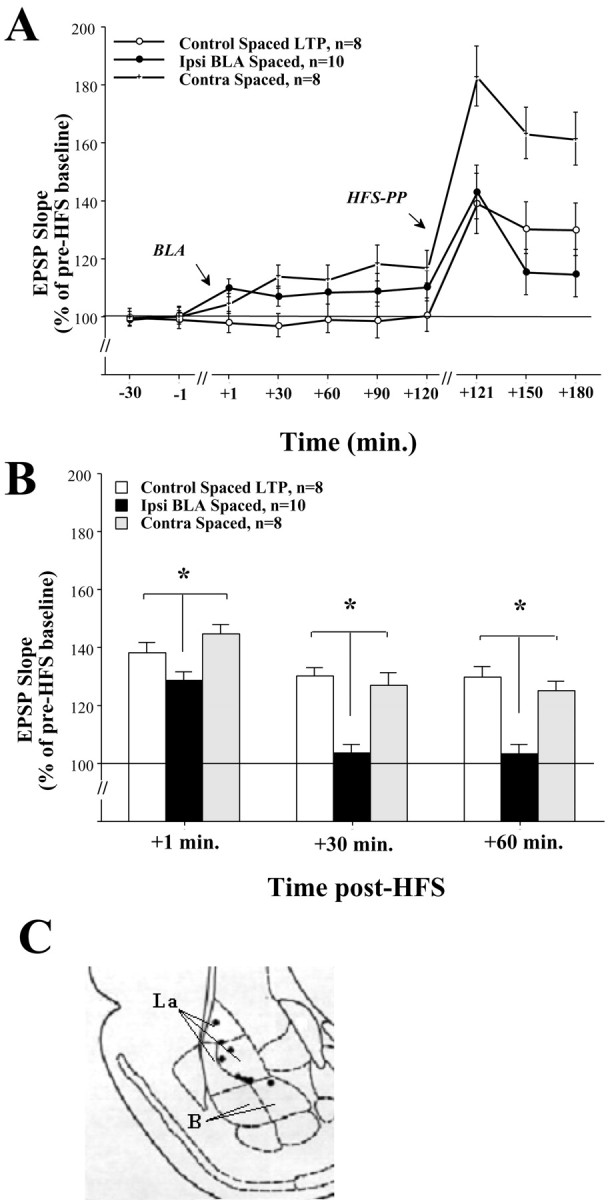
A, DG-EPSP slope after contralateral BLA stimulation. Shown is the DG-EPSP slope (in response to PP stimulation) for the duration of the experiment.B, Contralateral spaced activation does not suppress DG-LTP. Contralateral BLA spaced activation (n = 8) did not suppress DG-LTP as seen in the Ipsi BLA Spaced group at +1 min (*p < 0.01), +30 min (*p < 0.001), and +60 min post-HFS (*p < 0.001), and the Contra BLA Spaced group was not significantly different from the Control Spaced LTP group. Together with the results from Figure3A, this further supports the possibility that the ipsilateral and contralateral BLA effects on DG-LTP are mediated via different mechanisms. C, Schematic drawings of BLA electrode placements. Solid black circles indicate the locations of the Contra BLA Spaced group.
Using overall mixed ANOVA [groups × time (7 × 10)], we found a significant difference in EPSP slope levels between the groups (F(6,45) = 3.367; p = 0.008). However, there was an increase in EPSP slope levels after BLA/CeA activation in all the groups (except for the control spaced LTP group) before the application of HFS to the PP [time point of +120 min (Figs. 4A, 5A, 6A)]. Thus we calculated DG-LTP levels after HFS to the PP (time points +121, +150, and +180 min) as percentage of the pre-HFS baseline (time point +120 min) and not as percentage of the initial baseline (time point −1 min). Using overall mixed ANOVA [groups × time (7 × 3)], we found a significant difference in DG-LTP levels between the groups (F(6,46) = 9.624; p = 0.0001) that was further analyzed (Figs. 4B,5B, 6B).
Ipsilateral BLA spaced activation suppresses DG-LTP, whereas CeA spaced activation does not
Figure 4A shows DG-EPSP slope (in response to PP stimulation) for the duration of the experiment. The level of potentiation in the control spaced LTP group after HFS to the PP was significantly different from 100% at all times post-HFS [ttest for difference from baseline (100%): +1 min,t(7) = 3.7981, p < 0.01; +30 min, t(7) = 3.4494,p < 0.05; +60 min,t(7) = 3.3132; p < 0.05].
Figure 4B shows a significant difference between the groups at +30 min (one-way ANOVA:F(2,20) = 33.147; p < 0.0001) and +60 min post-HFS to the PP (F(2,20) = 20.977; p< 0.0001). Post hoc comparisons showed a significant difference between the ipsi BLA spaced group and the control spaced LTP and the CeA spaced groups at +30 min (control spaced LTP: p < 0.0001; CeA spaced: p < 0.0001) and at +60 min post-HFS to the PP (control spaced LTP:p < 0.0001; CeA spaced: p < 0.0001).
This confirms our previous results of BLA spaced activation suppressing DG-LTP (Akirav and Richter-Levin, 1999b). Moreover, it further suggests that under these conditions CeA spaced activation does not modulate DG-LTP. Note that although BLA and CeA spaced activation induced a shift in baseline EPSP (Fig. 4A), the level of LTP in the CeA group was similar to that in the control group (Fig.4B). This indicates that the lack of potentiation in the BLA spaced group was not caused by saturation of plasticity as a result of the shift in baseline EPSP before HFS.
The placements of the electrode tips located in the ipsi BLA spaced and the CeA spaced groups are shown in Figure 4C, (1)and (2), respectively.
Ipsilateral BLA spaced activation is mediated by NE and CORT, but not by 5-HT
Figure 5A shows DG-EPSP slope (in response to PP stimulation) for the duration of the experiment. Figure 5Bshows a significant difference between the groups at +30 and +60 min post-HFS (one-way ANOVA: +30 min:F(3,28) = 10.354, p < 0.0001; +60 min: F(3,28) = 8.995,p < 0.0001). Post hoc comparisons showed a significant difference between the ipsi BLA spaced group and the DSP-4 spaced and the Met spaced groups at both +30 min (DSP-4 spaced:p < 0.0001; Met spaced: p < 0.0001) and +60 min post-HFS (DSP-4 spaced: p < 0.0001; Met spaced: p < 0.01). There was no significant difference between the DSP-4 spaced group and the Met spaced group at any time point. This suggests that both NE and CORT may mediate the BLA spaced suppressing effect on DG-LTP.
As a control group for modulators depletion, another group of animals was depleted of 5-HT using PCPA. Post hoc comparisons showed a significant difference between the PCPA spaced group and the DSP-4 spaced and the Met spaced groups at both +30 min (DSP-4 spaced:p < 0.05; Met spaced: p < 0.01) and +60 min post-HFS (DSP-4 spaced: p < 0.05; Met spaced:p < 0.01). There was no significant difference between the PCPA spaced group and the ipsi BLA spaced group at any time point. This suggests that the BLA spaced suppressing effect on DG-LTP is probably not dependent on serotonergic activation.
The placements of the electrode tips located in the DSP-4 spaced, the Met spaced, and the PCPA spaced groups are shown in Figure5C, (1), (2), and (3), respectively.
Contralateral spaced activation does not suppress DG-LTP
Figure 6A shows DG-EPSP slope (in response to PP stimulation) for the duration of the experiment. Figure6B shows a significant difference between the groups at +1, +30, and +60 min post-HFS (one-way ANOVA: +1 min:F(2,23) = 5.851, p < 0.001; +30 min: F(2,23) = 17.406, p < 0.0001; +60 min:F(2,23) = 15.177, p < 0.0001). Post hoc comparisons showed a significant difference between the ipsi BLA spaced group and the control LTP and the contra BLA spaced groups at +1 min (control LTP:p = 0.053; contra BLA spaced: p < 0.01), +30 min (control LTP: p < 0.001; contra BLA spaced: p < 0.001) and +60 min post-HFS (control LTP:p < 0.001; contra BLA spaced: p < 0.001). There was no significant difference between the control LTP group and the contra BLA spaced group at any time point.
This shows that the contralateral spaced activation does not suppress DG-LTP, and together with the results from Figure 3A, further supports that there are differential mechanisms underlying the ipsilateral and the contralateral BLA effects on DG-LTP.
Note that although there is a shift in baseline EPSP in the contra BLA spaced group (Fig. 6A), the level of LTP in this group was similar to that of the control spaced LTP group. This indicates that the lack of potentiation in the BLA spaced group was not caused by saturation of plasticity as a result of the shift in baseline EPSP before HFS.
The placements of the electrode tips located in the contra spaced group are shown in Figure 6C. To control for a possible lateralization effect of the BLA, electrodes were placed in the right and left hemispheres alternately, but are shown only in the right hemisphere for the purpose of clarity.
DISCUSSION
The BLA affects hippocampal plasticity in a complex manner. Ipsilateral and contralateral BLA priming has an enhancing effect on hippocampal plasticity, whereas ipsilateral BLA spaced activation has an inhibitory influence. The ipsilateral and contralateral BLA activation seem to affect the hippocampus via differential mechanisms, and finally both NE and CORT seem to be required for BLA modulation (enhancement or suppression) of DG-LTP.
Phase 1: BLA priming
Ipsilateral BLA priming significantly enhanced DG-LTP levels, thus providing further support for our previous findings (Akirav and Richter-Levin, 1999a,b). In contrast, priming the CeA did not enhance DG-LTP, supporting reports showing that the CeA does not modulate hippocampal memory processes and plasticity (Ikegaya et al., 1994;Roozendaal and McGaugh 1996, 1997). Taken together, these findings strongly support the view that the CeA is not the exclusive output of amygdala complex and that the BLA may be part of a parallel processing system (Killcross et al., 1997; Amorapanth et al., 2000).
We hypothesized that NE mediates the BLA priming enhancing effect and CORT mediates the spaced inhibitory effect, and not vice versa. Indeed, NE-depleted rats showed no priming effect. These findings are in agreement with evidence showing that BLA activation of adrenergic mechanisms may induce hippocampal-dependent memory enhancement (Liang et al., 1990; Hatfield and McGaugh, 1999).
In contrast to our hypothesis, however, priming was absent also in the CORT-depleted rats. This finding corresponds to reports showing that administration of exogenous CORT in the appropriate temporal context, i.e., in close relation to training, potentiated memory for hippocampal-dependent tasks (Sandi et al., 1997; Roozendaal et al., 1999). In general, it has been suggested that glucocorticoid levels at the onset of an emotional event permissively mediate the cognitive stress response, whereas the subsequent stress-induced rise in glucocorticoid concentrations suppresses the cognitive response (Sapolsky et al., 2000). Additionally, because the blocking of priming by metyrapone was evident only 30 min post-HFS, it is possible that amygdala-induced increase in CORT levels is required for post post-tetanic potentiation mechanisms of LTP enhancement.
Furthermore, although priming of the contralateral BLA had similar enhancing effects on DG-LTP as priming of the ipsilateral BLA, contralateral priming was found to be NE and CORT independent, implying that different neural mechanisms underlie the ipsilateral and contralateral amygdala priming effects on hippocampal plasticity.
Phase 2: BLA spaced activation
Ipsilateral BLA spaced activation significantly suppressed DG-LTP levels, hence providing further support for our suggested biphasic model of amygdala modulation of hippocampal plasticity (Akirav and Richter-Levin, 1999b). CeA spaced activation did not suppress DG-LTP, further supporting the view that the CeA does not modulate hippocampal plasticity.
In accordance with our hypothesis that the inhibitory effects of the spaced activation of the amygdala on DG-LTP are mediated by CORT, inhibition was significantly suppressed in CORT-depleted rats. However, contrary to our expectations, NE depletion also prevented the inhibition of LTP by BLA spaced activation.
Contrary to the effects of ipsilateral spaced activation, contralateral BLA spaced activation did not suppress DG-LTP. Together with the above-described differences between the dependency of ipsilateral and contralateral priming effects on NE and CORT, these results indicate that the ipsilateral and contralateral effects of BLA on hippocampal plasticity are mediated via different mechanisms.
BLA-DG: possible pathways
BLA activity may affect hippocampal LTP through a number of pathways. The BLA is composed of the basal, lateral, and accessory basal nuclei that project to the parasubiculum and the entorhinal cortex (EC), which projects to the DG (Pikkarainen et al., 1999). It has been suggested (Ikegaya et al., 1997) that noradrenergic activity in the BLA potentiates NMDA receptor-mediated transmission in the amygdala and thus facilitates the induction of DG-LTP. Others have suggested that NMDA receptor activation is not required for BLA enhancement of DG-LTP and that this effect is triggered by synergistic actions of glutamatergic and nonglutamatergic mechanisms (Frey et al., 2001). All the same, the effect still may be triggered through or dependent on direct action of noradrenergic mechanisms in the BLA (Liang et al., 1986, 1990, 1995; Ferry and McGaugh, 2000; Frey et al., 2001).
Another possibility is the involvement of other brain structures that may be influenced by the BLA. The locus ceruleus may be activated to induce NE release in the hippocampus and contribute to the facilitation of LTP (Bliss et al., 1983; Neuman and Harley, 1983; Stanton and Sarvey, 1985). Glucocorticoids enter the brain readily and can directly influence hippocampal GRs to modulate LTP (McEwen and Sapolsky, 1995). Thus amygdala modulation of the hypothalamus may affect CORT release and, by this, hippocampal LTP (Price and Amaral, 1981).
The BLA projects to the CeA (Savander et al., 1995), which is a major output nucleus of the amygdala with direct connections to centers involved in NE and glucocorticoid secretion (Davis, 1992). However, we found that the CeA does not modulate DG-LTP. Similarly, behavioral and electrophysiological data show that whereas BLA lesions block modulatory effects on the hippocampus, CeA lesions are ineffective (Ikegaya et al., 1994; Roozendaal and McGaugh 1996, 1997). Thus, the CeA is clearly not sufficient to mediate the BLA effects on hippocampal plasticity.
The mediation of the stress hormones
Both NE and CORT were found to mediate the enhancing as well as suppressive effects of the BLA on DG-LTP. Noradrenergic activation of the BLA is required for the adrenal steroids to influence hippocampal memory storage (Quirarte et al., 1997; Roozendaal et al., 1999), and glucocorticoids seem to exert a permissive action on the efficacy of the noradrenergic system (de Kloet, 1991; Roozendaal, 2000; Roozendaal et al., 2002). It is currently unclear whether an interaction between these two modulatory systems or their parallel action is required. It may be that lack of either system could affect BLA modulation of hippocampal LTP to the same degree. Furthermore, it is not known whether the interaction occurs at the level of the amygdala, in the hippocampus, within a third region, or a combination of those possibilities.
Because both NE and CORT seem to be involved in the enhancing as well as the inhibitory effects of the BLA, it is intriguing to try to explain what may define whether an enhancement or an inhibition of LTP will take place. One possibility is the involvement of a third mediator that will define the outcome. Such a mediator could be acetylcholine (ACh). ACh has been suggested to mediate the transition of early into late phase LTP by BLA activation (Frey et al., 2001), and there are indications that NE effects on memory involve subsequent cholinergic activation in the amygdala (Introini-Collison et al., 1996). Furthermore, it has been suggested that ACh is involved in stress effects on hippocampal processing (Bhatnagar et al., 1997; Kaufer et al., 1998). Another mediator could be corticotropin-releasing factor (CRF). CRF is released from the hypothalamus in response to stress and leads to the secretion of the stress hormones (Lathe, 2001). CRF injected into the DG produced a dose-dependent and long-lasting enhancement in synaptic efficacy of these neurons (Wang et al., 1998), but sustained administration of CRF prevented the occurrence of LTP (Rebaudo et al., 2001).
Another possible explanation for the involvement of both NE and CORT is that the effects seen are time dependent, i.e., the effects of a brief exposure to these hormones are excitatory, whereas their prolonged presence in the spaced phase may lead to the inhibitory effect.
A third possibility, which is not mutually exclusive, could be that the effects depend on the exact ratio between the effects of the two hormones, i.e., both are required for the modulation, but the specific concentration of each will define the outcome.
Ipsilateral versus contralateral effects
Adding to this already complex picture is the possible interaction with the contralateral BLA. It has been shown (Ikegaya et al., 1994) that lesion of the ipsilateral but not of the contralateral BLA affected the induction of DG-LTP. In addition, it has been shown that inactivation of the ipsilateral but not of the contralateral BLA blocked the effects of a GR agonist in the hippocampus on an avoidance task (Roozendaal et al., 1999) and that a lesion of the ipsilateral BLA, but not the contralateral BLA, blocked the enhancing effect of cAMP analog infused into the EC on memory for an inhibitory avoidance task (Roesler et al., 2002). The authors suggested that this modulatory effect was mediated via ipsilateral neural pathways connecting the BLA and the EC, because if BLA modulation was mediated only by peripheral stress responses, then the ipsilateral and the contralateral BLA should have had similar effects on hippocampal memory (Roozendaal, 2000). Here, we found a more multifaceted picture. The contralateral BLA, when primed, enhanced DG-LTP, but this effect was NE and CORT independent, whereas the spaced contralateral BLA activation did not inhibit DG-LTP.
The basal nucleus and the accessory basal nucleus give rise to substantial projections to the contralateral basal nucleus and the accessory basal nucleus, respectively; the lateral nucleus does not project to the contralateral amygdala, but it projects to the ipsilateral basal nucleus (Pitkanen et al., 1995; Savander et al., 1997). It is possible that these hemisphere-crossing connections mediate the contralateral priming effect, although such an explanation would predict that the contralateral priming effect would be NE and CORT dependent, like ipsilateral priming. Our findings argue against this possibility.
Taken together, the results suggest the existence of two distinctive pathways: an ipsilateral neural pathway that requires the involvement of NE and CORT and a contralateral pathway that presumably acts through the mediation of another brain structure. The effects of this pathway are NE and CORT independent.
Summary
An emotional experience activates the BLA, which in turn modulates hippocampal-dependent memory in a complex manner via the mediation of the stress hormones NE and CORT. The complexity revealed here may be necessary for the establishment of an accurate and detailed memory of emotionally rich experiences. The mechanisms described can explain how it is possible that stressful experiences can both enhance and impair memories, depending on the particular details of the events. Although the specific pathways involved are still obscure and require further research, it can already be envisaged that abnormalities in the complexity with which the BLA is able to modulate hippocampal function are likely to contribute to stress-related affective disorders.
Footnotes
This research was supported by The Israel Science Foundation–The Charles H. Revson Foundation (582/00-1 to G.R.-L.). We thank Dr. Carolyn Harley for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Gal Richter-Levin, Laboratory of Behavioral Neuroscience, Department of Psychology, University of Haifa, Haifa 31905, Israel. E-mail:gal.r-l@psy.haifa.ac.il.
REFERENCES
- 1.Abe K. Modulation of hippocampal long-term potentiation by the amygdala: a synaptic mechanism linking emotion and memory. Jpn J Pharmacol. 2001;86:18–22. doi: 10.1254/jjp.86.18. [DOI] [PubMed] [Google Scholar]
- 2.Akirav I, Richter-Levin G. Priming stimulation in the basolateral amygdala modulates synaptic plasticity in the rat dentate gyrus. Neurosci Lett. 1999a;30:83–86. doi: 10.1016/s0304-3940(99)00488-7. [DOI] [PubMed] [Google Scholar]
- 3.Akirav I, Richter-Levin G. Biphasic modulation of hippocampal plasticity by behavioral stress and basolateral amygdala stimulation in the rat. J Neurosci. 1999b;1:10530–10535. doi: 10.1523/JNEUROSCI.19-23-10530.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amorapanth P, LeDoux JE, Nader K. Different lateral amygdala outputs mediate reactions and actions elicited by a fear-arousing stimulus. Nat Neurosci. 2000;3:74–79. doi: 10.1038/71145. [DOI] [PubMed] [Google Scholar]
- 5.Bhatnagar S, Costall B, Smythe JW. Hippocampal cholinergic blockade enhances hypothalamic-pituitary-adrenal responses to stress. Brain Res. 1997;22:244–248. doi: 10.1016/s0006-8993(97)00684-7. [DOI] [PubMed] [Google Scholar]
- 6.Bliss TVP, Goddard GV, Riives M. Reduction of long-term potentiation in the dentate gyrus of the rat following selective depletion of monoamines. J Physiol (Lond) 1983;334:475–491. doi: 10.1113/jphysiol.1983.sp014507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahill L, McGaugh JL. Mechanisms of emotional arousal and lasting declarative memory. Trends Neurosci. 1998;21:294–299. doi: 10.1016/s0166-2236(97)01214-9. [DOI] [PubMed] [Google Scholar]
- 8.Cahill L, Prins B, Weber M, McGaugh JL. Beta-adrenergic activation and memory for emotional events. Nature. 1994;20:702–704. doi: 10.1038/371702a0. [DOI] [PubMed] [Google Scholar]
- 9.Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 10.de Kloet ER. Brain corticosteroid receptor balance and homeostatic control. Front Neuroendocrinol. 1991;12:95–164. doi: 10.1016/j.yfrne.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 11.de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- 12.Diamond DM, Bennett MC, Engstrom DA, Fleshner M, Rose GM. Adrenalectomy reduces the threshold for hippocampal primed burst potentiation in the anesthetized rat. Brain Res. 1989;17:356–360. doi: 10.1016/0006-8993(89)90919-0. [DOI] [PubMed] [Google Scholar]
- 13.Diamond DM, Bennett MC, Fleshner M, Rose GM. Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus. 1992;2:421–430. doi: 10.1002/hipo.450020409. [DOI] [PubMed] [Google Scholar]
- 14.Diamond DM, Fleshner M, Rose GM. Psychological stress repeatedly blocks hippocampal primed burst potentiation in behaving rats. Behav Brain Res. 1994;30:1–9. doi: 10.1016/0166-4328(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 15.Diamond DM, Puls JM, Park CR, Rose MG. Differential effects of stress on hippocampal and amygdaloid LTP: insight into the neurobiology of traumatic memories. In: Holscher C, editor. Neuronal mechanisms of memory formation: concepts of long-term potentiation and beyond. Cambridge UP; Cambridge, UK: 2000. pp. 379–404. [Google Scholar]
- 16.Dunwiddie TV, Roberson NL, Worth T. Modulation of long-term potentiation: effects of adrenergic and neuroleptic drugs. Pharmacol Biochem Behav. 1982;17:1257–1264. doi: 10.1016/0091-3057(82)90130-7. [DOI] [PubMed] [Google Scholar]
- 17.Feldman S, Conforti N, Siegel RA. Adrenocortical responses following limbic stimulation in rats with hypothalamic deafferentations. Neuroendocrinology. 1982;35:205–211. doi: 10.1159/000123382. [DOI] [PubMed] [Google Scholar]
- 18.Ferry B, McGaugh JL. Role of amygdala norepinephrine in mediating stress hormone regulation of memory storage. Acta Pharmacol Sin. 2000;21:481–493. [PubMed] [Google Scholar]
- 19.Ferry B, Roozendaal B, McGaugh JL. Basolateral amygdala noradrenergic influences on memory storage are mediated by an interaction between β- and α1-adrenoceptors. J Neurosci. 1999;15:5119–5123. doi: 10.1523/JNEUROSCI.19-12-05119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey S, Bergado-Rosado J, Seidenbecher T, Pape HC, Frey JU. Reinforcement of early long-term potentiation (early-LTP) in dentate gyrus by stimulation of the basolateral amygdala: heterosynaptic induction mechanisms of late-LTP. J Neurosci. 2001;15:3697–3703. doi: 10.1523/JNEUROSCI.21-10-03697.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatfield T, McGaugh JL. Norepinephrine infused into the basolateral amygdala posttraining enhances retention in a spatial water maze task. Neurobiol Learn Mem. 1999;71:232–239. doi: 10.1006/nlme.1998.3875. [DOI] [PubMed] [Google Scholar]
- 22.Ikegaya Y, Saito H, Abe K. Attenuated hippocampal long-term potentiation in basolateral amygdala-lesioned rats. Brain Res. 1994;5:157–164. doi: 10.1016/0006-8993(94)91377-3. [DOI] [PubMed] [Google Scholar]
- 23.Ikegaya Y, Saito H, Abe K. High-frequency stimulation of the basolateral amygdala facilitates the induction of long-term potentiation in the dentate gyrus in vivo. Neurosci Res. 1995;22:203–207. doi: 10.1016/0168-0102(95)00894-7. [DOI] [PubMed] [Google Scholar]
- 24.Ikegaya Y, Nakanishi K, Saito H, Abe K. Amygdala beta-noradrenergic influence on hippocampal long-term potentiation in vivo. NeuroReport. 1997;29:3143–3146. doi: 10.1097/00001756-199709290-00027. [DOI] [PubMed] [Google Scholar]
- 25.Introini-Collison IB, Dalmaz C, McGaugh JL. Amygdala beta-noradrenergic influences on memory storage involve cholinergic activation. Neurobiol Learn Mem. 1996;65:57–64. doi: 10.1006/nlme.1996.0006. [DOI] [PubMed] [Google Scholar]
- 26.Izquierdo I, Medina JH. Correlation between the pharmacology of long-term potentiation and the pharmacology of memory. Neurobiol Learn Mem. 1995;63:19–32. doi: 10.1006/nlme.1995.1002. [DOI] [PubMed] [Google Scholar]
- 27.Jaim-Etcheverry G, Zieher LM. DSP-4: a novel compound with neurotoxic effects on noradrenergic neurons of adult and developing rats. Brain Res. 1980;28:513–523. doi: 10.1016/0006-8993(80)90049-9. [DOI] [PubMed] [Google Scholar]
- 28.Kaufer D, Friedman A, Seidman S, Soreq H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature. 1998;28:373–377. doi: 10.1038/30741. [DOI] [PubMed] [Google Scholar]
- 29.Kerr DS, Huggett AM, Abraham WC. Modulation of hippocampal long-term potentiation and long-term depression by corticosteroid receptor activation. Psychobiology. 1994;22:123–133. [Google Scholar]
- 30.Killcross S, Robbins TW, Everitt BJ. Different types of fear-conditioned behaviour mediated by separate nuclei within amygdala. Nature. 1997;24:377–380. doi: 10.1038/41097. [DOI] [PubMed] [Google Scholar]
- 31.Lathe R. Hormones and the hippocampus. J Endocrinol. 2001;169:205–231. doi: 10.1677/joe.0.1690205. [DOI] [PubMed] [Google Scholar]
- 32.Liang KC, Juler RG, McGaugh JL. Modulating effects of posttraining epinephrine on memory: involvement of the amygdala noradrenergic system. Brain Res. 1986;12:125–133. doi: 10.1016/0006-8993(86)91049-8. [DOI] [PubMed] [Google Scholar]
- 33.Liang KC, McGaugh JL, Yao HY. Involvement of amygdala pathways in the influence of post-training intra-amygdala norepinephrine and peripheral epinephrine on memory storage. Brain Res. 1990;5:225–233. doi: 10.1016/0006-8993(90)90400-6. [DOI] [PubMed] [Google Scholar]
- 34.Liang KC, Chen LL, Huang TE. The role of amygdala norepinephrine in memory formation: involvement in the memory enhancing effect of peripheral epinephrine. Chin J Physiol. 1995;38:81–91. [PubMed] [Google Scholar]
- 35.McEwen BS, Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 36.McGaugh JL. Involvement of hormonal and neuromodulatory systems in the regulation of memory storage. Annu Rev Neurosci. 1989;12:255–287. doi: 10.1146/annurev.ne.12.030189.001351. [DOI] [PubMed] [Google Scholar]
- 37.McGaugh JL. Memory—a century of consolidation. Science. 2000;14:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- 38.Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M. Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: an immunohistochemical and in situ hybridization study. Neurosci Res. 1996;26:235–269. doi: 10.1016/s0168-0102(96)01105-4. [DOI] [PubMed] [Google Scholar]
- 39.Neuman RS, Harley CW. Long-lasting potentiation of the dentate gyrus population spike by norepinephrine. Brain Res. 1983;22:162–165. doi: 10.1016/0006-8993(83)91106-x. [DOI] [PubMed] [Google Scholar]
- 40.Packard MG, Cahill L. Affective modulation of multiple memory systems. Curr Opin Neurobiol. 2001;11:752–756. doi: 10.1016/s0959-4388(01)00280-x. [DOI] [PubMed] [Google Scholar]
- 41.Pavlides C, Watanabe Y, McEwen BS. Effects of glucocorticoids on hippocampal long-term potentiation. Hippocampus. 1993;3:183–192. doi: 10.1002/hipo.450030210. [DOI] [PubMed] [Google Scholar]
- 42.Pavlides C, Watanabe Y, Magarinos AM, McEwen BS. Opposing roles of type I and type II adrenal steroid receptors in hippocampal long-term potentiation. Neuroscience. 1995;68:387–394. doi: 10.1016/0306-4522(95)00151-8. [DOI] [PubMed] [Google Scholar]
- 43.Pikkarainen M, Ronkko S, Savander V, Insausti R, Pitkanen A. Projections from the lateral, basal, and accessory basal nuclei of the amygdala to the hippocampal formation in rat. J Comp Neurol. 1999;11:229–260. [PubMed] [Google Scholar]
- 44.Pitkanen A, Stefanacci L, Farb CR, Go GG, LeDoux JE, Amaral DG. Intrinsic connections of the rat amygdaloid complex: projections originating in the lateral nucleus. J Comp Neurol. 1995;29:288–310. doi: 10.1002/cne.903560211. [DOI] [PubMed] [Google Scholar]
- 45.Price JL, Amaral DG. An autoradiographic study of the projections of the central nucleus of the monkey amygdala. J Neurosci. 1981;1:1242–1259. doi: 10.1523/JNEUROSCI.01-11-01242.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quirarte GL, Roozendaal B, McGaugh JL. Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci USA. 1997;9:14048–14053. doi: 10.1073/pnas.94.25.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rebaudo R, Melani R, Balestrino M, Izvarina N. Electrophysiological effects of sustained delivery of CRF and its receptor agonists in hippocampal slices. Brain Res. 2001;13:112–117. doi: 10.1016/s0006-8993(01)03160-2. [DOI] [PubMed] [Google Scholar]
- 48.Rey M, Carlier E, Talmi M, Soumireu-Mourat B. Corticosterone effects on long-term potentiation in mouse hippocampal slices. Neuroendocrinology. 1994;60:36–41. doi: 10.1159/000126717. [DOI] [PubMed] [Google Scholar]
- 49.Richter-Levin G, Akirav I. Amygdala-hippocampus dynamic interaction in relation to memory. Mol Neurobiol. 2000;22:11–20. doi: 10.1385/MN:22:1-3:011. [DOI] [PubMed] [Google Scholar]
- 50.Richter-Levin G, Segal M. Spatial performance is severely impaired in rats with combined reduction of serotonergic and cholinergic transmission. Brain Res. 1989;16:404–407. doi: 10.1016/0006-8993(89)91437-6. [DOI] [PubMed] [Google Scholar]
- 51.Richter-Levin G, Segal M, Sara S. An alpha 2 antagonist, idazoxan, enhances EPSP-spike coupling in the rat dentate gyrus. Brain Res. 1991;1:291–294. doi: 10.1016/0006-8993(91)90521-v. [DOI] [PubMed] [Google Scholar]
- 52.Robinson GB, Racine RJ. Long-term potentiation in the dentate gyrus: effects of noradrenaline depletion in the awake rat. Brain Res. 1985;28:71–78. doi: 10.1016/0006-8993(85)90303-8. [DOI] [PubMed] [Google Scholar]
- 53.Roesler R, Roozendaal B, McGaugh JL. Basolateral amygdala lesions block the memory-enhancing effect of 8-Br-cAMP infused into the entorhinal cortex of rats after training. Eur J Neurosci. 2002;15:905–910. doi: 10.1046/j.1460-9568.2002.01924.x. [DOI] [PubMed] [Google Scholar]
- 54.Roozendaal B. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000;25:213–238. doi: 10.1016/s0306-4530(99)00058-x. [DOI] [PubMed] [Google Scholar]
- 55.Roozendaal B, McGaugh JL. Amygdaloid nuclei lesions differentially affect glucocorticoid-induced memory enhancement in an inhibitory avoidance task. Neurobiol Learn Mem. 1996;65:1–8. doi: 10.1006/nlme.1996.0001. [DOI] [PubMed] [Google Scholar]
- 56.Roozendaal B, McGaugh JL. Basolateral amygdala lesions block the memory-enhancing effect of glucocorticoid administration in the dorsal hippocampus of rats. Eur J Neurosci. 1997;9:76–83. doi: 10.1111/j.1460-9568.1997.tb01355.x. [DOI] [PubMed] [Google Scholar]
- 57.Roozendaal B, Portillo-Marquez G, McGaugh JL. Basolateral amygdala lesions block glucocorticoid-induced modulation of memory for spatial learning. Behav Neurosci. 1996;110:1074–1083. doi: 10.1037//0735-7044.110.5.1074. [DOI] [PubMed] [Google Scholar]
- 58.Roozendaal B, Nguyen BT, Power AE, McGaugh JL. Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci USA. 1999;28:11642–11647. doi: 10.1073/pnas.96.20.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roozendaal B, Quirarte GL, McGaugh JL. Glucocorticoids interact with the basolateral amygdala beta-adrenoceptor–cAMP/cAMP/PKA system in influencing memory consolidation. Eur J Neurosci. 2002;15:553–560. doi: 10.1046/j.0953-816x.2001.01876.x. [DOI] [PubMed] [Google Scholar]
- 60.Sandi C, Loscertales M, Guaza C. Experience-dependent facilitating effect of corticosterone on spatial memory formation in the water maze. Eur J Neurosci. 1997;9:637–642. doi: 10.1111/j.1460-9568.1997.tb01412.x. [DOI] [PubMed] [Google Scholar]
- 61.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 62.Savander V, Go CG, LeDoux JE, Pitkanen A. Intrinsic connections of the rat amygdaloid complex: projections originating in the basal nucleus. J Comp Neurol. 1995;16:345–368. doi: 10.1002/cne.903610211. [DOI] [PubMed] [Google Scholar]
- 63.Savander V, Ledoux JE, Pitkanen A. Interamygdaloid projections of the basal and accessory basal nuclei of the rat amygdaloid complex. Neuroscience. 1997;76:725–735. doi: 10.1016/s0306-4522(96)00371-5. [DOI] [PubMed] [Google Scholar]
- 64.Seidenbecher T, Reymann KG, Balschun D. A post-tetanic time window for the reinforcement of long-term potentiation by appetitive and aversive stimuli. Proc Natl Acad Sci USA. 1997;18:1494–1499. doi: 10.1073/pnas.94.4.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stanton PK, Sarvey JM. Depletion of norepinephrine, but not serotonin, reduces long-term potentiation in the dentate gyrus of rat hippocampal slices. J Neurosci. 1985;5:2169–2176. doi: 10.1523/JNEUROSCI.05-08-02169.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang HL, Wayner MJ, Chai CY, Lee EH. Corticotrophin-releasing factor produces a long-lasting enhancement of synaptic efficacy in the hippocampus. Eur J Neurosci. 1998;10:3428–3437. doi: 10.1046/j.1460-9568.1998.00352.x. [DOI] [PubMed] [Google Scholar]