

Abstract
Here, we show that the p53 family member, p73, is necessary for survival and long-term maintenance of CNS neurons, including postnatal cortical neurons. In p73−/− animals, cortical neuron number is normal at birth but decreases significantly by postnatal day 14 (P14)–P16 because of enhanced apoptosis. This decrease continues into adulthood, when p73−/− animals have approximately one-half as many cortical cells as their wild-type littermates. Cortical neurons express the ΔNp73α protein, and overexpression of ΔNp73 isoforms rescues cortical neurons from diverse apoptotic stimuli. Thus, ΔNp73 isoforms are survival proteins in cortical neurons, and their deletion causes a gradual loss of cortical neurons in the weeks and months after birth. This decrease in CNS neuron number in p73−/− animals is not limited to the cortex; facial motor neuron number is decreased, and postnatal development of the olfactory bulb is greatly perturbed. These findings, together with our previous work showing that ΔNp73 is essential for survival of peripheral sympathetic neurons (Pozniak et al., 2000), indicate that p73 isoforms are essential survival proteins in CNS as well as PNS neurons, and that they likely play a role not only during developmental cell death but also in the long-term maintenance of at least some adult neurons.
Keywords: p73, p53, cortical neurons, facial motor neurons, olfactory bulb, neuronal survival, neuronal development, neuronal apoptosis, neuronal degeneration, camptothecin, PI3-kinase
Neurodegenerative disorders are characterized by an enhanced rate of neuronal apoptosis in the CNS, resulting in devastating loss of function. Although the intracellular mechanisms that regulate peripheral neuron survival have been intensively studied, less is known of the signaling mechanisms that determine the life versus death of CNS neurons. One protein known to play an important role in regulating neuronal survival in both the PNS and CNS is the p53 tumor suppressor protein (for review, see Miller et al., 2000; Morrison and Kinoshita, 2000). Overexpression of p53 is sufficient to cause the death of a variety of neurons (Slack et al., 1996; Xiang et al., 1996; Jordan et al., 1997; Miller et al., 2000;Morrison and Kinoshita, 2000), and studies of p53−/− animals have shown that it is essential for developmental death of sympathetic neurons (Aloyz et al., 1998) and injury-induced death of cortical and hippocampal neurons (Morrison et al., 1996; for review, see Miller et al., 2000; Morrison and Kinoshita, 2000). p53 fulfills this pivotal function by integrating diverse extracellular stimuli and subsequently regulating the neuronal apoptosis decision upstream of theBcl2 family, Apaf1, and caspases.
We have demonstrated recently that the apoptotic function of p53 in neurons is modulated by a second family member, p73 (Pozniak et al., 2000). Full-length isoforms of p73 (TAp73) are structurally similar to p53 and, like p53, act as transcription factors that can induce cellular apoptosis (Jost et al., 1997; Kaghad et al., 1997; Stiewe and Putzer, 2001). However, the predominant isoforms of p73 in vivo are truncated proteins that lack the N-terminal transactivation domain (ΔNp73) (Pozniak et al., 2000; Yang et al., 2000). These truncated ΔNp73 variants function as naturally occurring dominant-inhibitory proteins and can inhibit the transcriptional activity of both p53 and TAp73 (Yang et al., 2000; Fillippovich et al., 2001; Grob et al., 2001). In this regard, we demonstrated previously (Pozniak et al., 2000) that overexpression of ΔNp73 isoforms inhibited sympathetic neuron apoptosis caused by NGF withdrawal or p53 overexpression, and that developmental death of sympathetic neurons was enhanced in p73−/− animals. Because the only detectable isoform of p73 in sympathetic neurons was ΔNp73β, a molecule whose levels are upregulated by NGF (Pozniak et al., 2000), then these findings indicated that ΔNp73 functions in the developing PNS as an essential anti-apoptotic molecule, potentially by antagonizing the proapoptotic functions of p53.
These findings led us to hypothesize that p73 might also function as an essential prosurvival molecule in CNS neurons. To test this hypothesis, we have examined a number of neuronal populations, including cortical neurons and facial motor neurons in p73−/− animals. We report here that p73 is essential for development and long-term maintenance of normal neuron numbers in at least some CNS structures, including the cortex, olfactory bulb, and facial motor nucleus. Thus, ΔNp73 isoforms function as essential prosurvival molecules in both the CNS and PNS and are important not just during the period of developmental death but also for the maintenance of at least some populations of adult neurons.
MATERIALS AND METHODS
Histological and immunocytochemical analysis.Maintenance and genotyping of p73−/− animals were performed as described previously (Pozniak et al., 2000). For histological analysis, animals were perfused with 4% paraformaldehyde, and brains were cryoprotected, sectioned, and Nissl stained as described previously (Majdan et al., 1997). Immunostaining and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) were performed essentially as described previously (Majdan et al., 2001). Neuronal-specific nuclear protein (NeuN) immunocytochemistry (1:200;Chemicon, Temecula, CA) was performed using the Mouse-on-Mouse blocking kit (Vector Laboratories, Burlingame, CA) and a streptavidin-CY3-conjugated secondary antibody (1:2000; Jackson ImmunoResearch, West Grove, PA).
For quantitation of cortical thickness, sections were measured from the corpus callosum to the pial surface using image analysis. For quantitation of relative mean cell or neuron numbers, equivalent fields from three cortical levels (see Fig. 2e) were chosen, and the number of Nissl- or NeuN-stained cells was quantified in a cortex strip spanning 574 μm for postnatal day 1 (P1)–P3 and P14–P16 brains and 287 μm for adult brains. Cell density was calculated using the area and cell-count measurements. For quantitation of TUNEL, equivalent coronal sections at the caudal level were selected, and all of the TUNEL-positive cells within the cortex were counted. All digital image acquisition and analysis was performed with Northern Eclipse software (Empix) using a Sony (Tokyo, Japan) XC-75CE CCD video camera.
Fig. 2.
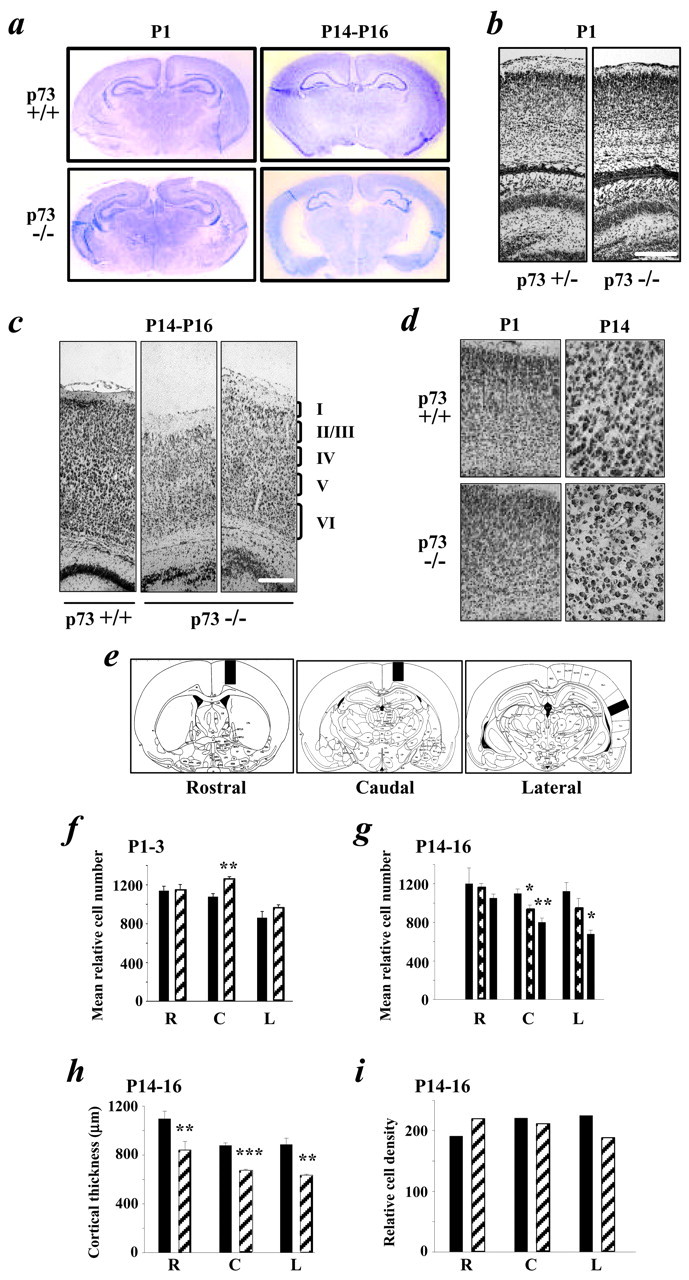
Absence of p73 leads to a loss of cortical neurons during the first 2 postnatal weeks. a, Photographs of coronal forebrain sections of P1–P3 and P14–P16 p73+/+ and p73−/− mice stained with cresyl violet. b,c, Photomicrographs of p73+/+ or p73+/− versus p73−/− Nissl-stained coronal sections of the cortex at P1 (b) and P14–P16 (c). The cortical layers are denoted on the right ofc. Scale bars: b, 200 μm;c, 150 μm. d, Higher magnification photomicrographs of Nissl-stained coronal sections showing that, at P1–P3, the gross structure of the cortex and cell density are similar in p73+/+ versus p73−/− animals and that, at P14–P16, there is no apparent increase in cell density. e, Relative cell number was determined by quantitating the total number of Nissl-stained cells in 574 μm (P1–P3 and P14–P16) or 287 μm (adult) strips at the rostral, caudal, and lateral levels shown in these schematic diagrams. f, g, Graphs showing the mean relative cell number at the rostral (R), caudal (C), and lateral (L) levels in coronal sections of the p73+/+ (black bars) and p73+/− (hatched bars) versus p73−/− (striped bars) cortex at P1–P3 (f) and P14–P16 (g). Asterisks are for statistical comparisons with the p73+/+ numbers. h,i, Graphs showing cortical thickness (h) and relative cell density (mean cell number per mean cell area) in cells per 1000 μm2(i) at the rostral, caudal, and lateral levels in p73+/+ (black bars) versus p73−/− (striped bars) cortex. In all graphs except i, results represent mean ± SEM. *p < 0.05; **p < 0.005; ***p < 0.001. Ini, no error bars are shown because the relative cell density was calculated and not directly measured.
Distribution of neurons in the brainstem and cerebellum was plotted using a system for image analysis consisting of a light microscope (BX40; Olympus Optical, Tokyo, Japan), equipped with an X-Y movement-sensitive stage (BioPoint XYZ; LEP, Hawthorne, NY), az-axis indicator (MT12 microcator; Heidenhain, Traunreut, Germany), and a video camera (DC200; Dage-MTI, Michigan City, IN) coupled to a computer containing Stereo Investigator software (Microbrightfield, Colchester, VT). This software allows the region of interest to be outlined at low magnification and cells to be plotted within these outlines after evaluation at high magnification (100× magnification; numerical aperture, 1.3) (Luk and Sadikot, 2001).
An unbiased stereological technique, the optical fractionator (West et al., 1996; Luk and Sadikot, 2001), was used to estimate neuron number and volume in the facial nucleus. The facial nucleus was seen over a rostrocaudal distance of 360 μm at P1 and 405 μm at P14 [equivalent to plates 16–21; atlas of Jacobowitz and Abbott (1998)for P1 and bregma −5.68 to −6.48 mm; atlas of Franklin and Paxinos (1997) for P14]. Every fourth 40- or 45-μm-thick section from P1 and P14 mice, respectively, was examined throughout the facial nucleus. Systematic random sampling of the facial nucleus was performed by randomly translating a grid with 100 × 100 μm squares onto the section of interest using the Stereo Investigator software. An optical dissector with a brick size of 30 × 30 × 5 μm with exclusion lines (1–3) was applied at each sampling site at the intersection of the grid lines. All randomly distributed computer-generated sampling sites were examined using the 100× objective. Large cells with a distinct nucleus that fell within the counting brick and not intersecting the exclusion lines were enumerated. Estimates of the total number of facial neurons were generated in each animal using the Stereo Investigator software.
The number of neurons in the lateral (dentate) deep cerebellar nucleus (DCN) was determined in a single section corresponding to bregma −5.80 mm in the adult mouse atlas of Franklin and Paxinos (1997). Profile counts were used, and glia were excluded based on cell size (<7 μm diameter). Statistics were performed using a two-tailed unpaired Student's t test.
Analysis of cortical neuron cultures. Primary cortical neuron cultures were obtained from embryonic day 16 (E16) to E17 mice as described previously (Wartiovaara et al., 2002). At 6 din vitro (DIV), cultures were harvested for biochemical analysis or infected with 100 multiplicities of infection of replication-deficient adenoviruses expressing Escherichia coli β-galactosidase (Toma et al., 2000) or green fluorescent protein (GFP)-tagged ΔNp73α or ΔNp73β (Pozniak et al., 2000). At 2 d after infection, one-half of the media were replaced by fresh media containing 10 μm camptothecin or 75 μm LY294002, and the neurons were incubated at 37°C for 24 hr. TUNEL was performed as described previously (Toma et al., 2000). For visualization of infected cells, the ΔNp73 adenoviruses express GFP from a second cistron (Pozniak et al., 2000), whereas β-galactosidase-infected neurons were immunostained as described previously (Wartiovaara et al., 2002). Immunocytochemistry for activated caspase-3 was performed as for β-galactosidase, using an antibody specific for the cleaved protein (1:1000; Cell Signaling Technology, Beverly, MA) and a CY3-conjugated anti-rabbit secondary antibody (1:400; Jackson ImmunoResearch). For quantitation, four to six random images of each treatment (per experiment) were captured and processed, using Northern Eclipse software (Empix).
Western blot analysis. Two-dimensional separation and Western blot analysis of endogenous p73 protein were performed as described previously (Pozniak et al., 2000), using the ER-15 (1:50; Neomarkers, Fremont, CA) p73 antibody and a secondary goat anti-mouse HRP antibody (1:5000; Amersham Biosciences, Arlington Heights, IL). Expression of exogenous full-length p73, ΔNp73α, and ΔNp73β was achieved using recombinant adenoviruses encoding these proteins to infect human embryonic kidney 293 cells, as described previously (Pozniak et al., 2000).
RESULTS
ΔNp73 is expressed by cortical neurons and rescues them from diverse apoptotic stimuli upstream of caspase-3 activation
To determine whether p73 might play a role in regulating survival of CNS neurons, as it does those in the PNS, we focused on cortical neurons. Initially, we characterized the expression of p73 isoform(s) in the developing cortex. Two-dimensional gel electrophoresis and Western blotting of lysates of E19 cortical tissue from p73+/+ and p73−/− animals (Fig. 1a) revealed that only the truncated, ΔNp73α isoform was expressed at detectable levels (Fig. 1a). Similar results were obtained when we analyzed highly enriched cultures of E17 cortical neurons that were maintained for 6 DIV (data not shown). On the basis of these findings, we then asked whether ΔNp73α functioned as an anti-apoptotic molecule in cortical neurons as it does in sympathetic neurons (Pozniak et al., 2000). To perform these experiments, cultured cortical neurons were infected with recombinant adenoviruses expressing ΔNp73α or ΔNp73β; these viruses also express GFP as a marker (Pozniak et al., 2000). As a control, neurons were infected with an adenovirus expressing β-galactosidase. Two days later, neurons were then exposed to one of two apoptotic stimuli: the DNA-damaging agent camptothecin, which causes apoptosis via a p53-dependent mechanism (Enokido et al., 1996), or the phosphatidylinositol 3 (PI3)-kinase inhibitor LY294002, which interrupts the essential PI3-kinase survival pathway (Hetman et al., 1999). TUNEL 1 d later revealed that overexpression of either ΔNp73 isoform was sufficient to protect cortical neurons from apoptosis induced by either of these treatments (Fig. 1b,c); >40% of β-galactosidase-expressing neurons were TUNEL positive after camptothecin or LY294002 treatment, whereas <5% of neurons expressing ΔNp73α or ΔNp73β were positive.
Fig. 1.
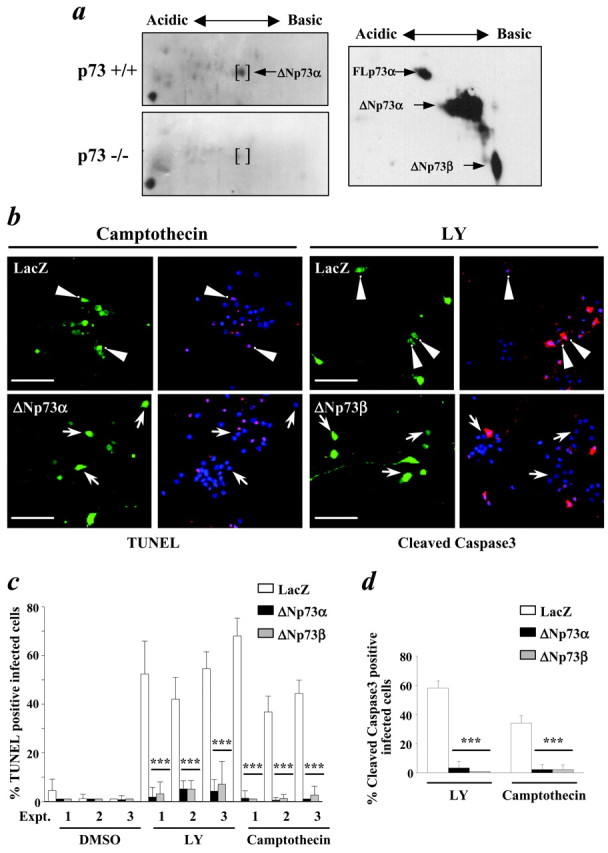
ΔNp73α, the predominant p73 isoform in the developing cortex, rescues cortical neurons from apoptosis induced by DNA damage or inhibition of the PI3-kinase survival pathway.a, Two-dimensional Western blot analysis of lysates of E19 cortex from p73+/+ versus p73−/− animals, probed with an antibody to all p73 isoforms (left two panels). Thebrackets indicate a spot found only in the p73+/+ cortex and that migrates at the same size and the same pI as ΔNp73α. The right panel is a similar analysis of exogenous full-length p73α (FLp73α), ΔNp73α, and ΔNp73β. b, Photomicrographs of postmitotic cortical neurons infected with recombinant adenoviruses expressing β-galactosidase (LacZ) or ΔNp73α, treated with 10 μm camptothecin or 75 μm LY294002 (LY) for 24 hr, and then analyzed by TUNEL (left four panels) or by immunostaining for the active, cleaved form of caspase-3 (right four panels).Arrowheads indicate β-galactosidase-positive cells that colocalize either with TUNEL or cleaved caspase-3 immunoreactivity, whereas the arrows indicate ΔNp73α-positive cells that are not positive for either TUNEL or caspase-3. Scale bars, 100 μm. c, Quantitation of TUNEL data similar to that shown in b. Results are shown from three independent experiments (Expt. 1,2, or 3) and show the percentage of infected cells that were TUNEL positive. d, Quantitation of cleaved caspase-3 immunoreactivity data similar to that shown in b. Results show the percentage of infected neurons positive for cleaved caspase-3. ***p < 0.001.
One way that ΔNp73 isoforms rescue cells from apoptosis is by interference with the apoptotic actions of p53. To determine whether ΔNp73 rescued cortical neurons from apoptosis upstream of caspase activation, as would be predicted if it functioned at the level of p53 (Cregan et al., 1999), we performed similar rescue experiments and then immunostained cortical neurons for activated caspase-3 (Fig.1b). These studies revealed that, as seen with TUNEL, both camptothecin and LY294002 led to caspase-3 activation in uninfected or β-galactosidase-infected neurons, whereas overexpression of either ΔNp73 isoform was sufficient to prevent this activation (Figs.1b,d). Thus, ΔNp73α is expressed by cortical neurons and is sufficient to inhibit their apoptosis in response to p53 activation or interruption of the PI3-kinase survival pathway at a level upstream of caspase-3 activation.
Enhanced apoptosis causes a decrease in cortical neuron number during the first 2 postnatal weeks in p73−/− animals
We have observed previously enhanced sympathetic neuron apoptosis in p73−/− mice during the period of naturally occurring sympathetic neuron death (Pozniak et al., 2000). To determine whether a similar phenomenon occurs in the CNS of p73−/− animals, we examined the forebrains of p73−/− versus p73+/+ littermates at two periods: P1–P3 and P14–P16. Nissl staining of coronal sections revealed that the forebrains of P1–P3 p73−/− animals were grossly normal in structure, although the hippocampus was disorganized (Fig.2a), as reported previously (Yang et al., 2000). Higher magnification analysis indicated that the organization of the cortical mantle was relatively normal in p73−/− animals (Fig. 2b,d) but that by P14–P16, the lateral ventricles were enlarged, and the cortical mantle was thinner (Fig. 2a,c), with no apparent increase in cortical cell density (Fig. 2c,d). Moreover, the brain weights of P14–P16 p73−/− animals were reduced by ∼20% relative to wild-type and heterozygous animals [mean values for p73+/+ brains, 0.36 gm (n = 2); p73+/− brains, 0.35 gm (n = 7); p73−/− brains, 0.29 gm (n = 3)].
To determine whether, as predicted by these findings, cortical cell numbers were decreased in the P14–P16 p73−/− cortex, we chose three different levels of the cortex relative to easily identifiable landmarks (Fig. 2e) to quantitate the total number of Nissl-stained cells in a strip 573 μm wide extending from the corpus callosum to the pia. This analysis demonstrated that, at P1–P3, the number of cortical cells in p73−/− versus p73+/+ animals was similar at all three cortical levels (Fig. 2f; Table1), with a small, statistically significant increase in relative cell number at the caudal level. In contrast, by P14–P16, the mean relative number of cortical cells was significantly reduced at both the caudal and lateral levels in p73−/− animals in comparison with wild-type littermates (Fig. 2g; Table 1). Interestingly, a statistically significant decrease in cell number was also observed in p73+/− cortex at the caudal level (Fig.2g). Quantitation of cortical thickness revealed that it too was reduced at all three levels in p73−/− animals (Fig.2h; Table 1), whereas cortical density was not altered (Fig.2i). Thus, P14–P16 p73−/− brains displayed decreased cortical thickness, and this decrease was attributable to a loss of cells in the first 2 postnatal weeks.
Table 1.
Quantification of relative cell, neuron number, and cortical thickness
| Age and genotype | Relative cell number | Relative neuron number | |||||
|---|---|---|---|---|---|---|---|
| Rostral | Caudal | Lateral | Rostral | Caudal | Lateral | ||
| P1–P3 | +/+ | 1072 ± 103 | 1062 ± 34 | 939 ± 85 | 947 ± 57 | 821 ± 14 | 903 ± 65 |
| −/− | 1098 ± 681-a | 959 ± 351-c | 917 ± 321-d | 827 ± 121-e | 885 ± 441-f | ||
| (+15%) | |||||||
| P14–P16 | +/+ | 1201 ± 164 | 1098 ± 47 | 1121 ± 91 | 1218 ± 33 | 1218 ± 73 | 921 ± 7 |
| +/− | 1163 ± 41 | 934 ± 451-g | 941 ± 107 | ||||
| (−15%) | |||||||
| −/− | 1050 ± 441-h | 799 ± 4581-i | 678 ± 411-i | 989 ± 451-k | 733 ± 471-l | 740 ± 241-m | |
| (−13%) | (−27%) | (−40%) | (−19%) | (−40%) | (−20%) | ||
| Adult | +/+ | 771 ± 18 | 793 ± 68 | 619 ± 6 | |||
| −/− | 437 ± 201-n | 448 ± 231-o | 404 ± 81-p | ||||
| (−43%) | (−44%) | (−35%) | |||||
| Cortical thickness (μm) | |||||||
| P14–P16 | +/+ | 1098 ± 60 | 864 ± 17 | 867 ± 57 | |||
| −/− | 837 ± 301-q | 661 ± 231-r | 627 ± 161-s | ||||
| (−24%) | (−25%) | (−30%) | |||||
Values represent the mean ± SEM. (%) represents cell loss (−) or increase (+) relative to p73+/+. p obtained using Student's t test.
p = 0.42;
p < 0.05;
p = 0.42;n = 3 for each genotype.
p + 0.33;
p = 0.37;
p = 0.42;n = 3 for each genotype.
p < 0.05;
p = 0.22;
p < 0.05;
p < 0.05; p73+/+,n = 3; p73+/−, n = 4; p73−/−,n = 5.
p < 0.005;
p < 0.005;
p < 0.005; p73+/+,n = 3; p73−/−, n = 4.
p < 0.005.
p < 0.005; p73+/+,n = 4; p73−/−, n = 6.
p < 0.005;n = 3 for each genotype.
p < 0.05;
p < 0.001;
p < 0.05; p73+/+,n = 4; p73−/−, n = 6.
To determine whether the Nissl-stained cortical cells lost between P1–P3 and P14–P16 were neurons, we performed a similar quantitative analysis after immunostaining with the panneuronal marker NeuN (Fig.3a–c). As seen with Nissl staining, NeuN immunostaining revealed that cortical thickness, density, and neuron number were grossly similar in the cortex of p73−/− and p73+/+ animals at P1–P3 (Fig. 3a). Quantitation of NeuN-positive neurons at the rostral, caudal, and lateral levels supported this conclusion (Fig. 3d; Table 1). In contrast, NeuN staining at P14–P16 confirmed that the cortical mantle was thinner (Fig. 3b,c), and quantitation of these immunostained sections for relative neuron number revealed a significant loss of neurons at all three levels of the cortex in the absence of p73 (Fig. 3e; Table 1).
Fig. 3.
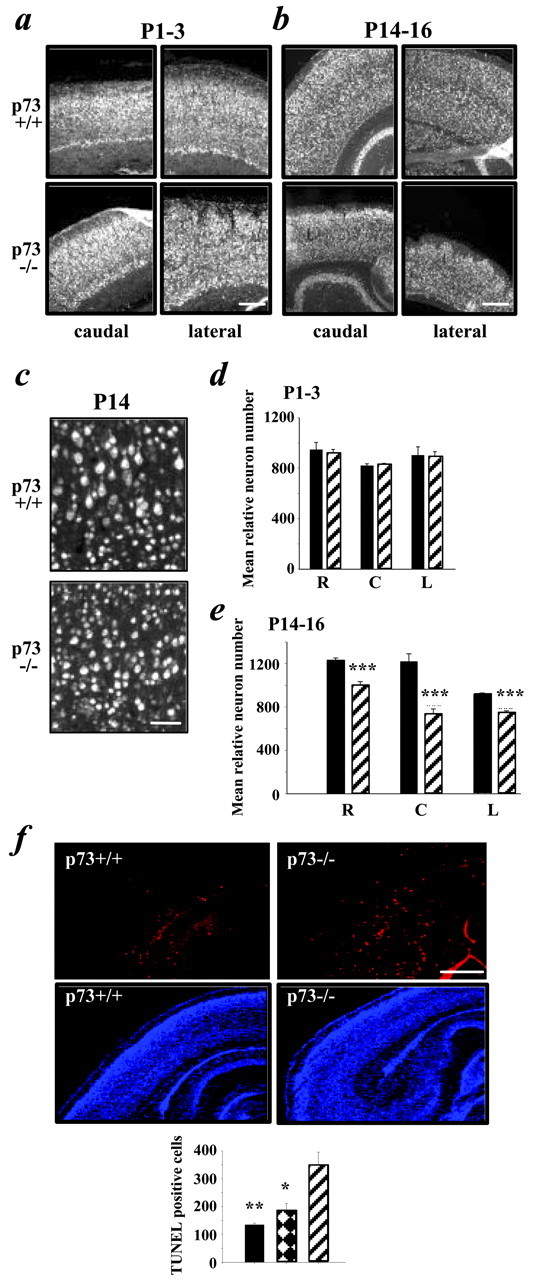
p73 is essential for maintenance of cortical neurons during the first 2 postnatal weeks. a–c, Photomicrographs of immunostaining for the neuron-specific protein NeuN on coronal sections of the p73+/+ versus p73−/− cortex at P1–P3 (a) and P14–P16 (b).c, Higher magnification photomicrograph taken at the caudal level at P14–P16. Scale bars: a,b, 200 μm; c, 80 μm.d, e, Graphs showing the mean relative number of NeuN-positive neurons at the rostral (R), caudal (C), and lateral (L) levels in coronal sections of the p73+/+ (black bars) versus p73−/− (striped bars) brain at P1–P3 (d) and P14–P16 (e). Results are mean ± SEM. ***p < 0.005. f, Apoptosis is enhanced in p73−/− cortex during the first postnatal week. Photomicrographs of coronal sections at the caudal level of P4–P6 p73+/+ versus p73−/− brains analyzed by TUNEL (red,top two panels) and counterstained with Hoechst (blue, bottom two panels) are shown. Thetwo right panels are photographs of the same field, as are the two left panels. Inset, Graph of the total number of TUNEL-positive cells found in the cortex at the caudal level of p73+/+ (black bar), p73+/− (hatched bar), and p73−/− (striped bar) animals. Asterisks indicate significance relative to the p73−/− numbers (*p < 0.05; **p < 0.005). Scale bar, 150 μm.
To determine whether the observed neuronal loss was caused by apoptosis, we performed TUNEL on coronal forebrain sections at P4–P6 (Fig. 3f), a time point immediately succeeding the time (P1–P3) when cortical neuron number was normal. To quantitate the results, TUNEL-positive cells were counted throughout the extent of the cortex at the caudal level. This quantitation revealed that apoptosis was increased approximately threefold in the cortex of p73−/− versus p73+/+ animals (mean ± SE: p73+/+, 135 ± 6; p73−/−, 347 ± 49; p < 0.006; n = 3 for both groups) (Fig. 3f). Thus, the absence of p73 leads to enhanced apoptosis and loss of cortical neurons in the first 2 postnatal weeks.
Cell number is further reduced in the mature p73−/− cortex
We then attempted to determine whether this loss of cortical neurons also occurred after P14–P16. Although most p73−/− animals die by P21 (Yang et al., 2000), a small proportion survive into adulthood. These adult p73−/− (aged 6–12 weeks) animals were somewhat smaller than age-matched littermate controls and displayed some gait abnormalities and weakness, although they were able to feed normally. These animals did not display any enlargement of the skull; their brains were smaller than those of control littermates, and the cortical hemispheres were translucent, consistent with significant tissue loss (see Figs. 4a,6a). Coronal sections at the level of the forebrain revealed a gross enlargement of the lateral ventricles and a significant thinning of the cortical mantle, particularly at the lateral levels (Fig. 4a,b). This thinning of the cortex was not accompanied by an increase in cell density (Fig. 4c). Of the seven animals analyzed, four displayed an extremely thin cortical mantle (Fig. 4a,b, middle panels), whereas three displayed a somewhat less dramatic phenotype (Fig.4b, bottom right panel). This variability might be attributable to differences in compensation by the related family member p63 (Yang et al., 1998), which is also expressed in the cortex as a ΔNp63 isoform (Govoni et al., 2001).
Fig. 4.
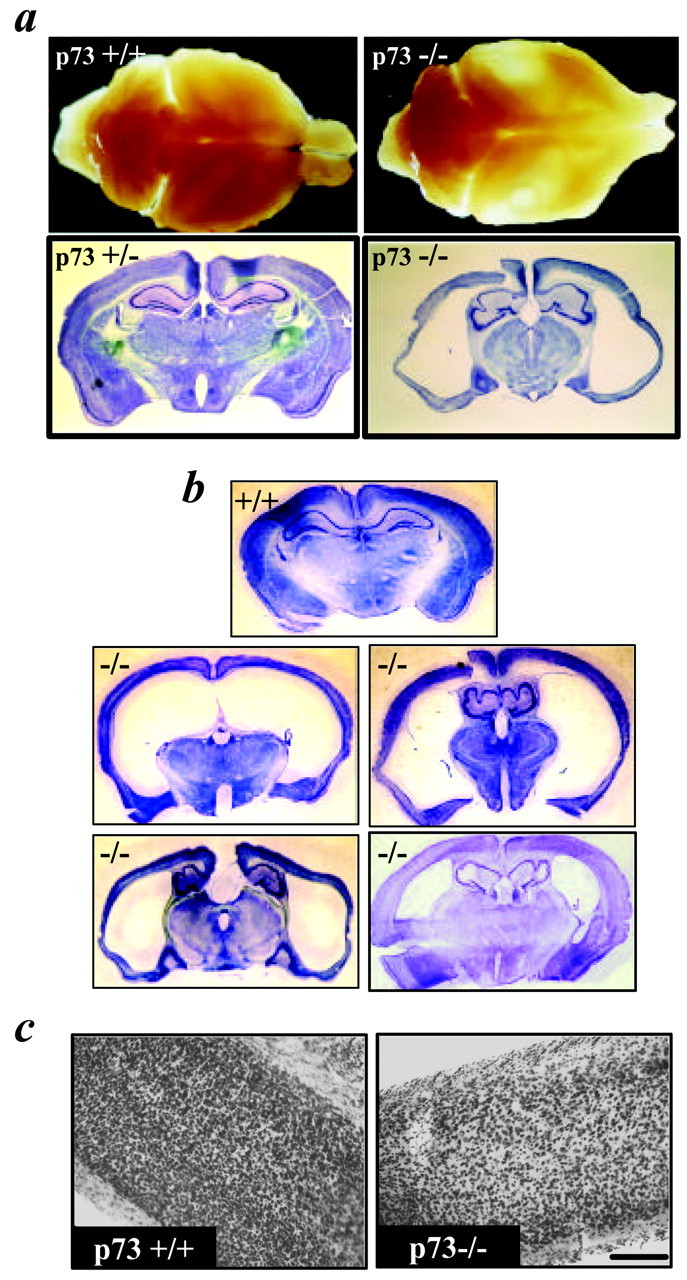
Morphological analysis of the adult p73−/− brain. a, Top two panels, Photographs of p73+/+ versus p73−/− brains. Brains were photographed on top of a light box to show the translucency of the p73−/− cortex.a, Bottom two panels, Photographs of Nissl-stained coronal sections from representative p73+/− and p73−/− brains. The p73−/− section derives from the brain shown in thetop right panel. b, Photographs of Nissl-stained coronal sections through the forebrain of four p73−/− brains. For comparison, the top panel is a coronal section at a similar level from an adult p73+/+ brain. Themiddle three brains were classified as severely affected (as was that shown in a), whereas the right bottom panel was classified as moderately affected.c, Photomicrograph of the Nissl-stained cortex from a p73+/+ versus p73−/− brain at the lateral level. Scale bar, 150 μm.
Fig. 6.
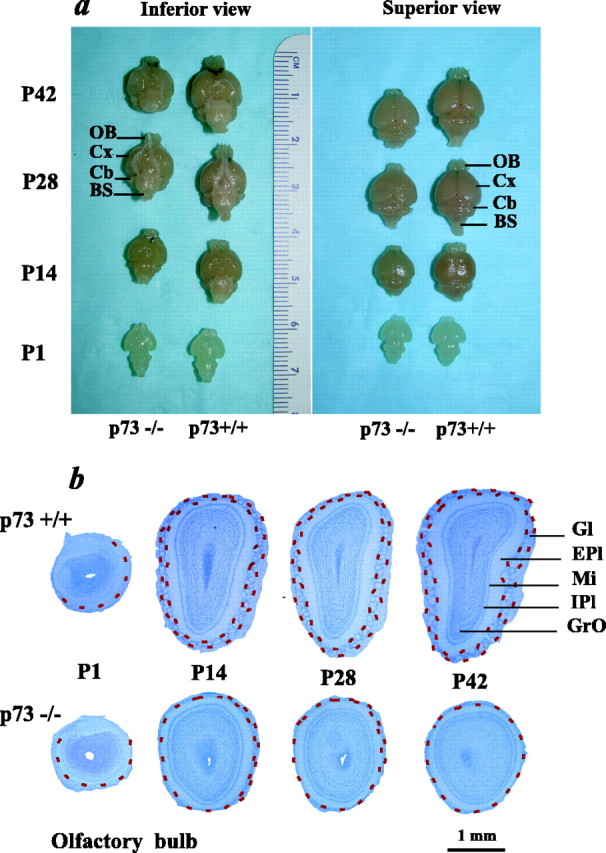
Perturbations in postnatal CNS development and maintenance in p73−/− animals are not limited to the cortex.a, Photographs of representative brains perfused and dissected from p73−/− versus p73+/+ animals at P1, P14, P28, and P42. The ruler is shown as a size reference. OB, Olfactory bulb; Cx, cortex; Cb, cerebellum;BS, brainstem. The inferior view shows the base of the brains, whereas the superior view shows the top of the brains. b, Photomicrographs of Nissl-stained coronal sections through the olfactory bulb of p73+/+ versus p73−/− mice at P1, P14, P28, and P42. Red dotted lines indicate the glomerular layer. Gl, Glomerular layer; EPl, external plexiform layer;Mi, mitral cell layer; IPl, internal plexiform layer; GrO, granule cell layer.
Higher magnification analysis revealed that the dramatic thinning of the cortex observed in p73−/− animals was apparently not accompanied by an increase in cell density but was instead attributable to the loss of Nissl-stained cells (Fig. 4c). NeuN immunostaining supported the conclusion that many of the Nissl-stained cells that were lost were neurons (Fig. 5a). Moreover, the p73−/− neurons that remained at this age appeared to be smaller in size as a population (Fig. 5a), although NeuN immunostaining did not allow us to distinguish whether this was attributable to a general decrease in cell size or to an enhanced loss of larger projection neurons. To quantitate these findings, we determined relative cell numbers in a 287-μm-wide strip at the caudal and lateral levels in six p73−/− animals, including animals that demonstrated both the severe and more moderate phenotypes (Fig.4b). This analysis showed that relative Nissl-stained cell numbers were decreased 43 and 44% at the caudal and lateral levels, respectively, in the p73−/− cortex (Fig. 5b; Table 1). For comparison, at P14–P16, relative cell numbers were 27 and 40% lower in p73−/− animals at these same two levels (Table 1). A similar quantitative analysis of NeuN-positive neurons at the lateral level of the adult cortex revealed that the relative neuron number was also decreased by ∼35% in p73−/− animals (Fig. 5c; Table 1). For comparison, at P14–P16, relative neuron numbers were 20% lower at this level in p73−/− animals (Table 1). Thus, cells are lost between P14–P16 and adulthood, indicating that p73 is necessary for the long-term maintenance of cortical cells, including neurons.
Fig. 5.

Cell and neuron number are further reduced in the adult p73−/− cortex. a, Photomicrographs of NeuN-stained neurons in the cortex at the lateral level of p73+/+ versus p73−/− animals. Micrographs increase in magnification fromleft to right. Scale bars:a, 200 μm; b, 80 μm;c, 20 μm. b, Quantitation of the number of Nissl-stained cells at the caudal and lateral levels of p73+/+ (black bars) versus p73−/− (striped bars) adult cortex. Note that the area quantitated was only 287 μm wide, as opposed to 584 μm wide for the earlier ages.c, Quantitation of the number of NeuN-stained neurons at the lateral level of the p73+/+ (black bars) versus p73−/− (striped bars) cortex. In both graphs, results are mean ± SEM. **p < 0.005.
p73 is essential for postnatal development of additional CNS structures, including the olfactory bulb
These studies, together with our previous studies on sympathetic neurons, indicated that p73 is essential for the postnatal maintenance of at least some CNS and PNS neurons. To determine whether these findings reflected a more general requirement for p73 during postnatal development, we systematically compared the brains of p73−/− versus p73+/+ animals at ages ranging from P1 to P42. Examination of the gross morphology of these brains (Fig.6a) led to a number of conclusions. First, the brains of p73−/− versus p73+/+ brains were approximately similar immediately after birth (Fig. 6a), as we had observed for the newborn cortex. Second, by P14, the p73−/− brains were already noticeably smaller than the p73+/+ brains (Fig.6a). Third, this decreased brain size was maintained after P14, with the largest difference in brain size detectable at P42, the latest time point examined (Fig. 6a).
This decrease in relative brain size was particularly obvious for forebrain structures, including, as predicted, the cortex, as well as the olfactory bulb. In particular, although the p73−/− olfactory bulb was relatively normal at birth, by P14, it was significantly decreased in size relative to the p73+/+ bulb (Fig. 6a). To examine this in more detail, brains were sectioned coronally and Nissl stained. This analysis (Fig. 6b) demonstrated that at birth, the size and morphology of the p73−/− versus p73+/+ olfactory bulbs were similar. In contrast, by P14, the p73−/− olfactory bulb was smaller, and the glomerular layer was reduced in size (Fig. 6b). By P42, the glomerular layer was almost nonexistent in p73−/− animals, and the entire olfactory bulb was much smaller (Fig.6a,b). In addition, cell density was apparently reduced, and the layers were less defined than in p73+/+ animals (Fig.6b). Thus, the postnatal development and maintenance of the olfactory bulb are severely perturbed in the absence of p73.
To determine whether p73 was also required for more caudal CNS structures, we examined two additional populations of neurons: brainstem facial motor neurons and cerebellar neurons of the lateral DCN. To examine facial motor neurons, we took serial sections throughout the extent of the facial nucleus in p73+/+ versus p73−/− animals at P1 and P14 and performed unbiased stereology using the optical fractionator method (West et al., 1996; Nemchinsky et al., 2000; Luk and Sadikot, 2001). Nissl staining of brainstem sections revealed that the structure of the facial nucleus was similar in p73+/+ and p73−/− animals (Fig.7a). However, stereological measurements revealed that the number of facial motor neurons was reduced by ∼29% in the P1 p73−/− facial nucleus (p73+/+, 5676 ± 111; p73−/−, 4021 ± 106; p < 0.001) (Table2), and that the total nucleus volume was also somewhat reduced (Table 2). At P14, facial motor neuron number was still decreased ∼26% in the p73−/− facial nucleus (p73+/+, 4960 ± 266; p73−/−, 3662 ± 109; p < 0.01) (Table 2). Thus, the number of facial motor neurons is reduced 25–30% at birth in p73−/− animals, and this reduction is maintained postnatally. Because the period of naturally occurring facial neuron death occurs primarily before birth in mice (Grieshammer et al., 1998), then one possible explanation for this deficit is that developmental cell death is enhanced in facial motor neurons, as observed previously in sympathetic neurons (Pozniak et al., 2000).
Fig. 7.
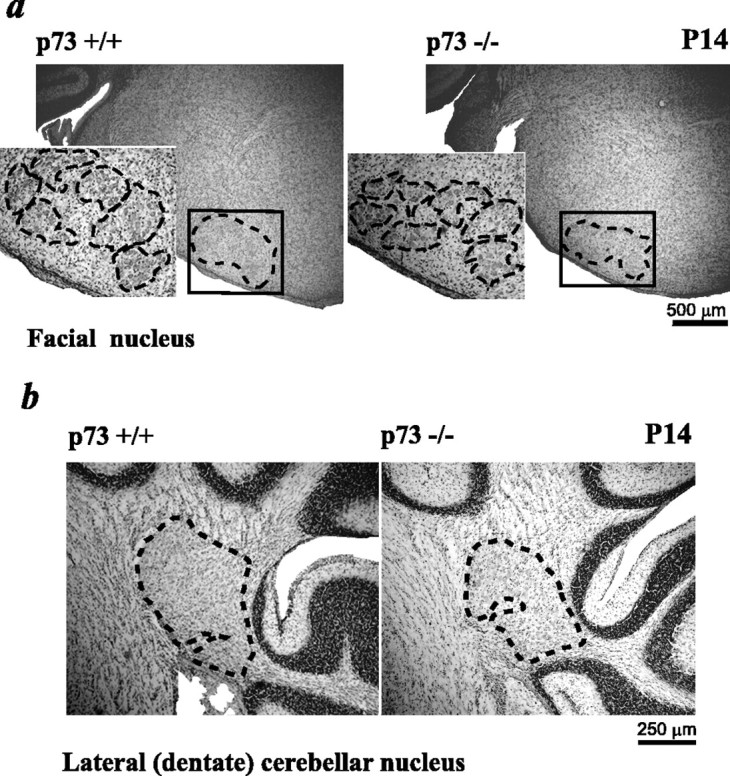
The morphology of the facial nuclei and cerebellum is similar in p73+/+ versus p73−/− animals, but neuron number is reduced in the absence of p73. a, Photomicrographs of Nissl-stained coronal sections through the brainstem at the level of the facial nuclei in P14 p73+/+ versus p73−/− animals. Theblack box outlines the region of analysis, and theblack dotted lines outline the facial nucleus. Theinsets show the facial nucleus at higher magnification, with the subdivisions of the nucleus outlined with black dotted lines. b, Photomicrographs of the lateral (dentate) cerebellar nucleus in coronal, Nissl-stained sections of the cerebellum. The position of the lateral DCN is outlined with black dotted lines.
Table 2.
Quantification of facial motor neurons and lateral deep cerebellar nucleus neurons
| Age and genotype | Neuron number | Nucleus volume (mm3) | |
|---|---|---|---|
| Facial motor neurons | |||
| P1 | +/+ | 5676 ± 111 | 0.079224 ± 0.0026 |
| −/− | 4021 ± 1062-a | 0.064719 ± 0.00212-b | |
| P14 | +/+ | 4960 ± 266 | 0.123 ± 0.0058 |
| −/− | 3662 ± 1092-c | 0.115 ± 0.00672-d | |
| (−26%) | |||
| Lateral (dentate) deep cerebellar neurons (relative number at one level) | |||
| P14 | +/+ | 362 ± 23 | |
| −/− | 243 ± 262-e | ||
| (−33%) | |||
Values represent the mean ± SEM. (%) represents cell loss relative to p73+/+. p values were obtained using Student's t test.
p < 0.001;
p < 0.05;
p < 0.01;
p < 0.05;
p < 0.05;n = 3 in all cases.
To determine whether a similar deficit occurred in other caudal populations of p73−/− neurons, we examined the cerebellum. At P14, the gross morphology of the cerebellum was approximately similar in p73+/+ versus p73−/− animals (Fig. 7b) (data not shown), with reference to both the granule cell layer and Purkinje cells. Moreover, the relative location of the deep cerebellar nuclei was also maintained in the absence of p73 (Fig. 7b) (data not shown). To determine whether numbers of neurons were reduced in any of these structures, we quantitated the number of neurons at a defined level in the lateral DCN (Fig. 7b). This analysis revealed that the relative neuron number at this level was reduced by ∼30% (p73+/+, 362 ± 23; p73−/−, 243 ± 26; p < 0.05), suggesting that, as seen with the facial nucleus, the position and morphology of the lateral cerebellar nucleus were appropriate, but numbers of neurons were decreased.
DISCUSSION
The intracellular signals important for the long-term maintenance of most CNS neurons are still only poorly understood. Here, we have identified a protein, ΔNp73, that is sufficient to promote survival of cortical neurons in response to diverse apoptotic stimuli and deletion of which causes the gradual loss of cortical neurons in the weeks and months after birth. We propose that this finding is important not only for our understanding of neuronal survival mechanisms but also for our understanding of neurodegenerative disorders, which are characterized by the accelerated but nonetheless gradual loss of neurons in the adult human brain.
Although a number of other prosurvival proteins, such as bcl-x (Motoyama et al., 1995), are essential for survival of newly born, embryonic neurons (Ranger et al., 2001), little is known about the proteins that mediate long-term neuronal survival. Two exceptions are the TrkB neurotrophin receptor and bcl-2; a targeted deletion of TrkB in postnatal cortical and hippocampal neurons led to eventual degeneration of a subpopulation of neurons (Xu et al., 2001), whereas bcl-2 deletion caused progressive degeneration of postnatal peripheral and motor neurons (Michaelidis et al., 1996). The data presented here showing that p73 is essential for survival of young postnatal and adult, but not embryonic cortical neurons, together with these previous studies, provide a compelling argument that the intracellular mechanisms mediating the survival of developing versus mature neurons may differ significantly in at least some populations of neurons.
Although it is difficult to definitively demonstrate that a phenotype observed in vivo reflects a direct mechanism, our data showing that cortical neurons express ΔNp73α and that ΔNp73α and ΔNp73β are highly potent survival molecules for cortical neurons strongly support the idea that the cortical neuron loss seenin vivo is cell autonomous. Thus, although a previous study suggested that the ventricular enlargement observed in the p73−/− brain was caused by problems with fluid homeostasis (Yang et al., 2000), four findings documented here indicate that it is a secondary consequence of ongoing cellular loss: (1) enhanced cortical apoptosis occurs at P4–P6, before gross ventricular enlargement; (2) classical hydrocephalus leads to brain and skull enlargement, whereas p73−/− brains are reduced in size and weight at all time points examined from P14 to P42; (3) cell density is constant or reduced in the p73−/− cortex, whereas true hydrocephalus causes increased cortical cell density; and (4) facial motor neurons, which are unlikely to be directly affected by hydrocephalus, are reduced in number after the period of naturally occurring cell death. Thus, in the absence of p73, ventricular enlargement occurs as neurons degenerate and tissue mass decreases, a phenomenon also observed in the degenerating human brain.
Studies reported here demonstrate that the enhanced postnatal neuron loss in the p73−/− cortex is caused by increased apoptosis. However, although we show that postnatal development and maintenance of the p73−/− olfactory bulb is greatly perturbed and that facial motor neurons are reduced in number, our studies do not establish the reason for these perturbations. With regard to the olfactory bulb, a period of developmental cell death occurs with a peak at P5 (Fiske and Brunjes, 2001), suggesting that the deficit in olfactory bulb size that occurs between P1 and P14 may reflect an enhancement of naturally occurring cell death. The apparent ongoing decrease in size and cell density that then occurs from P14 to P42 may be attributable to perturbed neuronal maintenance, much as we have documented in the cortex. However, at least three additional mechanisms could also account for this phenotype. First, afferent activity is known to be essential for olfactory neuron survival (Couper et al., 2000; Leo et al., 2000), and ΔNp73 is expressed in the olfactory epithelium and vomeronasal organ, as well as in the olfactory bulb (Yang et al., 2000). Thus, the gradual loss of the glomerular layer and the decreased olfactory bulb size may be a secondary consequence of the ongoing loss of olfactory sensory neurons in the epithelium, an idea supported by the finding that development of olfactory sensory neurons in the vomeronasal organ (but not the main olfactory epithelium) is perturbed in p73−/− animals (Yang et al., 2000). Second, ongoing adult neurogenesis via the rostral migratory stream contributes to maintenance of the olfactory bulb (Alvarez-Buylla and Garcia-Verdugo, 2002), and it is possible that p73 is important for adult neurogenesis. Finally, it is possible that ΔNp73 acts as a prosurvival protein not only for neurons but also for glial cells, and glial cell loss might contribute to tissue loss throughout the p73−/− nervous system. These alternatives are not mutually exclusive.
With regard to the decreased neuron number in the facial nucleus, we propose that this is attributable to enhanced naturally occurring cell death, much as we have seen for sympathetic neurons (Pozniak et al., 2000). In particular, developmental death primarily occurs embryonically in the mouse facial nucleus (Grieshammer et al., 1998), although some neuronal loss is seen postnatally, as confirmed here. Our finding that p73−/− facial motor neuron number is decreased 25–30% at birth, and that the magnitude of this decrease subsequently remains constant, is consistent with a model in which p73 is essential for trophic factor-induced survival of motor neurons during the period of target dependence, a model based on our data with sympathetic neurons. In this regard, Oppenheim et al. (2001) have reported that mice lacking cardiotrophin-1 show a similar decrease in facial motor neuron number at birth, a deficit that is caused by enhanced developmental motor neuron death in the absence of this trophic factor.
How does ΔNp73 maintain neuronal survival, either developmentally or in the mature nervous system? Because ΔNp73 can directly inhibit p53-mediated apoptosis (Yang et al., 2000; Fillippovich et al., 2001;Grob et al., 2001), and because p53 is responsible for injury-induced apoptosis of many populations of mature CNS neurons (for review, seeMiller et al., 2000; Morrison and Kinoshita, 2000), we propose that the proapoptotic and anti-apoptotic p53 family members together function as a key neuronal apoptotic checkpoint upstream of the Bcl2/Bax family (Miyashita and Reed, 1995; Cregan et al., 1999), Apaf1 (Fortin et al., 2001; Moroni et al., 2001), and the caspases (Cregan et al., 1999). In this model, neuronal life or death is determined by the relative balance between full-length members of the p53 family that are expressed in the nervous system, p53 and TAp63 (Govoni et al., 2001), versus the truncated family members, ΔNp73 and ΔNp63. Growth factors such as NGF would promote neuronal survival by upregulating levels of ΔNp73 (Pozniak et al., 2000), whereas insults such as trophic factor withdrawal or excitotoxicity would promote apoptosis by increasing p53 and/or potentially TAp63 levels (Sakhi et al., 1994;Morrison et al., 1996; for review, see Miller et al., 2000; Morrison and Kinoshita, 2000). In the absence of ΔNp73, this balance might be partially maintained by ΔNp63, but the threshold levels of p53/TAp63 required to “tip the balance” toward apoptosis would be decreased. Thus, minor insults that would normally be insufficient to cause apoptosis would, in the absence of p73, cause the gradual neuronal loss observed here. Such a threshold model would predict that loss of even a single allele of one of these genes would be sufficient to perturb neuronal apoptosis, a finding that has indeed been observed in p53+/− animals (Aloyz et al., 1998), and that is reported here, to some degree, for the p73+/− cortex.
Experiments reported here provide evidence that ΔNp73 is a potent prosurvival molecule for CNS neurons, and that its absence leads to an ongoing loss of postnatal cortical neurons and potentially other adult neurons. The finding that p73 deletion affects the long-term maintenance but not embryonic survival of cortical neurons argues that survival pathways differ at these two developmental stages in at least this one population of neurons. Moreover, our data with facial motor neurons provide additional support for the concept that some populations of neurons require p73 to survive the developmental cell death period, likely by acting downstream of neurotrophic factors. Thus, these findings identify an important prosurvival checkpoint with broad implications for the nervous system and provide a novel way of thinking about how genetic alterations may cause the accelerated but gradual loss of neurons that is characteristic of neurodegenerative disorders.
F.B.-H. and V.V.R. contributed equally to this paper.
This work was supported by research grants from the Canadian Institutes of Health Research (CIHR) (A.F.S. and F.D.M.). F.D.M. is a CIHR Senior Scientist, A.F.S. is supported by a Le Fonds de la Recherche en Santé de Québec Chercheur-Boursier, and both are Killam Scholars. C.D.P., F.B.-H., and A.F.L. are supported by CIHR studentships, F.B.-H. by a McGill Tomlinson studentship, and V.V.R. by a Savoy Foundation scholarship. We thank M. Ariey-Jouglard and A. Sylvestre for assistance with the p73 mouse colony, D. Kaplan for reading this manuscript, and members of the Miller and Kaplan laboratories for stimulating discussions.
Correspondence should be addressed to Freda Miller, Black 3403, Hospital for Sick Children Research Institute, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8. E-mail: fredam@sickkids.ca.
REFERENCES
- 1.Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p73 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarez-Buylla A, Garcia-Verdugo JM. Neurogenesis in adult subventricular zone. J Neurosci. 2002;22:629–634. doi: 10.1523/JNEUROSCI.22-03-00629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Couper LJM, Devine AH, Brunjes PC. Focal denervation alters cellular phenotypes and survival in the rat olfactory bulb: a developmental analysis. J Comp Neurol. 2000;425:409–421. doi: 10.1002/1096-9861(20000925)425:3<409::aid-cne6>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 4.Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enokido Y, Araki T, Tanaka K, Aizawa S, Hatanaka H. Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci. 1996;8:1812–1821. doi: 10.1111/j.1460-9568.1996.tb01325.x. [DOI] [PubMed] [Google Scholar]
- 6.Fillippovich I, Sorokina N, Gatei M, Haupt Y, Hobson K, Moallem E, Spring K, Mould M, McGuckin MA, Lavin MF, Khanna KK. Transactivation-deficient p73 alpha (p73Deltaexon2) inhibits apoptosis and competes with p53. Oncogene. 2001;20:514–522. doi: 10.1038/sj.onc.1204118. [DOI] [PubMed] [Google Scholar]
- 7.Fiske BK, Brunjes PC. Cell death in the developing and sensory-deprived rat olfactory bulb. J Comp Neurol. 2001;431:311–319. [PubMed] [Google Scholar]
- 8.Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS. Apaf1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol. 2001;155:207–216. doi: 10.1083/jcb.200105137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 10.Govoni G, Kaplan DR, Miller FD. Analysis of the p53 family member, p63, during neuronal development and apoptosis. Soc Neurosci Abstr. 2001;27:595.37. [Google Scholar]
- 11.Grieshammer U, Lewandoski M, Prevette D, Oppenheim RW, Martin GR, Pettmann B, Henderson CE. Muscle-specific cell ablation conditional upon Cre-mediated DNA recombination in transgenic mice leads to massive spinal and cranial motoneuron loss. Neuron. 1998;20:633–647. doi: 10.1006/dbio.1997.8859. [DOI] [PubMed] [Google Scholar]
- 12.Grob TJ, Novak U, Maisse C, Barcaroli D, Luthi AU, Pirnia F, Hugli B, Graber HU, De Laurenzi V, Fey MF, Melino G, Tobler A. Human deltaNp73 regulates dominant-negative feedback loop for TAp73 and p53. Cell Death Differ. 2001;8:1213–1223. doi: 10.1038/sj.cdd.4400962. [DOI] [PubMed] [Google Scholar]
- 13.Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 14.Jacobowitz DM, Abbott LC. Chemoarchitectonic atlas of the developing mouse brain. CRC; Boca Raton, FL: 1998. [Google Scholar]
- 15.Jordan J, Galindo MF, Prehn JG, Weichselbaum RR, Beckett M, Ghadge GD, Roos RP, Leiden JM, Miller RJ. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J Neurosci. 1997;17:1397–1405. doi: 10.1523/JNEUROSCI.17-04-01397.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jost CA, Marin MC, Kaelin WG., Jr p73 is a human p53 related protein that can induce apoptosis. Nature. 1997;389:191–194. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 17.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F, Caput D. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 18.Leo JM, Devine AH, Brunjes PC. Focal denervation alters cellular phenotypes and survival in the developing rat olfactory bulb. J Comp Neurol. 2000;417:325–336. doi: 10.1002/(sici)1096-9861(20000214)417:3<325::aid-cne6>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 19.Luk K, Sadikot AF. GABA promotes survival but not proliferation of the parvalbumin-immunoreactive interneurons in rodent neostriatum: an in vivo study with stereology. Neuroscience. 2001;104:93–103. doi: 10.1016/s0306-4522(01)00038-0. [DOI] [PubMed] [Google Scholar]
- 20.Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA. Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci. 1997;17:6988–6998. doi: 10.1523/JNEUROSCI.17-18-06988.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majdan M, Walsh GS, Aloyz R, Miller FD. TrkA mediates developmental sympathetic neuron survival in vivo by silencing an ongoing p75NTR-mediated death signal. J Cell Biol. 2001;155:1275–1286. doi: 10.1083/jcb.200110017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M, Thoenen H. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron. 1996;17:75–89. doi: 10.1016/s0896-6273(00)80282-2. [DOI] [PubMed] [Google Scholar]
- 23.Miller FD, Pozniak CD, Walsh G. Neuronal life and death: an essential role for the p53 family. Cell Death Differ. 2000;7:880–888. doi: 10.1038/sj.cdd.4400736. [DOI] [PubMed] [Google Scholar]
- 24.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 25.Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K. Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol. 2001;3:552–558. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- 26.Morrison RS, Kinoshita Y. The role of p53 in neuronal cell death. Cell Death Differ. 2000;7:868–879. doi: 10.1038/sj.cdd.4400741. [DOI] [PubMed] [Google Scholar]
- 27.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects from kainate-induced cell death. J Neurosci. 1996;16:1337–1347. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 29.Nemchinsky EA, Young WG, Yeung G, Shah RA, Gordon JW, Bloom FE, Morrison JH, Hof PR. Differential vulnerability of oculomotor, facial, and hypoglossal nuclei in G86R superoxide dismutase transgenic mice. J Comp Neurol. 2000;416:112–125. doi: 10.1002/(sici)1096-9861(20000103)416:1<112::aid-cne9>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 30.Oppenheim RW, Wiese S, Prevette D, Armanini M, Wang S, Houenou LJ, Holtmann B, Götz R, Pennica D, Sendtner M. Cardiotrophin-1, a muscle-derived cytokine, is required for the survival of subpopulations of developing motoneurons. J Neurosci. 2001;21:1283–1291. doi: 10.1523/JNEUROSCI.21-04-01283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 32.Ranger AM, Malynn BA, Korsmeyer SJ. Mouse models of cell death. Nat Genet. 2001;28:113–118. doi: 10.1038/88815. [DOI] [PubMed] [Google Scholar]
- 33.Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber SS. p53 induction is associated with neuronal damage in the central nervous system. Proc Natl Acad Sci USA. 1994;91:7525–7529. doi: 10.1073/pnas.91.16.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stiewe T, Putzer BM. p73 in apoptosis. Apoptosis. 2001;6:447–452. doi: 10.1023/a:1012433522902. [DOI] [PubMed] [Google Scholar]
- 36.Toma JG, El-Bizri H, Barnabé-Heider F, Aloyz R, Miller FD. Evidence that helix-loop-helix proteins collaborate with retinoblastoma tumor suppressor protein to regulate cortical neurogenesis. J Neurosci. 2000;20:7648–7656. doi: 10.1523/JNEUROSCI.20-20-07648.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wartiovaara K, Barnabé-Heider F, Miller FD, Kaplan DR. N-myc promotes survival and induces S-phase entry of postmitotic sympathetic neurons. J Neurosci. 2002;22:815–824. doi: 10.1523/JNEUROSCI.22-03-00815.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West MJ, Østergaard K, Andreasen OA, Finsen B. Estimation of the number of somatostatin neurons in the striatum: an in situ hybridization study using the optical fractionator method. J Comp Neurol. 1996;370:11–22. doi: 10.1002/(SICI)1096-9861(19960617)370:1<11::AID-CNE2>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 39.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt L. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2001;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- 41.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 42.Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, McKeon F, Caput D. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumors. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]