

Abstract
A neuropathological hallmark of Alzheimer's disease is the deposition of amyloid-β (Aβ) peptides in senile plaques in the hippocampus and cerebral cortex. Aβ is derived from larger integral membrane proteins termed amyloid precursor proteins (APP). We demonstrated previously that APP, synthesized by neurons in the entorhinal cortex, is transported via the perforant pathway to presynaptic terminals in the dentate gyrus. We reported that, although full-length APP and membrane-tethered, C-terminal APP derivatives (APP-CTFs) accumulate at terminal fields, the production of Aβ peptides at these sites was indeterminate. To test the hypothesis that APP-CTFs, generated from axonally transported APP, are further metabolized to Aβ peptides that are subsequently released and deposited proximal to nerve terminals, we created unilateral knife lesions of the perforant pathway of transgenic mice that exhibit hippocampal amyloid deposits. We observed pronounced reductions in amyloid burden in the ipsilateral dentate gyrus, findings that lead us to conclude that axonally transported APP gives rise to Aβ peptides that are released from presynaptic sites in the dentate gyrus and deposited in extracellular plaques. Moreover, our findings are consistent with the view that Aβ deposits are dynamic structures and that the perforant path lesion alters the equilibrium between Aβ production–deposition toward clearance as a consequence of blocked axonal transport of APP from the entorhinal cortex to terminal fields in the hippocampus.
Keywords: Alzheimer's disease, amyloid precursor protein, amyloid deposition, perforant pathway, hippocampus, synapse
Alzheimer's disease (AD) is characterized by the deposition of the amyloid-β (Aβ) peptides in the brain parenchyma and cerebrovasculature of affected individuals. Aβ are 39–43 amino acid peptides derived from amyloid precursor proteins (APP). Familial, early-onset forms of Alzheimer's disease (FAD) are caused by the expression of genes encoding mutant variants of APP and presenilins (PS1 and PS2) (Price and Sisodia, 1998; Selkoe, 2001); FAD-linked APP or presenilin variants cause disease by enhancing the levels, length, or fibrillogenic properties of Aβ peptides (Borchelt et al., 1996; Duff et al., 1996; Scheuner et al., 1996;Citron et al., 1997) and their subsequent deposition.
We and others have documented that neuronally expressed APP is subject to fast anterograde transport to nerve terminals (Koo et al., 1990; Ferreira et al., 1993; Sisodia et al., 1993; Amaratunga and Fine, 1995; Buxbaum et al., 1998; Kaether et al., 2000; Kamal et al., 2000,2001). We reported that APP, synthesized in the entorhinal cortex (EC), is transported via the perforant pathway to the hippocampus and dentate gyrus (Buxbaum et al., 1998). In our studies, a set of APP derivatives, including soluble APP and membrane-tethered APP C-terminal fragments (APP-CTFs) that harbor the entire Aβ sequence, accumulate at these presynaptic sites (Buxbaum et al., 1998). However, it has not been established whether the APP-CTFs, generated from axonally transported APP, are further metabolized to Aβ peptides and whether these peptides are subsequently released and deposited proximal to nerve terminals. To address this important issue, we exploited transgenic mice coexpressing FAD-linked APP and presenilin 1 variants that exhibit amyloid deposition throughout the neocortex and hippocampal formation (Borchelt et al., 1997).
To examine whether APP transported via the perforant pathway is a major contributor to accumulation of Aβ deposits in the hippocampus, we performed unilateral lesions of the perforant pathway of transgenic mice that express both the FAD-linked human PS1-ΔE9 variant (Lee et al., 1996) and a chimeric mouse–human APP Swedish (APPswe) (Borchelt et al., 1997) and assessed amyloid burden in the hippocampal formation after the lesion (Fishman et al., 2001). Our assumption, based on compelling evidence that amyloid deposition is in equilibrium with clearance (Holtzman et al., 1999; Schenk et al., 1999), was that we should be able to shift the equilibrium toward clearance simply by abrogating APP transport (and subsequent Aβ production–deposition) at terminal fields. Fulfilling this prediction, we now show that amyloid deposits in the ipsilateral (lesioned) hippocampus of FAD-linked transgenic mice are cleared within 1 month after the perforant pathway lesion. Importantly, clearance was most pronounced in a region of the ipsilateral hippocampus corresponding to the dentate gyrus. Finally, we assessed amyloid deposition in mice in which perforant pathway lesions were performed before the onset of amyloid deposition. Four months after the lesion, amyloid burdens in the lesioned and unlesioned hippocampus are essentially identical. Hence, it is likely that amyloid deposition in the ipsilateral hippocampus is the consequence of release of Aβ from reactive presynaptic terminals that have reinnervated the ipsilateral hippocampus.
MATERIALS AND METHODS
Transgenic mice. Mice coexpressing FAD mutant human PS1-ΔE9 and a chimeric mouse–human APP695 harboring a human Aβ domain and mutations (K595N, M596L) linked to Swedish FAD pedigrees (APPswe) have been described previously (Borchelt et al., 1996, 1997;Lee et al., 1997). The background strains for APPswe are {C3H/HeJ × C57BL/6J F3} × C57BL/6J n1, and PS1-ΔE9 are C3H/HeJ × C57BL/6J F3.
Perforant pathway lesion. Animals were deeply anesthetized using a mixture of ketamine (75 mg/kg) and xylazine (4 mg/kg; Henry Schein, Melville, NY) and placed on a Kopf stereotaxic apparatus adjusted to mice (David Kopf Instruments, Tujunga, CA). A wire knife assembly was used for precision perforant pathway lesion. Coordinates for knife cut lesion relative to bregma are as follows (in mm): anteroposterior, −4.6; mediolateral, ±3.3; dorsoventral, −4.5 to −3.5 (from dura), with the nose bar set at 3.5. An opening in the scalp at the above coordinates was performed using electric drill, and the dura was exposed. The assembly is set to the ventral extent of the lesion (−4.5 mm). After penetration, the knife was extended to form a hook that extends medially for 2 mm, the assembly was raised to the dorsal extent of the lesion (3.5 mm), and the knife was retracted, reextended, and again lowered to the ventral extent. The knife hook was raised and lowered five times to verify complete transection of the perforant pathway; each retraction and reextension was performed to ensure that flexible axons that were stretched by the knife hook were cut. To verify the accuracy of the lesion, a crystal of the fluorescent dye Fluoro-Ruby (Molecular Probes, Eugene, OR) were dissolved in saline and introduced into the injury site concomitantly with knife penetration. The injury site was subsequently visualized by Olympus Optical (Melville, NY) Fluoview confocal laser scanning microscope.
Histology. Animals were perfused transcardially under deep anesthesia with a saline solution, followed by fixative solution composed of 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1m phosphate buffer. The brains were removed and kept in fixative overnight, followed by flotation in 30% sucrose solution. Brains were sectioned horizontally (50 μm) using a frozen-stage equipped microtome and then placed at −20°C in cryoprotectant solution (glycerol and ethylene glycol-based PBS). Every sixth tissue section was subject to immunocytochemistry, as described below, and analyzed by confocal microscopy. Sections containing Fluoro-Ruby-labeled injury sites were analyzed using a MetaMorph imaging program (Universal Imaging Corporation, West Chester, PA). Equivalent sections of nonlesioned mice, were also analyzed in this manner.
Immunohistochemistry. Free floating horizontal brain sections (50 μm) were rinsed three times for 10 min each in Tris-buffered saline (TBS), blocked using blocking–permeabilizing solution (5% donkey serum and 0.25% Triton X-100) for 1–3 hr, and incubated with mouse anti-S100β (1:10,000; Swant, Bellinzona, Switzerland), goat anti-GFAP (1:5000; Dako, Cambridgeshire, UK), rabbit anti-neurofilament (NF-200 kDa) (1:500;Chemicon, Temecula, CA), mouse anti-Bassoon (1:1000; a gift from Dr. Craig C. Garner, University of Alabama at Birmingham, Birmingham, AL), mouse anti-human Aβ 6E10 (1:1000), or rabbit anti-Aβ42 antibodies (FCA3542; 1:1000; a gift from Dr. Frederic Checler, Centre National de la Recherche Scientifique, Valbonne, France) for 72 hr at 4°C. Tissues were then rinsed in blocking solution for 2 hr. Sections were incubated with secondary antibodies (1:250; Jackson ImmunoResearch, West Grove, PA) at room temperature. Sections were then washed three times for 10 minutes each in TBS and mounted on gelatin-coated slides using polyvinyl alcohol–1,4-diazabicyclo-[2.2.2]octane (PVA–DABCO; Sigma, St. Louis, MO) mounting solution.
Imaging and quantification of amyloid burden.Immunofluorescence in sections were visualized and imaged using anOlympus Optical Fluoview confocal laser scanning microscope. Images at each wavelength were collected separately, using a separate and specific excitation filter. Images were taken and recorded using a Fluoview 2.1 program. Fluorescent images were either used for quantification of amyloid burden or processed for collage by Dell (Round Rock, TX) OptiPlex GX1 running Photoshop 5.0 (Adobe Systems, Mountain View, CA). Z series of 10 μm depth were imaged from each section with 1 μm intervals between images. Volume of amyloid burden was quantified using Multiscan 500PS (Sony, Tokyo, Japan) equipped with MetaMorph 4.1 (Universal Imaging Corporation). Z series images were converted in all planes into binary images, after calibration of pixel size. Single components were considered noise and removed uniformly from all planes. Images were converted to gray scale by thresholding, and the burden volume was estimated.
RESULTS
Quantitative analysis of amyloid burden in the hippocampus of transgenic mice
To examine amyloid deposition and clearance in the hippocampus, we used transgenic mice that coexpress FAD-linked mutant human PS1-ΔE9 (Lee et al., 1997) and APPswe (Borchelt et al., 1996, 1997). The expression of mutant PS1-ΔE9 in the singly transgenic line S9 (Lee et al., 1997) is ∼1.5-fold higher than endogenous PS1, whereas expression of APPswe in the singly transgenic line C3-3 (Borchelt et al., 1996, 1997) is ∼2.5-fold over endogenous APP. In contrast to the singly transgenic C3-3 line, which exhibits sparse amyloid deposits at between 18 and 20 months of age (Borchelt et al., 1997), the doubly transgenic mice that coexpress APPswe and PS1-ΔE9 exhibit amyloid deposits, most prominently in the hippocampal formation, at the age of 5–6 months. Figure 1 shows representative images of brain sections from mice stained with the human Aβ-specific antibody 6E10 and visualized by indirect immunofluorescence using laser confocal microscopy. The leftand right panels depict Aβ antibody-immunoreactive deposits in the dentate gyrus and hippocampus, respectively, of transgenic mice at the age of 6 months (A), 8 months (B), and 13 months (C).
Fig. 1.
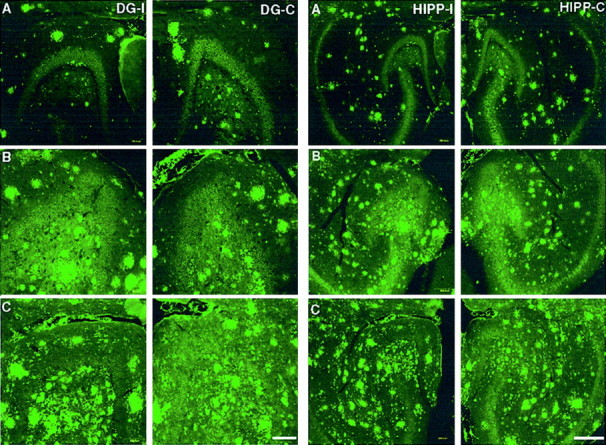
Representative images of amyloid immunoreactivity in the dentate gyrus (left panels; DG) and hippocampus (right panels; HIPP) of unlesioned transgenic mice (A, 6-month-old mouse;B, 8-month-old mouse; C, 13-month-old mouse). Amyloid immunoreactivity in the left side (DG-I,HIPP-I) was compared with that detected in the contralateral (right) side (DG-C,HIPP-C). Amyloid was detected by immunolabeling with 6E10 antibodies. No significant difference in amyloid immunoreactivity could be detected. Scale bars, 250 μm.
Whereas the extent of amyloid deposition in the hippocampus increases with age, as expected (Borchelt et al., 1997), there is no obvious differences in the Aβ burden between the left and right hemispheres in either the dentate gyrus or the hippocampus of nonlesioned mice. To quantify amyloid burden in the hippocampus and dentate gyrus of unlesioned transgenic mice, we performed a quantitative analysis of the volume of Aβ-immunoreactive structures. Z series of hippocampal images were obtained using a confocal microscope, and the volume of amyloid burden was analyzed using MetaMorph software. The hippocampal area that was analyzed includes the dentate gyrus, regio superior, and regio inferior, whereas the dentate gyrus subfield that was subject to analysis included the granule cell layer (stratum granulare) and the outer molecular layer (stratum moleculare). Morphometric quantification of Aβ immunoreactivity confirmed that there are no significant differences in amyloid deposition between the left and right hemispheres in animals between the ages of 5 and 13 months (Fig.2).
Fig. 2.

Quantitative analysis of amyloid burden in the hippocampus of unlesioned mice. For each individual animal analyzed, amyloid burden in the left side was compared with the burden in the right side in both the hippocampus (left) and the dentate gyrus (right). The ratio between the hemispheres is indicated. No difference in amyloid burden could be observed.
Perforant pathway transection leads to reduced amyloid burden in the hippocampus
Having established that amyloid deposition occurs to the same extent in both hemispheres of unlesioned animals, we then asked whether unilateral transection of the perforant pathway would impact on amyloid burden in the hippocampus. The rationale of this experiment is that, after transection, delivery of axonally transported APP from the entorhinal cortex to the terminal fields of the hippocampus would not occur, thus shifting the balance between Aβ production–deposition toward clearance. The perforant pathway provides the major neuronal input to the hippocampus from the EC and involves afferent projections from layers II and III of the EC that terminate in the outer molecular layer of granule cell dendrites in the dentate gyrus (Hjorth-Simonsen and Jeune, 1972; Tamamaki and Nojyo, 1993). Thus, transection of the perforant pathway through the presubiculum from its medial surface through the splenium of the corpus callosum completely disconnects the hippocampus from the entorhinal cortex.
We assessed amyloid burden by confocal microscopy of brain sections of lesioned mice 1 month after the transection. Figure3 shows representative images of amyloid immunoreactivity in both the ipsilateral (lesioned) and contralateral (unlesioned) dentate gyrus (left panels) or hippocampus (right panels) of 7 month (A), and two 10 month (B, C) animals. We observed a qualitative reduction of amyloid immunoreactivity in the ipsilateral hippocampus and dentate gyrus compared with the contralateral side.
Fig. 3.
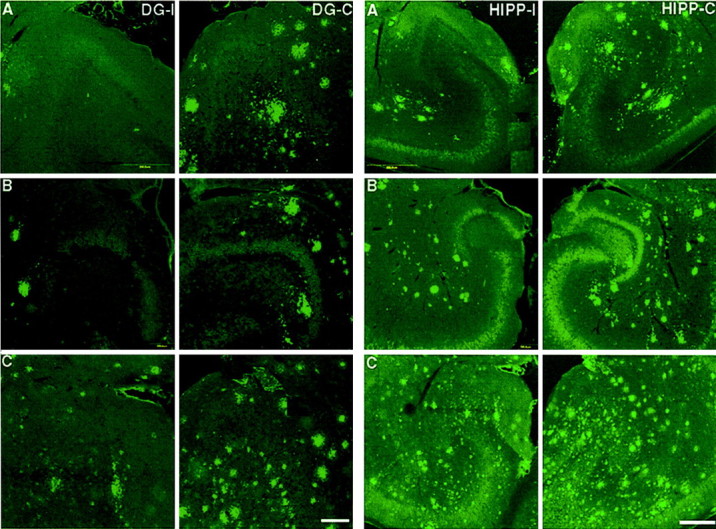
Representative images of amyloid immunoreactivity in the dentate gyrus (left panels; DG) and hippocampus (right panel; HIPP) of perforant pathway-lesioned mice (A, 7-month-old mouse;B, C, 10-month-old mice). Amyloid immunoreactivity in the ipsilateral (lesioned) side (DG-I, HIPP-I) was compared with that in the contralateral side (DG-C,HIPP-C) 1 month after perforant pathway lesion. Amyloid was detected by immunolabeling with 6E10 antibodies. Immunoreactivity of amyloid deposits is reduced in the ipsilateral hippocampus and dentate gyrus compared with the contralateral side. Scale bars, 250 μm.
To quantify the magnitude of change in amyloid burden 1 month after transection, we used morphometric approaches, as described in Materials and Methods. Figure 4 is a quantitative analysis of amyloid burden in the ipsilateral versus contralateral side of individual animals between the ages of 7 and 10 months (at the time of lesion). In all brain sections of lesioned mice, the amyloid burden in the entire ipsilateral hippocampus was approximately twofold to threefold lower than the burden in the entire contralateral hippocampus (Fig. 4). However, comparison of the amyloid burden of the dentate gyrus alone revealed that the reduction in amyloid burden on the ipsilateral side was between 5.5- and 10-fold lower than the contralateral side (Fig. 4). Parallel analysis of other brain areas that are not directly affected by the lesion, including the cortex, failed to show differences in amyloid burden between left and right hemispheres after the lesion (data not shown).
Fig. 4.

Quantitative analysis of amyloid burden in the hippocampus of perforant pathway-lesioned mice 1 month after lesion. Analysis revealed a significant reduction in amyloid burden in the ipsilateral hippocampus (left) and dentate gyrus (right) compared with the contralateral side (the ratio between the hemispheres is indicated). The reduction in amyloid burden in the dentate gyrus was most pronounced.
Although clearance of amyloid deposits after perforant pathway lesion occurs in a region-specific manner, it was conceivable that lesion-induced astroglial activation and subsequent inflammatory reactions may play a critical role in the process of amyloid clearance. To examine this issue, we introduced a stab wound lesion into the perforant pathway of transgenic mice, one that does not cause substantial transection of the axons that perforate the dentate gyrus. One month later, we quantified amyloid burden in brain sections of these mice. These studies revealed that there were no significant differences in amyloid burden between the ipsilateral and the contralateral hippocampi or dentate gyrus of the stab-wounded mice (Fig. 5). Thus, perforant pathway lesion-mediated clearance of amyloid deposition occurs in a highly selective, region-specific manner, consistent with the notion that axonal delivery of APP from the entorhinal cortex is responsible for Aβ production and deposition in the dentate gyrus.
Fig. 5.

Amyloid burden in the ipsilateral and contralateral hippocampus and dentate gyrus of stab wound-treated mice 1 month after treatment. Top panels show 6E10-labeled hippocampus of a stab wound-treated mouse. Bottom panelsshows quantitative analysis of amyloid burden in the ipsilateral (HIPP-I) and contralateral (HIPP-C) hippocampus and dentate gyrus of stab wound-treated mice 1 month after treatment. No significant difference in amyloid burden was found between the ipsilateral and the contralateral hippocampus or dentate gyrus in these animals. Scale bar, 250 μm.
Amyloid clearance attenuates astrogliosis and neuritic dystrophy
As has been reported previously, we observed the presence of both activated astrocytes and microglia in brain regions with amyloid deposition. In unlesioned animals, immunocytochemical staining with antibodies against S100β, the major low-affinity Ca+2 binding protein in astrocytes and microglial cells (Garbuglia et al., 1999; Adami et al., 2001), disclosed the presence of extensively ramified glial cells (Fig.6A,B). Higher-magnification confocal images revealed some overlap in Aβ immunoreactivity and S100β-positive glial processes (Fig.6C–E), suggesting that these cells are actively reacting to amyloid deposition. In contrast, in hippocampal sections from mice with perforant pathway lesions, S100β immunoreactivity was markedly reduced in the ipsilateral hippocampus, with few, albeit morphologically quiescent, glial cells that remained (Fig.6F). In contrast, reactive glia were prominent around amyloid deposits in the contralateral side (Fig. 6G). Hence, it is reasonable to assume that the presence of insoluble amyloid aggregates induces glial activation and that the activated phenotype of these cells returns to a quiescent state once the deposited Aβ peptides are no longer present. However, it should be noted that microglia and astrocytes have been shown to become activated in the terminal zones of lesioned entorhinal afferents to remove degenerating axons and terminals, and, hence, it is conceivable that clearance may, in part, be mediated by these lesion-activated cells.
Fig. 6.
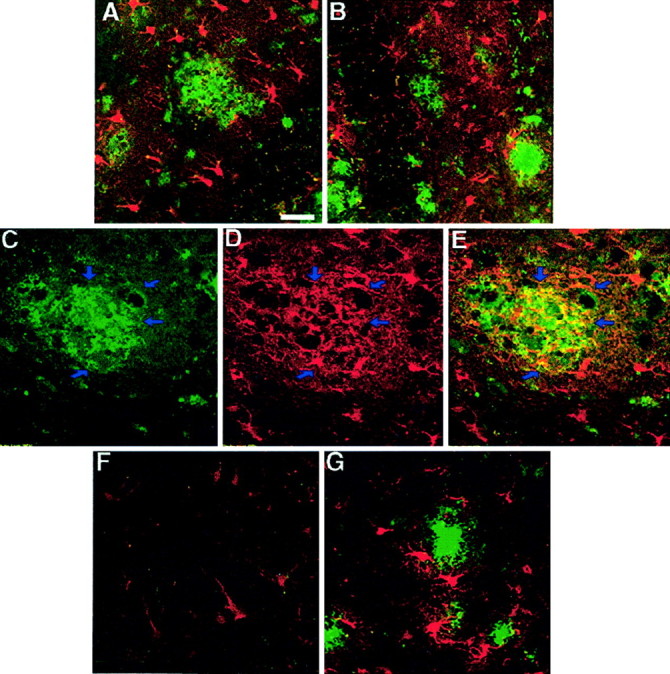
A, B, Reactive gliosis in the hippocampus and dentate gyrus of unlesioned mice as could be detected by immunostaining using S100β antibodies in dentate gyrus in the left hemisphere (A) and in the right one (B). S100β immunoreactivity (inred) was most pronounced around amyloid deposits (immunostained using 6E10 antibodies; in green). Some overlap between S100β-labeled glia (D) and amyloid deposits (6E10 antibodies; C) could be detected (E, see arrows). F,G, Reactive morphology of glia is reduced in the ipsilateral hippocampus (F) but not in the contralateral one (G) 1 month after perforant pathway lesion. Scale bar, 150 μm.
In addition to changes in glial morphology and immunoreactivity, we also observed marked alterations in the general organization of neurofilament-positive neuronal processes. The presence of dystrophic neurites around amyloid deposits has been observed in specimens from human AD subjects, as well as in transgenic mouse models (Vickers et al., 1996; Irizarry et al., 1997a,b; Holtzman et al., 2000; Pigino et al., 2001), but the mechanism regulating this process is unclear. It has been suggested that fibrillar plaques induce local neuritic alterations and the subsequent modifications of cytoskeletal proteins within associated neuronal processes (Dickson and Vickers, 2001). To examine possible changes in neurofilament morphology during the process of amyloid clearance, brain sections of lesioned and unlesioned mice were immunolabeled with antibodies specific for neurofilament H (NF-H). In the unlesioned (contralateral) hippocampus of the transgenic mice, we observed marked NF-H-positive neuritic dystrophy (Fig.7A,B). This phenotype was reversed to a normal pattern 1 month after perforant pathway lesion (Fig. 7C). Hence, lesion-induced clearance of amyloid deposition in the hippocampus results in the reversal of neuritic dystrophy and attenuation of astrocyte–glial activity.
Fig. 7.

Neurofilament morphology in the dentate gyrus–hippocampus as detected by immunolabeling 1 month after perforant pathway lesion. A, B, Contralateral hippocampus (B, high power).C, Ipsilateral hippocampus. Brain sections are double immunolabeled for neurofilament (in green) and β-amyloid (6E10 antibodies; in red). Neuritic atrophy could be detected in the contralateral side. In contrast, neurofilament morphology in the ipsilateral side seems to be recovered, at least in part. Scale bars, 250 μm.
Perforant pathway lesion-induced reinnervation and subsequent deposition of Aβ in the dentate gyrus
Previous studies have established that the terminal arbors of entorhinal fibers in the dentate gyrus undergo a cycle of deinnervation and subsequent reinnervation after perforant pathway lesions. Deinnervation is completed 7–10 d after perforant pathway lesion, a process that creates a zone of deinnervated postsynaptic elements that become receptive to new axonal growth. This process, termed reactive synaptogenesis, occurs progressively over the next 60–90 d (Frotscher et al., 1997; Turner et al., 1998). Sprouting and reactive synaptogenesis of intrinsic hippocampal fibers takes place in the of the outer two-thirds of the molecular layer of the dentate gyrus. In addition, the deinnervated field in the dentate gyrus also receives inputs from the contralateral entorhinal cortex and predominantly from cells in layer II that normally project to the ipsilateral dentate gyrus (Steward, 1976). The reinnervating cells are located in the dorsal half of the entorhinal cortex and concentrated in the medial most portion of layer II (Steward and Vinsant, 1978). Finally, it is highly likely that, in addition to intrinsic and contralateral afferents, cholinergic neurons of the septohippocampal pathway will also reinnervate the denervated zone, as has been described previously (Nadler et al., 1974, 1977). In any event, we hypothesized that, if Aβ deposition at terminal fields is the result of Aβ released from synaptic sites, then lesions of the perforant pathway in mice before the onset of deposition should lead to initial deinnervation and subsequent recruitment of reinnervating fibers to the outer molecular layers of the dentate gyrus. In turn, newly formed synaptic termini should provide a new “reservoir” from which Aβ is subsequently released. To examine the hypothesis that reinnervation of terminal zones could lead to presynaptic release and deposition of amyloid, we performed unilateral perforant pathway lesions in transgenic mice at 4 months of age, a time at which amyloid deposition is still undetectable. Four months later, these animals were killed, and brain sections were examined for the presence of Aβ deposits by indirect immunofluorescence using the human Aβ-specific antibody 6E10. As we predicted, quantitative analysis of amyloid burden in the dentate gyrus and hippocampus of these perforant pathway-lesioned mice revealed little, if any, difference between the ipsilateral (lesioned) and contralateral hemispheres (Fig. 8). In addition, we did not detect any difference in reactive gliosis between the ipsilateral side and the contralateral hemispheres (data not shown).
Fig. 8.

Quantitative analysis of amyloid burden in the hippocampus of mice after reinnervation of the outer molecular layer of the dentate gyrus. Mice were subject to perforant pathway lesion at 4 months of age. Four months later, brain sections were analyzed for amyloid burden. The ratio between the hemispheres is indicated. No difference in amyloid burden could be observed between the lesioned hippocampus and the unlesioned one.
To confirm that reactive synaptogenesis had occurred in the dendritic arbors of dentate neurons of these mice, brain sections were examined for the presence of Bassoon (tom Dieck et al., 1998), a protein that is specifically localized to presynaptic active zones (Zhai et al., 2001). Brain sections of animals killed 1 week, 1 month, or 4 months after lesion were analyzed. Bassoon immunoreactivity was readily detectable in the ipsilateral hippocampus of 4 month postlesioned mice but was markedly lower in brain sections of 1 month and barely detected in sections of 1 week postlesioned mice (Fig.9). These findings are consistent with the reported rate of reinnervation and synaptogenesis in the dentate gyrus after perforant pathway lesions (Turner et al., 1998). To examine the relationship between presynaptic structures and amyloid deposits in the lesioned hippocampus, we performed double labeling with antibodies specific for Bassoon and the highly fibrillogenic form of Aβ, termed Aβ42 (FCA3542) (Barelli et al., 1997). These studies revealed the presence of presynaptic elements in immediate proximity to amyloid deposits, findings we interpret to suggest that deposited Aβ is released from presynaptic sites and deposited in the extracellular milieu (Fig. 9E–G). Notably, we also observed colocalization of numerous Bassoon-immunoreactive structures with Aβ42 (Fig. 9G, arrows). These coimmunoreactive structures likely represent dystrophic terminals in proximity to plaques, findings that offer the notion that Aβ42 deposited in extracellular sites are generated at presynaptic terminals. Additional ultrastructural and immuno-EM studies will be required to confirm these observations. Essentially identical distributions of Bassoon and Aβ42 were observed in the contralateral hippocampus of lesioned mice or nonlesioned mice with amyloid deposits (data not shown).
Fig. 9.
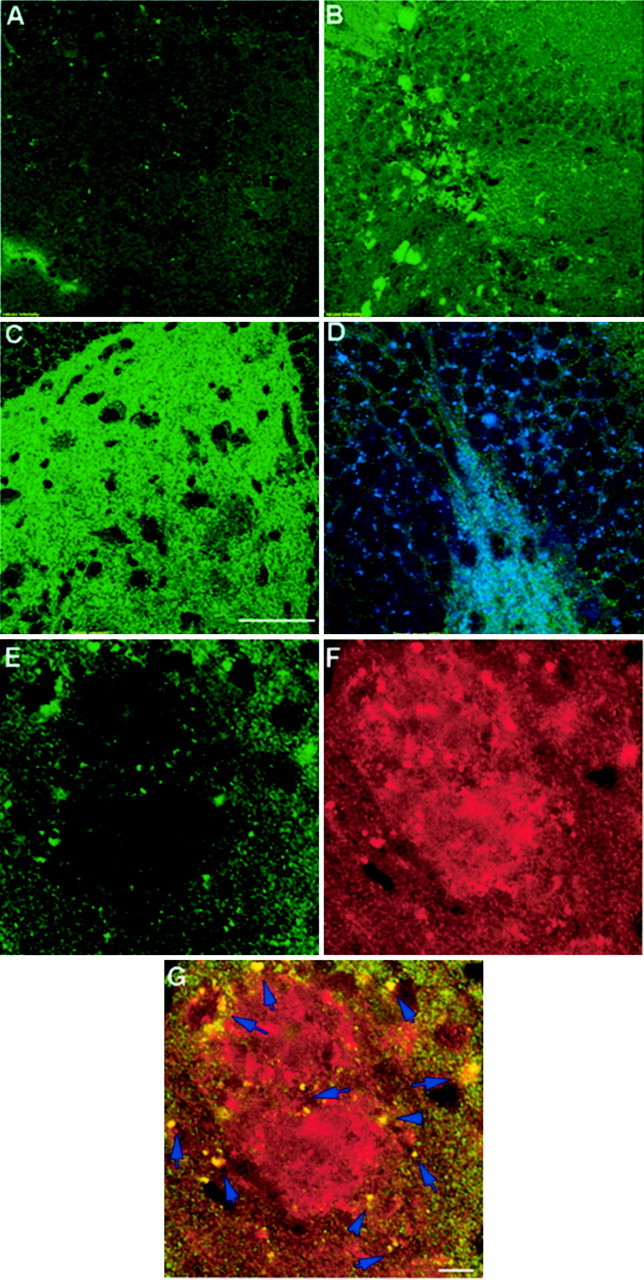
Reactive synaptogenesis in the outer molecular layer of mice 4 months after perforant pathway lesion. Brain sections were examined for the presence of the presynaptic marker Bassoon 1 week (A), 1 month (B), or 4 months (C, D) after lesion. Immunoreactivity for Bassoon was easily detected in the ipsilateral hippocampus of 4 month postlesion mice, less pronounced in brain sections of 1 month mice, and barely detected in sections of 1 week postlesioned mice. D, Bassoon and synapsin double-labeled synapses in the dentate gyrus of 4 month postlesion mice. E–G, Synapses could be detected, surrounding amyloid deposits. This close proximity may suggest that the source of deposited material is synapse released. E, Bassoon immunoreactivity in the dentate gyrus of 4 month postlesion mice.F, Amyloid immunoreactivity at the same area asE, as detected by FCA3542 antibodies. G, Merged image of Bassoon and amyloid immunoreactivities.Arrows in G indicate overlapping staining of immunoreactivity for Aβ42 and Bassoon. Scale bars, 250 μm.
DISCUSSION
The hippocampal formation is a critical neuroanatomical structure responsible for certain aspects of learning and declarative memory. These neuropsychological parameters are primarily diminished in AD and likely a reflection of extensive deposition of amyloid in senile plaques in the hippocampus (Braak et al., 1994; Braak and Braak, 1996). The hippocampus, and specifically, the granule cell dendrites in the dentate gyrus, receives major afferent projections from the EC, a region that receives significant cortical input and serves as a relay station to the hippocampal formation. Neuropathological studies have revealed that neurons in the EC and the hippocampus are particularly vulnerable in AD, with abundant amyloid deposition and neurofibrillary pathology in both regions (Van Hoesen et al., 1991; Braak and Braak, 1996; Gomez-Isla et al., 1996).
In previous efforts, we used a metabolic radiolabeling paradigm to document that APP synthesized by EC neurons are axonally transported via the perforant pathway to the hippocampal formation (Buxbaum et al., 1998). We also demonstrated that these APP species, and a set of membrane-tethered APP derivatives, termed APP-CTFs, accumulate in the dentate gyrus. However, we were unsuccessful in demonstrating that APP or APP-CTFs were further processed to Aβ peptides. Hence, it remained uncertain whether Aβ peptides that accumulate in amyloid deposits in the hippocampal formation were derived from APP that is synthesized locally or by catabolism of APP (or APP-CTFs) that are synthesized at distal sites and axonally transported to synaptic terminals. In an attempt to establish that APP and/or APP-CTFs could be processed to Aβ peptides in vivo, we used transgenic mice that express a chimeric mouse–human APP polypeptide harboring the familial Swedish double mutation (Borchelt et al., 1996). In brain of line C3-3, this polypeptide, termed APPswe, is expressed at approximately twofold higher levels than endogenous APP, and sandwich ELISA studies indicate that the steady-state levels of Aβ in brain of these animals is ∼20–25 pmol/gm (Borchelt et al., 1996) (D. Borchelt, personal communication). Unfortunately, we did not succeed in detecting Aβ in the hippocampus 2 hr after the injection of up to 1 mCi of [35S]methionine into the entorhinal cortex or in cortical tissue after a bolus injection of 1 mCi of [35S]methionine into the frontal cortex (data not shown) of these animals. Our calculations reveal that, if all newly synthesized APP is labeled in the 2 hr period and if 10% of APP molecules are metabolized to Aβ peptides, it would take nearly 1 year to detect the presence of radiolabeled Aβ by autoradiography. Moreover, the rapid turnover of newly generated Aβ (estimatedt1/2 of ∼1–2 hr)(Savage et al., 1998), likely served to further compromise our ability to detect radiolabeled Aβ species.
To examine the contribution of EC-derived, axonally transported APP in Aβ production and deposition in the hippocampus, we chose to exploit transgenic mice coexpressing FAD-linked APP (APPswe) and PS1 that exhibit amyloid deposits throughout the hippocampus and cortex by the age of ∼5–6 months. In these animals, the steady-state levels of Aβ42 peptides are slightly elevated, but these species contribute a small fraction (∼15–20%) to total Aβ levels in the brains of these animals. Our strategy involved unilateral transection of the perforant pathway and assessment of hippocampal amyloid deposition after disconnection of the EC and the terminal fields. Our rationale was that disconnection of the hippocampus from the entorhinal cortex might lead to clearance of preexisting deposits because the incoming entorhinal afferents would not be able to transport APP to terminal fields. In the present report, we provide several novel insights that are consistent with the notion that axonally transported APP is a principal contributor to amyloid deposition in the hippocampal formation. First, we document that, after unilateral lesions of the perforant pathway, amyloid burden in the ipsilateral hippocampus is diminished, and this reduction in amyloid burden is associated with a highly selective clearance in the dentate gyrus. Second, and in support of these latter findings, reactive gliosis and neuritic dystrophy are markedly reduced in the ipsilateral hippocampus compared with extensive neuropathology observed in the contralateral side. These findings offer the conclusion that occluding the transport of APP, APP-CTFs, or Aβ transport to terminal fields reduces amyloid burden simply by shifting the equilibrium from Aβ production and synaptic deposition toward clearance.
The mechanism of amyloid clearance is not presently clear but may involve active phagocytosis of deposited Aβ peptides by microglia or by the binding of Aβ peptides by the protease inhibitor α2 macroglobulin and clearance via the low-density lipoprotein receptor-related protein LRP (Narita et al., 1997; Shibata et al., 2000). With regard to the former model, we provided evidence for the codistribution of Aβ-immunoreactive structures and glial processes, similar to the description of the close apposition of microglia and congophilic amyloid plaques in the brains of APP23 transgenic mice that overexpress an FAD-linked human APP (Bornemann et al., 2001). Our studies support several previous conclusions that amyloid deposits are dynamic structures (Schenk et al., 1999; DeMattos et al., 2001, 2002) and may suggest that turnover of amyloid deposits is an ongoing process in the brain.
Finally, and to assess the contribution of synaptically released Aβ to extracellular amyloid deposition, in vivo, we performed unilateral lesions in transgenic mice at a time before the onset of deposition. In contrast to the massive reduction in amyloid burden in the ipsilateral hippocampus of mice with preexisting amyloid deposits 1 month after lesion, we failed to detect differences in amyloid burden between the ipsilateral and the contralateral dentate gyrus and hippocampus 4 months after lesions in mice in which amyloid deposition is not apparent before the lesion. These findings strongly suggest that reinnervation of the outer two-thirds of the molecular layer of the dentate gyrus by intrinsic hippocampal fibers, afferent fibers from the contralateral entorhinal cortex, and/or septal cholinergic neuron afferents leads to release of Aβ from synaptic termini and subsequent deposition in the extracellular space.
Although our data support our view that synaptically released Aβ are the species deposited in the extracellular space, several important questions remain. For example, it is not clear whether the deposited Aβ peptides are derived from full-length APP, APP-CTFs that are in transit along axons, or APP-related species that are subject to proteolytic processing at nerve terminals. Recent studies have provided evidence that Aβ peptides can be generated in vitro using membranes prepared from mouse sciatic nerve, and these peptides appear to be present within axonal membranes that undergo rapid anterograde transport (Kamal et al., 2001). Unfortunately, we have not been successful in detecting radiolabeled Aβ derived from neuronally synthesized and axonally transported APP in the perforant pathway of rats (Buxbaum et al., 1998) or the transgenic mice used in this study (see above; data not shown).
Second, we have not formally proven that Aβ deposited in the terminal fields of entorhinal afferents are derived from APP transported from entorhinal neurons. In this regard, we cannot rule out the possibility that Aβ production–secretion and subsequent deposition at terminal fields of entorhinal afferents might be a function of synaptic activity-dependent release of Aβ from dendritic spines of dentate granule cells. As a corollary, it could be argued that the perforant path lesion limits entorhinal-derived synaptic input and, in so doing, leads to a reduction in amyloid burden via normal clearance processes. However, our previous demonstration that radiolabeled APP-CTFs, the penultimate precursors of Aβ and Aβ-related peptides, are detected in the hippocampus after injections of [35S]methionine in the entorhinal cortex strongly argue that Aβ is generated from axonally transported APP. Finally, it should be noted that, although we see marked reductions in amyloid burden in the dentate gyrus, we also observed reductions, albeit limited, in amyloid burden in the hippocampal CA1 and CA3 subfields after perforant pathway lesions. Again, it could be argued that the perforant pathway lesion diminishes synaptic drive through the entire trisynaptic circuit, i.e., from granule cell afferents to CA3 mossy fibers and subsequently projections from CA3 projections via Shaeffer collaterals to dendritic terminals of CA1 pyramidal neurons. On the other hand, it is now well established that the dendritic arbors of CA1 and CA3 pyramidal neurons receive considerable innervation from ipsilateral afferents of layer III neurons in the entorhinal cortex (Steward and Scoville, 1976), in addition to projections from the septum and brainstem. Thus, this reduction in amyloid burden in the CA1–CA3 sector is not incompatible with our view that disconnection of afferents from layer II and layer III neurons from the hippocampus leads to clearance of amyloid deposits, specifically in the dendritic fields of granule cell neurons and CA1–CA3 pyramidal neurons, respectively. Recognizing the limitations of our conclusion that synaptic release of Aβ is a principal contributor to amyloid deposition, we add that our findings are consistent with the compellingin vivo demonstrations that, in diffuse plaques of AD patients and aged nonhuman primates, Aβ is present along neuronal dendrites and around the soma of neurons included in the plaques (Martin et al., 1991; Probst et al., 1991).
Footnotes
This work was supported by National Institutes of Health Grants AG-021494 (S.S.S.) and AG-20047-02 (D.A.P.) and the Ellison Medical Foundation and the Ruth Broad Medical Research Foundation (S.S.S.).
Correspondence should be addressed to Dr. Sangram S. Sisodia, Department of Neurobiology, Pharmacology, and Physiology, The University of Chicago, Chicago, IL 60637. E-mail:ssisodia@drugs.bsd.uchicago.edu.
REFERENCES
- 1.Adami C, Sorci G, Blasi E, Agneletti AL, Bistoni F, Donato R. S100B expression in and effects on microglia. Glia. 2001;33:131–142. [PubMed] [Google Scholar]
- 2.Amaratunga A, Fine RE. Generation of amyloidogenic C-terminal fragments during rapid axonal transport in vivo of beta-amyloid precursor protein in the optic nerve. J Biol Chem. 1995;270:17268–17272. doi: 10.1074/jbc.270.29.17268. [DOI] [PubMed] [Google Scholar]
- 3.Barelli H, Lebeau A, Vizzavona J, Delaere P, Chevallier N, Drouot C, Marambaud P, Ancolio K, Baxbaun JD, Khorkova O, Heroux J, Sahasrabudhe S, Martinez J, Warter JM, Mohr M, Checler F. Characterization of new polyclonal antibodies specific for 40 and 42 amino acid-long amyloid beta peptides: their age to examine the cell biology of presenting and the immunohistochemistry of sporadic Alzheimer's disease and cerebral amyloid angiopathy cases. Mol Med. 1997;3:695–707. [PMC free article] [PubMed] [Google Scholar]
- 4.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 5.Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 6.Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol. 2001;158:63–73. doi: 10.1016/s0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braak H, Braak E. Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E, Strothjohann M. Abnormally phosphorylated tau protein related to the formation of neurofibrillary tangles and neuropil threads in the cerebral cortex of sheep and goat. Neurosci Lett. 1994;171:1–4. doi: 10.1016/0304-3940(94)90589-4. [DOI] [PubMed] [Google Scholar]
- 9.Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St. George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 11.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- 13.Dickson TC, Vickers JC. The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer's disease. Neuroscience. 2001;105:99–107. doi: 10.1016/s0306-4522(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 14.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 15.Ferreira A, Caceres A, Kosik KS. Intraneuronal compartments of the amyloid precursor protein. J Neurosci. 1993;13:3112–3123. doi: 10.1523/JNEUROSCI.13-07-03112.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fishman CE, Cummins DJ, Bales KR, DeLong CA, Esterman MA, Hanson JC, White SL, Paul SM, Jordan WH. Statistical aspects of quantitative image analysis of beta-amyloid in the APP(V717F) transgenic mouse model of Alzheimer's disease. J Neurosci Methods. 2001;108:145–152. doi: 10.1016/s0165-0270(01)00381-8. [DOI] [PubMed] [Google Scholar]
- 17.Frotscher M, Heimrich B, Deller T. Sprouting in the hippocampus is layer-specific. Trends Neurosci. 1997;20:218–223. doi: 10.1016/s0166-2236(96)01018-1. [DOI] [PubMed] [Google Scholar]
- 18.Garbuglia M, Verzini M, Sorci G, Bianchi R, Giambanco I, Agneletti AL, Donato R. The calcium-modulated proteins, S100A1 and S100B, as potential regulators of the dynamics of type III intermediate filaments. Braz J Med Biol Res. 1999;32:1177–1185. doi: 10.1590/s0100-879x1999001000001. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hjorth-Simonsen A, Jeune B. Origin and termination of the hippocampal perforant path in the rat studied by silver impregnation. J Comp Neurol. 1972;144:215–232. doi: 10.1002/cne.901440206. [DOI] [PubMed] [Google Scholar]
- 21.Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997a;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997b;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaether C, Skehel P, Dotti CG. Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell. 2000;11:1213–1224. doi: 10.1091/mbc.11.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 27.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 28.Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee MK, Borchelt DR, Wong PC, Sisodia SS, Price DL. Transgenic models of neurodegenerative diseases. Curr Opin Neurobiol. 1996;6:651–660. doi: 10.1016/s0959-4388(96)80099-7. [DOI] [PubMed] [Google Scholar]
- 30.Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levey AI, Jenkins N, Copeland N, Price DL, Sisodia SS. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med. 1997;3:756–760. doi: 10.1038/nm0797-756. [DOI] [PubMed] [Google Scholar]
- 31.Martin LJ, Sisodia SS, Koo EH, Cork LC, Dellovade TL, Weidemann A, Beyreuther K, Masters C, Price DL. Amyloid precursor protein in aged nonhuman primates. Proc Natl Acad Sci USA. 1991;88:1461–1465. doi: 10.1073/pnas.88.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nadler JV, Matthews DA, Cotman CW, Lynch GS. Development of cholinergic innervation in the hippocampal formation of the rat. II. Quantitative changes in choline acetyltransferase and acetylcholinesterase activities. Dev Biol. 1974;36:142–154. doi: 10.1016/0012-1606(74)90197-3. [DOI] [PubMed] [Google Scholar]
- 33.Nadler JV, Cotman CW, Paoletti C, Lynch GS. Histochemical evidence of altered development of cholinergic fibers in the rat dentate gyrus following lesions. II. Effects of partial entorhinal and simultaneous multiple lesions. J Comp Neurol. 1977;171:589–604. doi: 10.1002/cne.901710410. [DOI] [PubMed] [Google Scholar]
- 34.Narita M, Holtzman DM, Schwartz AL, Bu G. Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J Neurochem. 1997;69:1904–1911. doi: 10.1046/j.1471-4159.1997.69051904.x. [DOI] [PubMed] [Google Scholar]
- 35.Pigino G, Pelsman A, Mori H, Busciglio J. Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci. 2001;21:834–842. doi: 10.1523/JNEUROSCI.21-03-00834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 37.Probst A, Langui D, Ipsen S, Robakis N, Ulrich J. Deposition of beta/A4 protein along neuronal plasma membranes in diffuse senile plaques. Acta Neuropathol. 1991;83:21–29. doi: 10.1007/BF00294426. [DOI] [PubMed] [Google Scholar]
- 38.Savage MJ, Trusko SP, Howland DS, Pinsker LR, Mistretta S, Reaume AG, Greenberg BD, Siman R, Scott RW. Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J Neurosci. 1998;18:1743–1752. doi: 10.1523/JNEUROSCI.18-05-01743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 40.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 41.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 42.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steward O. Reinnervation of dentate gyrus by homologous afferents following entorhinal cortical lesions in adult rats. Science. 1976;194:426–428. doi: 10.1126/science.982024. [DOI] [PubMed] [Google Scholar]
- 45.Steward O, Scoville SA. Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentata of the rat. J Comp Neurol. 1976;169:347–370. doi: 10.1002/cne.901690306. [DOI] [PubMed] [Google Scholar]
- 46.Steward O, Vinsant SL. Identification of the cells of origin of a central pathway which sprouts following lesions in mature rats. Brain Res. 1978;147:223–243. doi: 10.1016/0006-8993(78)90837-5. [DOI] [PubMed] [Google Scholar]
- 47.Tamamaki N, Nojyo Y. Projection of the entorhinal layer II neurons in the rat as revealed by intracellular pressure-injection of neurobiotin. Hippocampus. 1993;3:471–480. doi: 10.1002/hipo.450030408. [DOI] [PubMed] [Google Scholar]
- 48.tom Dieck S, Sanmarti-Vila L, Langnaese K, Richter K, Kindler S, Soyke A, Wex H, Smalla KH, Kampf U, Franzer JT, Stumm M, Garner CC, Gundelfinger ED. Bassoon, a novel zinc-finger CAG/glutamine-repeat protein selectively localized at the active zone of presynaptic nerve terminals. J Cell Biol. 1998;142:499–509. doi: 10.1083/jcb.142.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turner DA, Buhl EH, Hailer NP, Nitsch R. Morphological features of the entorhinal-hippocampal connection. Prog Neurobiol. 1998;55:537–562. doi: 10.1016/s0301-0082(98)00019-7. [DOI] [PubMed] [Google Scholar]
- 50.Van Hoesen GW, Hyman BT, Damasio AR. Entorhinal cortex pathology in Alzheimer's disease. Hippocampus. 1991;1:1–8. doi: 10.1002/hipo.450010102. [DOI] [PubMed] [Google Scholar]
- 51.Vickers JC, Chin D, Edwards AM, Sampson V, Harper C, Morrison J. Dystrophic neurite formation associated with age-related beta amyloid deposition in the neocortex: clues to the genesis of neurofibrillary pathology. Exp Neurol. 1996;141:1–11. doi: 10.1006/exnr.1996.0133. [DOI] [PubMed] [Google Scholar]
- 52.Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]