

Abstract
Hypersensitivity resulting from nerve injury or morphine tolerance/hyperalgesia is predicted to involve similar cellular and molecular mechanisms. One expected but incompletely explored mechanism is the activation of central neuroimmune responses associated with these conditions. To begin to address this, we undertook three separate studies: First, we determined the acute antinociceptive action of morphine, the rate of development of opioid tolerance, and withdrawal-induced hyperalgesia/allodynia in nerve-injured and sham-operated rats using noxious (thermal and mechanical) and non-noxious (mechanical allodynia) behavioral paradigms. Second, we investigated the impact of chronic morphine treatment on spinal glial activation and cytokine expression after L5 spinal nerve transection or sham surgery. Third, we examined the consequences of spinal administration of cytokine inhibitors on the development of morphine tolerance and morphine withdrawal-induced hyperalgesia and allodynia. Results demonstrated that after nerve injury, the antinociceptive effect of acute morphine was significantly decreased, and the rate of development of tolerance and opioid withdrawal-induced hyperalgesia/allodynia was significantly enhanced compared with that after sham surgery. Chronic administration of morphine to sham-operated rats activated spinal glia and upregulated proinflammatory cytokines [interleukin (IL)-1β, IL-6, and tumor necrosis factor-α]. This neuroimmune activation was further enhanced in nerve-injured rats after chronic morphine treatment. Spinal inhibition of proinflammatory cytokines restored acute morphine antinociception in nerve-injured rats and also significantly reversed the development of morphine tolerance and withdrawal-induced hyperalgesia and allodynia in nerve-injured or sham-operated rats. Targeting central cytokine production and glial activation may improve the effectiveness of morphine and reduce the incidence of morphine withdrawal-induced hyperalgesia and allodynia in neuropathic pain conditions.
Keywords: neuropathy, morphine tolerance, hyperalgesia, glia, interleukin-1β, interleukin-6, tumor necrosis factor-α
Neuropathic pain is associated with severe, chronic sensory disturbances characterized by spontaneous pain, increased responsiveness to painful stimuli (hyperalgesia), and pain perceived in response to normally non-noxious stimuli (allodynia). Pain associated with neuropathy is difficult to treat, often being only partially relieved by high doses of opioids in humans (Cherny et al., 1994). A complete understanding of decreased opioid efficacy in the treatment of neuropathic pain has eluded the research community. Moreover, the literature on the analgesic effect and development of tolerance to opioids in neuropathic pain is contradictory. Several studies have demonstrated that neuropathy-induced hyperalgesia leads to a decrease in antinociception and to early development of tolerance to morphine (Mao et al., 1995; Christensen and Kayser, 2000). On the contrary, Backonja et al. (1995) and Catheline et al. (1996) reported that peripheral nerve injury increased the antinociceptive potency and delayed the development of tolerance to morphine. From these behavioral studies, it appears that the efficacy of morphine in attenuating hyperalgesia and/or allodynia is largely dependent on the animal model, the behavioral measure, and the route of drug administration.
Neuronal plasticity associated with hyperalgesia and morphine tolerance has similar cellular and molecular mechanisms, suggesting predictable interactions between hyperalgesia and morphine tolerance (Mao et al., 1995; Mayer et al., 1999). In addition to neurons and neurotransmitters, the role of non-neuronal cells, such as glia, and their secretory products in the development of hyperalgesia has been studied recently. Both microglia, the intrinsic macrophages of the CNS, and astrocytes release a variety of proinflammatory cytokines [interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α], which play a role in mediating or maintaining hyperalgesia and allodynia (DeLeo and Yezierski, 2001; Milligan et al., 2001; Watkins et al., 2001a,b). Similar to that in nerve injury, glial activation is also reported in the course of development of morphine tolerance (Song and Zhao, 2001), and neural effects of opioids, including analgesia, are altered by cytokines (Peterson et al., 1998; Gul et al., 2000; Rady and Fujimoto, 2001).
Although proinflammatory cytokines may modulate the analgesic effects of morphine, and chronic morphine treatment activates spinal glial cells, it is not known whether long-term morphine administration in a neuropathic pain rat model affects spinal neuroimmune activation. Considering that opioids have diverse effects on the peripheral immune system, it is important to know the consequences of opioid treatment on CNS-related immune responses. One can expect an enhanced disease process or worsening of withdrawal in the course of opioid treatment during neuropathic conditions. In view of this, the present study had a threefold design: (1) to evaluate and compare the antinociceptive action of acute morphine and the rate of development of morphine tolerance and withdrawal-induced hyperalgesia/allodynia in sham-operated and L5 spinal nerve-injured rats using noxious and non-noxious behavioral paradigms, (2) to explore possible effects of chronic morphine on spinal glial activation using immunocytochemistry and cytokine expression using RNase protection assays (RPAs) and ELISA in nerve-injured and sham-operated rats, and (3) to determine the effect of spinal inhibition of IL-1β, IL-6, and TNF-α on the acute analgesic action of morphine, development of opioid tolerance, and withdrawal-induced hyperalgesia/allodynia in sham-operated and nerve-injured rats.
MATERIALS AND METHODS
Animals
Male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) weighing 175–200 gm at the start of surgery were used. The animals were allowed to habituate to the housing facilities for ≥1 week before the experiments began. Behavioral studies were performed in a quiet room between the hours of 9:00 and 11:00 A.M. The Institutional Animal Care and Use Committee at Dartmouth College approved the procedures in this study. Efforts were made to limit distress and use the minimum number of animals necessary to achieve statistical significance as set forth by International Society for the Study of Pain guidelines (Covino et al., 1980).
Surgery
The unilateral peripheral mononeuropathy was produced according to the method described previously by Colburn et al. (1999). Briefly, rats were anesthetized with halothane in O2carrier (induction, 4%; maintenance, 2%). A small incision to the skin overlying L5-S1 was made followed by retraction of the paravertebral musculature from the vertebral transverse processes. The L6 transverse process was partially removed, exposing the L4 and L5 spinal nerves. The L5 spinal nerve was identified, lifted slightly, and transected. The wound was irrigated with saline and closed in two layers with 3-0 polyester suture (facial plane) and surgical skin staples.
Behavioral tests
The antinociceptive and antiallodynic properties of morphine were evaluated within 45–60 min after injection. Opioid withdrawal-induced hyperalgesia and allodynia were recorded 16 hr after the last injection of morphine. Mechanical sensitivity to non-noxious stimuli was measured by applying 2 and 12 gm von Frey filaments (Stoelting, Wood Dale, IL) on the plantar surface of the ipsilateral hind paw, as described previously (Colburn et al., 1999). The number of paw withdrawals in three sets of 10 stimulations for each set to this normally non-noxious stimulus determined mechanical allodynia. Mechanical nociceptive thresholds were evaluated using an Analgesy-Meter (Ugo Basile, Comerio, Italy), as explained by Stein et al. (1990). Rats were gently held, and incremental pressure (maximum of 250 gm) was applied onto the dorsal surface of the ipsilateral hind paw. The pressure required to elicit paw withdrawal, the paw-pressure threshold (PPT), was determined. Thermal nociceptive thresholds were evaluated by the hot water tail-flick test (Bian et al., 1999), which consisted of immersing the tail in water maintained at 49°C and recording the latency to a rapid flick. A 15 sec cutoff time was used. In the noxious test paradigms, the mean of three consecutive measurements made, separated by 10 sec, was determined and expressed as a percentage of maximal possible effect (%MPE).
Experimental design
Evaluation of the acute analgesic effect of morphine in sham-operated or nerve-injured rats. On postoperative days 6 or 11, when behavior reached steady state, both sham-operated and L5 nerve-transected rats were administered a 1–10 mg/kg intravenous injection of morphine via tail vein under halothane anesthesia (n = 6 rats per group). The analgesic effect was evaluated using the hot water tail-flick and paw-pressure Analgesy Meter. The threshold response of animals to noxious stimuli before administration of morphine served as the basal latency.
Evaluation of development of morphine tolerance and withdrawal-induced hyperalgesia and allodynia in sham-operated and L5 nerve-transected rats. Rats were treated with subcutaneous injections of either saline or morphine (10 mg/kg) (Sigma, St. Louis, MO). The injections were given twice daily at 8:00–9:00 A.M. and 4:00–5:00 P.M. for 5 d, beginning on day 6 and ending on day 10 after surgery to induce opioid tolerance. Development of analgesic and antiallodynic tolerance to chronic morphine was recorded on days 1, 3, and 5 (i.e., postsurgery days 6, 8, and 10) of the morphine treatment. Chronic morphine withdrawal-induced hyperalgesia or allodynia in these animals was assessed 16 hr after the last injection of morphine (i.e., on postsurgery day 11). Behavior recorded on day 6 before the beginning of morphine treatment served as the basal latency (n = 8 rats per group).
Qualitative assessment of glial fibrillary acidic protein and OX-42 immunoreactivity in lumbar spinal cord. On day 11 after the recording of morphine withdrawal-induced hyperalgesia and allodynia, animals were anesthetized and transcardially perfused with 0.1m PBS, pH 7.4, followed by 4% paraformaldehyde in PBS. Lumbar spinal cord sections were harvested and processed as described previously (Colburn et al., 1999). Immunohistochemistry was performed on 20 μm free-floating L5 spinal cord sections. A monoclonal antibody to OX-42 (1:2 working dilution from William F. Hickey, Dartmouth Hitchcock Medical Center) was used to label the expression of CR3/CD11b on activated microglia. A polyclonal antibody to glial fibrillary acidic protein (GFAP) (1:20,000 working dilution; Dako, Carpinteria, CA) was used to label astrocytes (n= 4 rats per group).
Tissue collection for quantifying cytokine mRNA and proteins. To quantify cytokine mRNA and protein levels, a separate group of animals than those discussed in the aforementioned paragraph B was killed by CO2 asphyxiation followed by decapitation immediately after behavioral testing on day 11 after surgery. Inserting an 18 gauge needle into the caudal end of the vertebral column and flushing the spinal cord out with ice-cold PBS achieved spinal cord isolation. The spinal cord was flash frozen immediately on dry ice and stored at −80°C until homogenization. L5 lumbar spinal cord was removed from the intact frozen cord at the time of quantifying mRNA and protein.
RNase protection assay. Assessment of temporal cytokine mRNA expression in the L5 lumbar spinal cord was performed using a Ribonuclease MultiProbe RPA system (PharMingen, San Diego, CA). Total RNA from L5 lumbar spinal cord was isolated by the TRIzol extraction method (Invitrogen, Carlsbad, CA). Total RNA (15 μg) was hybridized to 32P-labeled antisense RNA probes transcribed using the rat cytokine-1 (rCK-1) multiprobe template set [including IL-1α/β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-10, TNF-α/β, interferon (IFN)-γ, L32, and glycera-ldehyde-3-phosphate dehydrogenase], resulting in double-stranded target RNA. After RNase digestion, protected RNA and probe were resolved on a denaturing polyacrylamide gel and visualized by overnight autoradiography. Semiquantitative image analysis was used to compare mRNA levels based on band intensities for each cytokine; the intensity of each band was measured using NIH Image software and assigned an arbitrary unit based on the measured intensity levels. Image intensity for the housekeeping gene (L32) and background levels were used to normalize cytokine measurements and were compared with the relative levels of mRNA. The relative mean level of cytokine mRNA in different groups of rats (saline- or morphine-treated in sham or L5 nerve-transected rats) was determined, and levels were normalized by those for normal animals and reported as ratios to normal (n = 4 rats per group).
Protein estimation by ELISA. Standard ELISA was performed for quantitative determination of IL-1β, IL-6, and TNF-α protein. L5 lumbar spinal cord homogenization was prepared as explained previously (Sweitzer et al., 2001b). In brief, weighed sections of L5 spinal cord were homogenized in homogenization buffer consisting of a protease inhibitor (Boehringer Mannheim, Mannheim, Germany) using a Power Gen 125 tissue tearer (Fisher Scientific, Suwanee, GA). Samples were spun at 20,000 × g for 30 min at 4°C. Supernatant was aliquoted and stored at −80°C for future protein quantification. IL-1β, TNF-α (R & D Systems, Minneapolis, MN), and IL-6 (Biosource, Camarillo, CA) protein concentrations were determined using the quantitative sandwich enzyme immunoassay according to the manufacturer's directions. IL-1β, IL-6, and TNF-α protein quantification was determined by comparing samples to the standard curve generated from the respective kits (n = 4 rats per group).
Evaluation of the inhibition of spinal proinflammatory cytokines (IL-1β, IL-6, and TNF-α) on acute analgesic action of morphine, development of morphine tolerance, and opioid withdrawal-induced hyperalgesia in sham or L5 nerve-transected rats. Separate groups of rats (either sham operated or nerve transected) received either saline or morphine and were treated once daily (at 11:00 A.M. to 12:00 P.M.) with a cocktail consisting of a fixed-dose combination of IL-1 receptor antagonist (IL-1ra; 100 μg/rat), soluble TNF receptor (sTNFR; 30 μg/rat) (both were kind gifts from Amgen, Thousand Oaks, CA), and goat anti-rat IL-6-neutralizing antibody (0.08 μg/rat; R & D Systems). This cocktail (in PBS) was administered via direct lumbar puncture under brief halothane anesthesia at a volume of 15 μl/rat on postoperative days 6–10. On day 11, after recording morphine withdrawal-induced hyperalgesic and allodynic behavior, these rats were treated with morphine (2 mg/kg, i.v.) via the tail vein, and the antihyperalgesic activities were evaluated. The threshold latency recorded before the intravenous administration of morphine (day 11) was used as the basal latency (n = 6 rats per group). The selection of IL-1ra, sTNFR, and anti IL-6 antibody doses was based on our previous reports (Arruda et al., 2000; Sweitzer et al., 2001b).
Statistical analysis
Values are expressed as means ± SEM and were analyzed for significance with one-way ANOVA followed by a post hocBonferroni analysis using STATA 5.0 (STATA Corp., College Station, TX).p values of <0.05 were considered significant. To access morphine analgesia, the data of noxious mechanical and thermal tests were converted to a percentage of MPE using the following formula: %MPE = (WT − CT)/(CO − CT) × 100, where WT equals withdrawal latency (in seconds) or threshold (in grams) after morphine/saline treatment, and CT equals the cutoff value (i.e., 250 gm for mechanical test and 15 sec for the tail-flick test).
RESULTS
General results
The mean paw-withdrawal pressure to noxious mechanical stimuli or tail-flick latency to thermal stimuli between unoperated and sham-operated rats showed no significant difference. However, in L5 nerve-transected rats, mean PPT and tail-flick latency were significantly decreased compared with the sham-operated or control group of animals, indicating development of mechanical and thermal hyperalgesia in L5 nerve-transected animals. Similarly, nerve-injured rats also developed mechanical allodynia to both 2 and 12 gm of mechanical stimuli (Table 1). The development of mechanical and thermal hyperalgesia and mechanical allodynia increased in a time-dependent manner after nerve transection and reached steady state between days 6 and 11 after surgery. This steady-state period was used to study the acute analgesic effect of morphine, the development of morphine tolerance, and its withdrawal-induced hyperalgesia in neuropathic rats.
Table 1.
Neuropathy (by L5 spinal nerve transection) induced hyperalgesia and allodynia in rats
| n | Thermal hyperalgesia (tail-flick latency in seconds) | Mechanical hyperalgesia (paw-pressure threshold in grams) | Mechanical allodynia (number of paw withdrawals per 30 stimuli) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Day 6 | Day 11 | ||||||||
| Day 6 | Day 11 | Day 6 | Day 11 | 2 gm | 12 gm | 2 gm | 12 gm | ||
| Control | 8 | 5.5 ± 0.14 | 5.3 ± 0.2 | 104.5 ± 3 | 102.5 ± 2.7 | 0 | 0 | 0 | 0.1 ± 0.2 |
| Sham | 8 | 5.4 ± 0.13 | 5.2 ± 0.3 | 93 ± 4.5 | 95.6 ± 3.8 | 0.15 ± 0.1 | 0.1 ± 0.2 | 0.2 ± 0.1 | 0.3 ± 0.3 |
| L5Tx | 8 | 3.2 ± 0.1*,1-160 | 3.08 ± 0.2*,1-160 | 50 ± 5.5*,1-160 | 46.9 ± 2.8*,1-160 | 8.3 ± 0.8*,1-160 | 14.9 ± 2*,1-160 | 7.9 ± 1.2*,1-160 | 15.2 ± 1*,1-160 |
Behavioral response to noxious or non-noxious stimuli in unoperated (control), sham-operated (sham), and L5 spinal nerve-transected (L5Tx) rats was recorded on postoperative days 6 and 11 (steady state). Values are means ± SEM.
p < 0.05 versus control;
F1-160: p < 0.05 versus sham-operated rats (Bonferroni test).
Neuropathy decreased the acute analgesic effect of morphine
When tested on days 6 or 11 after the surgical procedure (steady state), administered morphine (1–10 mg/kg, i.v.) produced significant and dose-dependent antinociception to both noxious thermal and mechanical stimuli in sham-operated rats. In nerve-injured rats, lower doses of morphine (1 and 2 mg/kg) failed to elicit any antinociceptive effect in either noxious test paradigm. However, with increasing doses (5 and 10 mg/kg), morphine retained its antinociceptive effect in nerve-injured rats. A cumulative dose–response curve of morphine in nerve-injured rats showed a clear rightward shift in its antinociceptive effect, indicating decreased analgesic properties of morphine in nerve-injured rats (Fig. 1). Although morphine produced a dose-dependent antiallodynic effect in L5 nerve-transected animals, it was not compared directly with sham-operated rats; the sham group did not demonstrate tactile allodynia, preventing evaluation of the antiallodynic activity of morphine in these rats.
Fig. 1.

Decreased antinociceptive activity of acute morphine in L5 nerve-transected rats. Behavioral response to noxious thermal (tail-flick test) and mechanical (paw-pressure test) stimuli was recorded on postoperative days 6 or 11. Antinociceptive activity of acute morphine (M; 1–10 mg/kg) was recorded 45 min after its intravenous administration and expressed as a percentage of MPE. A rightward shift in the dose–response curve of morphine in L5 nerve-transected rats (L5Tx) compared with sham-operated rats indicates decreased antinociceptive activity of morphine in neuropathic conditions. Values are mean ± SEM (n = 6). *p < 0.05 versus basal latency (recorded before morphine administration);+p < 0.05 versus sham-operated rats (Bonferroni test).
Neuropathy enhanced the development of morphine tolerance
In a daily injection paradigm, either sham-operated or nerve-injured rats tested on day 1 of morphine treatment showed no significant difference in antinociceptive action. These animals showed an almost complete analgesic effect to the first dose of morphine (10 mg/kg, s.c.) in both thermal and mechanical test paradigms. On day 3, the percentage of MPE of morphine was reduced to 45 and 19% in the tail-flick test and to 42 and 3% in the paw-pressure test in sham-operated and neuropathic rats, respectively. On day 5 of the morphine treatment, both groups of animals showed complete development of antinociceptive tolerance. A significant decrease in the antinociceptive action of morphine was observed on day 3 in nerve-injured rats compared with sham-operated rats. This indicates an early development of antinociceptive tolerance in these animals (Fig. 2). In the present experimental conditions, unlike antihyperalgesic action, complete tolerance to antiallodynic activity of morphine was not observed. However, there was a significant decrease in the antiallodynic activity of morphine (for 12 gm of stimuli) on day 5 compared with day 1 treatment (Fig. 2), suggesting a slower rate of tolerance development to non-noxious mechanical stimuli. Development of tolerance to the antiallodynic activity of morphine in nerve-injured rats was not compared with sham-operated rats, because they did not develop allodynia to a non-noxious mechanical stimulus.
Fig. 2.
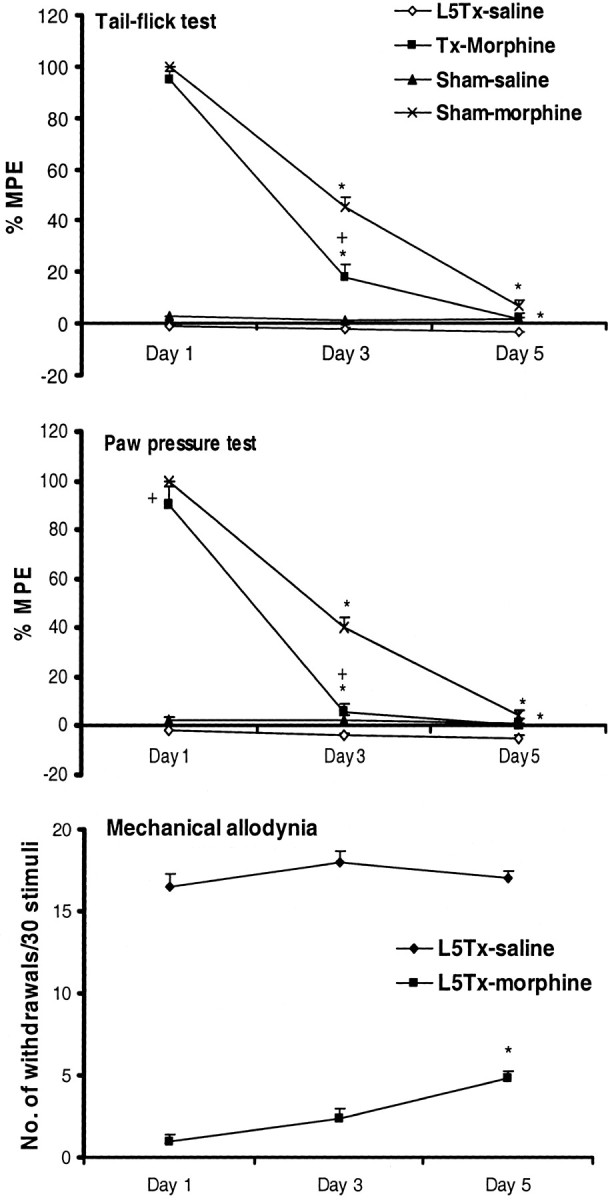
L5 nerve transection enhances the development of morphine tolerance to analgesic and antiallodynic actions. Chronic morphine (10 mg/kg, s.c., twice daily for 5 d) treatment was initiated on postoperative day 6 to L5 nerve-transected (L5Tx) and sham-operated rats. Antinociceptive and antiallodynic activity of morphine in these rats was accessed on days 1, 3, and 5 of the treatment (i.e., postoperative days 6, 8, and 10). Behavior recorded before the beginning of morphine treatment represents baseline responses. Analgesic activity of morphine against noxious thermal and mechanical stimuli was expressed as a percentage of MPE, whereas antiallodynic activity was expressed as average numbers of paw withdrawals to 30 stimuli of 12 gm of von Frey filament. A significant decrease in the antinociceptive action of morphine (both in the tail-flick and in the paw-pressure tests) in L5 nerve-transected rats (L5Tx-morphine) compared with sham rats (Sham-morphine) on day 3 of the treatment indicates early development of morphine tolerance in neuropathic conditions. Values are mean ± SEM (n = 8–10). *p < 0.05 versus antinociceptive or antiallodynic activity of morphine observed on the first day of its treatment;+p < 0.05 versus morphine-treated, sham-operated (Sham-morphine) rats (Bonferroni test). Note that sham-operated rats did not develop tactile allodynia, preventing evaluation of the antiallodynic activity of morphine in these animals.
Neuropathy enhanced morphine withdrawal-induced hyperalgesia and allodynia
Chronic administration of morphine (10 mg/kg, s.c., twice daily for 5 d) to sham-operated or neuropathic rats led to withdrawal-induced thermal and mechanical hyperalgesia and mechanical allodynia when recorded 16 hr after the last injection. Morphine withdrawal-induced hyperalgesia and allodynia were significantly greater in the nerve-injured rats compared with sham-operated rats (Table 2). Unlike the saline-treated neuropathic rats, in which the development of mechanical allodynia and hyperalgesia was primarily restricted to the ipsilateral paw, chronic morphine treatment showed the development of hyperalgesia and allodynia even in the contralateral paw (data not shown).
Table 2.
Chronic morphine withdrawal-induced hyperalgesia and allodynia in sham-operated and L5 nerve-transected rats
| n | Sham-operated rats | L5 nerve-transected rats | ||||
|---|---|---|---|---|---|---|
| Saline | Morphine | Saline | Morphine | |||
| Thermal hyperalgesia (tail-flick latency in seconds) | 8 | 5.3 ± 0.2 | 3.7 ± 0.2* | 3.2 ± 0.2 | 2.8 ± 0.3 | |
| Mechanical hyperalgesia (paw-pressure threshold in grams) | 8 | 98.5 ± 4.2 | 69.4 ± 2.4* | 48.2 ± 3.0 | 35.3 ± 1.8*,2-160 | |
| 2 gm | 0 | 3.9 ± 0.5* | 7.9 ± 0.6 | 14.3 ± 2.1*,2-160 | ||
| Mechanical allodynia (number of paw withdrawals per 30 stimuli) | 8 | 12 gm | 0.1 ± 0.2 | 6.9 ± 0.7* | 15.4 ± 0.9 | 20.1 ± 0.8*,2-160 |
Pretreatments consisted of two daily injections of morphine (10 mg/kg, s.c.) or saline administered on postoperative days 6–10. Withdrawal-induced hyperalgesia and allodynia were recorded 16 hr after the last injection of morphine or saline (i.e., on day 11). Data are expressed as mean ± SEM.
p < 0.05 versus respective saline-treated rats;
F2-160: p < 0.05 compared with chronic morphine-treated, sham-operated rats (Bonferroni test).
Chronic morphine treatment enhanced OX-42 and GFAP immunoreactivity in lumbar spinal cord of sham-operated and neuropathic rats
As shown in Figures 3, Aand C, and 4, A and C, immunoreactive OX-42 and GFAP were significantly elevated in the lumbar spinal cord in nerve-injured rats compared with sham-operated rats on day 11 after surgery. Acute administration of morphine did not enhance OX-42 or GFAP immunoreactivity in the lumbar spinal cord of control or sham-operated rats (data not shown), but after chronic treatment, a significant increase in OX-42 and GFAP immunoreactivity in the L5 lumbar dorsal horn of the spinal cord of these animals was observed (Figs.3B, 4B). Chronic administration of morphine to nerve-injured rats further increased OX-42 and GFAP immunoreactivity in the L5 lumbar spinal cord of these animals (Figs.3D, 4D). Unlike the saline-treated neuropathic rats, in which increased expression of OX-42 and GFAP was primarily observed in the ipsilateral side, chronic morphine treatment demonstrated increases in glial activity in both ipsilateral and contralateral dorsal horns (data not shown).
Fig. 3.
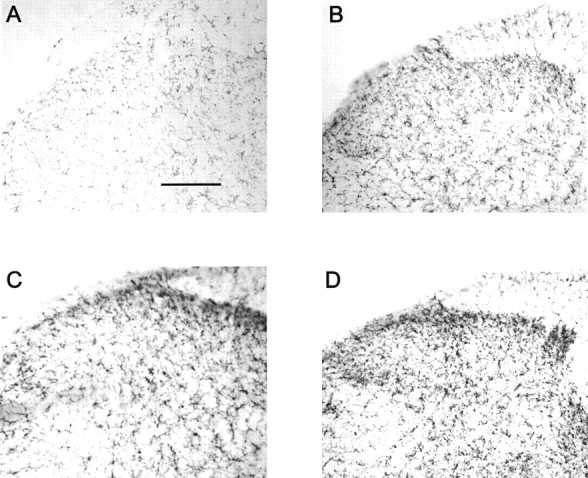
Increased microglial activation by chronic morphine treatment in sham-operated and neuropathic rats. Chronic administration of morphine to either sham-operated rats (B) or L5 nerve-transected rats (D) showed enhanced OX-42 immunostaining in the dorsal horn of the L5 lumbar spinal cord compared with saline-treated, sham-operated rats (A) or neuropathic rats (C), respectively. Scale bar, 150 μm (n = 4 per group).
Fig. 4.
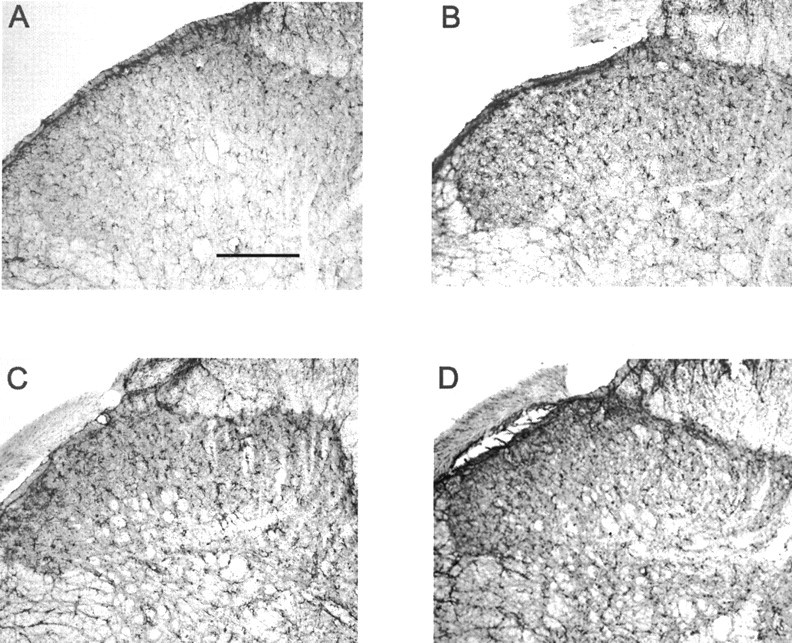
Increased astrocyte activation by chronic morphine treatment in sham-operated and neuropathic rats. Chronic administration of morphine to either sham-operated rats (B) or L5 nerve-transected rats (D) showed enhanced GFAP immunostaining in the dorsal horn of the L5 lumbar spinal cord compared with saline-treated, sham-operated rats (A) or neuropathic rats (C), respectively. Scale bar, 150 μm (n = 4 per group).
Chronic morphine treatment enhanced transcription of proinflammatory cytokines in L5 lumbar spinal cord of sham-operated and neuropathic rats
Constitutive expression of mRNA for IL-1α/β, TNF-α/β, and IL-6 was observed in L5 lumbar spinal cord of sham-operated, saline-treated rats. After an L5 spinal nerve transection (on postoperative day 11), significant increases in mRNA levels of IL-1α/β, TNF-α/β, and IL-6 were observed compared with sham-operated animals. Chronic administration of morphine (10 mg/kg, s.c., twice daily for 5 d) to sham-operated rats significantly increased the mRNA levels for IL-1β, IL-6, and TNF-α compared with saline-treated animals, whereas the levels of IL-1α and TNF-β (lymphotoxin) were unaffected. Chronic treatment of morphine to L5 nerve-transected rats further enhanced the level of IL-1β, TNF-α, and TNF-β compared with saline-treated, nerve-transected rats (Fig. 5 and Table 3). Under the present experimental conditions, other cytokines included in the RPA kit (rCK-1) were not consistently detected.
Fig. 5.
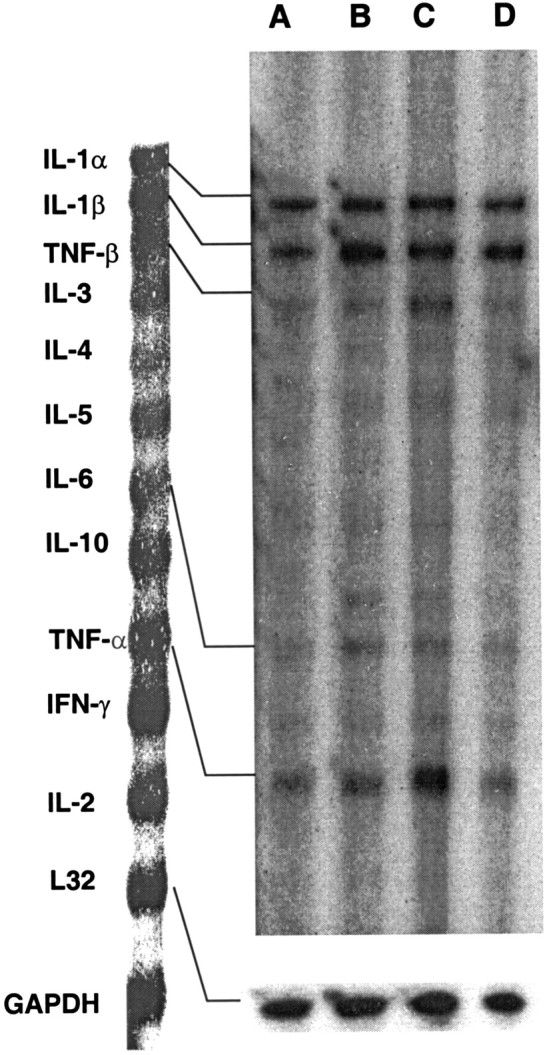
Representative RNase protection assay depicting cytokine mRNA expression for IL-1α, IL-1β, IL-6, TNF-α, and TNF-β in L5 lumbar spinal cord of saline-treated, sham-operated rats (A) and neuropathic rats (B), chronic morphine-treated neuropathic rats (C), and sham-operated rats (D) (n = 4 per group). Note that mRNA for IL-2, IL-4, IL-5, IL-10, and IFN-γ was inconsistent or not readable in nontransected animals at present experimental conditions. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase.
Table 3.
mRNA levels of IL-1α, IL-1β, TNF-β, IL-6, and TNF-α in L5 lumbar spinal cord after peripheral neuropathy, chronic morphine treatment, or a combination of both
| Treatment | n | IL-1α | IL-1β | TNF-β | IL-6 | TNF-α |
|---|---|---|---|---|---|---|
| Sham morphine | 4 | 1.13 ± 0.14 | 1.6 ± 0.083-150 | 1.3 ± 0.09 | 1.73 ± 0.063-150 | 1.4 ± 0.113-150 |
| L5Tx saline | 4 | 1.59 ± 0.073-150 | 2.6 ± 0.073-150 | 2.11 ± 0.093-150 | 3.0 ± 0.093-150 | 2.25 ± 0.133-150 |
| L5Tx morphine | 4 | 1.75 ± 0.083-150,3-160 | 3.1 ± 0.063-150,3-160 | 3.18 ± 0.113-150,3-160 | 3.09 ± 0.113-150 | 3.06 ± 0.063-150,3-160 |
A multiprobe ribonuclease assay was performed using total mRNA from lumbar spinal cord of different groups of rats isolated on day 11 after L5 nerve transection (L5Tx) or sham surgery to detect the expression of regulatory molecules. The value for the normalized quantity for each band was obtained by dividing with theL32 housekeeping gene control. Individual mRNA concentration in terms of fold activation was calculated as the ratio of their expression compared with sham saline-treated animals, in which normal values are weighed as 1. Values are mean ± SEM (n = 4).
F3-150: p < 0.05 versus sham saline group;
F3-160: p < 0.05 versus L5Tx saline-treated group (Bonferroni test).
Chronic morphine treatment enhanced proinflammatory cytokine protein levels in L5 lumbar spinal cord of sham-operated and neuropathic rats
Results from the analysis of L5 lumbar spinal cord homogenates demonstrated that L5 spinal nerve transection produced significant increases in IL-1β, IL-6, and TNF-α protein levels compared with saline-treated, sham-operated rats. In sham-operated rats, chronic administration of morphine increased protein levels of IL-1β and IL-6 compared with saline-treated rats. Although levels of TNF-α increased in chronic morphine-treated rats, these values were not statistically significant. In L5 spinal nerve-transected rats, chronic administration of morphine significantly enhanced the IL-1β and TNF-α protein levels, whereas increased levels of IL-6 in these animals were not statistically significant compared with saline-treated, nerve-transected rats (Table 4).
Table 4.
Proinflammatory cytokine level in L5 lumbar spinal cord after peripheral neuropathy, chronic morphine treatment, or a combination of both
| Treatment | n | IL-1β (pg/mg total protein) | IL-6 (pg/mg total proteins) | TNF-α (pg/mg total proteins) |
|---|---|---|---|---|
| Sham saline | 4 | 43.5 ± 5.4 | 197.3 ± 17 | 25.8 ± 2.8 |
| Sham morphine | 4 | 73.5 ± 9.24-150 | 359 ± 62.44-150 | 36.5 ± 5.6 |
| L5Tx saline | 4 | 215 ± 20.74-150 | 720.5 ± 56.54-150 | 115.3 ± 8.24-150 |
| L5Tx morphine | 4 | 301.8 ± 16.24-150,4-160 | 782.8 ± 28.74-150 | 169 ± 10.94-150,4-160 |
L5 lumbar spinal cords of sham- or L5 nerve-transected (L5Tx) rats, treated chronically with either saline or morphine, were harvested on postoperative day 11 (immediately after behavioral testing) to detect the protein levels of IL-1β, IL-6, and TNF-α by standard ELISA. Values are mean ± SEM (n = 4).
F4-150: p < 0.05 versus sham saline group;
F4-160: p < 0.05 versus L5Tx saline-treated group (Bonferroni test).
Intrathecally administered IL-1ra, sTNFR, and anti-IL-6 antibody restored morphine analgesia and reversed the development of morphine tolerance and withdrawal-induced hyperalgesia in neuropathic and sham-operated rats
Chronic administration of a fixed-dose combination of IL-1ra, sTNFR, and anti-IL-6 antibody to sham-operated rats on postoperative days 6–10 did not modulate the analgesic actions of acute morphine (2 mg/kg, i.v.). However, in L5 spinal nerve-transected rats, chronic administration of IL-1ra, sTNFR, and anti-IL-6 antibody significantly restored the analgesic actions of acute morphine. Chronic administration of morphine (10 mg/kg, s.c., twice daily) for 5 d (postoperative days 6–10) induced complete analgesic tolerance in both sham and nerve-transected rats as evidenced by their inability to exhibit an analgesic effect to the administered morphine (2 mg/kg, i.v.) tested on postoperative day 11. However, chronic administration of IL-1ra, sTNFR, and anti-IL-6 antibody during the induction of morphine tolerance to both sham and L5 nerve-transected rats showed significant antinociceptive action to administered morphine (2 mg/kg, i.v.) on postoperative day 11 (Fig. 6). Chronic administration of IL-1ra, sTNFR, and anti-IL-6 antibody during induction of morphine tolerance significantly reversed the development of opioid withdrawal-induced hyperalgesia and allodynia in sham and L5 nerve-transected rats (Table 5).
Fig. 6.

Inhibition of spinal proinflammatory cytokines restores acute analgesic action of morphine in neuropathic- and morphine-tolerant rats. Sham-operated and L5 nerve-transected rats received chronic saline (A); chronic IL-1ra, sTNFR, and anti IL-6 antibody (B); chronic morphine (C); and chronic morphine plus IL-1ra, sTNFR, and anti IL-6 antibody (D) treatment on postoperative days 6–10. Acute antinociceptive action of morphine (2 mg/kg, i.v.) in these rats was evaluated against noxious thermal (tail-flick test) and mechanical (paw-pressure test) stimuli on postoperative day 11 (for details, see Materials and Methods). Data show that chronic inhibition of proinflammatory cytokines restores the acute antinociceptive actions of intravenously administered morphine in saline- or morphine-tolerant, nerve-transected and morphine-tolerant, sham-operated rats. Values are mean (%MPE) ± SEM (n = 5). *p < 0.05 versus group A nerve-transected rats; +p < 0.05 versus group C sham or nerve-transected rats (Bonferroni test).
Table 5.
Effect of chronic administration of IL-1ra, sTNFR, and anti-IL-6 antibody on the development of morphine withdrawal-induced hyperalgesia and allodynia in sham-operated or L5 nerve-transected rats
| n | Sham-operated rats | L5 nerve-transected rats | ||||
|---|---|---|---|---|---|---|
| Untreated | Treated | Untreated | Treated | |||
| Thermal hyperalgesia (tail-flick latency in seconds) | 8 | 3.7 ± 0.1 | 4.4 ± 0.25-150 | 2.8 ± 0.13 | 4.1 ± 0.35-150 | |
| Mechanical hyperalgesia (paw-pressure threshold in grams) | 8 | 69.4 ± 2.4 | 86.7 ± 55-150 | 35.3 ± 2 | 61.5 ± 65-150 | |
| 2 gm | 3.9 ± 0.5 | 1.2 ± 0.75-150 | 14.3 ± 2 | 7.8 ± 25-150 | ||
| Mechanical allodynia (number of paw withdrawals per 30 stimuli) | 8 | 12 gm | 6.9 ± 0.7 | 3.5 ± 0.65-150 | 20.1 ± 1 | 11.4 ± 2.45-150 |
Pretreatments consisted of two daily injections of morphine (10 mg/kg s.c.), administered on postoperative days 6–10. IL-1ra, sTNFR, and anti-IL-6 antibody (treated) or saline (untreated) was administered intrathecally once daily during induction of morphine tolerance (i.e., on postoperative days 6–10). Withdrawal-induced hyperalgesia and allodynia were recorded 16 hr after the last injection of morphine or saline (i.e., on day 11). Data are expressed as mean ± SEM.
F5-150: p < 0.05 versus respective untreated group (Bonferroni test).
DISCUSSION
The main findings of this study show that persistent pain induced by an L5 spinal nerve transection in rats decreased the acute analgesic action of morphine, enhanced the development of analgesic tolerance to chronic morphine treatment, and aggravated opioid withdrawal-induced hyperalgesia and allodynia. Similar to what has been observed after peripheral nerve injury, chronic morphine treatment led to glial activation and enhanced proinflammatory cytokine expression in the L5 lumbar spinal cord of normal or sham-operated rats. Chronic morphine treatment to neuropathic rats further enhanced nerve injury-associated increases in spinal neuroimmune activation. In addition, a decrease in the acute antinociceptive response of morphine in neuropathic rats, development of morphine tolerance, and withdrawal-induced hyperalgesia and allodynia in nerve-injured or sham-operated rats were partially reversed by neutralizing or antagonizing proinflammatory cytokines.
Both previous studies (Mao et al., 1995; Nichols et al., 1997; Fundytus et al., 2001) and our present study clearly show that the acute antinociceptive effect of morphine against noxious thermal and mechanical stimuli decreased in nerve-injured rats. Although nerve-injured rats did not respond to lower doses of morphine but did respond with increasing doses, the threshold latency to both noxious thermal and mechanical stimuli was significantly increased compared with predrug latency. This suggests that the analgesic effect of an opioid is reduced during neuropathic pain, and that one can obtain adequate analgesia by increasing the dose of morphine. Repeated administration of morphine for a period of 5 d developed a complete antinociceptive tolerance in both sham-operated and nerve-injured rats. However, a significant decrease in the antinociceptive effect in nerve-injured rats compared with that of sham-operated rats on day 3 of morphine treatment suggests an earlier development of antinociceptive tolerance in these animals. This observation is in agreement with the findings of Christensen and Kayser (2000), who showed enhanced development of antinociceptive tolerance to systemic morphine in nerve-injured rats. However, in contrast to our observation, Christensen and Kayser (2000) reported that unlike the noxious mechanical stimuli, antinociceptive tolerance to thermal stimuli was not developed in nerve-injured rats. This observed difference in our studies could be attributable to differences in the methodological approach to evaluate the development of morphine tolerance or could be attributable to differences in the behavioral paradigm used to evaluate the thermal hyperalgesia in neuropathic rats. We did not observe complete tolerance to the antiallodynic activity of morphine (unlike the development of complete tolerance to antihyperalgesia). This indicates that within the same model of neuropathic pain, pathophysiological mechanisms mediating the abnormal reactions to noxious and innocuous stimuli are different. The various animal models used may have a fundamentally different pathophysiology (Bars et al., 2001), and the sensitivity to opioids may vary accordingly (McCormack et al., 1998).
Other interesting results observed in this study were enhanced opioid withdrawal-induced hyperalgesia and allodynia in nerve-injured rats compared with sham-operated rats. Chronic administration of opioids followed by its abrupt withdrawal or by naloxone treatment induced a hypersensitivity state characterized by hyperalgesia and allodynia in mice and rats (Devulder et al., 1996; Li et al., 2001a,b;Nozaki-Taguchi and Yaksh, 2002). Results showing an enhanced rate of development of antinociceptive tolerance and withdrawal-induced hyperalgesia and allodynia during chronic morphine treatment to neuropathic rats suggest cross-interaction between hypersensitivity mechanisms operating during the process of neuropathic pain and chronic opioid treatment.
Central neuroimmune activation and neuroinflammation have been postulated to mediate and/or modulate the pathogenesis of persistent pain states. Proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, induce a long-term alteration of synaptic transmission in the CNS and play a critical role in the development and maintenance of neuropathic pain (DeLeo and Yezierski, 2001; Sweitzer et al., 2001a). In the CNS, the major contributors of cytokine release are glia. Astrocytes and microglia can produce cytokines on activation (Kreutzberg, 1996; Aloisi, 2001; Dong and Benveniste, 2001). Nerve injury or peripheral inflammation has been reported to activate glial cells and increase the proinflammatory cytokine levels in the CNS (DeLeo and Yezierski, 2001; Watkins et al., 2001a,b). In line with this, the present study showed increased microglial and astrocytic activity, as evidenced by increases in OX-42 and GFAP immunoreactivity in the L5 lumbar spinal cord of nerve-injured rats. Similarly, mRNA for proinflammatory cytokines (IL-1β, IL-6, and TNF-α) and lymphotoxin (TNF-β) and protein levels of IL-1β, IL-6, and TNF-α were also increased in the lumbar spinal cord of L5 nerve-transected rats.
Central or peripheral administration of IL-1β, IL-6, and TNF-α is known to induce hyperalgesia and allodynia in rats (Oka and Hori, 1999). Raffa et al. (1993) postulated that cytokines might interact with opioid receptors and modulate its actions. Gul et al. (2000)and Rady and Fujimoto (2001) showed a reduction in the analgesic effect of morphine after exogenous administration of IL-1β. In the present study, chronic administration of a fixed-dose combination of anti-IL-6 antibody, sTNFR, and IL-1ra to nerve-transected animals partially restored the acute antinociceptive effect of morphine.
Both opioid tolerance and neuropathic pain conditions share features of diminished μ-opioid analgesia and abnormal pain (hyperalgesia and allodynia). These common features have led to suggestions of common mechanisms in nerve injury or chronic opioid-induced hyperalgesia (Mayer et al., 1999; Przewlocki and Przewlocka, 2001). Recent evidence suggests that glial cells might possibly modulate chronic opioid actions. Chronic morphine treatment activates spinal and cortical astrocyte activity (Beitner-Johnson et al., 1993; Song and Zhao, 2001), and inhibition of this activation by glial inhibitors partially reversed the development of morphine tolerance (Song and Zhao, 2001). Activated glia release excitatory amino acids (EAA), nitric oxide (NO), and proinflammatory cytokines (Kreutzberg, 1996;Aloisi, 2001; Dong and Benveniste, 2001). Although the role of EAA and NO in the development of morphine tolerance and opioid withdrawal-induced hyperalgesia has been extensively studied, the contribution of proinflammatory cytokines in this mechanism has not yet been reported. In the present study, we showed that chronic morphine treatment enhanced mRNA and protein levels of IL-1β and IL-6 in the lumbar spinal cord of sham or normal rats. In the nerve-injured rats, chronic administration of morphine further enhanced glial activation and proinflammatory cytokine levels in L5 lumbar spinal cord, suggesting synergistic or additive interaction in this process. The increased proinflammatory response in chronic morphine-treated rats paralleled the behavioral hypersensitivity to noxious and non-noxious stimuli observed during the withdrawal period. Furthermore, the ability of a fixed-dose combination of IL-6 antibody, sTNFR, and IL-1ra administered during induction of morphine tolerance to substantially reverse antinociceptive and antiallodynic tolerance, morphine withdrawal-induced hyperalgesia, and allodynia in sham-operated and nerve-transected rats suggests the possible involvement of proinfla-mmatory cytokines in the development of opioid tolerance and withdrawal-induced hyperalgesia and allodynia in these rats. Failure of these specific cytokine inhibitors to modulate the acute analgesic effect of morphine in nonoperated animals suggests that a compensatory mechanism may be in place that leads to activation of spinal proinflammatory neuroimmune responses during chronic morphine treatment. Supporting this, acute administration of morphine to normal rats did not enhance OX-42/GFAP immunostaining (Song and Zhao, 2001) (present observation), nor did it enhance the mRNA or protein levels of IL-1β, IL-6, and TNF-α in the lumbar spinal cord (data not shown).
Activation of spinal proinflammatory cytokines by chronic opioid treatment may be caused by its direct interaction with immune cells of the CNS, or it could be mediated through other mediators, such as dynorphin. Both nerve injury and chronic opioid treatment increase spinal dynorphin levels (Chao and Basbaum, 1989; Dubner and Ruda, 1992;Rattan and Tejwani, 1997). Neutralizing dynorphin activity in these conditions blocks nerve injury-induced hyperalgesia, restores spinal morphine antinociception (Nichols et al., 1997; Wegert et al., 1997), and prevents development of opioid tolerance in rats (Vanderah et al., 2000). It was shown recently that the antianalgesic action of dynorphin against morphine is mediated through IL-1β, suggesting the possible role of cytokines in antianalgesic actions of diverse agents (Laughlin et al., 2000; Rady and Fujimoto, 2001). The presence of opioid receptors and the ability of morphine to prime microglia for enhanced production of TNF-α (Chao et al., 1994; Peterson et al., 1998) suggest a possible direct interaction of morphine with glial cells. Similarly, chronic administration of morphine augments the production of proinflammatory cytokines by macrophages (Wang et al., 2002). Other possibilities for chronic morphine-induced activation of proinflammatory immune responses could be the involvement of mitogen-activated protein (MAP) kinase or protein kinase C (PKC) pathways. MAP kinase and PKC are intriguing, because they are key players in the intracellular signaling cascade leading to the development of morphine tolerance, neuropathic pain, and production of proinflammatory immune responses (Kontny et al., 1998; Ji et al., 1999;Mayer et al., 1999; Rausch et al., 2000; Fundytus et al., 2001; Ma et al., 2001; Watkins et al., 2001b).
These observations suggest that opioid therapy for chronic neuropathic pain should be used cautiously, especially in immunocompromised cases. The use of agents that selectively inhibit glial activation and/or central proinflammatory cytokines may overcome such deleterious effects and spare the beneficial effect of opioids during long-term treatment in neuropathic pain conditions. The present results indicate that reduction of the antinociceptive effect of morphine in nerve-injured rats, development of antinociceptive tolerance, and development of withdrawal-induced hyperalgesia and allodynia in sham or nerve-injured rats involves spinal neuroimmune activation. It can be anticipated that further understanding of the neuroimmune mechanisms underlying hyperalgesia, opioid tolerance, and their interactions would advance and improve clinical management of opioid therapy in the management of chronic pain syndromes.
Footnotes
This work was supported by National Institute of Drug Abuse Grant DA11276 (J.A.D.). We thank Tracy Wynkoop for editorial assistance, Dr. William F. Hickey for antibodies and glial expertise, and Amgen for the generous supply of IL-1ra and sTNFR.
Correspondence should be addressed to Joyce A. DeLeo, Department of Anesthesiology, HB 7125, Dartmouth Hitchcock Medical Center, Lebanon, NH 03756. E-mail: Joyce.A.DeLeo@Dartmouth.edu.
REFERENCES
- 1.Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 2.Arruda JL, Sweitzer S, Rutkowski MD, DeLeo JA. Intrathecal anti-IL-6 antibody and IgG attenuates peripheral nerve injury-induced mechanical allodynia in the rat: possible immune modulation in neuropathic pain. Brain Res. 2000;879:216–225. doi: 10.1016/s0006-8993(00)02807-9. [DOI] [PubMed] [Google Scholar]
- 3.Backonja MM, Miletic G, Miletic V. The effect of continuous morphine analgesia on chronic thermal hyperalgesia due to sciatic constriction injury in rats. Neurosci Lett. 1995;96:61–64. doi: 10.1016/0304-3940(95)11844-m. [DOI] [PubMed] [Google Scholar]
- 4.Bars DL, Gozariu M, Cadden SW. Animal models of nociception. Pharmacol Rev. 2001;53:597–652. [PubMed] [Google Scholar]
- 5.Beitner-Johnson D, Guitart X, Nestler EJ. Glial fibrillary acidic protein and the mesolimbic dopamine system: regulation by chronic morphine and Lewis-Fisher strain differences in the rat ventral tegmental area. J Neurochem. 1993;61:1766–1773. doi: 10.1111/j.1471-4159.1993.tb09814.x. [DOI] [PubMed] [Google Scholar]
- 6.Bian D, Ossipov MH, Ibrahim M, Raffa RB, Tallarida RJ, Malan TP, Jr, Lai J, Porreca F. Loss of antiallodynic and antinociceptive spinal/supraspinal morphine synergy in nerve-injured rats: restoration by MK-801 or dynorphine antiserum. Brain Res. 1999;831:55–63. doi: 10.1016/s0006-8993(99)01393-1. [DOI] [PubMed] [Google Scholar]
- 7.Catheline G, Kayser V, Guilbaud G. Further evidence for a peripheral component in the enhanced antinociceptive effect of systemic morphine in mononeuropathic rats: involvement of κ-, but not δ-opioid receptors. Eur J Pharmacol. 1996;315:135–143. doi: 10.1016/s0014-2999(96)00629-2. [DOI] [PubMed] [Google Scholar]
- 8.Chao CC, Gekker G, Sheng WS, Hu S, Monica T, Peterson PK. Priming effect of morphine on the production of tumor necrosis factor-α by microglia: implications in respiratory burst activity and human immunodeficiency virus-1 expression. J Pharmacol Exp Ther. 1994;269:198–203. [PubMed] [Google Scholar]
- 9.Chao HJ, Basbaum AI. Ultrastructural analysis of dynorphin B-immunoreactive cells and terminals in the superficial dorsal horn of the deafferented spinal cord of the rat. J Comp Neurol. 1989;281:193–205. doi: 10.1002/cne.902810204. [DOI] [PubMed] [Google Scholar]
- 10.Cherny NI, Thaler HT, Friedlander-Klar H, Lapin J, Foley KM, Houde R, Portenoy RK. Opioid responsiveness of cancer pain syndrome caused by neuropathic or nociceptive mechanisms: a combined analysis of controlled, single-dose studies. Neurology. 1994;44:857–861. doi: 10.1212/wnl.44.5.857. [DOI] [PubMed] [Google Scholar]
- 11.Christensen D, Kayser V. The development of pain-related behavior and opioid tolerance after neuropathy-inducing surgery and sham surgery. Pain. 2000;88:231–238. doi: 10.1016/S0304-3959(00)00334-1. [DOI] [PubMed] [Google Scholar]
- 12.Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- 13.Covino BG, Dubner R, Gybels J, Losterlitz HW, Liebeskind JC, Sternbach RA, Vylicky L, Yamamura H, Zimmerman M. Ethical standards for investigations of experimental pain in animals. Pain. 1980;9:141–143. doi: 10.1016/0304-3959(80)90002-0. [DOI] [PubMed] [Google Scholar]
- 14.DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 15.Devulder J, Bohyn P, Castille F, De Laat M, Rolly G. A case of uncommon withdrawal symptoms after a short period of spinal morphine administration. Pain. 1996;64:589–591. doi: 10.1016/0304-3959(95)00187-5. [DOI] [PubMed] [Google Scholar]
- 16.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 17.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 18.Fundytus ME, Yashpal K, Chabot JG, Osborne MG, Lefebvre CD, Dray A, Henry JL, Coderre TJ. Knockdown of spinal metabotropic glutamate receptor 1 (mGluR(1)) alleviates pain and restores opioid efficacy after nerve injury in rats. Br J Pharmacol. 2001;132:354–367. doi: 10.1038/sj.bjp.0703810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gul H, Yildiz O, Dogrul A, Yesilyurt O, Isimer A. The interaction between IL-1β and morphine: possible mechanism of the deficiency of morphine-induced analgesia in diabetic mice. Pain. 2000;89:39–45. doi: 10.1016/S0304-3959(00)00343-2. [DOI] [PubMed] [Google Scholar]
- 20.Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- 21.Kontny E, Ziolkowska M, Ryzewska A, Maslinski W. Protein kinase C dependent pathway is critical for the production of proinflammatory cytokines (TNF-α, IL-1β, IL-6). Cytokines. 1998;11:839–848. doi: 10.1006/cyto.1998.0496. [DOI] [PubMed] [Google Scholar]
- 22.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 23.Laughlin TM, Bethea JR, Yezierski RP, Wilcox GL. Cytokine involvement in dynorphin-induced allodynia. Pain. 2000;84:159–167. doi: 10.1016/s0304-3959(99)00195-5. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Angst MS, Clark JD. A murine model of opioid-induced hyperalgesia. Brain Res Mol Brain Res. 2001a;86:56–62. doi: 10.1016/s0169-328x(00)00260-6. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Angst MS, Clark JD. Opioid-induced hyperalgesia and incisional pain. Anesth Analg. 2001b;93:204–209. doi: 10.1097/00000539-200107000-00040. [DOI] [PubMed] [Google Scholar]
- 26.Ma W, Zheng WH, Powell K, Jhamandas K, Quirion R. Chronic morphine exposure increases the phosphorylation of MAP kinases and the transcription factor CREB in dorsal root ganglion neurons: an in vitro and in vivo study. Eur J Neurosci. 2001;14:1091–1104. doi: 10.1046/j.0953-816x.2001.01731.x. [DOI] [PubMed] [Google Scholar]
- 27.Mao J, Price DD, Mayer DJ. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995;61:353–364. doi: 10.1016/0304-3959(95)00022-K. [DOI] [PubMed] [Google Scholar]
- 28.Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci USA. 1999;96:7731–7736. doi: 10.1073/pnas.96.14.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCormack K, Prather P, Chapleo C. Some new insight into the effects of opioids in phasic and tonic nociceptive test. Pain. 1998;78:79–98. doi: 10.1016/S0304-3959(98)00146-8. [DOI] [PubMed] [Google Scholar]
- 30.Milligan ED, O'Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema R, Holguin A, Martin D, Maier SF, Watkins LR. Intrathecal HIV-1 envelope glycoprotein gp120 enhanced pain states mediated by spinal cord proinflammatory cytokines. J Neurosci. 2001;21:2808–2819. doi: 10.1523/JNEUROSCI.21-08-02808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nichols ML, Lopez Y, Ossipov MH, Bian D, Porreca F. Enhancement of the anti allodynic and anti nociceptive efficacy of spinal morphine by antisera to dynorphin A (1–13) or MK801 in a nerve-ligation model peripheral neuropathy. Pain. 1997;69:317–322. doi: 10.1016/S0304-3959(96)03282-4. [DOI] [PubMed] [Google Scholar]
- 32.Nozaki-Taguchi N, Yaksh TL. Spinal and peripheral mu opioids and the development of secondary tactile allodynia after thermal injury. Anesth Analg. 2002;94:968–974. doi: 10.1097/00000539-200204000-00036. [DOI] [PubMed] [Google Scholar]
- 33.Oka T, Hori T. Brain cytokines and pain. In: Watkins LR, Maier SF, editors. Cytokines and pain. Birkhauser Verlag; Boston: 1999. pp. 183–203. [Google Scholar]
- 34.Peterson PK, Molitor TW, Chao CC. The opioid-cytokine connection. J Neuroimmunol. 1998;83:63–69. doi: 10.1016/s0165-5728(97)00222-1. [DOI] [PubMed] [Google Scholar]
- 35.Przewlocki R, Przewlocka B. Opioids in chronic pain. Eur J Pharmacol. 2001;429:79–91. doi: 10.1016/s0014-2999(01)01308-5. [DOI] [PubMed] [Google Scholar]
- 36.Rady JJ, Fujimoto JM. Confluence of antianalgesic action of diverse agents through brain interleukin1β in mice. J Pharmacol Exp Ther. 2001;299:659–665. [PubMed] [Google Scholar]
- 37.Raffa RB, Mathiasen JR, Kimball ES, Vaught JL. The combined immunological and antinociceptive defects of beige-J mice: the possible existence of a “mu-repression.”. Life Sci. 1993;52:1–8. doi: 10.1016/0024-3205(93)90281-7. [DOI] [PubMed] [Google Scholar]
- 38.Rattan AK, Tejwani GA. Effect of chronic treatment with morphine, midazolam and both together on dynorphin (1–13) level in the rat. Brain Res. 1997;754:239–244. doi: 10.1016/s0006-8993(97)00084-x. [DOI] [PubMed] [Google Scholar]
- 39.Rausch O, Newton RA, Bingham S, Macdonald R, Case CP, Sanger GJ, Lawson SN, Reith AD. Nerve injury-associated kinase: a sterile 20-like protein kinase up-regulated in dorsal root ganglia in a rat model of neuropathic pain. Neuroscience. 2000;101:767–777. doi: 10.1016/s0306-4522(00)00392-4. [DOI] [PubMed] [Google Scholar]
- 40.Song P, Zhao Z-Q. The involvement of glial cells in the development of morphine tolerance. Neurosci Res. 2001;39:281–286. doi: 10.1016/s0168-0102(00)00226-1. [DOI] [PubMed] [Google Scholar]
- 41.Stein C, Gramsch C, Herz A. Intrinsic mechanisms of antinociception in inflammation: local opioid receptors and β-endorphin. J Neurosci. 1990;10:1292–1298. doi: 10.1523/JNEUROSCI.10-04-01292.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sweitzer SM, Arruda JL, DeLeo JA. The cytokine challenge: methods for the detection of central cytokines in rodent models of persistent pain. In: Kruger L, editor. Methods in pain research. CRC; New York: 2001a. pp. 109–132. [Google Scholar]
- 43.Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001b;103:529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- 44.Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dorgul A, Zhong C, Zhang E, Malan PT, Jr, Ossipov MH, Lai J, Porreca F. Dynorphin promotes abnormal pain and spinal opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–7079. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Charboneau R, Balasubramanian S, Barke RA, Loh HH, Roy S. The immunosuppressive effects of morphine treatment are partially dependent on corticosterone and mediated by the μ-opioid receptor. J Leukoc Biol. 2002;71:782–790. [PubMed] [Google Scholar]
- 46.Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001a;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 47.Watkins LR, Milligan ED, Maier SF. Spinal cord glia: new player in pain. Pain. 2001b;93:201–205. doi: 10.1016/S0304-3959(01)00359-1. [DOI] [PubMed] [Google Scholar]
- 48.Wegert S, Ossipov MH, Nicholas ML, Bian D, Vanderah TW, Malan TP, Jr, Porreca F. Differential activities of intrathecal MK801 or morphine to alter responses to thermal and mechanical stimuli in normal or nerve-injured rats. Pain. 1997;71:57–64. doi: 10.1016/s0304-3959(97)03337-x. [DOI] [PubMed] [Google Scholar]