

Abstract
We demonstrated previously that amyloid precursor protein (APP) is anterogradely transported from the entorhinal cortex (ERC) to the dentate gyrus via axons of the perforant pathway. In the terminal fields of these inputs, APP undergoes proteolysis to generate C-terminal fragments containing the entire amyloid β peptide (Aβ) domain. The present study was designed to test the hypothesis that APP derived from ERC neurons is the source of the Aβ peptide deposited in the hippocampal dentate gyrus in Alzheimer's disease (AD) and in transgenic mice with Aβ amyloidosis. We used mice harboring two familial AD-linked genes (human APP Swedish and presenilin1-ΔE9), in which levels of Aβ (especially Aβ42) are elevated, leading to the formation of amyloid plaques, and lesioned the ERC to interrupt the transport of APP from ERC to hippocampus. Our results show that, on the side of ERC lesion, numbers of APP-immunoreactive dystrophic neurites and Aβ burden were significantly reduced by ∼40 and 45%, respectively, in the dentate gyrus compared with the contralateral side. Reductions in APP and Aβ were more substantial in the molecular layer of the dentate, i.e., a region that contains the ERC terminals, and were associated with a parallel decrease in total APP and Aβ measured by Western blot and ProteinChip immunoassays. Silver and thioflavine staining confirmed the reduction of amyloid plaques on the side of deafferentation. These results are consistent with the hypothesis that ERC may be the primary source of amyloidogenic Aβ in the dentate gyrus, and they suggest an important role of corticocortical and corticolimbic forward connections in determining patterns of amyloid deposition in AD.
Keywords: Alzheimer's disease, Aβ, APP, axonal transport, entorhinal cortex, perforant pathway, senile plaques
Senile plaques are a pathological hallmark of Alzheimer's disease (AD), and their principal component is amyloid-β (Aβ), a unique 4 kDa peptide derived by proteolysis of the amyloid precursor protein (APP). The origin of Aβ has been the subject of some debate, with some investigators suggesting sources in the periphery or serum, whereas others favor a neural source (Buxbaum et al., 1998). Synthesized in the cell bodies of neurons, APP is anterogradely transported within axons to nerve terminals in both the PNS and CNS (Koo et al., 1990; Morin et al., 1993; Sisodia et al., 1993; Buxbaum et al., 1998), in which it appears to be processed to amyloidogenic fragments in the endocytic recycling pathway (Nordstedt et al., 1993; Koo and Squazzo, 1994; Ikin et al., 1996;Marquez-Sterling et al., 1997). The localization of APP processing in distal axons/synapses suggests that terminals are the source of Aβ present in senile plaques. This concept is supported by descriptive studies of AD, in which there appears to be a predilection of plaques for the terminal fields of corticocortical pathways (Rogers and Morrison, 1985; Braak and Braak, 1990; Arnold et al., 1991; Beach and McGeer, 1992; Hof and Morrison, 1994).
The allocortical hippocampus is especially burdened with lesions, including senile plaques, in AD (Braak and Braak, 1990; Hof and Morrison, 1994; Sheng et al., 1995; Su and Ni, 1998). A major corticocortical input to the mammalian hippocampus is provided via the perforant pathway, which contains axons originating in neurons of the entorhinal cortex (ERC) and terminating in the outer two-thirds of the molecular layer of the dentate gyrus (Scheff, 1989). We established previously that, in the perforant pathway, APP is transported as part of the fast anterograde component to terminals in the dentate gyrus; at these sites, APP undergoes proteolysis to generate C-terminal fragments containing the entire Aβ domain, which may be the penultimate precursors of synaptic Aβ (Buxbaum et al., 1998).
Because the projections of ERC to the dentate gyrus are not directly reciprocated, the ERC–dentate projection is an ideal site to study the hypothesis of propagation of amyloid pathology via forward corticocortical connections in AD (Hof and Morrison, 1994). If ERC is a major source of APP and secretases interacting to form Aβ in the dentate gyrus, the removal of ERC and its connections with the hippocampus would be predicted to reduce APP-immunoreactive dystrophic neurites [APP(+) neurites] and amyloid burden in the dentate gyrus. This hypothesis can now be tested experimentally in transgenic (Tg) mice, which faithfully reproduce the amyloid pathology encountered in AD. These animals are engineered to overexpress the human APP Swedish (APPswe) mutation or combinations of APPswe with other familial AD-linked mutations (Games et al., 1995; Borchelt et al., 1997; Price and Sisodia, 1998; Price et al., 1998; Rockenstein et al., 2001).
In the present study, we use ERC lesions to remove the neocortical inputs to hippocampus and demonstrate significant decrements in amyloid pathology in the dentate gyrus of APPswe/presenilin 1-ΔE9 [APPswe/PS1-ΔE9] mice with established amyloid deposits. Our results are consistent with the hypothesis that ERC afferents are the principal source of APP processed to generate Aβ deposits in the dentate gyrus and that patterns of termination of corticocortical connections may determine the distribution of senile plaques in cortex in AD.
MATERIALS AND METHODS
Animals and experimental design. Eighteen APPswe/PS1-ΔE9 Tg mice (aged 9.1 ± 0.2 months), generated as described previously (Borchelt et al., 1997), were randomly assigned to three groups: one group (lesion) received ERC lesions (n = 11), a second group (sham) received craniotomy with pial disruption (n = 4), and a third group was left intact (n = 3). All experiments were performed according to protocols approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions.
Entorhinal lesions cause extensive reorganization of the neuropil in the molecular layer of the dentate gyrus, including proliferation of terminals from commissural projections of the contralateral hippocampus, crossed projections of the contralateral entorhinal cortex, and projections arising in the medial septum. These terminals fill in the void caused by the elimination of the ipsilateral entorhinal terminals in a process that is termed reactive synaptogenesis and proceeds along a well characterized time course (Deller and Frotscher, 1997). ERC terminals are probably completely eliminated by 6 d after lesion (Steward et al., 1990). Sprouting of commissural projections resulting in functional synapses starts at 9 d after lesion and is fully developed by 15 d (West et al., 1975). Crossed entorhinal projections sprout after the first week after lesion, and an intense proliferation of new synapses is seen in 8–12 d (Steward and Loesche, 1977). Thus, by 2 weeks after lesion, most of the reactive synaptogenesis seems to have taken place. All of our experimental subjects with entorhinal lesions or sham surgeries were allowed to survive 1 month after surgery. In selecting this survival time, we took into account the time it takes for the reactive synaptogenesis to conclude and allowed an additional 2 weeks for the reorganized afferent connectivity to be established.
Some genetic alterations that cause AD-like pathologies in Tg mice, including PS1 mutations, appear to result in changes in the synaptic reorganization of the dentate gyrus after ERC lesions (White et al., 2001; Kadish et al., 2002). This effect raises concerns that wild-type mice may not be appropriate controls in the present study, because they have a different baseline response to ERC lesions. To address this problem, we compared measures in the dentate gyrus ipsilateral to the lesion with those contralateral to ERC lesion, such that each animal served as its own control.
ERC lesions and preparation of tissues. Mice were anesthetized and placed in a Kopf small animal stereotactic frame (David Kopf Instruments, Tujunga, CA) equipped with a modified mouse nasal device that allows for the use of gas anesthesia (oxygen/nitrous oxide/enflurane, 33:66:1) and extra-auricular side bars that prevent damage to the airways. The posterior ERC was aspirated after direct visualization with a dorsal craniotomy at the cerebello-hemispheric border (immediately rostral to the lambda suture) using the anterior aspect of the petrosal crest as a bony landmark for the caudal end of the hemisphere. Sham operations involved a craniotomy and an incision of dura pia immediately rostral to the lambda suture. All animals were killed 1 month after surgery by intracardial perfusion–fixation (histochemistry and immunohistochemistry) or decapitation (Western blotting and ProteinChip analysis).
Perfusion–fixation was performed with a brief PBS flush, followed by 4% freshly depolymerized paraformaldehyde. Brains were postfixed in the same fixative overnight, embedded in paraffin, and sectioned (10 μm) in the coronal plane. For protein/peptide studies, brains were quickly removed from calvaria and sliced with a 1.5 mm brain slicer at the coronal plane; hippocampi were dissected on wet ice under an operating microscope.
Verification of ERC lesions. Sections through the ERC from paraffin-embedded tissues (see below, Immunohistochemistry for amyloid proteins and synaptophysin) were stained with cresyl violet to assess the placement and extent of the ERC lesion. Only cases with successful ablations of the posterior ERC were included in this study (Fig.1A,B). The deafferentation of the dentate gyrus was confirmed with AChE histochemistry using a silver intensification of the Tsuji reaction in a preliminary group of animals that were perfused as the present series and then processed for microtome sectioning. The signature pattern of disconnection from the ERC is a drastic change from an even, low-intensity AChE fiber staining in the molecular layer in normal mice (Fig. 1C) to the formation of an intense band of staining in the outer molecular layer in animals with ERC or perforant path lesions (Scheff, 1989) (Fig. 1D).
Fig. 1.

Location and efficacy of ERC lesions implemented in this study. A, B, Photomicrographs of cresyl violet-stained sections through the ERC of Tg mice that underwent sham surgeries (A) or ERC lesions (B). Note the normal anatomy of ERC inA and the extent and specificity of ERC ablation (arrow) in a representative lesion case (B). C, D, AChE-stained sections through the dentate gyrus of Tg mice with sham surgeries (C) or ERC lesions (D). Note the typical change in the AChE staining pattern after ERC lesions, i.e., the diminution of the intense AChE (+) band in the supragranular region and the increased density (sprouting) of AChE fibers toward the outer half of the molecular cell layer as a result of synaptogenesis in septal cholinergic afferents.G, Granule cell layer; M, molecular cell layer. Scale bars: A, B, 400 μm;C, D, 40 μm.
Silver and thioflavine histochemistry. Silver staining was performed on paraffin sections through the hippocampus according to the method of Hirano (Nakano and Hirano, 1987). Thioflavine staining was performed on adjacent sections (Guntern et al., 1992).
Immunohistochemistry for amyloid proteins and synaptophysin.Mice were perfused with PBS, followed by 4% paraformaldehyde. Brains were postfixed in 4% paraformaldehyde overnight, embedded in paraffin, and sectioned (10 μm) in the coronal plane. Immunohistochemical staining was performed according to methods described in detail previously (Griffin et al., 1993). Briefly, paraffin sections were deparaffinized in xylene, rehydrated in serial concentrations of ethanol solutions, and then permeabilized in 0.5% Triton X-100 for 10 min, followed by 0.2 N HCl for 20 min. Endogenous peroxidase was blocked with 3% H2O2 in methanol for 30 min. Primary antibodies [mouse anti-human Aβ4G8 (Signet, Dedham, MA), mouse anti-human APP695 (Zymed, South San Francisco, CA), and mouse anti-human synaptophysin (Dako, Carpinteria, CA)] were diluted in Tris-buffered saline (TBS) containing 2% normal goat serum (Aβ4G8, 1:300; APP695, 1:100; synaptophysin, 1:20) and were used in overnight incubations at room temperature (RT). Linking antibody (goat anti-mouse IgG; ICN Biomedicals, Costa Mesa, CA) was diluted 1:50 in TBS with 2% normal goat serum and used in 30 min incubations at RT. After a wash in PBS, sections were incubated with mouse peroxidase anti-peroxidase (Sternberger Monoclonals, Lutherville, MD) for 30 min at RT. Immunoreactive structures were developed using a standard DAB chromogen reaction. Double-label immunohistochemistry for APP (using the mouse anti-human APP695 antibody diluted 1:100) and Aβ (using the Aβ4G8 antibody diluted 1:300) was performed on deparaffinized sections with the aid of commercially available kits (K1359; Dako) essentially as described previously (Sheng et al., 2001).
Quantitative assessment of APP(+) dystrophic neurites, Aβ burden, and synapse density in dentate gyrus. To assess the burden of dentate gyrus with APP(+) neurites and amyloid deposits, sections at coronal planes corresponding to ∼2 mm posterior to bregma were used for image analysis. APP(+) neurites and Aβ deposits in plaques in the dentate gyrus were captured with a CCD video camera attached to a Dell (Round Rock, TX) computer and measured by NIH Image software (version 1.65). The burden of dentate gyrus with APP(+) neurites and Aβ deposits was expressed per animal and per brain side (ipsilateral and contralateral to lesion) as proportional occupancy of the dentate gyrus by APP- or Aβ-immunoreactive structures in plaques (total cross-sectional dentate area covered by immunoreactivity/entire cross-sectional dentate gyrus area, both expressed in square micrometers). APP and Aβ plaque burden were assessed separately for the entire dentate gyrus and for the molecular and polymorph layer/CA3. This distinction allowed for separate observations on the following: impact of ERC lesions on the dentate gyrus as a whole; direct influences on the primary field of denervation (i.e., on the molecular layer, which contains the terminals from the lesioned ERC); and the impact of denervation on polymorph layer/CA3 (i.e., an area that contains the axons–collaterals–terminals of denervated neurons and is thereby situated one synapse away from the degenerated perforant pathway terminals). To standardize among different animals with varying baseline intensities of APP and Aβ immunoreactivities in the various anteroposterior planes, burden of APP and Aβ on the side of the lesion were expressed as percentiles of APP and Aβ burden in the contralateral dentate gyrus (total dentate and molecular and polymorph layers) on the same section. Group averages were generated from the lesion, sham, and intact groups, and they were studied statistically with ANOVA, followed by Fisher's ad hoc test to detect specific differences between groups. Synaptic density in the molecular and polymorph layer of the dentate gyrus was also measured, in lesioned animals only, on the side ipsilateral and the side contralateral to lesion. Density was expressed, as in the case of amyloid markers, as the proportional occupancy of the molecular and polymorph layers by normal-appearing synaptophysin (+) puncta. Densities on the lesioned side, as well as the side contralateral to lesion, from different animals were combined to generate averages, which were then compared between the two groups, using a Student'st test.
Western blotting. Dentate gyrus ipsilateral and contralateral to lesion were dissected under an operating microscope from coronal 1-mm-thick slices of fresh brain stained with nonalcoholic hematoxylin. Effort was made to avoid the Ammon's horn, although some medial CA3 was included. Tissue samples were homogenized in T-PER (Pierce, Rockford, IL) containing a protease inhibitor cocktail (Boehringer Mannheim, Mannheim, Germany), and total protein levels were measured with the Micro BCA protein assay reagent kit (Pierce). Twenty micrograms of protein were loaded onto 4–12% NuPAGE precast gels (Invitrogen, Carlsbad, CA) and electrophoresed along with molecular weight markers (Amersham Biosciences, Arlington Heights, IL). Protein was transferred to the nitrocellulose membrane BA-S 85 (Schleicher & Schuell, Keene, NH). Blot was blocked in 0.05m TBS, pH 7.4, containing 5% nonfat powdered milk and then incubated in antibodies APP695 or Aβ4G8, both diluted 1:500 in TBS containing 5% nonfat powdered milk, overnight at 4°C. After washing in TBS, blots were incubated in HRP-linked donkey anti-mouse IgG (Amersham Biosciences), diluted 1:2000 in TBS containing 5% nonfat powdered milk for 1 hr at room temperature. Blots were then treated with the SuperSignal Chemiluminescent Substrate (Pierce) and exposed to Kodak-XAR film (Eastman Kodak, Rochester, NY). Film was digitized and analyzed by NIH Image software (version 1.65) for amount of electrophoresed APP and Aβ per sample. Differences in levels of APP and Aβ between the dentate gyrus ipsilateral and contralateral to lesion were assessed with a Student's t test.
Aβ ProteinChip assay. These assays used the ProteinChip β-Amyloid Multipeptide kit (Ciphergen Biosystems, Fremont, CA), which included the preactivated ProteinChip array PS20. Monoclonal antibody Aβ6E10 or control bovine IgG was coupled to PS20 chip by adding 2 μl of a stock solution (0.5 mg/ml in PBS) to each spot and incubating overnight at 4°C in a humid chamber. Unreacted sites were blocked with 0.5 m ethanolamine, pH 8.0, for 30 min and then washed for 30 min with PBS containing 0.5% Triton X-100 (T-PBS) and then for 5 min with PBS. Protein prepared from tissue samples (20 μg) or an Aβ peptide mixture (600 fmol) were spotted on the array with the aid of a bioprocessor. Chip was incubated at room temperature for 2 hr. Subsequently, chip was washed with T-PBS and then with PBS, rinsed with water, and allowed to dry. A saturated solution of α-cyano-4-hydroxy-cinnamic acid (CHCA) was further diluted 1:50 in 50% acetonitrile and 0.5% trifluoroacetic acid; 0.5 μl of diluted CHCA were added to the array spots, and chips were dried at room temperature. Chips were read on surface-enhanced laser desorption ionization-time of flight mass spectrometer (SELDI) (Ciphergen Biosystems).
RESULTS
ERC lesions decrease amyloid plaque burden in the dentate gyrus, especially the molecular layer
All APPswe/PS1-ΔE9 Tg mice showed numerous amyloid plaques in hippocampus and neocortex, with a region-specific distribution. In the hippocampus, there were a large number of plaques in the dentate gyrus and Ammons's horn, including the molecular and polymorph dentate layers (Figs.2-4). On double-labeled immunohistochemical preparations, all Aβ deposits were intimately associated with dystrophic neurites enriched in APP (Fig. 3E,F).
Fig. 2.

Photomicrographs of ERC lesion effects on Aβ deposits in the dentate gyrus. A, B, Sections through the hippocampus of a Tg mouse with sham surgery (A) and a Tg mouse with ERC ablation (B) stained with Aβ4G8. A1,A2, B1, and B2 represent magnifications of the demarcated areas in A andB. There is strong Aβ immunoreactivity in the form of plaques in dentate gyrus and the CA3 field. Note the decreased Aβ load in the dentate gyrus (especially in the molecular layer of dentate gyrus) on the side ipsilateral to lesion (B1) compared with the side contralateral to lesion (B2) and sham (A1, A2). MDG, Molecular layer of dentate gyrus; PoDG, polymorph layer of dentate gyrus. Scale bars: A, B, 800 μm;A1, A2, B1,B2, 100 μm.
Fig. 3.

Effects of ERC lesions on APP(+) dystrophic neurites. Sections through the dentate gyrus of sham (A,B) and lesioned (C, D) Tg mice contralateral and ipsilateral (B, D) to sham or ERC lesion were stained with APP695. APP(+) dystrophic neurites in plaques are significantly decreased on the side of ERC lesions, especially in the molecular layer of the dentate gyrus (D). E and F show that APP immunoreactivity (red) colocalizes with Aβ deposits (yellow) in slightly different compartments of amyloid plaques in double-labeled immunohistochemical preparations. MDG, Molecular layer of dentate gyrus;PoDG, polymorph layer of dentate gyrus. Scale bars:A–D, 100 μm; E, F, 50 μm.
Fig. 4.

Bar graphs representing Aβ (A,C) and APP(+) neurite (B,D) load in the dentate gyrus of intact, sham, and ERC-lesioned Tg mice. A and B represent values from total dentate, and C and Drepresent values from the polymorph and molecular layers separately in ERC-lesioned Tg mice. Proportional occupancy (burden) of dentate by Aβ- and APP-immunoreactive structures in plaques on the lesioned (sham, ERC lesions) or the right-sided dentate gyrus (intact) was expressed as a percentage of Aβ and APP burden on the contralateral side (the latter set as 100%). *p < 0.05; **p < 0.0001; by ANOVA. Significance is caused by a difference between sham and lesion or intact and lesion but not sham and intact.
ERC lesions caused a significant decrease in numbers of Aβ deposits and APP(+) dystrophic neurites (Figs. 2, 3). In intact mutant mice, Aβ burden and APP(+) neurites were the same in the dentate gyrus of the two hemispheres, as shown by an identical areal occupancy of Aβ and APP on the two sides; in sham-operated mice, Aβ and APP burden were also comparable in the two dentate gyri (Figs.2A, 3A,B). In contrast, Aβ and APP burden in the dentate gyrus of ERC-lesioned mice were significantly decreased on the lesioned side (Figs.2B, 3C,D); the areal occupancy of Aβ was 45 ± 7%, and the occupancy of APP(+) neurites was 39 ± 12% of the contralateral dentate gyrus (Fig.4A,B). ERC lesions were also associated with a significant decrease in silver- and thioflavine-S-stained plaques in the dentate gyrus ipsilateral to ERC lesions (Fig. 5).
Fig. 5.

Effects of ERC lesions on amyloid plaques visualized with traditional histochemical methods such as silver (A, B) and thioflavine (C–F). Lesioned side is on theright. There is a trend for a reductions of both silver- and thioflavine-stained plaques in the molecular layer of the dentate gyrus. There are no obvious differences in the polymorph cell layer.MDG, Molecular layer of dentate gyrus;PoDG, polymorph layer of dentate gyrus. Scale bars:A–D, 100 μm; E, F, 50 μm.
When APP(+) neurites and Aβ burden were assessed separately in the molecular and polymorph layers, APP(+) neurites and Aβ deposits appeared to be reduced in both layers; the Aβ burden was significantly reduced only in the molecular layer (40 ± 4% of the contralateral side), whereas APP(+) neurite reduction was more significant in the molecular than in the polymorph layer (23 ± 4 and 57 ± 8%, respectively, of the contralateral side) (Fig.4C,D).
ERC lesions decrease levels of APP and Aβ in the dentate gyrus
On Western blots of the dentate gyrus, APP was detected as a ∼105 kDa band, and Aβ was detected antibody as a ∼4 kDa band (Fig. 6A). Quantitative analysis of the digitized film images showed a 74% reduction in APP and an 84% reduction in total Aβ after ERC lesions (Fig.6B) (both p < 0.05). These decrements of the two markers in the deafferented dentate gyrus were not attributable to decreased numbers of synapses, because synaptic density was similar at the primary site of denervation (the molecular layer of the dentate gyrus) ipsilateral and contralateral to lesion by counts of densities of synaptophysin (+) puncta (Fig.6C). This observation is consistent with the idea that the process of reactive synaptogenesis had been completed in the dentate gyrus of our experimental subjects.
Fig. 6.
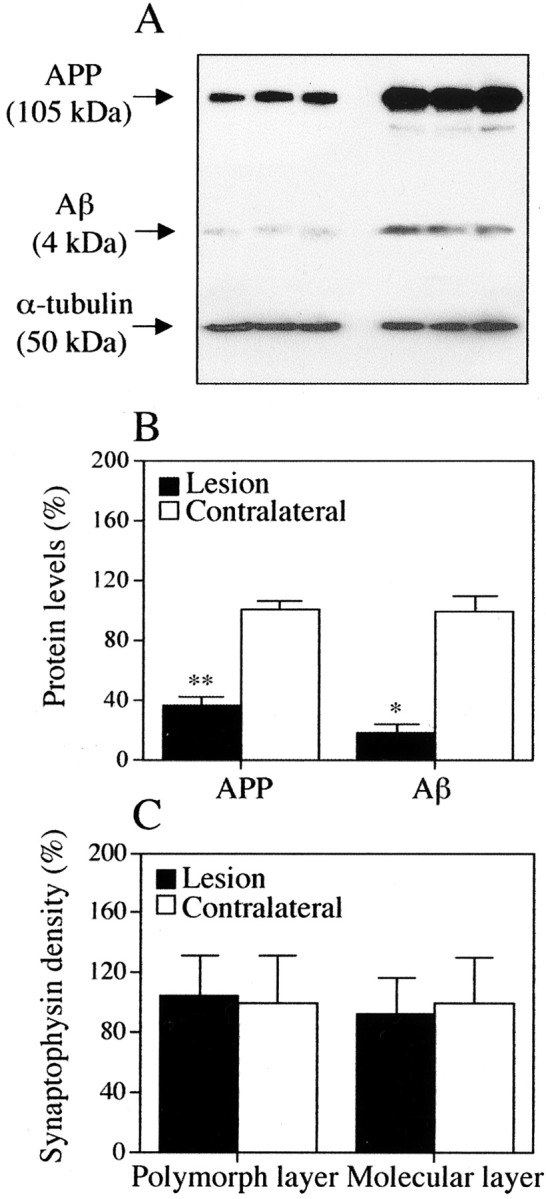
APP and Aβ protein levels in dentate gyrus after ERC lesions. A, B, Western blots showing APP (top) and Aβ (middle) levels in homogenates of dentate gyrus ipsilateral (left lanes) and contralateral (right lanes) to ERC lesions. α-Tubulin (bottom) was run as loading control.B, Bar graphs of APP and total Aβ protein levels in ipsilateral and contralateral to ERC lesion by Western blot analysis. *p < 0.05; **p < 0.005 (comparison between lesion and contralateral side). C, Measurements of normal synapse density in the molecular and polymorph layers of the dentate gyrus 1 month after ERC lesions show no difference between lesioned and contralateral side. This is evidence that APP and Aβ proteins studied in A andB originated approximately in the same number of synapses in the two sides.
On ProteinChip assays, Aβ1-40 (mass value of 4329) and Aβ1-42 (mass value of 4513) were detected in supernatants of dentate gyrus ipsilateral (Fig. 7A) and contralateral (Fig. 7B) to the lesion. Based on SELDI readings, there was a 30% reduction in Aβ1-40 and a 40% reduction in Aβ1-42 in the dentate gyrus after ERC lesions. Aβ1-42 reduction was statistically significant (p < 0.05) (Fig.7C).
Fig. 7.

ProteinChip assays for Aβ1-40 and Aβ1-42 in the dentate gyrus ipsilateral (A) and contralateral (B) to an ERC lesion. Aβ1-40 peak corresponds to a mass value of 4329; Aβ1-42 peak has a mass value of 4513. C, Bar graph of Aβ1-40 and Aβ1-42 protein levels ipsilateral and contralateral to ERC lesion by ProteinChip analysis. *p < 0.005 (comparison between lesion and contralateral side).
DISCUSSION
Our experiments demonstrate that the interruption of a model corticocortical pathway, i.e., the projection from ERC to the dentate gyrus, results in amelioration of amyloid plaque burden in animals genetically predisposed to AD-like amyloidosis. Our observations are consistent with the idea that ERC is the major source of APP in the molecular layer of the dentate gyrus (Buxbaum et al., 1998) and support the notion that plaques have a preference for the terminal fields of corticocortical pathways (Rogers and Morrison, 1985; Braak and Braak, 1990; Arnold et al., 1991; Beach and McGeer, 1992; Hof and Morrison, 1994; Koliatsos, 1996).
Specifically, the association between APP(+) neurites and Aβ deposits in plaques in the dentate gyrus of APPswe/PS1-ΔE9 mice and similar reductions in both APP and Aβ immunoreactivities after ERC lesions support our previous suggestion that Aβ in the dentate is derived from APP synthesized in ERC neurons and processed in the terminal fields to generate amyloidogenic C-terminal peptides (Buxbaum et al., 1998). Aβ is generated from APP through cleavage by a β- or γ-secretase activity (Price and Sisodia, 1998; Price et al., 1998;Cai et al., 2001). The β-site APP cleavage enzyme 1 is the neuronal β-secretase that is more active in the presence of the APPswe mutation, whereas PS1 mutations selectively activate γ-secretase to generate greater amounts of Aβ42 (Octave et al., 2000; Vassar and Citron, 2000; Cai et al., 2001; Selkoe, 2001). The APPswe/PS1 Tg mice used in the present study have higher levels of all Aβ species (attributable to APPswe) and a preferential increase in Aβ42 (attributable to PS1-ΔE9) in the brain (Borchelt et al., 1996). ERC lesions cause a significant decrease in both total Aβ and Aβ42 levels in the dentate gyrus of these mice. Although we cannot rule out that ERC lesions may have an effect on the activity of secretases, the parallel reduction of APP after these lesions suggests strongly that a primary mechanism for the amelioration of Aβ synthesis/deposition is the decreased availability of APP (perhaps along with transported secretases) to terminal fields.
An important question is whether the apparent reduction in Aβ in the dentate gyrus after ERC lesions is the direct consequence of decreased APP transported to the dentate or whether there is also a contribution by more complex processes. Such processes may be related to reactive synaptogenesis (West et al., 1975; Steward and Loesche, 1977; Scheff, 1989; Steward et al., 1990; Deller and Frotscher, 1997) or to the reduced synaptic drive in the perforant pathway and a hypothetical reduction in APP/Aβ synthesized locally in the hippocampus. If ERC lesions exert their effects primarily by depriving the dentate gyrus of a major source of APP, these effects should be either restricted to the primary field of denervation, i.e., the molecular layer, or maximal in the molecular layer, despite a wider distribution into the hippocampal trisynaptic circuit. This spread of the effect might include the polymorph layer, which is one synapse away from the deafferentation site. Our data show that ERC lesions cause a much greater decrease in APP(+) neurites and amyloid deposits in the molecular than in the polymorph layer and that the reduction in Aβ deposits is only significant in the molecular layer. This pattern supports the idea that a reduction in APP present in ERC terminals plays a major role in the decrease of Aβ in the molecular layer. This conclusion is supported by our Western blot findings showing that levels of APP continued to be low in the dentate gyrus ipsilateral to ERC lesion 1 month after surgery, despite the fact that total synaptic density [as assessed by numbers of synaptophysin (+) puncta] had returned to normal. By 1 month after lesion, the process of reactive synaptogenesis is complete, and a continued reduction in immunoreactive APP indicates a selective role of associational, as distinguished from commissural and subcortical, connections as a source of APP and Aβ in corticocortical pathways.
In conclusion, this investigation demonstrates that the removal of a corticocortical associational input in a model of Aβ amyloidosis reduces total APP and Aβ and amyloid plaque burden in terminal fields. Our findings also suggest that associational inputs may play particularly significant roles in determining patterns of Aβ deposition in the cortex and limbic system of individuals with AD.
Footnotes
This work was supported by National Institutes of Health Grant AGO5146 and National Institute on Aging Grants T32 and NS07435. Dr. David Borchelt provided APPswe/PS1-ΔE9 Tg mouse founders, and Susan Bora offered expert technical assistance.
Correspondence should be addressed to Dr. V. E. Koliatsos, The Johns Hopkins University School of Medicine, Neuropathology Division, Ross Building, Room 558, 720 Rutland Avenue, Baltimore, MD 21205. E-mail: koliat@jhmi.edu.
REFERENCES
- 1.Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 2.Beach TG, McGeer EG. Senile plaques, amyloid beta-protein, and acetylcholinesterase fibres: laminar distributions in Alzheimer's disease striate cortex. Acta Neuropathol. 1992;83:292–299. doi: 10.1007/BF00296792. [DOI] [PubMed] [Google Scholar]
- 3.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate A beta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 4.Borchelt DR, Ratovitski T, Van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Braak E. Morphology of Alzheimer disease. Fortschr Med. 1990;108:621–624. [PubMed] [Google Scholar]
- 6.Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Aβ peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 8.Deller T, Frotscher M. Lesion-induced plasticity of central neurons: sprouting of single fibres in the rat hippocampus after unilateral entorhinal cortex lesion. Prog Neurobiol. 1997;53:687–727. doi: 10.1016/s0301-0082(97)00044-0. [DOI] [PubMed] [Google Scholar]
- 9.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 10.Griffin WST, Stanley LC, Yeralan O, Rovnaghi CR, Marshak DR. Methods for the study of cytokine in human neurodegenerative disease. Methods Neurosci. 1993;17:268–287. [Google Scholar]
- 11.Guntern R, Bouras C, Hof PR, Vallet PG. An improved thioflavine S method for staining neurofibrillary tangles and senile plaques in Alzheimer's disease. Experientia. 1992;48:8–10. doi: 10.1007/BF01923594. [DOI] [PubMed] [Google Scholar]
- 12.Hof PR, Morrison JH. The cellular basis of cortical disconnection in Alzheimer's disease and related dementing conditions. In: Katzman R, Bick KL, editors. Alzheimer's disease. Raven; New York: 1994. pp. 197–229. [Google Scholar]
- 13.Ikin AF, Annaert WG, Takei K, De Camilli P, Jahn R, Greengard P, Buxbaum JD. Alzheimer amyloid protein precursor is localized in nerve terminal preparations to Rab5-containing vesicular organelles distinct from those implicated in the synaptic vesicle pathway. J Biol Chem. 1996;271:31783–31786. doi: 10.1074/jbc.271.50.31783. [DOI] [PubMed] [Google Scholar]
- 14.Kadish I, Pradier L, van Groen T. Transgenic mice expressing the human presenilin 1 gene demonstrate enhanced hippocampal reorganization following entorhinal cortex lesions. Brain Res Bull. 2002;57:587–594. doi: 10.1016/s0361-9230(01)00751-1. [DOI] [PubMed] [Google Scholar]
- 15.Koliatsos VE. Biological therapies for Alzheimer's disease: focus on trophic factors. Crit Rev Neurobiol. 1996;10:205–238. doi: 10.1615/critrevneurobiol.v10.i2.40. [DOI] [PubMed] [Google Scholar]
- 16.Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- 17.Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marquez-Sterling NR, Lo AC, Sisodia SS, Koo EH. Trafficking of cell-surface β-amyloid precursor protein: evidence that a sorting intermediate participates in synaptic vesicle recycling. J Neurosci. 1997;17:140–151. doi: 10.1523/JNEUROSCI.17-01-00140.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morin PJ, Abraham CR, Amaratunga A, Johnson RJ, Huber G, Sandell JH, Fine RE. Amyloid precursor protein is synthesized by retinal ganglion cells, rapidly transported to the optic nerve plasma membrane and nerve terminals, and metabolized. J Neurochem. 1993;61:464–473. doi: 10.1111/j.1471-4159.1993.tb02147.x. [DOI] [PubMed] [Google Scholar]
- 20.Nakano I, Hirano A. Atrophic cell processes of large motor neurons in the anterior horn in amyotrophic lateral sclerosis: observation with silver impregnation method. J Neuropathol Exp Neurol. 1987;46:40–49. doi: 10.1097/00005072-198701000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993;268:608–612. [PubMed] [Google Scholar]
- 22.Octave JN, Essalmani R, Tasiaux B, Menager J, Czech C, Mercken L. The role of presenilin-1 in the gamma-secretase cleavage of the amyloid precursor protein of Alzheimer's disease. J Biol Chem. 2000;275:1525–1528. doi: 10.1074/jbc.275.3.1525. [DOI] [PubMed] [Google Scholar]
- 23.Price DL, Sisodia SS. Mutant genes in familial Alzheimer's disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 24.Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer's disease: genetic studies and transgenic models. Annu Rev Genet. 1998;32:461–493. doi: 10.1146/annurev.genet.32.1.461. [DOI] [PubMed] [Google Scholar]
- 25.Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E. Early formation of mature amyloid-β protein deposits in a mutant APP transgenic model depends on levels of Aβ1–42. J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 26.Rogers J, Morrison JH. Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer's disease. J Neurosci. 1985;5:2801–2808. doi: 10.1523/JNEUROSCI.05-10-02801.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheff SW. Synaptic reorganization after injury: the hippocampus as a model system. In: Seil FJ, editor. Neuronal regeneration and transplantation. Liss; New York: 1989. pp. 137–156. [Google Scholar]
- 28.Selkoe DJ. Presenilin, Notch, and the genesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:11039–11041. doi: 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheng JG, Mrak RE, Griffin WS. Microglial interleukin-1 α expression in brain regions in Alzheimer's disease: correlation with neuritic plaque distribution. Neuropathol Appl Neurobiol. 1995;21:290–301. doi: 10.1111/j.1365-2990.1995.tb01063.x. [DOI] [PubMed] [Google Scholar]
- 30.Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, Griffin WS. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steward O, Loesche J. Quantitative autoradiographic analysis of the time course of proliferation of contralateral entorhinal efferents in the dentate gyrus denervated by ipsilateral entorhinal lesions. Brain Res. 1977;125:11–21. doi: 10.1016/0006-8993(77)90356-0. [DOI] [PubMed] [Google Scholar]
- 33.Steward O, Torre ER, Phillips LL, Trimmer PA. The process of reinnervation in the dentate gyrus of adult rats: time course of increases in mRNA for glial fibrillary acidic protein. J Neurosci. 1990;10:2373–2384. doi: 10.1523/JNEUROSCI.10-07-02373.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su Y, Ni B. Selective deposition of amyloid-beta protein in the entorhinal-dentate projection of a transgenic mouse model of Alzheimer's disease. J Neurosci Res. 1998;53:177–186. doi: 10.1002/(SICI)1097-4547(19980715)53:2<177::AID-JNR6>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 35.Vassar R, Citron M. Aβ-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 36.West JR, Deadwyler S, Cotman CW, Lynch G. Time-dependent changes in commissural field potentials in the dentate gyrus following lesions of the entorhinal cortex in adult rats. Brain Res. 1975;97:215–233. doi: 10.1016/0006-8993(75)90446-1. [DOI] [PubMed] [Google Scholar]
- 37.White F, Nicoll JAR, Roses AD, Horsburgh K. Impaired neuronal plasticity in transgenic mice expressing human apolipoprotein E4 compared to E3 in a model of entorhinal cortex lesions. Neurobiol Dis. 2001;8:611–625. doi: 10.1006/nbdi.2001.0401. [DOI] [PubMed] [Google Scholar]