

Abstract
Cannabinoid receptors and their endogenous ligands have been recently identified in the brain as potent inhibitors of neurotransmitter release. Here we show that, in a rat model of Parkinson's disease induced by unilateral nigral lesion with 6-hydroxydopamine (6-OHDA), the striatal levels of anandamide, but not that of the other endocannabinoid 2-arachidonoylglycerol, were increased. Moreover, we observed a decreased activity of the anandamide membrane transporter (AMT) and of the anandamide hydrolase [fatty acid amide hydrolase (FAAH)], whereas the binding of anandamide to cannabinoid receptors was unaffected. Spontaneous glutamatergic activity recorded from striatal spiny neurons was higher in 6-OHDA-lesioned rats. Inhibition of AMT byN-(4-hydroxyphenyl)-arachidonoylamide (AM-404) or by VDM11, or stimulation of the cannabinoid CB1 receptor by HU-210 reduced glutamatergic spontaneous activity in both naïve and 6-OHDA-lesioned animals to a similar extent. Conversely, the FAAH inhibitors phenylmethylsulfonyl fluoride and methyl-arachidonoyl fluorophosphonate were much more effective in 6-OHDA-lesioned animals. The present study shows that inhibition of anandamide hydrolysis might represent a possible target to decrease the abnormal cortical glutamatergic drive in Parkinson's disease.
Keywords: anandamide, CB1 receptor, dopamine, excitatory amino acids, glutamate, Parkinson's disease, striatum
Cannabinoids, the main psychoactive constituents of marijuana, exert a wide range of effects in the CNS through the activation of CB1 receptors (Gerard et al., 1991; Westlake et al., 1994; Pertwee, 1997; Schlicker and Kathmann, 2001). These receptors are densely expressed in the striatum (Herkenham et al., 1991; Matsuda et al., 1993; Tsou et al., 1998), a brain region involved in motor processes, cognition, and motivation (Calabresi et al., 1997; Berke and Hyman, 2000). The effects of CB1 receptor activation by cannabinoids include loss of concentration, impairment of memory, enhancement of sensory perception, and euphoria (Dewey, 1986;Howlett, 1995; Ameri, 1999).
The wide CB1 receptor distribution in the striatum (Herkenham et al., 1991) provides the opportunity for functional interactions of endogenous cannabinoids, such as anandamide (AEA) and 2-arachidonoylglycerol (2-AG), with the dopaminergic nigrostriatal pathway. In fact, a recent evidence in freely moving rats has shown that AEA release is enhanced by quinpirole, a D2-like dopamine (DA) receptor agonist (Giuffrida et al., 1999), and that AEA transport inhibition reverses DA D2-like receptor responses (Beltramo et al., 2000). Moreover, chronic treatment with D2 antagonists results in an increased expression of CB1 receptor mRNA in the striatum (Mailleux and Vanderhagen, 1993). Finally, activation of both D2 and CB1 receptors decreases adenylyl cyclase activity through the same Gi-protein (Meschler and Howlett, 2001), which in turn stimulates mitogen-activated protein kinases (Boulaboula et al., 1995). This pathway is involved in cell survival and synaptic plasticity (Coogan et al., 1999; Otani et al., 1999; Calabresi et al., 2001; Sweatt, 2001), supporting the idea of a close interaction between DA and (endo)cannabinoids in these critical functions.
It has been reported that corticostriatal glutamatergic transmission is enhanced after DA denervation induced by 6-hydroxydopamine (6-OHDA) injection (Calabresi et al., 1993, 2000; Schwartig and Huston, 1996; Centonze et al., 2001; Tang et al., 2001). The pharmacological inhibition of such an hyperactivity may, therefore, be beneficial in Parkinson's disease (PD). Here we show that spontaneous EPSCs (sEPSCs) were increased in frequency and amplitude in 6-OHDA-lesioned rats and were modulated by manipulation of the endocannabinoid system. In these animals we found increased levels of AEA, but not of 2-AG, paralleled by an abnormal downregulation of anandamide membrane transporter (AMT) and fatty acid amide hydrolase (FAAH) activity, without significant changes in AEA binding to CB1 receptors. We suggest that targeting a specific step of these biochemical processes might provide a novel approach to treat the abnormal striatal glutamatergic activity observed in PD.
MATERIALS AND METHODS
6-OHDA lesions. All the experiments were conducted in conformity with the European Communities Council Directive of November 1986 (86/609/EEC). Wistar rats were injected unilaterally with 6-OHDA (8 μg/4 μl of saline containing 0.1% ascorbic acid) rostral to the substantia nigra under stereotaxic coordinates: anterior, 3.7 mm anterior; ventral, 2.2 mm dorsal to the interaural line; lateral, 2.2 mm from the midline. Twenty days later, the rats were tested with a subcutaneous injection of 0.05 mg/kg apomorphine, and the contralateral turns were recorded with automatic rotometers for 3 hr. Only those rats consistently making at least 200 contralateral turns were used for our studies. After brain dissection, we confirmed that the nigrostriatal pathway was lesioned. This was established by noting a >95% loss of DA neurons in the substantia nigra compacta and the almost complete absence of DA terminals in the striatum. This was detected by the immunoperoxidase technique using a monoclonal antibody for tyrosine hydroxylase. Rats were used 2–3 months after the 6-OHDA lesion. As control animals (naive) we used sham-operated rats of similar ages injected with saline not containing 6-OHDA.
Biochemistry. The endogenous levels of anandamide and of 2-arachidonoylglycerol in the striatum were determined by gas chromatography-electron impact mass spectrometry, as recently reported (Maccarrone et al., 2001). The binding of [3H]AEA and of [3H]CP55,940 to striatal membrane fractions was determined by rapid filtration assays, as described (Maccarrone et al., 2000). Apparent dissociation constant (Kd) and maximum binding (Bmax) values were calculated through nonlinear regression analysis, using Prism 3 program (GraphPad Software, San Diego, CA). The uptake of [3H]AEA by the AEA membrane transporter (AMT) was assayed in synaptosomes prepared from the striatum as described (Maccarrone et al., 2001). The hydrolysis of [3H]AEA by the FAAH (E.C. 3.5.1.4) activity was assayed in striatum extracts by reversed phase-HPLC, as reported (Maccarrone et al., 2000). The kinetic constants of AMT and FAAH activity, i.e., apparent Michaelis–Menten constant (Km) and maximum velocity (Vmax), were calculated through nonlinear regression analysis, using Prism 3. The activity of phospholipase D (PLD; E.C. 3.1.4.4) was assayed by measuring the release of [14C]ethanolamine from 1,2-dioleoyl-3-phosphatidyl-[2-14C]ethanolamine, according to Moesgaard et al. (2000). [3H]AEA (223 Ci/mmol) and [3H]CP55,940 (126 Ci/mmol) were from NEN DuPont de Nemours (Köln, Germany); 1,2-dioleoyl-3-phosphatidyl[2-14C]ethanolamine (55 mCi/mmol) was from Amersham Pharmacia Biotech (Uppsala, Sweden). Biochemical data are expressed as the mean ± SD, and statistical analysis was performed by the Student's t test (ST), through the InStat 3 program (GraphPad).
Electrophysiology. Preparation and maintenance of rat corticostriatal slices have been previously described (Calabresi et al., 1993, 2001). Briefly, corticostriatal coronal slices 190- to 200-μm-thick were prepared from 2- to 3-month-old Wistar rats (naive and 6-OHDA-lesioned) and kept in artificial CSF (ACSF) whose composition was (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, and 25 NaHCO3. ACSF temperature was maintained at 35°C, and it was gassed with O2 and CO2 (95:5%). For whole-cell patch-clamp recordings, electrodes (4–5 MΩ) were filled with a solution containing (in mm): K+-gluconate (125), NaCl (10), CaCl2, (1.0), MgCl2 (2.0), 1,2-bis (2-aminophenoxy) ethane-N,N,N,N-tetra-acetic acid (0.5), HEPES (19), GTP (0.3), and Mg-ATP (1.0), adjusted to pH 7.3 with KOH. Striatal spiny neurons were clamped at −80 to −85 mV, close to their resting membrane potential. Spontaneous and evoked glutamatergic activity, as well as passive membrane properties, were recorded from striatal spiny neurons and monitored using an Axopatch 200B and 1D amplifiers, and Clampex 8.1 software (Axon Instruments, Union City, CA). Afterwards, sEPSCs and miniature EPSCs (mEPSCs) were analyzed offline by MiniAnalysis 5.4.1 software (Synaptosoft, Decatur, GA). To evoke EPSCs, bipolar electrodes were placed on corticostriatal fibers. In the experiments on paired-pulse facilitation (pulse interval, 40–60 msec) stimuli were delivered at 0.1 Hz. Spontaneous mEPSCs were recorded in the presence of 1 μm tetrodotoxin (TTX). The frequency of mEPSCs was sensibly slower than that of sEPSCs. However, longer samples were analyzed to achieve a statistically significant number of events before and after drug application.
All recordings were performed in the presence of 3 μmbicuculline to avoid the contamination of sEPSCs or mEPSCs by a GABAA-mediated component. Striatal medium spiny neurons were selected by means of infrared videomicroscopy (ZeissAxioskop, Jena, Germany) and a digital camera (Cohu, San Diego, CA).
For data presented as the mean ± SEM, statistical analysis was performed using the ST. When comparing two cumulative distributions, the Kolmogorov–Smirnov (KS) test was used. The significance level was established at p < 0.05 for both tests.
Drugs were applied by dissolving them to the desired final concentration in the saline solution perfusing the slice.N-(4-hydroxyphenyl) arachidonoylamide (AM-404),N-arachidonoylethanolamine (anandamide), and phenylmethylsulfonyl fluoride (PMSF) were from Sigma (Milano, Italy); bicuculline, TTX, VDM11, and HU-210 were from Tocris-Cookson (Bristol, UK);N-[2-(4-chloro-phenyl)ethyl]-1,3,4,5-tetrahydro-7,8-dihydroxy-2H-2-benzazepine-2-carbothioamide (capsazepine) was from Calbiochem (La Jolla, CA); methyl-arachidonoyl fluorophosphonate (MAFP) was from Cayman Chemicals (Ann Arbor, MI); andN-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxy amide (SR-141716) was a kind gift from Sanofi Recherche (Montpellier, France).
RESULTS
Rearrangement of striatal cannabinoid system in parkinsonian rats
The level of endogenous AEA was threefold higher in the striatum of 6-OHDA-lesioned rats, compared with naive animals, whereas endogenous 2-AG was unaffected (Table 1). Similar changes were recently reported in reserpine-treated rats, although the mechanism responsible for AEA enhancement was not investigated (Di Marzo et al., 2000). Here, we found that the activity of both AMT (Hillard et al., 1997; Piomelli et al., 1999) and AEA-hydrolyzing enzyme FAAH (Deustch and Chin, 1993; Désarnaud et al., 1995; Cravatt et al., 1996; Maccarrone et al., 1998) decreased in parkinsonian animals (Table 1, Fig.1A,B). The affinity of AMT for AEA was identical in naive and parkinsonian rats (Km = 383 ± 45 and 351 ± 49 nm, respectively; n = 4;p > 0.05; ST test), whereas the apparentVmax (181 ± 9 and 81 ± 4 pmol/min per milligram of protein, respectively) was significantly lower in the latter group (n = 4; p < 0.01; ST test). Remarkably, the affinity of AMT for AEA was very close to that reported for the transporter in several cellular models (for review, see Hillard and Jarrahian, 2000). Analogously, theVmax of FAAH decreased in parkinsonian rats (580 ± 48 and 1687 ± 165 pmol/min per milligram of protein, respectively; n = 4; p < 0.01; ST test), whereas the Km for AEA was unaffected (10.5 ± 1.7 and 12.4 ± 2.2 μm, respectively; n = 4;p > 0.05; ST test).
Table 1.
Endogenous levels of AEA and 2-AG, activity of PLD, of the AMT, and of the FAAH, and binding of AEA and CP55,940 in the striatum of rats
| Endogenous levels of AEA1-a | Endogenous levels of 2-AG1-a | PLD activity1-b | AMT activity1-c | FAAH activity1-d | Binding of AEA1-e | Binding of CP55,9401-f | |
|---|---|---|---|---|---|---|---|
| Naive | 250 ± 20 | 1250 ± 120 | 320 ± 32 | 90 ± 10 | 470 ± 50 | 150 ± 15 | 114 ± 11 |
| 6-OHDA lesioned | 750 ± 80* | 1300 ± 125 | 350 ± 35 | 35 ± 4* | 180 ± 20* | 167 ± 16 | 130 ± 12 |
Expressed as picomoles per milligram of protein.
Expressed as picomoles per minute per milligram of protein (substrate was 10 μm1,2-dioleoyl-3-phosphatidyl-[2-14C]ethanolamine).
Expressed as picomoles per minute per milligram of protein (substrate was 400 nm[3H]AEA).
Expressed as picomoles per minute per milligram of protein (substrate was 5 μm[3H]AEA).
Expressed as femtomoles per milligram of protein (ligand was 200 pm[3H]AEA).
Expressed as femtomoles per milligram of protein (ligand was 200 pm[3H]CP55,940).
p <0.01 compared with control (n= 4).
Fig. 1.

Biochemical changes of the cannabinoid system in experimental parkinsonism. A, The activity of the AEA membrane transporter AMT is reduced in 6-OHDA-lesioned animals compared with naive. B, The activity of the AEA hydrolase FAAH is lower in parkinsonian animals. C, The binding of [3H]CP55,940 is not changed after the treatment with 6-OHDA. For details on Km,Vmax, Kd,and Bmax, see Results.
The binding of the synthetic cannabinoid [3H]CP55,940 to CB1 receptors was not affected after 6-OHDA denervation (Table 1, Fig. 1C), showing a Kd = 358 ± 80 and 315 ± 73 pm, and aBmax = 350 ± 35 and 346 ± 35 fmol/mg protein, in naive and parkinsonian rats, respectively (n = 4; p > 0.05 for both; ST test). These Kd values are close those previously reported for the binding of [3H]CP55,940 to rat striatal membranes (for review, see Pertwee, 1997). Interestingly, also the binding of [3H]AEA was not affected by 6-OHDA denervation (Table 1), neither was the activity of phospholipase D (PLD), which remained almost identical in naive and 6-OHDA-lesioned striata (Table 1). PLD was assayed under conditions found to be optimal for the N-acyl-phosphatidylethanolamines (NAPE)-hydrolyzing PLD (Moesgaard et al., 2000), but a radiolabeled phosphatidylethanolamine was used instead of radiolabeled NAPEs, which are not commercially available. This is noteworthy, because NAPE-hydrolyzing PLD activity is considered the checkpoint in AEA synthesis, although the lack of specific inhibitors for this enzyme makes it difficult to assess conclusively its contribution to AEA metabolism (Hansen et al., 2000). Thus these results, taken together, suggest that the higher level of AEA in the striatum of parkinsonian rats might be attributable to a decreased cleavage rather than an increased synthesis. On the other hand, the synthesis of 2-AG by phospholipases A1 and C, and/or its degradation by monoacylglycerol lipase (Sugiura and Waku, 2000), do not seem to be affected by 6-OHDA denervation.
Spontaneous glutamatergic activity in naïve and 6-OHDA-lesioned rats
sEPSCs were recorded from corticostriatal rat slices by means of whole-cell patch-clamp techniques. Spiny neurons from 6-OHDA-lesioned rats showed in average a higher sEPSCs frequency and amplitude (see Fig. 3) compared with naive rats (1.9 ± 0.8 vs 6.2 ± 1.2 Hz; n = 30; p < 0.01; ST test; and 13.7 ± 2.1 vs 22.3 ± 5.5 pA;n = 28; p < 0.01; ST test, respectively), whereas the intrinsic membrane properties of these neurons were not affected (Calabresi et al., 1993). This synaptic activity was suppressed by 10 μm CNQX, an AMPA receptor blocker. Conversely, it was not affected by 3 μm bicuculline, a GABAAreceptor antagonist (not shown).
Fig. 3.
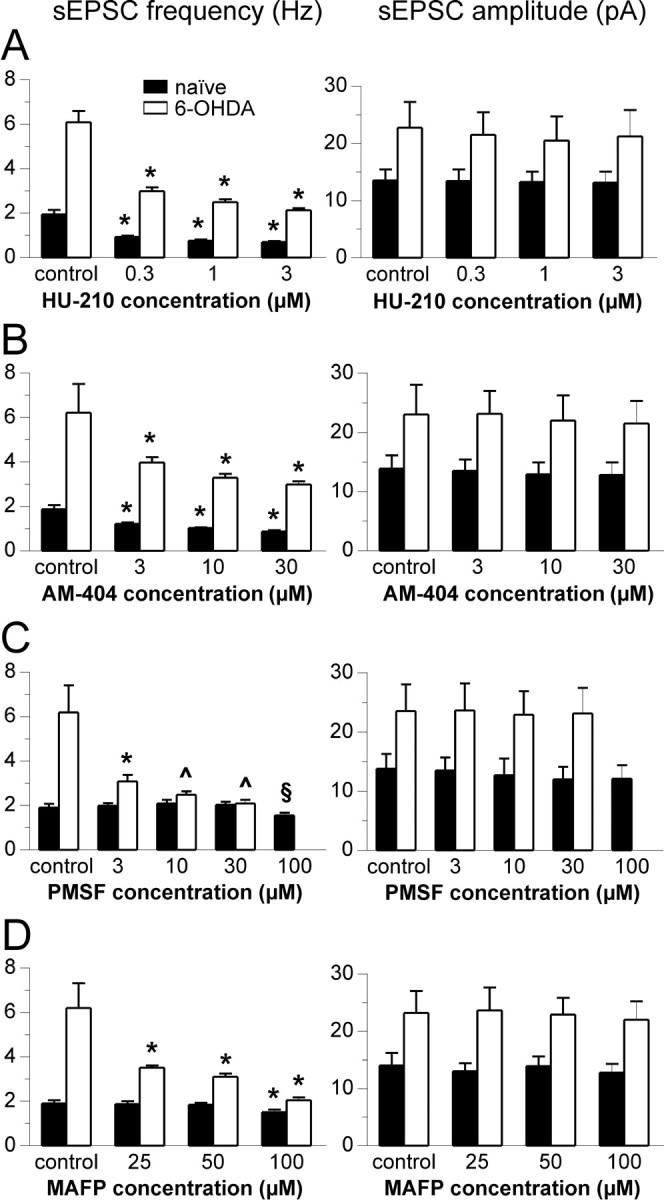
Effect of cannabinoids on glutamatergic sEPSC frequency and amplitude. Histograms in A–D summarize the effect of the different concentrations of HU-210 (A), AM 404 (B), PMSF (C), and MAFP (D) on sEPSC frequency (left) and amplitude (right) of all experiments. Note that the average control frequency (expressed in Hertz) and amplitude is higher in 6-OHDA-lesioned rats. Both HU-210 and AM-404 significantly reduce sEPSC frequency at all doses, without affecting their amplitude. Conversely, PMSF is effective in naive animals only at the higher dose (100 μm), whereas parkinsonian rats are more sensitive to low doses (3 μm) of this compound. Similar results were obtained with MAFP, which is active at 100 nm on naive animals, whereas at 25 nm it is active on 6-OHDA-lesioned ones. Also PMSF and MAFP do not affect sEPSCs amplitude. (*p < 0.01 compared with predrug control; ^p < 0.01 compared with predrug control, and p > 0.05 between groups; §p < 0.05 compared with predrug control; all values on graphs A–C, except ^, are p < 0.01 between groups; ST test;n = 4 for each condition.)
Activation of CB1 cannabinoid receptor reduces striatal glutamatergic spontaneous activity
Activation of CB1 cannabinoid receptors has been reported to reduce glutamate release in the striatum through a presynaptic mechanism (Gerdeman and Lovinger, 2001; Huang et al., 2001).
In our preparation, 10 min application of HU-210, a CB1 receptor agonist (Pertwee, 1997), reduced sEPSCs frequency in a dose-dependent manner in both 6-OHDA-lesioned and naive rats. Figure2A shows single experiments in which this effect was evident, whereas all the experiments in naive and 6-OHDA-lesioned animals are pooled in Figure3A. Interestingly, the average sEPSCs amplitude was not affected. Figure4A shows the normalized effect of HU-201 on sEPSCs frequency that was similar in the two groups.
Fig. 2.

Cannabinoids reduce the frequency of glutamatergic sEPSCs in striatal spiny neurons of naive and parkinsonian (6-OHDA) rats. A–D, Cumulative probability plots of glutamatergic sEPSCs recorded from single striatal neurons of naive (left) and a 6-OHDA-lesioned (right) rats. Electrophysiological traces show spontaneous striatal glutamatergic activity before (top) and after (bottom) drug administration. Application of 1 μm HU-210 (A) reduces significantly sEPSCs frequency (expressed as interevent interval) in both neurons (p < 0.01 for both; KS test). A similar effect is obtained with 10 μm AM-404 (B) in both naive (p< 0.05; KS test) and 6-OHDA-lesioned (p < 0.01; KS test) animals. Conversely, 10 μm PMSF (C) is ineffective on the naive neuron (p > 0.05; KS test) but reduces sEPSC frequency (p < 0.01; KS test) in the 6-OHDA-lesioned cell. Similarly, 25 nm MAFP was effective in decreasing sEPSC frequency only in the cell from a parkinsonian rat (p < 0.01; KS test) but not in the naive one (p > 0.05; KS test).
Fig. 4.

Pharmacology of cannabinoid-mediated reduction of striatal glutamatergic activity. A–C, Normalized dose–response curves of the sEPSC frequency inhibition by HU-210 (A), AM-404 (B), and PMSF (C). Only the effect of PMSF is significantly different in the two experimental groups (*p < 0.01 compared with predrug control, and p > 0.05 between groups; §p > 0.05 compared with predrug control; ^p < 0.05 compared with predrug control, and p > 0.01 between groups; ST test; n = 4 for each point). D, The inhibition of sEPSCs frequency by 10 μm AM-404 (p < 0.01 compared with control, ST test) shows that this effect was long-lasting and occluded further depression by 1 μm HU-210. This effect was similar in naive and parkinsonian rats (p > 0.05 between groups, ST test; n = 3 for each group).
Endogenous anandamide reduces striatal glutamatergic spontaneous activity
Blockade of AEA uptake by 10 min application of AM-404 (Piomelli et al., 1999; Beltramo et al., 2000) significantly reduced the frequency of glutamatergic sEPSCs in a dose-dependent manner in both naive and 6-OHDA-lesioned rats. Single experiments are shown in Figure2B, whereas all data are pooled in Figure3B. Also AM-404 did not significantly affect sEPSCs amplitude. The normalized effect of AM-404 on sEPSCs frequency was similar in the two groups (Fig. 4B).
Figure 4D shows the time course of 10 μm AM-404 effect. This figure also shows that AM-404 occludes a further reduction of synaptic activity by 1 μm HU-210. These effects, when normalized, were similar in naive and 6-OHDA-lesioned animals. Similar results were obtained with 10 μm VDM11 (n= 4; data not shown), another selective inhibitor of AMT (De Petrocellis et al., 2001).
Application in naïve animals of PMSF, an inhibitor of FAAH (Ueda et al., 2000; Wyley et al., 2000), caused a significant reduction of sEPSCs frequency only at high doses (100 μm), whereas MAFP, a potent inhibitor of FAAH (Deutsch et al., 1997; Ueda et al., 2000), was effective at 100 nm (Fig. 3D). Surprisingly, 6-OHDA treatment revealed a potent inhibitory effect of PMSF even at very low concentrations (3–30 μm), and MAFP was effective even at 25 nm (see Fig. 2, C and D, for single experiments and Fig. 3, C and D, for average data on both compounds). Again, neither PMSF nor MAFP had significant effects on sEPSCs amplitude either in naïve or in 6-OHDA-lesioned rats. Figure 4C shows the normalized effect of PMSF on sEPSCs frequency in both groups.
Pharmacology of the endocannabinoid effect
We also investigated the possibility that the effect of AEA on synaptic spontaneous activity was specifically mediated by CB1 receptors. Because AEA is quickly hydrolyzed by FAAH, in these experiments we coadministered 1 μm AEA and a low dose of PMSF (10 μm). Slices were preincubated in PMSF for 10 min before their coapplication with AEA. Although 3–30 μmPMSF per se was not able to affect sEPSC frequency in naive rats (Figs.2C, 3C, 4C, 5A), AEA plus PMSF was effective in reducing glutamatergic activity in both naive and 6-OHDA-lesioned animals (Fig.5A).
Fig. 5.

Specific activation of CB1 receptor reduces sEPSC frequency. A, Activation of CB1 receptor by AEA reduced glutamatergic spontaneous activity in spiny neurons from both naive and 6-OHDA-lesioned rats (*p < 0.01; ST test;n = 4 for each condition). A low dose of PMSF was added to anandamide to avoid the rapid degradation by endogenous FAAH (see Results for details). B, The histogram shows that blockade of CB1 receptor by 1 μm SR-141716 prevents the inhibitory action of AM-404 or PMSF, whereas blockade of VR1 vanilloid receptor has no effect (*p < 0.01; ST test; n = 4 for each condition).
We also used SR-141716, a specific CB1 receptor antagonist (Pertwee, 1997), to further investigate the pharmacological site of action of endogenous cannabinoids. Although SR-141716 per se did not alter glutamatergic spontaneous activity, 10 min of pretreatment with this drug blocked the effect of 10 μm AM-404 or 10 μm PMSF in both naive and 6-OHDA-lesioned animals (Fig.5B).
It has been reported that AEA can also activate other receptors rather than CB1, for example the vanilloid VR1 receptor (Zygmunt et al., 1999;De Petrocellis et al., 2001). Thus, we tested whether the preincubation of the slices for 10 min in 1 μm capsazepine, a vanilloid receptor antagonist (Zygmunt et al., 1999), could prevent the effect of AM-404 and PMSF. Capsazepine alone did not alter glutamatergic spontaneous activity. Moreover, it failed to alter the effect of AM-404 and PMSF on glutamatergic spontaneous activity, both in naive and 6-OHDA animals (Fig. 5B).
All the compounds that we used did not significantly alter per se the membrane properties (resting membrane potential and input resistance) of the recorded neurons. See Table2 for details.
Table 2.
The average resting membrane potential (RMP, mV) and the input resistance (IR, M) of the recorded striatal spiny neurons are reported
| Predrug control | 3 μmHU-210 | 30 μm AM-404 | 100 μm PMSF | 1 μmAEA | 1 μm SR-141716 | 1 μm capsaicin | 10 μmVDM11 | 100 nm MAFP | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RMP | IR | RMP | IR | RMP | IR | RMP | IR | RMP | IR | RMP | IR | RMP | IR | RMP | IR | RMP | IR | |
| Naive | −82 ± 3 | 195 ± 43 | −83 ± 2 | 209 ± 35 | −82 ± 1 | 210 ± 50 | −84 ± 2 | 198 ± 51 | −82 ± 3 | 195 ± 42 | −84 ± 4 | 201 ± 50 | −81 ± 2 | 217 ± 54 | −84 ± 6 | 202 ± 39 | −82 ± 4 | 205 ± 41 |
| 6-OHDA | −81 ± 2 | 204 ± 49 | −84 ± 3 | 216 ± 41 | −83 ± 3 | 222 ± 45 | −82 ± 2 | 215 ± 48 | −84 ± 2 | 197 ± 44 | −83 ± 1 | 195 ± 47 | −83 ± 3 | 208 ± 29 | −83 ± 5 | 199 ± 41 | −83 ± 4 | 203 ± 39 |
Each value ± SD corresponds to at least four recorded cells. Both RMP and IR were not significantly changed after the application of each drug (p >0.05, ST test). To simplify reading, only the maximal concentration is shown.
Presynaptic effect of CB1 receptor activation
We have performed a set of experiments to address the specific presynaptic effect of CB1 receptor activation. Evoked EPSCs were elicited by cortical fibers stimulation. Paired pulses (40–60 msec interval) were delivered at 0.1 Hz, and EPSCs were recorded throughout the whole experiment. Application of 1 μm HU-210 for 10 min reduced evoked EPSCs amplitude, increasing paired-pulse facilitation (Fig.6A).
Fig. 6.

Endocannabinoid CB1 receptor activation modulates glutamatergic activity with a presynaptic mechanism in both naive and parkinsonian rats. A, The CB1 agonist HU-210 (1 μm) reduces evoked postsynaptic currents. Traces are from single neurons of a naive (left) and a 6-OHDA-lesioned (right) animal, before (top traces) and after (bottom traces) the application of 1 μm HU-210. Note that the inhibition of evoked EPSCs by this compound increases paired-pulse facilitation, as shown also in the histogram (*p < 0.01; ST test;n = 4 for each group). B, In the presence of 1 μm TTX, action potential-independent release of glutamate generates mEPSCs, whose frequency and amplitude are both reduced compared with sEPSCs. Electrophysiological traces show spontaneous striatal glutamatergic activity recorded from single striatal spiny neurons of a naive (left) and a 6-OHDA-leisoned (right) rat, before (top) and after (bottom) the administration of 1 μm HU-210. Application of this compound did not further reduce mEPSC frequency (left histogram) or amplitude (right histogram) in both naive and parkinsonian animals (p > 0.05; ST test; n = 4 for each group).
We also analyzed the action potential-independent mEPSPs recorded in the presence of 1 μm TTX. Although sEPSCs result from neurotransmitter release elicited by action potentials, mEPSCs are supposed to arise from the spontaneous fusion of neurotransmitter-containing vesicles to the presynaptic terminal membrane. This latter phenomenon is independent of the activation of presynaptic voltage-dependent ion channels. The efficacy of TTX was assessed by observing the disappearance of evoked EPSCs. In these recording conditions, amplitude and frequency of mEPSCs were lower compared with sEPSCs (Fig. 6B, seehistograms). This effect was similar in naive and 6-OHDA-lesioned animals. Application of 1 μmHU-210 did not affect either the frequency or the amplitude of mEPSCs (Fig. 6B, traces and histograms). These results suggest that activation of CB1 receptors by HU-210 specifically reduces glutamate release from presynaptic terminals mediated by action potentials, whereas it does not affect action potential-independent release of this neurotransmitter.
DISCUSSION
Main findings
In the present study we show that experimental parkinsonism causes complex plastic changes of the endocannabinoid system. In particular, we have obtained four novel findings in the striatum of 6-OHDA-denervated rats: (1) the levels of endogenous AEA are increased; (2) the activity of the AEA membrane transporter, as well as that of the AEA hydrolase FAAH, is reduced; (3) the level of CB1 receptor and the binding of AEA to this receptor is not affected; and (4) the pharmacological inhibition of FAAH produces a much stronger depression of striatal glutamatergic activity compared with naive rats. This latter finding, taken together with the observation that denervated striata express a dramatic overactivity of glutamatergic transmission, suggests that targeting the FAAH might be beneficial in experimental parkinsonism. Accordingly, ionotropic glutamate receptors antagonists improve experimental PD symptoms (Chase and Oh, 2000).
The binding of AEA to CB1 receptor induces inhibition of N- and P/Q-type Ca2+ channels (Mackie and Hille, 1992; Pertwee, 1997; Twitchell et al., 1997), inhibition of forskolin-induced cAMP accumulation, and stimulation of A-type and inward-rectifying K+ channels (Henry and Chavkin, 1995; Childers and Deadwyler, 1996). Accordingly, a negative modulation of corticostriatal glutamatergic transmission by cannabinoids has been recently shown (Gerdeman and Lovinger, 2001;Huang et al., 2001). CB1 receptor agonists, such as exogenous AEA, HU-210, or WIN 55,212–2, inhibit synaptic release of glutamate in the rat striatum through a complex presynaptic mechanism involving Gi/o-protein-coupled modulation of N-type Ca2+ channels (Pertwee, 1997; Huang et al., 2001).
AEA is released from membraneN-arachidonoyl-phosphatidyl-ethanolamines of depolarized neurons through a phospholipase D (Hansen et al., 2000), and its activity at CB1 receptors depends on its life span in the extracellular space, which is regulated by AEA degradation (Deutsch and Chin, 1993). This normally occurs in a two-step process, including: (1) cellular uptake by the AMT (Hillard and Jarrahian, 2000), and (2) intracellular degradation by the enzyme FAAH (Ueda et al., 2000). Also the other endogenous agonist of CB1 receptors, 2-AG, is taken up by a membrane transporter, which is poorly characterized but seems to share some properties with AMT (Bisogno et al., 2001). 2-AG is then hydrolyzed by a monoacylglycerol lipase (Sugiura and Waku, 2000), and in addition by FAAH under some circumstances (Ueda et al., 2000). The synthesis of 2-AG is controlled by phospholipase A1 and/or by phospholipase C (Sugiura and Waku, 2000), following a pathway different from that of AEA (Hansen et al., 2000). Interestingly, we have found that 2-AG levels, unlike those of AEA, were not altered by experimental parkinsonism.
Abnormal endocannabinoid function in experimental parkinsonism
As already reported (Calabresi et al., 1993; Tang et al., 2001), spontaneous glutamatergic activity recorded in the striatum of parkinsonian rats is increased. This effect reflects the loss of D2 receptor-mediated control of corticostriatal transmission (Cepeda et al., 2001). Interestingly, D2 and CB1 receptors share the same signal transduction pathway and closely cooperate in the negative regulation of striatal excitatory transmission (Meschler and Howlett, 2001). Thus, the finding that endogenous levels of AEA are higher in parkinsonian rats may reflect a compensatory mechanism to control the cortical glutamatergic drive to the striatum. However, this mechanism seems not to be sufficient in 6-OHDA-lesioned rats, because spontaneous excitatory activity is still higher in these animals. In fact, only the further increase of endogenous AEA tone, achieved by AMT or FAAH blockade, restores the normal corticostriatal function. In agreement with the present study, Hansen et al. (2001) observed changes in endocannabinoid homeostasis in a rat model of altered glutamatergic neurotransmission, causing widespread neurodegeneration. In particular, they measured an increase of AEA, but not of 2-AG and CB1 receptor density.
It is not clear from our data why parkinsonian rats are more sensitive to FAAH inhibition than naive rats, whereas the other pharmacological tools acting on the endocannabinoid system have the same effects in both groups. We can speculate that, because FAAH is less active in 6-OHDA-lesioned rats, this enzyme is more vulnerable to inhibition by PMSF. However, also AMT is reduced in these animals, thus we should also expect an increased sensitivity to AM-404. It is clear, therefore, that further studies are necessary to address these issues. Presumably, FAAH activity plays a major role in determining AEA levels in the striatum of 6-OHDA-lesioned animals, in accordance to a recent report (Cravatt et al., 2001) showing that mice lacking FAAH have a 15-fold augmented level of AEA in the brain.
Presynaptic modulation of glutamate release by endocannabinoids
In good agreement with previous studies, all the electrophysiological experiments we have performed indicate that CB1 receptor activation reduces glutamate release through a presynaptic mechanism (Gerdeman and Lovinger, 2001; Huang et al., 2001). First, endogenous AEA, as well as HU-210, reduces sEPSC frequency without affecting their amplitude. Second, evoked EPSCs amplitude is reduced by HU-210 increasing paired-pulse facilitation. Third, mEPSCs are not affected by this drug in terms of both frequency and amplitude. Taken together, these data suggest that presynaptic action potentials are required to observe the inhibitory effect of CB1 receptor activation, according to Huang et al. (2001). However, a different mechanism, not involving presynaptic action potential-dependent Ca2+ increases, has been suggested byGerdeman and Lovinger (2001). The goal of the present study was the analysis of complex plastic changes induced by DA-denervation in the endocannabinoid system, rather than the characterization of the molecular mechanisms underlying endocannabinoid-mediated inhibition of glutamatergic synaptic transmission. Nevertheless, we feel that this issue needs further investigations.
In conclusion, although further work seems to be necessary to better characterize the changes in striatal synaptic transmission after 6-OHDA denervation, here we propose that drugs modulating the cleavage of endogenous cannabinoids might represent a novel pharmacological approach in the therapy of Parkinson's disease.
Footnotes
This work was supported by a Schizophrenia Finalized Project (Istituto di Ricovero e Cura a Carattere Scientifico “S. Lucia”) to P.C., and two Ministero dell'Università e della Ricerca Scientifica e Tecnologica, Consiglio Nazionale delle Ricerche Biotechnology Programs (L. 95/95) to G.B. and A.F.-A. We thank Dr. J.-L. Gaïarsa (Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Marseille, France) for his kind advice on spontaneous activity study, Prof. R. Pertwee (University of Aberdeen, UK) for his helpful discussion on anandamide binding, Dr. A. Cartoni for the analysis of endogenous cannabinoid levels, and M. Tolu for his technical assistance.
Correspondence should be addressed to Dr. Paolo Calabresi, Dipartimento di Neuroscienze, Università di Roma “Tor Vergata,” Via Montpellier 1, 00133 Roma, Italy, E-mail:paolo.calabresi@uniroma2.it or Dr. Mauro Maccarrone, Dipartimento di Medicina Sperimentale e Scienze Biochimiche, Università di Roma “Tor Vergata,” Via Montpellier 1, 00133 Roma, Italy, E-mail:maccarrone@med.uniroma2.it.
REFERENCES
- 1.Ameri A. The effects of cannabinoids on the brain. Prog Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 2.Beltramo M, Rodríguez de Fonseca F, Navarro M, Calignano A, Gorriti MA, Grammatikopoulos G, Sadile AG, Giuffrida A, Piomelli D. Reversal of dopamine D2 receptor responses by anandamide transport inhibitor. J Neurosci. 2000;20:3401–3407. doi: 10.1523/JNEUROSCI.20-09-03401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berke JD, Hyman SE. Addiction, dopamine and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 4.Bisogno T, Maccarrone M, De Petrocellis L, Jarrahian A, Finazzi-Agrò A, Hillard C, Di Marzo V. The uptake by cells of 2-arachidonoylglycerol, an endogenous agonist of cannabinoid receptors. Eur J Biochem. 2001;268:1982–1989. doi: 10.1046/j.1432-1327.2001.02072.x. [DOI] [PubMed] [Google Scholar]
- 5.Boulaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G, Casellas P. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312:637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calabresi P, Mercuri NB, Sancesario G, Bernardi G. Electrophysiology of dopamine-denervated striatal neurons. Implications for Parkinson's disease. Brain. 1993;116:433–452. [PubMed] [Google Scholar]
- 7.Calabresi P, De Murtas M, Bernardi G. The neostriatum beyond the motor function: experimental and clinical evidence. Neuroscience. 1997;78:39–60. doi: 10.1016/s0306-4522(96)00556-8. [DOI] [PubMed] [Google Scholar]
- 8.Calabresi P, Centonze D, Bernardi G. Electrophysiology of dopamine in normal and denervated striatal neurons. Trends Neurosci. 2000;23:S57–S63. doi: 10.1016/s1471-1931(00)00017-3. [DOI] [PubMed] [Google Scholar]
- 9.Calabresi P, Gubellini P, Picconi B, Centonze D, Pisani A, Bonsi P, Greengard P, Hipskind RA, Borrelli E, Bernardi G. Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J Neurosci. 2001;21:5110–5120. doi: 10.1523/JNEUROSCI.21-14-05110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Centonze D, Picconi B, Gubellini P, Bernardi G, Calabresi P. Dopaminergic control of synaptic plasticity in the striatum. Eur J Neurosci. 2001;13:1071–1077. doi: 10.1046/j.0953-816x.2001.01485.x. [DOI] [PubMed] [Google Scholar]
- 11.Cepeda C, Hurst RS, Altemus KL, Flores-Hernández J, Calvert CR, Jokel ES, Grandy DK, Low MJ, Rubinstein M, Ariano MA, Levine MS. Facilitated glutamatergic transmission in the striatum of D2 dopamine receptor-deficient mice. J Neurophysiol. 2001;85:659–670. doi: 10.1152/jn.2001.85.2.659. [DOI] [PubMed] [Google Scholar]
- 12.Chase TN, Oh JD. Striatal dopamine- and glutamate-mediated dysregulation in experimental parkinsonism. Trends Neurosci. 2000;23:S86–S91. doi: 10.1016/s1471-1931(00)00018-5. [DOI] [PubMed] [Google Scholar]
- 13.Childers SR, Deadwyler SA. Role of cyclic AMP in the actions of cannabinoid receptors. Biochem Pharmacol. 1996;52:819–827. doi: 10.1016/0006-2952(96)00419-4. [DOI] [PubMed] [Google Scholar]
- 14.Coogan AN, O'Leary DM, O'Connor JJ. P42–44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- 15.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Giulia NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 16.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agrò A, Di Marzo V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J Biol Chem. 2001;276:12856–12863. doi: 10.1074/jbc.M008555200. [DOI] [PubMed] [Google Scholar]
- 18.Désarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes: identification and partial characterization. J Biol Chem. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 19.Deutsch DG, Chin SA. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- 20.Deutsch DG, Omeir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A. Methyl arachidonyl fluorophosphonate: a potent irreversible inhibitor of anandamide amidase. Biochem Pharmacol. 1997;53:255–260. doi: 10.1016/s0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- 21.Dewey WL. Cannabinoid pharmacology. Pharmacol Rev. 1986;38:151–178. [PubMed] [Google Scholar]
- 22.Di Marzo V, Hill MP, Bisogno T, Crossman AR, Brotchie JM. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson's disease. FASEB J. 2000;14:1432–1438. doi: 10.1096/fj.14.10.1432. [DOI] [PubMed] [Google Scholar]
- 23.Gerard GCM, Mollereau C, Vassart G, Parmentier M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem J. 1991;279:54–61. doi: 10.1042/bj2790129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerdeman G, Lovinger DM. CB1 cannabinoid receptor inhibit synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- 25.Giuffrida A, Parsons LH, Kerr TM, Rodríguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–362. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- 26.Hansen HS, Moesgaard B, Hansen HH, Petersen G. N-acylethanolamines and precursor phospholipids: relation to cell injury. Chem Phys Lipids. 2000;108:135–150. doi: 10.1016/s0009-3084(00)00192-4. [DOI] [PubMed] [Google Scholar]
- 27.Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, Manzanares J, Ikonomidou C, Schmid HH, Fernandez-Ruiz JJ, Hansen HS. Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem. 2001;78:1415–1427. doi: 10.1046/j.1471-4159.2001.00542.x. [DOI] [PubMed] [Google Scholar]
- 28.Henry DJ, Chavkin C. Activation of inward rectifying potassium channels (GIRK1) by co-expressed rat brain cannabinoid receptors in Xenopus oocytes. Neurosci Lett. 1995;186:91–94. doi: 10.1016/0304-3940(95)11289-9. [DOI] [PubMed] [Google Scholar]
- 29.Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptor in the brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hillard CJ, Jarrahian A. The movement of N-arachidonoyle-thanolamine (anandamide) across cellular membranes. Chem Phys Lipids. 2000;108:123–134. doi: 10.1016/s0009-3084(00)00191-2. [DOI] [PubMed] [Google Scholar]
- 31.Hillard CJ, Edgemont WS, Jarrahian A, Campbell WB. Accumulation of N-arachidonylethanolamide (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- 32.Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 33.Huang C-C, Lo S-W, Hsu K-S. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol (Lond) 2001;523(3):731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maccarrone M, van der Stelt M, Rossi A, Veldink GA, Vliegenthart JFG, Finazzi-Agrò A. Anandamide hydrolysis by human cells in culture and brain. J Biol Chem. 1998;273:32332–32339. doi: 10.1074/jbc.273.48.32332. [DOI] [PubMed] [Google Scholar]
- 35.Maccarrone M, Lorenzon T, Bari M, Melino G, Finazzi-Agrò A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J Biol Chem. 2000;275:31938–31945. doi: 10.1074/jbc.M005722200. [DOI] [PubMed] [Google Scholar]
- 36.Maccarrone M, Attinà M, Bari M, Cartoni A, Ledent C, Finazzi-Agrò A. Anandamide degradation and N-acylethanolamines level in wild-type and CB1 cannabinoid receptor knockout mice of different ages. J Neurochem. 2001;78:339–348. doi: 10.1046/j.1471-4159.2001.00413.x. [DOI] [PubMed] [Google Scholar]
- 37.Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mailleux P, Vanderhagen JJ. Dopaminergic regulation of cannabinoid receptor mRNA levels in the rat caudate-putamen: an in situ hybridization study. J Neurochem. 1993;61:1705–1712. doi: 10.1111/j.1471-4159.1993.tb09807.x. [DOI] [PubMed] [Google Scholar]
- 39.Matsuda LA, Bonner TI, Lolait SJ. Localization of cannabinoid receptor mRNA in the rat brain. J Comp Neurol. 1993;327:535–550. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- 40.Meschler JP, Howlett CA. Signal transduction interactions between CB1 cannabinoid and dopamine receptors in the rat and monkey striatum. Neuropharmacology. 2001;40:918–926. doi: 10.1016/s0028-3908(01)00012-0. [DOI] [PubMed] [Google Scholar]
- 41.Moesgaard B, Petersen G, Jaroszewski JW, Hansen HS. Age dependent accumulation of N-acyl-ethanolamine phospholipids in ischemic rat brain. A 31P NMR and enzyme activity study. J Lipid Res. 2000;41:985–990. [PubMed] [Google Scholar]
- 42.Otani S, Auclair N, Desce J-M, Roisin M-P, Crépel F. Dopamine receptors and group I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci. 1999;19:9788–9802. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 44.Piomelli D, Beltramo M, Glasnapp S, Lin SY, Goutopoulos A, Xie XQ, Makriyannis A. Structural determinants for recognition and translocation by the anandamide transporter. Proc Natl Acad Sci USA. 1999;96:5802–5807. doi: 10.1073/pnas.96.10.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci. 2001;22:565–572. doi: 10.1016/s0165-6147(00)01805-8. [DOI] [PubMed] [Google Scholar]
- 46.Schwartig RKW, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Prog Neurobiol. 1996;50:275–331. doi: 10.1016/s0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- 47.Sugiura T, Waku K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem Phys Lipids. 2000;108:89–106. doi: 10.1016/s0009-3084(00)00189-4. [DOI] [PubMed] [Google Scholar]
- 48.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 49.Tang K-C, Low MJ, Grandy DK, Lovinger DM. Dopamine-dependent synaptic plasticity in striatum during in vivo development. Proc Natl Acad Sci USA. 2001;98:1255–1260. doi: 10.1073/pnas.031374698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 51.Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 52.Ueda N, Puffenbarger RA, Yamamoto S, Deutsch DG. The fatty acid amide hydrolase (FAAH). Chem Phys Lipids. 2000;108:107–121. doi: 10.1016/s0009-3084(00)00190-0. [DOI] [PubMed] [Google Scholar]
- 53.Westlake TM, Howlett AC, Bonner TI, Matsuda LA, Herkenham M. Cannabinoid receptor binding and messenger RNA expression in human brain: an in vitro receptor autoradiographic and in situ hybridization histochemistry study of normal aged and Alzheimer's brain. Neuroscience. 1994;63:637–652. doi: 10.1016/0306-4522(94)90511-8. [DOI] [PubMed] [Google Scholar]
- 54.Wyley JL, Dewey MA, Jefferson RG, Winckler RL, Bridgen DT, Willoughby KA, Martin BR. Influence of phenylmethylsulfonyl fluoride on anandamide brain levels and pharmacological effects. Life Sci. 2000;67:1573–1583. doi: 10.1016/s0024-3205(00)00749-9. [DOI] [PubMed] [Google Scholar]
- 55.Zygmunt PM, Petersson J, Andersson DA, Chuang H-H, Sorgård M, Di Marzo V, Julius D, Högestätt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]