

Abstract
Glial fibrillary acidic protein (GFAP), the principal intermediate filament (IF) protein of mature astrocytes in the CNS, plays specific roles in astrocyte functions. GFAP has multiple phosphorylation sites at its N-terminal head domain. To examine the role of phosphorylation at these sites, we generated a series of substitution mutant mice in which phosphorylation sites (Ser/Thr) were replaced by Ala, in different combinations.Gfaphm3/hm3 mice carrying substitutions at all five phosphorylation sites showed extensive decrease in both filament formation and amounts of GFAP.Gfaphm1/hm1 andGfaphm2/hm2 mice, which carry substitutions at three of five sites and in different combinations, showed differential phenotypes. AlthoughGfaphm3/hm3 mice retained GFAP filaments in Bergmann glia in the cerebellum, the (Gfaphm3/hm3:Vim−/−) mice lacked GFAP filaments. Pulse-chase experiments of cultured astrocytes indicated that the Hm3-GFAP encoded byGfaphm3 was unstable particularly in the absence of vimentin, another IF protein. These results revealed the role of phosphorylation in turnover of GFAP and a synergistic role of GFAP and vimentin in the dynamics of glial filaments. The data further suggest that each of the phosphorylated sites has a distinct impact on the dynamics of GFAP.
Keywords: glial fibrillary acidic protein, GFAP, vimentin, phosphorylation, intermediate filament, astrocyte, filament formation, degradation, knock-in mice, gene targeting
There is a large family of intermediate filament (IF) proteins, the expression of which has a high degree of cell-type specificity. Glial fibrillary acidic protein (GFAP) is the principal IF protein of mature astrocytes in the CNS. Cumulative evidence from studies using GFAP null mutant mice underscored the importance of this protein in astrocyte functions (for review, seePekny 2001) (Gomi et al., 1995; Pekny et al., 1995, 1999; Liedtke et al., 1996; McCall et al., 1996; Shibuki et al., 1996; Nawashiro et al., 1998; Nawashiro et al., 2000; Anderova et al., 2001; Tanaka et al., 2001). The underlying mechanisms remain to be determined.
IF proteins are composed of the N-terminal head, central rod, and C-terminal tail domains (for review, see Steinert and Roop, 1988; Fuchs and Weber, 1994). Multiple amino acid residues in the head domain of GFAP, similar to other family members, are phosphorylated site specifically in vitro by several protein kinases, such as protein kinase A, protein kinase C, Ca2+/calmodulin-dependent protein kinase II, Rho kinase, and cdc2 kinase (Inagaki et al., 1990; Tokui et al., 1990; Tsujimura et al., 1994; Kosako et al., 1997). Although the head domain of GFAP is highly polymorphic among species, amino acid residues to be phosphorylated are conserved, thus implying the physiological importance of phosphorylation of GFAP. It has been suggested that phosphorylation at residues in the head domain inhibits polymerization and depolymerizes GFAP filaments in vitro (Inagaki et al., 1990). Transient overexpression using phosphorylation site-substitution mutants suggested that phosphorylation of GFAP by Rho kinase contributes to cytokinesis (Yasui et al., 1998).
We examined previously the distribution of phosphorylated GFAP in the mouse brain, and we suggested a role for phosphorylation in nondividing astrocytes (Takemura et al., 2002). To gain insight into the role of phosphorylation in vivo, we generated and characterized mutant mice with mutations introduced at GFAP phosphorylation sites.
MATERIALS AND METHODS
Generation of mutant mice
Substitution mutations were introduced at the Gfaplocus by gene targeting using embryonic day 14 (E14) embryonic stem (ES) cells, as described previously (Takemura et al., 2002). Substituted sequences were either obtained from the mutated human GFAP cDNA clones (a kind gift from Dr. M. Inagaki, Aichi Cancer Center Research Institute, Nagoya, Japan) (Yasui et al., 1998) or created by site-directed mutagenesis using PCR. A phosphoglycerate kinase promoter-driven neomycin resistance gene cassette (pgk-neo; a gift from Michael A. Rudnicki, McMaster University, Hamilton, Canada) (McBurney et al., 1991) flanked by loxP sites was excised using the Cre-loxP system after homologous recombination in one of the following: transient expression of Cre recombinase in targeted ES clones by electroporation of the plasmid pCre-Pac (Taniguchi et al., 1998) or intercross of mutant mice carryingloxP-pgk-neo-loxP and the ubiquitous Cre expression transgenic mouse line CAG-Cre (Sakai and Miyazaki, 1997). Genotypes of the mice were determined by PCR (Takemura et al., 2002). Substitutions introduced were verified by sequencing of the genomic DNA fragments amplified using a set of primers: 5′-GGGGTTCCTTGATCAATG-3′ and 5′-TGGCTCGAAGCTGGTTCAGT-3′. Substitution mutant mice and vimentin-deficient mice (Colucci-Guyon et al., 1994) were crossed to generate double-mutant mice.
Immunohistochemistry
Brains of mice were fixed by intracardiac perfusion with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, paraffin embedded, and sectioned (4–6 μm). Tissue sections were dewaxed and rehydrated.
Immunohistochemistry for GFAP or vimentin. Tissue sections in PBS were autoclaved for 5 min at 120°C and then were incubated for 1 hr at room temperature or overnight at 4°C with mouse monoclonal antibody against GFAP (1:100; GF12.24; Progen, Heidelberg, Germany) or vimentin (4H4; a kind gift from Dr. M. Inagaki). The primary antibodies were detected using Vectastain ABC kits (Vector Laboratories, Burlingame, CA) according to the recommendation of the manufacturer, followed by staining with 3,3′-diaminobenzidine tetrahydrochloride (Vector Laboratories). After halting the reaction, the sections were counterstained with hematoxylin (Merck, Darmstadt, Germany).
Immunohistochemistry for S100B. The rehydrated sections were treated in 0.1% trypsin in PBS for 30 min at 37°C and then were incubated overnight at 4°C with mouse monoclonal antibody against S100B (1:3000; SH-B1; Sigma, St. Louis, MO).
Northern blot analyses
Total mouse brain RNA was isolated from wild-type and mutant mice using RNeasy mini kits (Qiagen, Valencia, CA) according to the recommendation of the manufacturer. The RNA was electrophoresed on formaldehyde gels and transferred to nylon membrane (NEN, Boston, MA). Hybridization was done using as a probe 0.8 kb NheI digested fragment from mouse Gfap cDNA.
Quantification of GFAP from mouse brains
Mouse brains were homogenized in ice-cold buffer consisting of 50 mm phosphate buffer, pH 7.4, 2 mm EDTA, 2 mm EGTA, and a protease inhibitor cocktail (“Complete”; Roche Products, Hertforshire, UK). An equal volume of 2× SDS-sample buffer (0.25 m Tris-Cl, pH 6.8, 10% SDS, 20% glycerol, 10% 2-mercaptoethanol, and 0.02% bromophenol blue) was added. Protein (3 μg) was subjected to SDS-PAGE (10% polyacrylamide gel) and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA). After blocking with 5% skim milk in Tris-buffered saline, the membrane was immersed in primary antibodies against GFAP [1:500 (GF12.24; Progen) or 1:10,000 (Dako, Glostrup, Denmark)] or α-actin (1:2000; MAB1501; Chemicon, Temecula, CA), followed by immersion in peroxidase-conjugated rat anti-mouse IgG (1:5000; Zymed,San Francisco, CA) or anti-rabbit IgG (1:5000; Zymed). Signals were detected using the ECL (Amersham Biosciences, Little Chalfont, UK). Densitometry to quantify GFAP was done using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/nih-image/).
Preparation of astrocyte-enriched primary cultures and immunocytochemistry
Cerebral astrocyte cultures were prepared from E18.5 embryos, as described previously (Kato et al., 1979). After dissection and trypsin digestion of cerebral cortices, the dissociated cells were suspended in DMEM (Invitrogen, San Diego, CA) supplemented with 10% fetal bovine serum (FBS) (JRH Biologicals, Lenexa, KS) and antibiotics and then plated onto cell culture flasks (Falcon, Becton Dickinson, Rutherford, NJ).
Cells grown on glass chamber slides (Nalge Nunc International, Naperville, IL) were fixed in 3.7% formaldehyde in PBS for 10 min at room temperature and permeabilized with methanol for 5 min at −30°C. The cells were then incubated for 2 hr at room temperature with an anti-GFAP antibody (1:500; Dako). After washing in PBS, slides were incubated for 1 hr at room temperature with Alexa546 dye-conjugated secondary antibodies diluted at 1:1000 (Molecular Probes, Eugene, OR).
Pulse-chase experiments
Metabolic labeling with 35S-Met/Cys (100 μCi/ml, 15 min) was done in DMEM lacking Met/Cys (Invitrogen) and containing 10% FBS. Labeled cells were either processed immediately or cultured in normal medium for various chase periods (0, 1, 24, and 48 hr). The cells were scraped off with 171 mmNaCl, 6 mm sodium phosphate, pH 7.4, 600 mmKCl, 0.5% (w/v) Triton X-100, 1 mm EDTA, and protease inhibitor cocktail (“Complete;” Roche). Fractionation into cytosolic and residual cytoskeletal fractions was done by centrifugation: 15,000 × g for 15 min at 4°C. The cytosolic fractions were used for immunoprecipitation with an anti-GFAP antibody (Dako). Immunoprecipitates and cytoskeletal fractions were subjected to SDS-PAGE. Quantification of35S-GFAP was done using a Fujifilm (Tokyo, Japan) BAS-2500 image analyzer. Amounts of GFAP were quantified by immunoblotting using an anti-pan GFAP antibody GF12.24 and ECL.
Generation of antibodies against the human GFAP head domain
Antibodies specific to humanized GFAP were raised in a rabbit using a synthetic peptide corresponding to human GFAP head domain H2N-CGGLAPGRRLGPGT-CONH coupled to keyhole limpet hemocyanin. Antigen solution was injected every 28 d using the RIBI adjuvant system (Ribi ImmunoChem Research, Hamilton, MT). The rabbit was bled 3 months after the initial injection.
RESULTS
Generation of GFAP phosphorylation site-substitution mutant mice
The N-terminal head domain of GFAP contains multiple phosphorylation sites (Thr7, Ser8, Ser13, Ser17, and Ser38 in the numbering system for human GFAP). Our evidence shows that the head domain of human GFAP is compatible with mice in terms of phosphorylation (Takemura et al., 2002). To examine the role of GFAP phosphorylation in vivo, we developed three lines of knock-in mice: Gfap hm1, Gfap hm2, and Gfap hm3 knock-in mice, which express mutated GFAP carrying the human head domain sequence containing different combinations of substitution of Ala for Ser/Thr at the phosphorylation sites Hm1-GFAP (Ala for Thr7, Ser13, and Ser38), Hm2-GFAP (Ser13, Ser17, and Ser38), or Hm3-GFAP (Thr7, Ser8, Ser13, Ser17, and Ser38) (Fig.1A). To generate mutant mice, we replaced exon 1 of the Gfap gene with the humanized exon by gene targeting, as reported previously (Takemura et al., 2002) (Fig. 1B). The linearized targeting vector was electroporated into ES cells, and homologous recombinants were identified by Southern blot hybridization. TheloxP-pgk-neo-loxP gene cassette integrated into the genome was excised by transient expression of Cre recombinase in targeted ES cells or by crossing the germline transmittants with Cre transgenic mice (Sakai and Miyazaki, 1997). Homozygotes born from intercrosses of the resulting heterozygotes of each line grew normally and showed no obvious abnormalities. Northern blot analyses of total brain RNA using mouse GFAP cDNA and mouse β-actin cDNA fragments as a probe showed that relative amounts of Gfap mRNA in mutant mice were equal to those in the wild-type mice (Fig.1C).
Fig. 1.

Generation of Gfap substitution mutant mice. A, Schemes showing GFAP phosphorylation sites and mutant forms of GFAP. N-terminal amino acid sequences of human, mouse, and chimeric GFAP (Hwt-GFAP) are aligned. Combinations of GFAP phosphorylation sites and substitution mutations are aligned below. Only substituted sites are indicated. Asterisksindicate phosphorylation sites identified. B, Knock-in strategy of the human head domain sequence into the mouseGfap locus. Open boxes denote exons 1–9. The gray line in the exon 1 represents the human sequence containing mutations in phosphorylation sites. Filled triangles denote loxP sites. Thepgk-neo cassette (neo) was removed by the Cre-loxP system to give rise to theGfaphmx allele. hmxrepresents hm1, hm2, orhm3. A, ApaI.C, Northern blot analysis of the total brain RNA from adult wild-type (wt),Gfaphm1/hm1 (hm1),Gfaphm2/hm2 (hm2),Gfaphm3/hm3 (hm3),Vim−/− (v−), andGfaphm3/hm3:Vim−/−(hm3:v−) mouse brains using the 0.8 kb fragment of mouse Gfap cDNA as a probe compared with β-actin mRNA.
To verify the mutations in these mice, genomic sequences encoding the head domain were PCR amplified and sequenced. We also examined reactivity of the optic nerves fromGfaphm1/hm1,Gfaphm2/hm2, andGfaphm3/hm3 to anti-phospho-GFAP monoclonal antibodies, as described previously (Takemura et al., 2002). The optic nerves from Gfaphm3/hm3 mice showed no reactivity to the antibodies tested, i. e., TMG7 (for Thr7), YC10 (for Ser8), and KT13 (for Ser13) (data not shown). The optic nerves from Gfaphm1/hm1 andGfaphm2/hm2 mice were not reactive to TMG7 and KT13, and KT13, respectively (data not shown). These results confirmed that these mice carried the intended substitutions.
Distribution of GFAP filaments in mutant brains
When brain sections from adult wild-type and mutant animals were processed for immunohistochemistry, we found no significant differences regarding distribution and density of the cells labeled with anti-S100B antibodies (Fig.2C,F,I,L; Table 1), which suggested the normal development of astrocytes in all substitution mutants. In the immunohistochemistry for GFAP, we found no differences between wild-type and Gfaphwt/hwt mouse brains, as noted previously (Takemura et al., 2002). However, in theGfaphm3/hm3 mouse brain, we did observe a significant decrease in GFAP immunoreactivity in most brain regions, including hippocampus, midbrain, olfactory bulb, and neocortex (Fig.2J). Filamentous structures of GFAP were markedly lost in long processes of astrocytes. However, GFAP immunoreactivity in Bergmann glial processes in the cerebellum was indistinguishable between wild-type and Gfaphm3/hm3 mice (Fig. 2K), and the decrease was moderate in pons and subventricular zone (data not shown). Peri-vascular astrocytes in the hippocampus and other brain areas tended to have strong immunoreactivities of GFAP (Fig. 2J). InGfaphm1/hm1mouse brains, we found no significant differences in GFAP immunoreactivity from wild types (Fig.2D, Table 1). InGfaphm2/hm2 mice, there was a slight decrease in intensity of GFAP staining and in the numbers of astrocytes bearing GFAP-positive processes, in some brain areas (Fig.2G, Table 1). Cellular processes labeled with anti-GFAP antibodies tended to be short inGfaphm2/hm2 mice. These observations suggest the following: (1) phosphorylation of GFAP affects glial filament formation, (2) its role may vary among subsets of astrocytes, and (3) each of the phosphorylation sites may have distinct roles in terms of filament formation.
Fig. 2.
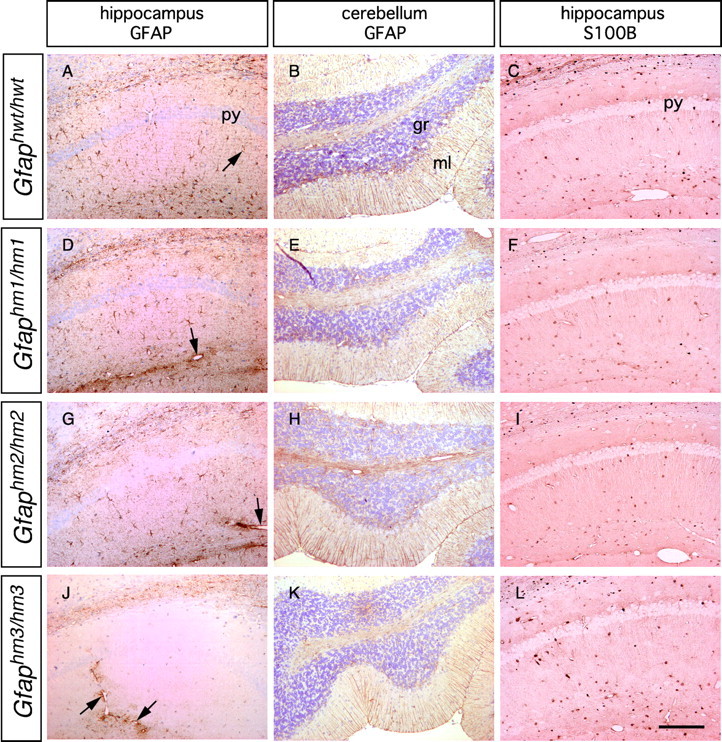
Immunohistochemical detection of GFAP and S100B in mutant mouse brains. Sections of the hippocampus and cerebellum from adult animal brains were stained with anti-GFAP or anti-S100B antibody. A, B,D, E, G, H,J, and K were counterstained with diluted hematoxylin. Note that GFAP immunoreactivity was greatly reduced in the hippocampus but not in the cerebellum ofGfaphm3/hm3 mice.Arrows indicate GFAP signals surrounding blood vessels. S100B immunoreactivity was not changed in the hippocampi of all genotypes (C, F, I,L). py, Pyramidal cell layer;gr, granule cell layer; ml, molecular layer. Scale bar, 200 μm.
Table 1.
Number of astrocytes bearing S100B-positive or GFAP-positive processes in corresponding hippocampal sections of mutant mice
| Mice | S100B-positive astrocytes | GFAP-positive process bearing astrocytes | ||
|---|---|---|---|---|
| Number1-a | pvalue1-b | Number1-a | pvalue1-b | |
| Wild type | 25.9 ± 2.8 | 24.1 ± 2.0 | ||
| Gfaphwt/hwt | 25.8 ± 2.9 | 0.9384 | 24.5 ± 2.3 | 0.6741 |
| Gfaphm1/hm1 | 26.7 ± 3.9 | 0.6025 | 22.2 ± 5.7 | 0.2242 |
| Gfaphm2/hm2 | 26.0 ± 2.3 | 0.9310 | 12.7 ± 2.1 | <0.0001 |
| Gfaphm3/hm3 | 26.6 ± 2.4 | 0.2208 | 0.4 ± 0.7 | <0.0001 |
Cells were counted in 700 × 300 μm areas arbitrarily set in the CA1 region only if a nucleus was present in the section. The sections were from two (S100B) or three (GFAP) animals, and five sections of each animal were analyzed. Data are presented as the mean ± SD.
Compared with wild type byt test.
Compensation of Hm3-GFAP by vimentin
Subventricular zone astrocytes and Bergmann glia express vimentin, which assembles together with GFAP into glial filaments. We predicted that vimentin masked the deficits of Hm3-GFAP in these subsets, although the mutations did not alter expression of vimentin, as was similar to the case for GFAP null mutant mice (Gomi et al., 1995). To test this possibility, we generated double-mutant mice that had deficits in GFAP phosphorylation (Gfaphm3/hm3) and vimentin deficiency (Vim−/−). The double-mutant (Gfaphm3/hm3:Vim−/−) mice grew normally and showed no obvious anatomical abnormality. Decrease in GFAP immunoreactivity was more severe inGfaphm3/hm3:Vim−/−mice than Gfaphm3/hm3 mice in other brain areas, including pons and subventricular zone astrocytes. The immunoreactivity was almost lost in Bergmann glial processes ofGfaphm3/hm3:Vim−/−mice (Fig. 3N) but not so in any other control littermates (Fig.3B,F,J). No differences in anti-S100B staining were noted among genotypes (Fig.3D,H,L,P). These observations mean that the presence of vimentin, in part, compensates for deficits in Hm3-GFAP in glial filament formation.
Fig. 3.
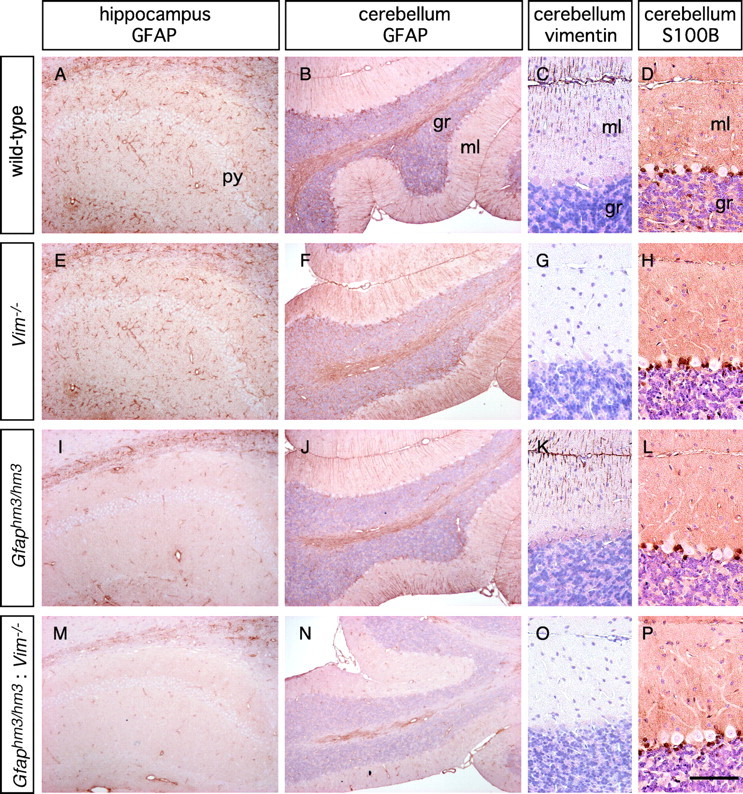
Vimentin-dependent Hm3-GFAP filament formation in the mouse brain. Sections of the hippocampus and cerebellum from adult animal brains of indicated genotypes were stained with anti-GFAP, anti-vimentin, or anti-S100B antibody.B–D, F–H, J–L, andN–P were counterstained with diluted hematoxylin. Note that Hm3-GFAP does not form filaments in the cerebellar cortex in the absence of vimentin. py, Pyramidal cell layer;gr, granule cell layer; ml, molecular layer. Scale bar: A, B, E,F, I, J, M,N, 200 μm; C, D,G, H, K, L,O, P, 100 μm.
Amounts of GFAP were decreased inGfaphm1/hm1andGfaphm3/hm3 but not inGfaphm2/hm2 mice
Results of immunohistochemical analyses showed that the quantity of GFAP changed in brains of mutant mice. We estimated amounts of GFAP by immunoblotting of total protein samples prepared from whole brains of these mice. The same blots were subjected to analysis with an anti-actin antibody to normalize the amounts of loaded proteins. Immunoblots with a pan-GFAP antibody detected lower molecular weight products in substitution mutants in addition to intact GFAP. As discussed below, the lower molecular weight products were proteins lacking N-terminal sequences. The ratio of truncated/intact products was small in Gfaphm1/hm1 (7.6% of total) and Gfaphm2/hm2 (8.5% of total) mice but relatively large (21.2% of total) inGfaphm3/hm3 mice. The amount of intact GFAP in Gfaphm2/hm2 andVim−/− mice was equivalent to that in wild-type mice. However, consistent with immunohistochemical data, GFAP was significantly decreased in Gfaphm3/hm3mice and was further decreased inGfaphm3/hm3:Vim−/−mice (Fig. 4). Significant decrease in GFAP was also observed in Gfaphm1/hm1mice, although there was no detectable abnormality in immunohistochemical analyses using anti-GFAP antibodies. These results suggested that phosphorylation affected the stability of GFAP in vivo and that different phosphorylation sites may have distinct roles in the dynamics of GFAP.
Fig. 4.

Effects of the mutations on GFAP amounts in brains. A, Immunoblot with anti-GFAP or actin antibody using total protein from whole brains of adult wild-type (wt), Gfaphm1/hm1(hm1), Gfaphm2/hm2(hm2), Gfaphm3/hm3(hm3), Vim−/−(v−), andGfaphm3/hm3:Vim−/−(hm3:v−) mice. B, Amount of GFAP in mutant mice brains. GFAP was quantified by densitometric analysis, using actin as an internal standard. Open andfilled bars represent total amounts of GFAP, including lower molecular weight products and only those of intact products, respectively. Indicated are the means ± SEM (wild types,n = 9; other genotypes, n = 3). *p < 0.05; **p < 0.001;NS, not significant; compared with wild type byt test.)
Glial filaments in primary cultured astrocytes
To further characterize the substituted GFAP in glial filament formation, we prepared primary cultured astrocytes from wild-type,Gfaphm3/hm3,Gfaphm3/hm3:Vim−/−, and Vim−/− E18.5 embryonic brains. Immunocytochemistry for GFAP revealed thatGfaphm3/hm3 astrocytes had no obvious abnormality compared with the wild-type cells in which GFAP filaments were clear, fine, and arrayed in parallel. In contrast, GFAP filaments in Vim−/− astrocytes were sparse, somewhat irregularly arrayed, and sometimes disrupted, as noted by others (Galou et al., 1996) (Fig.5C). Astrocytes fromGfaphm3/hm3:Vim−/−mice had the severest phenotypes. In addition to abnormalities observed in Vim−/− astrocytes, dot-like aggregate structures appeared inGfaphm3/hm3:Vim−/−astrocytes (Fig. 5D). These results support the notion of a compensatory role for vimentin regarding deficits of Hm3-GFAP in filament formation.
Fig. 5.

Abnormal aggregate formation of Hm3-GFAP in vimentin-deficient astrocytes. Primary cultured astrocytes were prepared from wild-type (A),Gfaphm3/hm3 (B),Vim−/− (C), andGfaphm3/hm3:Vim−/−(D) embryos. Glial filaments were immunostained with anti-GFAP antibody. The filamentous structures were sparse and irregularly arrayed in the absence of vimentin (asterisks in C and D). Dot-like structures were observed inGfaphm3/hm3:Vim−/−astrocytes (arrowheads in D). Scale bar, 50 μm.
Cultured astrocytes as well as reactive astrocytes in injured brains express nestin as the third component of astroglial IF. Immunohistochemistry using anti-nestin antibodies revealed unaltered nestin filaments in Gfaphm3/hm3 astrocytes but a disappearance in Vim−/− astrocytes (data not shown). Previous work (Eliasson et al., 1999) (E. Colucci-Guyon and C. Babinet, unpublished observation) demonstrated that vimentin but not GFAP is required for polymerization of nestins to form filaments. The results in this study confirmed these previous observations.
Metabolism of Hm3-GFAP
We compared the turnover of35S-methionine pulse-labeled wild-type, Hwt-GFAP and Hm3-GFAP in primary cultured astrocytes. After 15 min of pulse labeling, cells were cultured for different periods in nonradioactive medium. The labeled cells were fractionated into cytoskeletal and cytosolic components. Cytoskeletal fractions and immunoprecipitates with anti-GFAP antibodies from cytosolic fractions were analyzed using SDS-PAGE and then autoradiography (Fig.6). The same samples were also subjected to immunoblots using anti-pan GFAP antibodies to assess total amounts of GFAP. Because significant differences between wild-type and Hwt-GFAP were not observed, data from both samples were combined for statistical analysis. Turnover of Hm3-GFAP tended to be more rapid than that of wild-type GFAP (Fig. 7A), albeit with no statistical significance. In both types of cultures, amounts of radioactive GFAP in cytoskeletal fractions were larger at 1 hr after labeling than at 0 hr. In contrast, amounts of GFAP immunoprecipitated from cytosolic fractions were smaller at 1 hr after labeling than at 0 hr. These results suggested that a large population of newly synthesized GFAP was integrated into glial filaments over a 1 hr period. When we analyzed the influence of substitution mutation in the absence of vimentin, we observed a huge difference between Hm3-GFAP and wild-type GFAP. In the absence of vimentin, turnover of Hm3-GFAP was rapid compared with that of wild-type GFAP in both cytoskeletal and cytosolic fractions (Figs. 6, 7). The ratios of de novosynthesized GFAP to original GFAP in cytosolic and in cytoskeletal fractions (Fig. 7B) were higher inGfaphm3/hm3:Vim−/−astrocytes than in Vim−/− astrocytes immediately after pulse labeling, and these differences were negligible in 24 hr (Fig. 7B). Thus, Hm3-GFAP apparently had deficits at post-translational but probably not at translational stages. Lower molecular weight products (similar to Fig.4 data) were detected in cultured astrocytes from substitution mutant astrocytes (Fig. 6K). To examine the character of these products, we prepared immunoblots using antisera specific to the human GFAP head domain. Lower bands were not detected using anti-human head domain antibodies, which means that lower molecular weight products were head domain-truncated ones. We used data on intact products in the analysis mentioned above (Fig. 7). However, it should be noted that the same conclusions were obtained when the truncated products were integrated into the analyses.
Fig. 6.

Pulse-chase experiments of GFAP. Primary cultured astrocytes prepared from wild-type (A,F), Gfaphwt/hwt(B, G),Gfaphm3/hm3 (C,H), Vim−/−(D, I), andGfaphm3/hm3:Vim−/−(E, J) embryos were labeled for 15 min with 35S-Met/Cys, followed by immediate harvesting or chasing for 1, 24, or 48 hr. Autoradiographs of Triton X-100-insoluble cytoskeletal fractions and immunoprecipitates from Triton X-100-soluble cytosolic fractions with anti-GFAP antibody are shown inA–E and F–J. K, Lower molecular weight product detected by the GFAP antibody (asterisk) was not detected using a head domain-specific antibody. Immunoprecipitates fromGfaphm3/hm3 astrocytes were analyzed using autoradiography (lane 1), immunoblot with antibody to GFAP head domain (lane 2), or immunoblot with GF 12.24 as pan-GFAP antibody (lane 3).
Fig. 7.

Rapid turnover of Hm3-GFAP in the absence of vimentin. Data from pulse-chase experiments using primary cultured astrocytes of control (diamonds; n = 10), Gfaphm3/hm3(squares; n = 6),Vim−/− (triangles;n = 6), orGfaphm3/hm3:Vim−/−(circles; n = 6) genotypes were plotted. Relative radioactivities of GFAP in cytoskeletal fractions (open symbols) and cytosolic fractions (filled symbols) in the presence (left) or absence (right) of vimentin were shown in A. Ratios of radioactive GFAP to total GFAP estimated by immunoblots were compared betweenVim−/− andGfaphm3/hm3:Vim−/−in cytosolic (B, left) and cytoskeletal (B, right) fractions. Error bars represent SEM. NS, Not significant; *p < 0.02; **p < 0.0001; repeated-measures ANOVA
DISCUSSION
To gain insight into the role of GFAP phosphorylation in the brain, we generated three lines of knock-in mice, all of which expressed mutated forms of GFAP carrying the human head domain sequence containing different combinations of substitution of Ala for Ser/Thr at phosphorylation sites. These mutations differentially affected the stability of the proteins and of filament formation, indicating the role of GFAP phosphorylation in dynamics of glial filaments.
The Gfaphm1/hm1 mouse has substitution mutations at Rho kinase-dependent phosphorylation sites. This combination of mutations was reported to impair segregation of glial filaments into daughter cells in cytokinesis (Yasui et al., 1998). However, we did not observe histological abnormalities caused by impairment of cytokinesis inGfaphm1/hm1 mice or inGfaphm2/hm2 andGfaphm3/hm3 mice, even in the subventricular zone in which astrocytes are constitutively generated throughout their life span. Furthermore, we observed no detectable abnormalities in the development of astrocytes, as evaluated using S100B immunohistochemistry. Together with the previous findings that phospho-GFAP immunoreactivities are enriched in nondividing astrocytes (Takemura et al., 2002), our results suggest that phosphorylation of GFAP might play a role that is independent of cell division in the brain.
We observed severe reductions in GFAP immunoreactivity, indicating poor filament formation, and in amounts of GFAP in the brains ofGfaphm3/hm3 mice, which carry substitutions in all phosphorylation sites at the head domain. Because the mutation in Gfaphm3/hm3 mice did not alter the levels of Gfap mRNA (Fig. 1C), either the slowing of translation and/or accelerating degradation might lead to a decrease in amounts of GFAP. However, it should be noted that GFAP immunoreactivity was not affected in cerebellar Bergmann glia and some other subsets of astrocytes in Gfaphm3/hm3mice. Thus, it was unlikely that the decrease of GFAP was caused by the general translational problem of the mutated gene. Bergmann glia and other cells that showed significant GFAP-immunoreactivities in theGfaphm3/hm3 mice coexpress vimentin. Vimentin, as well as GFAP, belongs to the type III IF protein subfamily and can form glial filaments together with GFAP. This suggested that vimentin in part compensated for deficits in Hm3-GFAP in filament formation and stability. To test this possibility, we prepared double-mutant (Gfaphm3/hm3:Vim−/−) mice by crossing. These double mutants showed a marked loss of immunoreactivities in Bergmann cells and a loss or decrease in other subsets of astrocytes, including perivascular and subventricular astrocytes. Consistent with data on immunohistochemistry, immunoblot analysis indicated an additional decrease in amounts of GFAP in the double mutants compared with Gfaphm3/hm3mice (Fig. 4). Although sample numbers are limited, we also observed a clear decrease in GFAP immunoreactivities in Bergmann glia inGfaphm1/hm1:Vim−/−andGfaphm2/hm2:Vim−/−mice (data not shown). These results clearly indicate a cooperative role between vimentin and GFAP in vivo, as has been suggested previously (Eliasson et al., 1999).
Cooperation between vimentin and GFAP was further emphasized in cell culture studies of double-mutant astrocytes. Most cultured astrocytes coexpress vimentin and GFAP. Hm3-GFAP formed filaments indistinguishable from those in wild-type astrocytes, in the presence of vimentin (Fig. 7A). On the contrary, in the absence of vimentin, wild-type GFAP formed slightly irregularly shaped and sometimes truncated filaments (Fig. 5C), as reported previously (Galou et al., 1996). Hm3-GFAP in the double-mutant astrocytes formed truncated or granular structures. Pulse-chase experiments revealed that cytosolic Hm3-GFAP was clearly stabilized in the presence of vimentin, probably by forming a heterodimer or a hetero-oligomer (Fig. 7). Analysis of insoluble fractions suggested that the Hm3-GFAP also had a longer half-life in heteropolymers (Fig.7B). These observations provide evidence for the proteolysis-protective role of interaction between vimentin and GFAP.
It has been reported that two types of proteolytic mechanisms are involved in the degradation of IF proteins. There is evidence that ubiquitination is involved in degradation of keratin 8 and 18 and that phosphorylation of keratin 8 regulates ubiquitination-mediated turnover of keratins (Ku et al., 2000). Although we do not have direct evidence for the ubiquitination of GFAP, similar mechanisms may regulate turnover the glial filaments. Lee et al. (2000) detected 51 and 48 kDa GFAP molecules in cultured rat astrocytes. Based on pharmacological analysis, they suggested that the calcium-dependent protease calpain I is involved in generating smaller 48 kDa GFAP molecules. As shown in Figures 4 and 6, N-terminal truncated GFAP molecules were evident in the brain and cultured astrocytes of theGfaphm3/hm3 mouse. These results suggest that phosphorylation may also regulate the calpain I-dependent degradation of GFAP.
Interestingly, Gfaphm1/hm1 andGfaphm2/hm2 mice showed milder and differential phenotypes. Gfaphm1/hm1 mice, which carried substitutions at Thr7, Ser13, and Ser38, showed a decrease in amounts of GFAP but unaltered immunoreactivity in histochemistry. On the contrary,Gfaphm2/hm2 mice, which carried substitutions at Ser13, Ser17, and Ser38, did not show significant decreases in amounts of GFAP but did show abnormality in immunohistochemistry. These results suggest that Thr7 and Ser17 and perhaps each of other phosphorylation sites has a differential impact on distinct dynamic states of GFAP, i.e., degradation, filament formation, and probably also intracellular transportation. Combinations of these distinct phospho-residues might determine the entire dynamics of GFAP.
The present findings suggest that phosphorylation contributes to stabilization of GFAP in both cytosolic and cytoskeletal fractions. Data on in vitro reconstruction experiments suggested that phosphorylation disassembles GFAP filaments and dephosphorylated forms have a greater potential to assemble (Inagaki et al., 1990). Based on these observations, we propose a model for the role of phosphorylation of GFAP in the dynamics of glial filaments (Fig.8). The maintenance and dynamic behavior of IFs are thought to be regulated by equilibrium between soluble and filament fractions (Vikstrom et al., 1992; Paramio et al., 1997). Phosphorylation will shift the equilibrium to the soluble form and protect it from proteolytic degradation. Phosphorylation of soluble subunits will maintain them as a larger pool and thus facilitate reforming IFs, but decrease in the soluble pool will lead to the breakdown of IFs. In the case of keratin 8, it has been reported that the turnover of phosphorylation is extremely rapid compared with stability of the protein (Paramio, 1999). This kinetics should be beneficial for reforming IF networks and thus for plasticity of cell shapes. It is interesting to note that the modulation of Rho kinase activities alters cell shapes of N2a cells and also the phosphorylation state of vimentin (Nakamura et al., 2000); thus, we assume that similar mechanisms are applicable regarding the dynamics of vimentin. We suggest that the phosphorylation of GFAP (and perhaps also of vimentin) regulates the structural plasticity of glial filaments and eventually functions of astrocytes.
Fig. 8.

A proposed model for the role of GFAP phosphorylation. GFAP phosphorylation may play a dual role. The phosphorylation protects GFAP from proteolysis and contributes to shift soluble-filament equilibrium to the left. The larger pool of soluble subunits protected by phosphorylation and partial depolymerization of filamentous subunits triggered by phosphorylation will synergistically accelerate the structural plasticity of glial filaments.
Footnotes
This work was supported in part by a grant from the Ministry of Welfare (S.I.). M.T. is supported by the Junior Research Associate Program at RIKEN. We thank M. Inagaki for providing human GFAP cDNA and anti-phospho-GFAP and anti-vimentin monoclonal antibodies, M. Hooper, M. Rudnicki, S. Aizawa, and J.-I. Miyazaki for providing E14 ES cell line, pgk-neo, pMC1-DTA, and CAG-Cre transgenic mice, respectively, and T. Yamanaka for assistance in the early stage of this work. We thank all members of our laboratories and M. Murate for helpful discussions and technical advice, K. Inoue, T. Iwasato, H. Nishiyama, and H. Kanki for critical comments on this manuscript, and M. Ohara for editing and language assistance. M.T. thanks K. Ito for continuous encouragement.
Correspondence should be address to Dr. Shigeyoshi Itohara, Laboratory for Behavioral Genetics, RIKEN Brain Science Institute, 2-1 Hirosawa, Wako 351-0198, Japan. E-mail: sitohara@brain.riken.go.jp.
H. Gomi's present address: Institute for Molecular and Cellular Regulation, Gunma University, 3-39-15 Showa, Maebashi 371-8512, Japan.
REFERENCES
- 1.Anderova M, Kubinova S, Mazel T, Chvatal A, Eliasson C, Pekny M, Sykova E. Effect of elevated K+, hypotonic stress, and cortical spreading depression on astrocyte swelling in GFAP-deficient mice. Glia. 2001;35:189–203. doi: 10.1002/glia.1084. [DOI] [PubMed] [Google Scholar]
- 2.Colucci-Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell. 1994;79:679–694. doi: 10.1016/0092-8674(94)90553-3. [DOI] [PubMed] [Google Scholar]
- 3.Eliasson C, Sahlgren C, Berthold CH, Stakeberg J, Celis JE, Betsholtz C, Eriksson JE, Pekny M. Intermediate filament protein partnership in astrocytes. J Biol Chem. 1999;274:23996–24006. doi: 10.1074/jbc.274.34.23996. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- 5.Galou M, Colucci-Guyon E, Ensergueix D, Ridet JL, Gimenez y Ribotta M, Privat A, Babinet C, Dupouey P. Disrupted glial fibrillary acidic protein network in astrocytes from vimentin knockout mice. J Cell Biol. 1996;133:853–863. doi: 10.1083/jcb.133.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomi H, Yokoyama T, Fujimoto K, Ikeda T, Katoh A, Itoh T, Itohara S. Mice devoid of the glial fibrillary acidic protein develop normally and are susceptible to scrapie prions. Neuron. 1995;14:29–41. doi: 10.1016/0896-6273(95)90238-4. [DOI] [PubMed] [Google Scholar]
- 7.Inagaki M, Gonda Y, Nishizawa K, Kitamura S, Sato C, Ando S, Tanabe K, Kikuchi K, Tsuiki S, Nishi Y. Phosphorylation sites linked to glial filament disassembly in vitro locate in a non-alpha-helical head domain. J Biol Chem. 1990;265:4722–4729. [PubMed] [Google Scholar]
- 8.Kato T, Chiu TC, Lim R, Troy SS, Turriff DE. Multiple molecular forms of glia maturation factor. Biochim Biophys Acta. 1979;579:216–227. doi: 10.1016/0005-2795(79)90100-4. [DOI] [PubMed] [Google Scholar]
- 9.Kosako H, Amano M, Yanagida M, Tanabe K, Nishi Y, Kaibuchi K, Inagaki M. Phosphorylation of glial fibrillary acidic protein at the same sites by cleavage furrow kinase and Rho-associated kinase. J Biol Chem. 1997;272:10333–10336. doi: 10.1074/jbc.272.16.10333. [DOI] [PubMed] [Google Scholar]
- 10.Ku NO, Omary MB, Liao J, Price D. Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J Cell Biol. 2000;149:547–552. doi: 10.1083/jcb.149.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee YB, Du S, Rhim H, Lee EB, Markelonis GJ, Oh TH. Rapid increase in immunoreactivity to GFAP in astrocytes in vitro induced by acidic pH is mediated by calcium influx and calpain I. Brain Res. 2000;864:220–229. doi: 10.1016/s0006-8993(00)02180-6. [DOI] [PubMed] [Google Scholar]
- 12.Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron. 1996;17:607–615. doi: 10.1016/s0896-6273(00)80194-4. [DOI] [PubMed] [Google Scholar]
- 13.McBurney MW, Sutherland LC, Adra CN, Leclair B, Rudnicki MA, Jardine K. The mouse Pgk-1 gene promoter contains an upstream activator sequence. Nucleic Acids Res. 1991;19:5755–5761. doi: 10.1093/nar/19.20.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCall MA, Gregg RG, Behringer RR, Brenner M, Delaney CL, Galbreath EJ, Zhang CL, Pearce RA, Chiu SY, Messing A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc Natl Acad Sci USA. 1996;93:6361–6366. doi: 10.1073/pnas.93.13.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura Y, Hashimoto R, Amano M, Nagata K, Matsumoto N, Goto H, Fukusho E, Mori H, Kashiwagi Y, Kudo T, Inagaki M, Takeda M. Localized phosphorylation of vimentin by rho-kinase in neuroblastoma N2a cells. Genes Cells. 2000;5:823–837. doi: 10.1046/j.1365-2443.2000.00372.x. [DOI] [PubMed] [Google Scholar]
- 16.Nawashiro H, Messing A, Azzam N, Brenner M. Mice lacking GFAP are hypersensitive to traumatic cerebrospinal injury. NeuroReport. 1998;9:1691–1696. doi: 10.1097/00001756-199806010-00004. [DOI] [PubMed] [Google Scholar]
- 17.Nawashiro H, Brenner M, Fukui S, Shima K, Hallenbeck JM. High susceptibility to cerebral ischemia in GFAP-null mice. J Cereb Blood Flow Metab. 2000;20:1040–1044. doi: 10.1097/00004647-200007000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Paramio JM. A role for phosphorylation in the dynamics of keratin intermediate filaments. Eur J Cell Biol. 1999;78:33–43. doi: 10.1016/S0171-9335(99)80005-3. [DOI] [PubMed] [Google Scholar]
- 19.Paramio JM, Casanova ML, Alonso A, Jorcano JL. Keratin intermediate filament dynamics in cell heterokaryons reveals diverse behaviour of different keratins. J Cell Sci. 1997;110:1099–1111. doi: 10.1242/jcs.110.9.1099. [DOI] [PubMed] [Google Scholar]
- 20.Pekny M. Astrocytic intermediate filaments: lessons from GFAP and vimentin knock-out mice. Prog Brain Res. 2001;132:23–30. doi: 10.1016/S0079-6123(01)32062-9. [DOI] [PubMed] [Google Scholar]
- 21.Pekny M, Leveen P, Pekna M, Eliasson C, Berthold CH, Westermark B, Betsholtz C. Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J. 1995;14:1590–1598. doi: 10.1002/j.1460-2075.1995.tb07147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pekny M, Eliasson C, Siushansian R, Ding M, Dixon SJ, Pekna M, Wilson JX, Hamberger A. The impact of genetic removal of GFAP and/or vimentin on glutamine levels and transport of glucose and ascorbate in astrocytes. Neurochem Res. 1999;24:1357–1362. doi: 10.1023/a:1022572304626. [DOI] [PubMed] [Google Scholar]
- 23.Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- 24.Shibuki K, Gomi H, Chen L, Bao S, Kim JJ, Wakatsuki H, Fujisaki T, Fujimoto K, Katoh A, Ikeda T, Chen C, Thompson RF, Itohara S. Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron. 1996;16:587–599. doi: 10.1016/s0896-6273(00)80078-1. [DOI] [PubMed] [Google Scholar]
- 25.Steinert PM, Roop DR. Molecular and cellular biology of intermediate filaments. Annu Rev Biochem. 1988;57:593–625. doi: 10.1146/annurev.bi.57.070188.003113. [DOI] [PubMed] [Google Scholar]
- 26.Takemura M, Nishiyama H, Itohara S. Distribution of phosphorylated glial fibrillary acidic protein in the mouse central nervous system. Genes Cells. 2002;7:295–307. doi: 10.1046/j.1365-2443.2002.00513.x. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka H, Katoh A, Oguro K, Shimazaki K, Gomi H, Itohara S, Masuzawa T, Kawai N. Disturbance of hippocampal long-term potentiation after transient ischemia in GFAP deficient mice. J Neurosci Res. 2001;67:11–20. doi: 10.1002/jnr.10004. [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi M, Sanbo M, Watanabe S, Naruse I, Mishina M, Yagi T. Efficient production of Cre-mediated site-directed recombinants through the utilization of the puromycin resistance gene, pac: a transient gene-integration marker for ES cells. Nucleic Acids Res. 1998;26:679–680. doi: 10.1093/nar/26.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tokui T, Yamauchi T, Yano T, Nishi Y, Kusagawa M, Yatani R, Inagaki M. Ca2+-calmodulin-dependent protein kinase II phosphorylates various types of non-epithelial intermediate filament proteins. Biochem Biophys Res Commun. 1990;169:896–904. doi: 10.1016/0006-291x(90)91977-z. [DOI] [PubMed] [Google Scholar]
- 30.Tsujimura K, Tanaka J, Ando S, Matsuoka Y, Kusubata M, Sugiura H, Yamauchi T, Inagaki M. Identification of phosphorylation sites on glial fibrillary acidic protein for cdc2 kinase and Ca2+-calmodulin-dependent protein kinase II. J Biochem. 1994;116:426–434. doi: 10.1093/oxfordjournals.jbchem.a124542. [DOI] [PubMed] [Google Scholar]
- 31.Vikstrom KL, Lim SS, Goldman RD, Borisy GG. Steady state dynamics of intermediate filament networks. J Cell Biol. 1992;118:121–129. doi: 10.1083/jcb.118.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasui Y, Amano M, Nagata K, Inagaki N, Nakamura H, Saya H, Kaibuchi K, Inagaki M. Roles of Rho-associated kinase in cytokinesis; mutations in Rho-associated kinase phosphorylation sites impair cytokinetic segregation of glial filaments. J Cell Biol. 1998;143:1249–1258. doi: 10.1083/jcb.143.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]