

Abstract
Targeted deletion of the gene encoding the neuronal and neuroendocrine secreted polypeptide VGF (nonacronymic) produces a lean, hypermetabolic mouse. Consistent with this phenotype, VGF mRNA levels are regulated in the hypothalamic arcuate nucleus in response to fasting. To gain insight into the site(s) and mechanism(s) of action of VGF, we further characterized VGF expression in the hypothalamus. Double-label studies indicated that VGF and pro-opiomelanocortin were coexpressed in lateral arcuate neurons in the fed state, and that VGF expression was induced after fasting in medial arcuate neurons that synthesize neuropeptide Y (NPY). Like NPY, VGF mRNA induction in this region of the hypothalamus in fasted mice was inhibited by exogenous leptin. In leptin-deficient ob/ob and receptor-mutant db/db mice, VGF mRNA levels in the medial arcuate were elevated. To identify neural pathways that are functionally compromised by Vgf ablation, VGF mutant mice were crossed with obeseAy/a (agouti) andob/ob mice. VGF deficiency completely blocked the development of obesity inAy/a mice, whereas deletion of Vgf in ob/ob mice attenuated weight gain but had no impact on adiposity. Hypothalamic levels of NPY and agouti-related polypeptide mRNAs in both double-mutant lines were dramatically elevated 10- to 15-fold above those of wild-type mice. VGF-deficient mice were also found to resist diet- and gold thioglucose-induced obesity. These data and the susceptibility of VGF mutant mice to monosodium glutamate-induced obesity are consistent with a role for VGF in outflow pathways, downstream of hypothalamic and/or brainstem melanocortin 4 receptors, that project via the autonomic nervous system to peripheral metabolic tissues and regulate energy homeostasis.
Keywords: VGF, neurotrophin, hypothalamus, obesity, agouti, POMC, NPY, leptin, melanocortin
Hypothalamic neural pathways in the CNS regulate feeding and energy expenditure. The state of peripheral fat stores is relayed to the brain via circulating hormones such as leptin, an adipocyte-synthesized protein that transduces its signal through transport and binding to the leptin receptor system in the hypothalamus (Ahima et al., 2000; Schwartz et al., 2000). Here satiety-inducing melanocortin pathways decrease food intake through the interplay of two peptides that compete for binding to the melanocortin 4 receptor (MC4-R), α-melanocyte stimulating hormone (α-MSH), and its antagonist agouti-related polypeptide (AGRP). Effects of these leptin-responsive circuits on energy balance are mediated by projections within the hypothalamus to the brainstem, spinal cord, and cerebral cortex, and ultimately to autonomic pathways that innervate peripheral metabolic tissues (Ahima et al., 2000).
Analyses of several obese mouse models indicate that leptin and the melanocortin pathway play critical roles in the regulation of energy balance. Mice with mutations in the leptin (ob/ob) or leptin receptor (db/db) genes develop early onset obesity that is associated with a reduced metabolic rate, increased food intake, diabetes, and decreased fertility (Friedman and Halaas, 1998). Mice with defective signaling in the melanocortin pathway, caused by targeted deletion of the hypothalamic MC4-R or its agonist α-MSH [pro-opiomelanocortin (POMC) knock-out] or by overexpression of the melanocortin receptor antagonists agouti (Ay/a) or the agouti-related protein [AGRP or agouti-related transcript (ART)], develop a maturity onset obesity syndrome that is associated with hyperphagia, hyperinsulinemia, and hyperglycemia (Barsh, 1999; Salton et al., 2000a). Inbreeding of these obese mice with other strains that harbor additional genetic mutations, such as mahogany (Gunn et al., 1999;Nagle et al., 1999) or the neuropeptide Y (NPY) knock-out (Erickson et al., 1996), and analysis of the phenotypes of the resulting offspring has been an extremely useful technique to better define the molecular components of the leptin and melanocortin pathways.
Targeted deletion of the neurotrophin-regulated gene product called VGF (nonacronymic) produces a lean, hypermetabolic, and hyperactive mouse (Hahm et al., 1999). This gene encodes a secreted polypeptide that is expressed throughout the CNS and PNS, selectively in neurons in which it can be rapidly induced by neurotrophins, as well as in several endocrine and neuroendocrine tissues (Salton et al., 2000b). VGF expression is regulated in the brain by activity and injury, in the hypothalamic arcuate nucleus in response to fasting, and in the dorsal motor nucleus of the vagus (DMV) and nucleus tractus solitarius (NTS) in response to gastric manipulation (Salton et al., 2000b). The phenotype of the VGF mutant mouse suggests that this protein or one of its processed peptides regulates energy balance; however, the precise bioactive fragment(s) and mechanism of action of VGF have remained elusive. To identify potential site(s) of VGF action, we examined VGF expression in the hypothalamus and determined which central and peripheral neural pathways are impacted by targeted deletion of the Vgf gene. We report that Vgf gene ablation blocks the development of obesity from select environmental and genetic causes, suggesting that VGF functions in outflow pathways, downstream of hypothalamic or brainstem melanocortin receptors, to regulate energy expenditure.
MATERIALS AND METHODS
Mouse strains used. Targeted deletion of the mouseVgf gene and initial characterization of VGF mutant mice has been described previously (Hahm et al., 1999). Chimeric VGF knock-out males were directly crossed to 129/SvJ or repetitively backcrossed to C57BL/6 strains for 10 generations; homozygous VGF-deficient offspring of F10 and F1 heterozygotes on either background were phenotypically indistinguishable. For the experiments described here, mixed background VGF mutant mice from F2 and F3 generations were used. To generate double-mutant mice, fertile heterozygousVgf+/Vgf− female mice or bilaterally ovariectomized C57BL/6 × 129/SvJ females (The Jackson Laboratory, Bar Harbor, ME) that had been grafted withVgf−/Vgf− ovaries were used. These were mated with Ay/a (agouti) males or with fertile ob−/ob− males, both on C57BL/6 backgrounds (obtained from The Jackson Laboratory), that had been rescued by a course of intraperitoneal leptin. Recombinant murine leptin (20 μg/gm body weight), generously provided by Amgen Inc. (Thousand Oaks, CA), was delivered to ob−/ob− males twice daily for 7 d and then approximately every other day for 30–60 d.
Fasting and leptin replacement study. Wild-type anddb/db mice were fasted for 48 hr and killed (fasted group). For the leptin replacement study, ad libitum fed and fasting mice were injected intraperitoneally every 12 hr with saline or leptin (0.5 μg of recombinant murine leptin/gm body weight; R & D Systems, Minneapolis, MN). The final, fifth injection was administered 30 min before the mice were killed, 2 hr after lights on.
Chemical lesioning. Mice, 3–4 months of age, were administered a single intraperitoneal injection of gold thioglucose (GTG) (0.8 mg/gm body weight) (Bergen et al., 1998), after which mice were weighed at regular intervals and food intake was measured. Two weeks after GTG (or saline) injection, mice were killed and tissues were removed for analysis. Separate groups of wild-type and VGF mutant mice were killed 3 d after GTG injection, and brains were removed and examined histologically by Nissl staining (Hahm et al., 1999) to confirm that hypothalamic lesions developed in mice of each genotype.
Monosodium glutamate (MSG) injections were performed by minor modification of previous methods (Pizzi and Barnhart, 1976). Mice received the following daily subcutaneous injections starting at postnatal day 2 (P2) until P12: 2.5, 2.8, 3.2, 3.4, 3.6, 3.8, 4.0, 4.2, 4.4, 4.6, and 4.8 mg MSG per gram of body weight (in PBS). Mice were weighed weekly and killed at 9 months of age, hypothalamic RNAs were isolated, and organ and tissue weights were determined.
Northern blot, blood, and serum analyses. Northern blot analysis was performed as described previously (Mizuno et al., 1996b), using probes to NPY (Mizuno et al., 1996a), POMC (Mizuno et al., 1998), and AGRP (Mizuno and Mobbs, 1999); relative mRNA levels were determined by densitometric analysis of film autoradiograms.
Mice were anesthetized with avertin, and blood samples were collected by cardiac puncture. Blood glucose levels were determined using a one touch profile meter (Lifescan Inc., Milpitas, CA). Serum insulin and leptin levels were determined by radioimmunoassay (ICN Biomedicals, Inc., Costa Mesa, CA) or ELISA (Crystal Chem Inc., Chicago, IL).
Energy balance and body composition analyses. Food consumption was measured daily using either a liquid diet or a weighable solid pellet delivery system (Bio-Serv, Frenchtown, NJ) over 5 consecutive days and averaged. For the high-fat diet studies, mice were fed for 5 weeks with standard chow or high-calorie chow (5.396 kcal/gm) with a high fat (35.5%) and high carbohydrate (35.4%) content (#F2685; Bio-Serv). Body carcass analysis for total lipid, protein, and water was performed as described previously (Chung et al., 1998; Hahm et al., 1999).
Immunohistochemical characterization of VGF-containing neurons in the arcuate nucleus. Adult male Sprague Dawley rats (280–300 gm; Taconic Farms, Germantown, NY) were divided into three groups. To study the colocalization of VGF and α-MSH in the arcuate nucleus, animals (n = 3) were anesthetized with sodium pentobarbital (50 mg/kg body weight, i.p.) and stereotaxically injected intracerebroventricularly with 100 μg of colchicine in 6 μl of 0.9% saline 20 hr before perfusion fixation of the brain. To study the colocalization of VGF and NPY in the arcuate nucleus of fed and fasted rats, rats were implanted with a 22 gauge stainless steel guide cannula (Plastics One Inc., Roanoke, VA) into the lateral cerebral ventricle under stereotaxic control (coordinates from bregma: anteroposterior, −0.8; lateral, 1.2; dorsoventral, 3.2) through a burr hole in the skull. The cannula was secured to the skull with three stainless steel screws and dental cement and temporarily occluded with a dummy cannula. Bacitracin ointment was applied daily to the interface of the cement and the skin. Animals were weighed daily, and any animal showing signs of illness or weight loss after the third postoperative day was removed from the study and killed. One week after intracerebroventricular cannulation, the animals were divided into two groups. In the first group (n = 3), the animals were fed ad libitum. In the second group (n = 3), animals were fasted for 64 hr. Both groups were treated with 100 μg of colchicine in 6 μl of 0.9% saline through a 28 gauge needle that extended 1 mm below the guide cannula, 20 hr before perfusion fixation of the brain.
All colchicine-treated animals were deeply anesthetized with sodium pentobarbital and perfused transcardially with 20 ml of 0.01m PBS, pH 7.4, containing 15,000 U/l heparin sulfate, 150 ml of 2% paraformaldehyde/4% acrolein in 0.01 m phosphate buffer (PB), pH 7.4, followed by 30 ml of 2% paraformaldehyde in the same buffer. The brains were removed and cryoprotected in 30% sucrose in PBS at 4°C overnight and then frozen on dry ice.
Immunofluorescence was performed essentially as described previously (Fekete et al., 2000a). Coronal sections (25 μm thick) were incubated in mixtures of either sheep anti-α-MSH (1:5000) (Elias et al., 1998) or sheep anti-NPY antiserum (1:1000) (Peninsula Laboratories, Belmont, CA) and rabbit anti-VGF antisera (1:400) (Salton et al., 1995), rinsed in PBS, incubated in Texas Red-conjugated donkey anti-sheep IgG and FITC-conjugated donkey anti-rabbit IgG (both 1:40; Jackson ImmunoResearch, West Grove, PA), and analyzed by Zeiss (Thornwood, NY) Axioskop 2 epifluorescent microscopy.
Double-label in situ hybridization for POMC and VGF. Double-label in situ hybridization was performed as described previously (Elias et al., 1999; Fekete et al., 2000b). Sprague Dawley rats (280–300 gm) were divided into two groups. One-half of the animals (n = 4) had ad libitum access to food. The other group (n = 4) was fasted for 64 hr before the perfusion. Under sodium pentobarbital anesthesia (50 mg/kg body weight, i.p.), animals were perfused by intracardiac perfusion with 20 ml of 0.01 mPBS, pH 7.4, containing 15,000 U/l heparin sulfate followed by 150 ml of 4% paraformaldehyde in PBS. The brains were removed and postfixed by immersion in the same fixative for 2 hr at room temperature. Tissue blocks containing the hypothalamus were cryoprotected in 20% sucrose in PBS at 4°C overnight and then frozen on dry ice. Serial 20-μm-thick coronal sections through the rostrocaudal extent of the arcuate nucleus were cut on a cryostat (Reichert-Jung 2800 Frigocut-E; Leica Microsystems Inc., Bannockburn, IL), collected in freezing solution (30% ethylene glycol; 25% glycerol; 0.05 m PB) and stored at −20°C until used. Serial sections were washed in 2× SSC, acetylated with 0.25% acetic anhydride in 0.9% triethanolamine for 20 min, and then treated in graded solutions of acetone (50, 70, 90, and 100%), chloroform, and a descending series of acetone (100, 90, 70, and 50%) for 5 min each. After additional washes in 2×, 3×, and 4× SSC for 5 min each, the sections were hybridized with the mixture of digoxigenin-labeled cRNA probe for POMC and [35S]UTP-labeled cRNA probe for VGF.
The hybridization was performed for 16 hr at 56°C in 200 μl polypropylene tubes in a hybridization buffer (50% formamide, 2× SSC, 10% dextran sulfate, 0.5% SDS, and 250 μg/ml denatured salmon sperm DNA) that contained the digoxigenin-labeled POMC probe diluted 1:50 and 6× 105 cpm of35S-labeled VGF cRNA probe. The slides were washed in 1× SSC for 15 min and then treated with 25 μg/ml RNase for 1 hr at 37°C. After additional washes in 0.1× SSC (four times for 15 min) at 65°C, sections were washed in PBS, treated with a mixture of 0.5% Triton X-100 and 0.5% H2O2 for 15 min, and then treated with 2% bovine serum albumin (BSA) in PBS for 20 min to reduce the nonspecific antibody binding. The sections were incubated with sheep anti-digoxigenin-peroxidase Fab fragments (1:100; Boehringer Mannheim Corp., Indianapolis, IN) in 1% BSA in PBS for 2 d at 4°C. The sections were then rinsed in PBS and incubated in 0.1% biotinylated tyramide and 0.01% H2O2 in PBS for 10 min to intensify the hybridization signal. After additional washes, the sections were incubated in 7-amino-4-methylcoumarin-3-acetic acid (AMCA) Avidin D (1:250; Vector Laboratories, Burlingame, CA) and mounted on Superfrost/Plus glass slides (Fisher Scientific Co., Pittsburgh, PA). Slides were dipped into Kodak NTB2 autoradiography emulsion (Eastman Kodak, Rochester, NY), and the autoradiograms were developed after 7 d of exposure at 4°C. The specificity of hybridization was confirmed using sense probes, which resulted in the absence of specific hybridization in the paraventricular nucleus (PVN). To investigate regulation of VGF mRNA levels by fasting, freshly dissected brains from 48 hr fasted andad libitum fed C57BL/6, ob/ob, anddb/db mice were rapidly frozen. Matching sections through the hypothalami were processed for in situ hybridization, and 32P-labeled single-stranded mouse VGF cDNA probes were used as described previously (Mizuno et al., 1998;Hahm et al., 1999).
Real-time reverse transcriptase-PCR analysis. Total RNA was isolated using TRIzol (Invitrogen, Gaithersburg, MD), and first-strand cDNA synthesis was completed using the Superscript Choice System (Invitrogen). RNA was hybridized for 10 min at 70°C with 100 pmol/μl oligo-dT24, and first-strand synthesis was performed at 42°C for 60 min using Superscript II reverse transcriptase (RT). Primers for real-time PCR were first examined by standard PCR (94°C for 30 sec, 55°C for 1 min, and 72°C for 1 min × 30 cycles) and agarose gel electrophoresis for correct product size and absence of primer/dimer formation. Real-time PCRs (95°C for 15 sec, 60°C for 30 sec, and 72°C for 30 sec × 40 cycles) were performed in an ABI-prism 7700 sequence detector (Applied Biosystems, Foster City, CA). Product accumulation was measured in real-time, and mean cycle threshold (Ct), the cycle when product is first detectable, was determined for replicate samples (n = 4–8 independent reactions per primer pair and cDNA sample) run on the same plate. Different cDNA samples were normalized using primer sets to the housekeeping gene products glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and actin. Primers were as follows: GAPDH, 5′-CTTGCTCAGTGTCCTTGCTG-3′ and 5′-TGCGACTTCAACAGCAACTC-3′; actin, 5′-AGGTGACAGCATTGCTTCTG-3′ and 5′-GCTGCCTCAACACCTCAAC-3′; NPY, 5′-AGCAGAGGACATGGCCAGAT-3′ and 5′-AAATCAGTGTCTCAGGGCTGGA-3′; NPY Y5R, 5′-GAGAAGCACCTAACCGTTCCAG-3′ and 5′-TGAGGGAACGCTTGACTCTCAT-3′; AGRP, 5′-TGACTGCAATGTTGCTGAGTTGTG-3′ and 5′-TAGGTGCGACTACAGAGGTTCGTG-3′; POMC, 5′-GCCCTCCTGCTTCAGACCTC-3′ and 5′-CGTTGCCAGGAAACACGG-3′; cocaine amphetamine-regulated transcript (CART), 5′-CTACTCTGCCGTGGATGATGC-3′ and 5′-GGACTTCTTGCAACGCTTCG-3′; prohormone convertase 1 (PC1), 5′-AAGGGATGAGCAGGTACAAGGA-3′ and 5′-GCTGAGCTTTGCACTTGGAGA-3′; MC4-R, 5′-GTGGGCGTTATGAATTGACATG-3′ and 5′-TCTGATTTCGGCCACTACAGAA-3′; galanin, 5′-AAGAGAGGTTGGACCCTGAACAG-3′ and 5′-TCAAAGCAGAGAACAGAGGATTGG-3′.
Statistical analyses. With the exception of the real-time PCR results, which are presented as mean ± SD, data are presented as mean ± SEM. Data were analyzed by two-way ANOVA. When indicated by the appropriate p value (p < 0.05), groups were compared by Tukey–Kramer or Fisher's PLSD post hoc tests. Ap value of <0.05 was considered significant; pand n values are noted in the figures and/or accompanying legends.
RESULTS
Immunohistochemical localization of VGF in the hypothalami of fed and fasted rodents
Previous studies indicated that VGF mRNA was induced in the hypothalamic arcuate nucleus by fasting (Hahm et al., 1999). To determine whether VGF was synthesized in POMC-expressing cells, we mapped the distribution of neurons in the hypothalamus that make VGF mRNA or polypeptide and either POMC mRNA or α-MSH peptide using double-label in situ hybridization or immunohistochemistry, respectively, and compared the patterns in the fed and fasted mouse and rat.
In fed rats, using double-label immunofluorescence, VGF was present in 94% of α-MSH neurons in the retrochiasmatic area, 84% in the anterior portion of the arcuate nucleus, 69% in the midarcuate nucleus, and 56% in the caudal arcuate nucleus (Fig.1A–C). Using double-label in situ hybridization for VGF and POMC mRNAs, fed animals were found to have an intense VGF signal in the retrochiasmatic area and lateral arcuate nucleus that corresponded to the location of the α-MSH-containing cells (Fig.1D). No VGF hybridization signal was detected in the medial arcuate, where NPY-containing neurons are found. According to double-label immunofluorescence, VGF was present in <10% of the NPY-containing cells throughout the arcuate nucleus and retrochiasmatic area (Fig. 1F). In summary, in the fed state, VGF and POMC gene expression was found to significantly overlap in the lateral arcuate nucleus.
Fig. 1.

Colocalization of VGF with α-MSH and NPY in the arcuate nucleus of fed and fasted rats. Low-power micrographs of double-labeled immunofluorescence preparations (A–C) demonstrate the colocalization (yellow) of VGF (green) and α-MSH (red) in the retrochiasmatic region (A) and in the rostral (B) and caudal (C) part of the arcuate nucleus of fed colchicine-treated rats (n = 3). Low-power micrographs (D, E) show the colocalization (yellow) of POMC mRNA (labeled with AMCA, pseudocolored red) and VGF mRNA (dark-field image of the silver grains, pseudocolored green) in the arcuate nucleus of fed (D) (n= 4) and fasted (E) (n = 4) rats. In colchicine-treated fed rats (F), VGF immunoreactivity (green) and NPY immunoreactivity (red) are localized in different populations of arcuate neurons. In contrast, ∼50% of NPY-immunoreactive neurons (yellow), localized dorsally, cocontained VGF in the arcuate nucleus of fasted colchicine-treated rats (G) (n = 3).
NPY is a potent stimulator of food intake, as is AGRP (or ART), an α-MSH antagonist that binds to the MC4-R, and these two orexigenic peptides are coexpressed in hypothalamic medial arcuate neurons (Elias et al., 1998, 1999). Expression of hypothalamic NPY and AGRP mRNAs increase and POMC mRNA decreases with fasting (Hahn et al., 1998;Mizuno et al., 1998, 1999). To investigate in which hypothalamic neurons VGF expression is regulated during fasting, we colocalized POMC and VGF mRNAs and NPY and VGF polypeptides after food deprivation for 64 hr (Fig. 1E,G). We noted a marked reduction in VGF signal in the retrochiasmatic area and lateral arcuate nucleus in POMC-containing neurons. Overall, the total density of silver grains in the entire arcuate (medial and lateral groups) resulting from hybridization to VGF was less in fasting animals than in fed animals by ∼50%. However, we also observed increased VGF hybridization in the medial part of the arcuate nucleus that is not seen in the fed animals. Using double-label immunofluorescence, this increase was confined to NPY-producing neurons and was dorsal to the NPY neurons seen in the fed animals (Fig. 1G). In summary then, in the fasted state, VGF and NPY expression was induced in a population of medial arcuate neurons, but colocalization of VGF and POMC decreased.
Fasting-induced increases in VGF mRNA levels in the arcuate nucleus are inhibited by leptin
VGF expression in the arcuate nucleus of the hypothalamus increases with fasting, a state generally associated with decreased leptin levels. We therefore investigated whether exogenous leptin modulates hypothalamic VGF mRNA levels in wild-type mice and examined hypothalamic VGF mRNA expression in leptin-deficient (ob/ob) and leptin-resistant (db/db) mice. Mice were ad libitum fed or fasted as described in Materials and Methods, and a subset of these mice was injected with leptin or with saline. VGF mRNA was localized in the hypothalamus by in situ hybridization, essentially as described previously (Hahm et al., 1999). VGF mRNA levels were induced in fasted compared with fed wild-type mice in the medial arcuate nucleus and somewhat decreased in regions lateral to this, as described previously (Hahm et al., 1999) and shown above in the rat, and the patterns of hybridization noted in the leptin-injected fasted mice were very similar to the fed controls (Fig.2A–D). In leptin-deficient ob/ob or leptin receptor-mutantdb/db mice, hypothalamic VGF mRNA levels in the fed state resembled those of fasted wild-type mice, particularly in the arcuate nucleus (Fig. 2E–H). Although VGF expression is regulated by leptin in the hypothalamus of wild-type mice, VGF does not appear to be an integral component of leptin signaling pathways, because VGF mutant mice respond to exogenous leptin with weight loss, decreased feeding, increased hypothalamic POMC expression, and decreased adipose leptin expression (Fig. 2I).
Fig. 2.
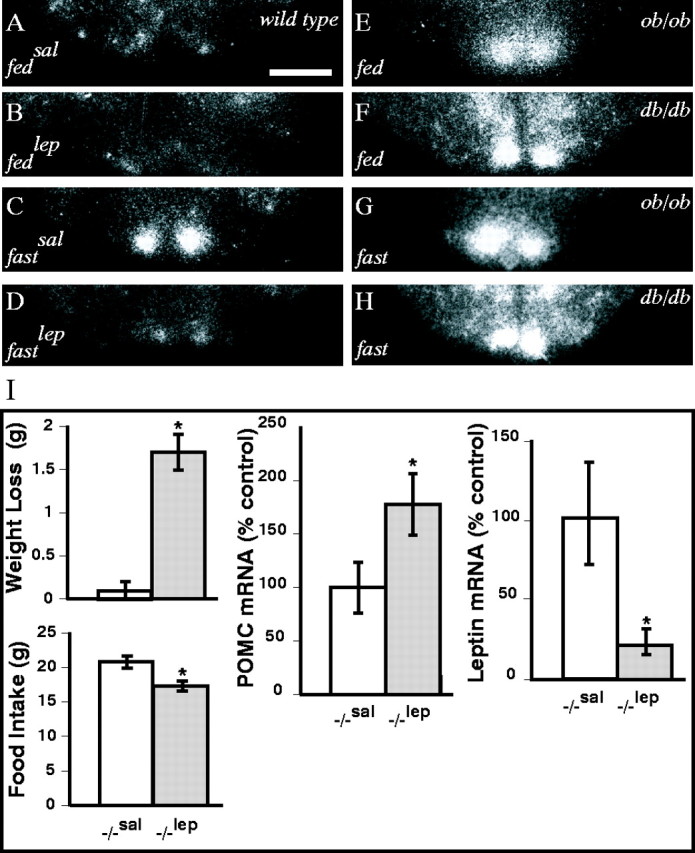
Hypothalamic expression of VGF is regulated by leptin. VGF expression was examined by in situhybridization in the hypothalami of ad libitum fed and 48 hr fasted wild-type, ob/ob, and db/dbmice (A–H). Wild-type mice were injected with saline (sal) or leptin (lep) as indicated (A–D). Photomicrographs of film autoradiograms, representative of coronal sections examined from three independent animals for each treatment group, are shown. Scale bar, 100 μm. VGF mutant mice respond to exogenous leptin (I). VGF-deficient mice were treated with leptin (20 μg/gm body weight, twice daily i.p. for 2 d), and weight loss, feeding, hypothalamic POMC mRNA, and adipose leptin mRNA were measured. Histograms identified by asterisks are significantly different from one another (mean ± SE;p < 0.05; ANOVA with Tukey–Kramer post hoc comparisons; n = 4–6 mice per treatment group).
VGF is required for the development of diet-induced obesity
Wild-type mice fed high-calorie diets (35.5% fat, 35.4% carbohydrate, 20% protein; 5.396 kcal/gm) for 5 weeks were all found to have substantially increased body weights, adiposity, and leptin levels in comparison with mice fed regular diets (Fig.3). Ablation of the Vgf gene blocked the metabolic effects of the high-fat diet (Fig.3A), and although a small but significant increase in fat pad weight was noted in knock-out mice fed the high-calorie diet (Fig.3B), no significant changes in adipose leptin mRNA (Fig.3C) or circulating leptin levels (data not shown) were observed.
Fig. 3.
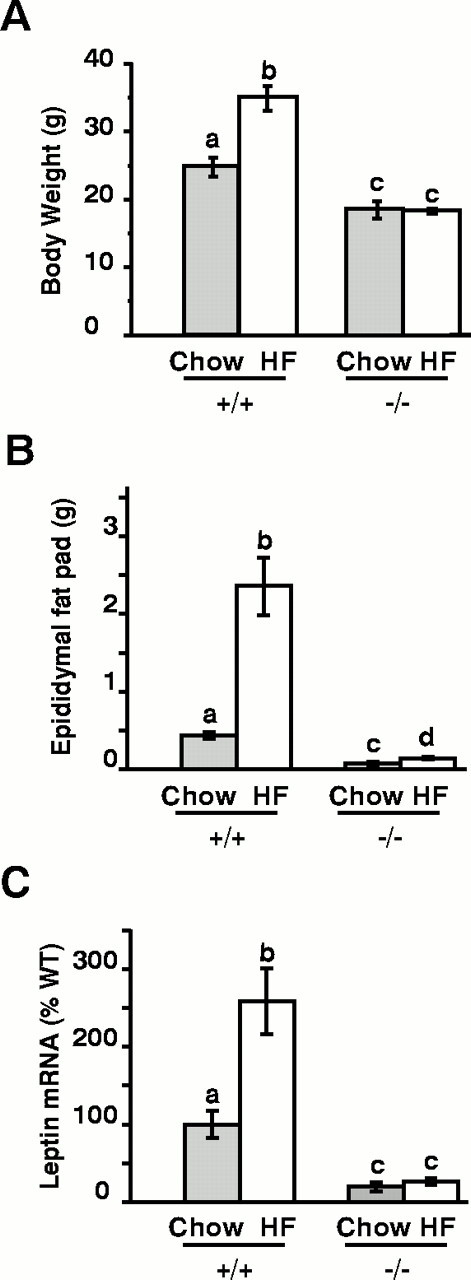
VGF mutant mice resist diet-induced obesity. Wild-type (WT) (+/+) and VGF mutant (−/−) mice were placed either on regular laboratory diets (Chow) or high-fat diets (HF) for 5 weeks. Mice were then weighed (A) and anesthetized, adipose tissues were removed and weighed (B), and total RNA was isolated and leptin mRNA levels were quantified by Northern blotting (C). Histograms identified by differentletters are significantly different from one another (mean ± SE; p < 0.05; ANOVA with Tukey–Kramer post hoc comparisons;n = 9–15 mice per treatment group).
Ablation of the Vgf gene blocks GTG- but not MSG-induced obesity
An alternative environmentally induced form of obesity is that caused by lesions in the ventromedial hypothalamic area (Elmquist et al., 1999). A particularly informative form of hypothalamic obesity is that produced by GTG, which is taken up by and is toxic to glucose-sensitive neurons primarily in the periarcuate area of the ventromedial hypothalamus (Bergen et al., 1998). As with diet-induced obesity, targeted deletion of the Vgf gene completely prevented the increase in body weight and hyperphagia produced by GTG treatment (Fig. 4A,B). This reduction in food intake in GTG-treated VGF mutant mice was also quite remarkable, because ablation of the Vgf gene does not influence food intake in wild-type mice. Thus, although GTG treatment led to the formation of a characteristic hypothalamic lesion by Nissl staining in all of the injected wild-type and VGF knock-out mice that were examined (n = 3 for each group) (data not shown), none of the GTG-injected VGF-deficient mice developed obesity.
Fig. 4.
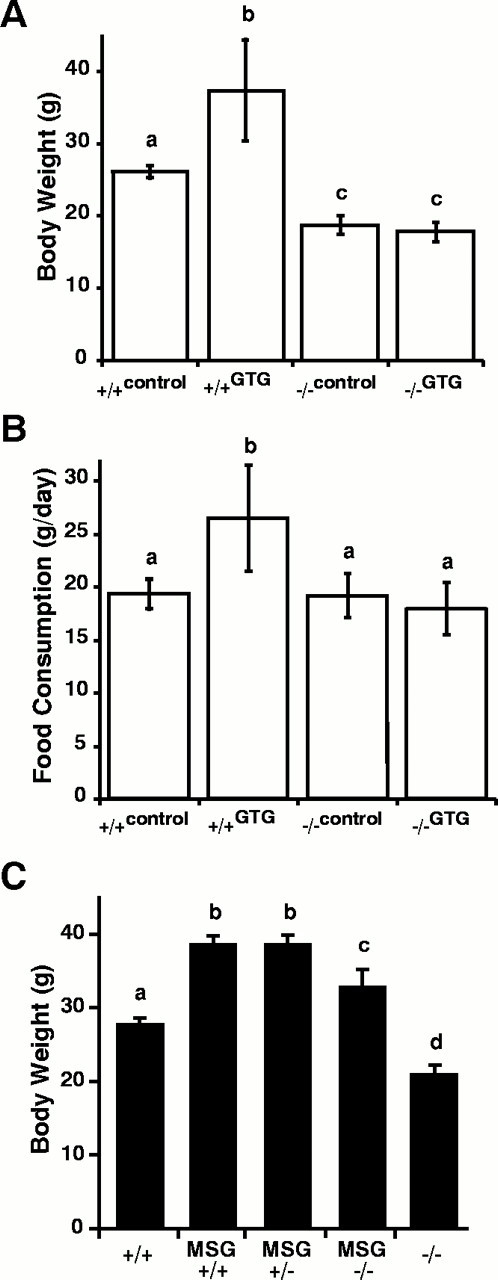
VGF mutant mice resist obesity caused by GTG- but not MSG-induced chemical lesions. Wild-type (+/+) and VGF-deficient (−/−) mice were treated with GTG, and body weights (A) and food consumption (B) were determined (mean ± SE) at 16 weeks of age. Wild-type and heterozygous and homozygous VGF-deficient mice were treated with MSG, and body weights were measured (mean ± SE) at 36 weeks of age (C). Histograms identified by different letters are significantly different from one another (p < 0.05; ANOVA with Tukey–Kramerpost hoc comparisons; n = 4–6 mice per treatment group).
Repetitive daily injections of MSG administered to neonatal mice from P2–P12 result in obesity (Pizzi and Barnhart, 1976), most likely through damage to the hypothalamus (Bergen et al., 1998; Morris et al., 1998) and the sympathetic nervous system (Morris et al., 1998;Tsukahara et al., 1998). NPY mRNA is virtually eliminated in the arcuate nucleus (Bergen et al., 1998), NPY content is reduced in several hypothalamic regions (Morris et al., 1998), and hypothalamic POMC mRNA levels decrease (Bergen et al., 1998). In addition, decreased glucose transporter protein-4 (GLUT4) transporter levels in the brown adipose tissue (BAT) of MSG-lesioned mice (Morris et al., 1998) and a decreased response of uncoupling protein (UCP) mRNA levels in BAT to acute cold exposure (Tsukahara et al., 1998) suggest a possible impairment of hypothalamic sympathetic input to brown fat that could disrupt thermogenesis and lead to increased adiposity. We therefore assessed whether Vgf gene ablation affected obesity caused by MSG treatment. In contrast to all the other forms of obesity examined, targeted deletion of the Vgf gene had little influence on the ability of MSG treatment to increase body weight (Fig. 4C). These results suggest that VGF functions in the hypothalamic/autonomic outflow pathways that innervate peripheral metabolic tissues such as BAT or white adipose tissue (WAT), and that in the VGF mutant mice, toxic damage to these pathways blocks the development of the hypermetabolic, lean phenotype.
VGF is required for the development of obesity inAy/a agouti but notob/ob mice
Agouti-mediated obesity results from ectopic overexpression of the agouti polypeptide, a melanocortin receptor blocker that decreases normal satiety signaling by α-MSH, a peptide that is processed from the POMC precursor (Fan et al., 1997; Barsh, 1999). To determine whether VGF might play a functional role in the hypothalamic melanocortin system, we generated agouti mice, agouti mice that were either heterozygous or homozygous for the targeted VGF mutation, and VGF mutant mice. Ablation of the Vgf gene inAy/a mice completely suppressed the obese agouti phenotype, including increased adiposity and body weight (Fig. 5), and blocked increased linear growth [Ay/a, 9.86 ± 0.18 cm (n = 8);Ay/a,Vgf−/Vgf−, 8.01 ± 0.18 (n = 3);Vgf−/Vgf−, 7.88 ± 0.1 (n= 5)]. Targeted deletion of the Vgf gene, however, did not block the effects of the Ay allele on coat color, unlike mutations in the mahogany gene, which encodes a transmembrane form of attractin that interacts with the agouti protein and melanocortin receptor to modify coat color (Gunn et al., 1999;Nagle et al., 1999). These rather dramatic differences in phenotype between Ay/a andAy/a,Vgf−/Vgf− mice suggested that VGF was required for the development of agouti-induced maturity onset obesity, and that VGF might therefore function in pathways downstream to the MC4-R that project via the autonomic nervous system to peripheral metabolic tissues.
Fig. 5.

VGF is required for the development of obesity in agouti mice. Representative agouti (Ay/a) and double-mutant (Vgf−/Vgf−,Ay/a) mice are shown (A). Body weight (B) and body composition (C) measurements for the indicated genotypes are shown (mean ± SE). Histograms identified by differentletters are significantly different from one another (B) (p < 0.05; ANOVA with Tukey–Kramer post hoc comparisons;n = number of mice analyzed of the indicated genotype).
Because VGF expression was upregulated in the hypothalami ofob/ob and db/db mice (Fig. 2), we investigated whether VGF was required for the development of obesity in leptin-deficient ob/ob mice. Mice with mutations in either the lepob or Vgf genes and double mutants (ob/ob,Vgf−/Vgf−) were generated (Fig. 6A). Targeted deletion of the Vgf gene completely blocked the effects of leptin deficiency on hyperphagia and attenuated body weight gain in double-mutant mice (Fig. 6). The effect that ablation of theVgf gene has on reduction of food intake in ob/obmice is particularly striking, because VGF deficiency does not influence food intake in wild-type mice (Fig. 6D). However, ablation of the Vgf gene did not prevent the development of increased adiposity (Fig. 6C) or reduced body temperature in ob/ob mice [ob/ob, 36.4 ± 0.6°C (n = 12);ob/ob,Vgf−/Vgf−, 36.8 ± 0.6°C (n = 6); wild type, 38.4 ± 0.5°C (n = 9); Vgf−/Vgf−, 38.1 ± 0.5°C (n = 6)]. These results demonstrate that products of the Vgf gene selectively influence specific aspects of metabolic regulation.
Fig. 6.

VGF is required for the development of hyperphagia but not increased adiposity in ob/ob mice. Representative males of the following genotypes are shown: VGF mutant (Vgf−/Vgf−), double-mutant (Vgf−/Vgf−,ob/ob),ob/ob, and wild type (A). Body weight (B), body composition (C), and daily food intake (D) were measured for the indicated genotypes (mean ± SE). Histograms identified by differentletters are significantly different from one another (B, D) (p < 0.05; ANOVA with Tukey–Kramer post hoc comparisons;n = number of mice analyzed of the indicated genotype).
Superinduction of hypothalamic NPY and AGRP expression in double-mutant ob/ob,Vgf−/Vgf−, andAy/a,Vgf−/Vgf− mice
Feeding and metabolic rate are controlled by a number of hypothalamic neuropeptides, and their relative levels of expression are, in addition to weight and body fat stores, a sensitive indicator of the animal's state of energy balance. To determine whether the observed failure of the obese phenotype to develop inAy/a,Vgf−/Vgf− mice was associated with changes in hypothalamic gene expression, real-time RT-PCR analysis was used to quantify mRNA levels for a number of neuropeptides and receptors in VGF knock-out, agouti,ob/ob, and double-mutant mice. Hypothalamic RNAs from each genotype were pooled (n = 2–3), and cDNAs were synthesized as described in Materials and Methods. Each primer pair was used to amplify hypothalamic cDNA by standard PCR methods, and agarose gel electrophoresis was used to ensure that single DNA fragments of the predicted size were obtained. Input amounts of cDNA used in each reaction were normalized by quantifying the housekeeping gene products actin and GAPDH, and the levels of the following mRNAs were quantified: POMC, NPY, AGRP, galanin, CART, NPY Y5 receptor, MC4-R, and PC1.
In Table 1, the results of real-time PCR analysis of cDNAs from wild-type,Ay/a, VGF mutant, and double-mutantAy/a,Vgf−/Vgf− mice are shown. Similarity in the mRNA levels between VGF mutant and double-mutantAy/a,Vgf−/Vgf− was noted, in contrast to the values obtained for obese agouti mice. In addition, NPY and AGRP mRNA levels were substantially higher inAy/a, Vgf−/Vgf− mice in comparison with VGF mutant mice. Real-time PCR analysis of cDNAs from wild-type,ob/ob, VGF-deficient, and double-mutantob/ob,Vgf−/Vgf− mice was also performed. VGF mutant mice have low leptin levels, and ob/ob mice express no leptin, so despite differences in weight and adiposity, similarities in hypothalamic AGRP and NPY mRNA levels were noted (Table 1). Double-mutant ob/ob,Vgf−/Vgf− mice were found, like double-mutantAy/a,Vgf−/Vgf− mice, to have even higher levels of AGRP and NPY mRNAs than eitherob/ob or VGF-deficient mice. In summary, we noted dramatic increases in AGRP and NPY mRNA levels in both double-mutant strains, which together with elevated levels in VGF mutant mice and the lack of hyperphagia in each of these three strains point to functional abnormalities in downstream neural pathways secondary to VGF deficiency.
Table 1.
Real-time RT-PCR analysis of hypothalamic neuropeptide gene expression using the comparative Ct method
| Vgf+/+,a/a Ct (fold change) | Vgf−/−,Ay/a Ct (fold change) | Vgf−/−,a/a Ct (fold change) | Vgf+/+,Ay/a Ct (fold change) | |
|---|---|---|---|---|
| POMC | 21.9 ± 0.1 (1.00) | 23.3 ± 0.2 (0.38) | 23.6 ± 0.1 (0.31) | 21.9 ± 0.2 (1.01) |
| CART | 20.5 ± 0.1 (1.00) | 20.4 ± 0.2 (1.09) | 21.3 ± 0.1 (0.56) | 19.0 ± 0.2 (2.89) |
| NPY | 22.1 ± 0.2 (1.00) | 18.7 ± 0.2 (10.6) | 21.3 ± 0.3 (1.72) | 23.2 ± 0.1 (0.47) |
| AGRP | 22.4 ± 0.2 (1.00) | 18.5 ± 0.2 (14.5) | 20.4 ± 0.2 (4.09) | 23.9 ± 0.2 (0.34) |
| GAL | 21.2 ± 0.1 (1.00) | 21.0 ± 0.1 (1.19) | 21.0 ± 0.2 (1.16) | 21.1 ± 0.1 (1.06) |
| GAPDH | 16.5 ± 0.0 (1.00) | 16.3 ± 0.0 (1.00) | 16.4 ± 0.1 (1.07) | 16.8 ± 0.1 (0.81) |
| Actin | 16.4 ± 0.1 (1.00) | 16.4 ± 0.0 (1.00) | 16.5 ± 0.1 (0.93) | 16.4 ± 0.0 (1.00) |
| Vgf+/+,ob+/+ Ct (fold change) | Vgf−/−,ob−/− Ct (fold change) | Vgf−/−,ob+/+ Ct (fold change) | Vgf+/+,ob−/− Ct (fold change) | |
|---|---|---|---|---|
| NPY | 25.9 ± 0.2 (1.00) | 22.5 ± 0.1 (10.6) | 24.3 ± 0.1 (2.92) | 24.8 ± 0.3 (2.04) |
| AGRP | 26.5 ± 0.4 (1.00) | 22.6 ± 0.2 (15.0) | 24.7 ± 0.2 (3.35) | 24.5 ± 0.4 (3.87) |
| GAL | 25.3 ± 0.3 (1.00) | 25.1 ± 0.2 (1.17) | 25.1 ± 0.1 (1.12) | 24.9 ± 0.2 (1.29) |
| GAPDH | 18.2 ± 0.06 (1.00) | 18.3 ± 0.1 (0.95) | 18.0 ± 0.2 (1.16) | 18.3 ± 0.1 (0.94) |
Relative levels of POMC, AGRP, NPY, CART, galanin (GAL), GAPDH, and actin mRNAs are expressed as Ct values. Relative fold increases in parentheses were determined by first normalizing mean Ct values with reference to mRNAs encoded by the housekeeping genes GAPDH and actin. Mean Ct ± SD values were determined from four to six independent, simultaneously run reactions, and each cDNA sample was synthesized from a pool containing two to three independent hypothalamic RNA isolates. Expression in the wild type was defined as 1. Similar results were obtained in at least three independent real-time PCR experiments.
DISCUSSION
Suppression of the obese phenotype of ob/ob or agouti mice through genetic inbreeding with other strains has been used to identify several molecular components of the leptin and melanocortin pathways. We found that inbreeding ofAy/a (agouti) and VGF-deficient mice suppressed maturity onset obesity. In contrast, VGF was not required for the development of obesity in response to leptin deficiency, but ablation of the Vgf gene did block the development of hyperphagia in ob/ob mice. The inverse result, partial reversal of the ob/ob obesity syndrome but no effect on the development of obesity in leptin-resistantAy/a mice, has been observed previously by genetic removal of NPY (Erickson et al., 1996; Hollopeter et al., 1998). VGF is therefore required for the development of diet-induced obesity and obesity caused by decreased activity in the melanocortin satiety pathway.
Where in the nervous system might VGF be functioning to regulate energy expenditure? Previous studies have indicated that VGF is expressed in peripheral sympathetic and parasympathetic neurons, widely expressed throughout the brain, and regulated in the hypothalamus in response to feeding (for review, see Salton et al., 2000b) (areas expressing VGF are indicated by the speckled pattern in Fig.7). Here we have shown that VGF is primarily coexpressed with POMC in the hypothalamic arcuate nucleus in the fed state, and that in the fasted state, VGF colocalization with POMC decreases concomitant with VGF induction in populations of neurons that express NPY and AGRP (Fig. 1). Recent studies using mice transgenic for a cAMP response element (CRE)-lacZ construct indicate that fasting activates reporter expression in the arcuate nucleus in a subpopulation of neurons that express NPY but not POMC (Shimizu-Albergine et al., 2001). Because several reports suggest that CRE-binding protein (CREB) is likely to be a critical regulator ofVgf gene expression in vitro and in vivo (Hawley et al., 1992; Bonni et al., 1995; D'Arcangelo et al., 1996; Di Rocco et al., 1997; Luc and Wagner, 1997), this subpopulation of arcuate neurons that increase expression of NPY in response to fasting are likely to be the same neurons that we have shown upregulate VGF (Fig. 1). It is also of interest that leptin administration to fasted CRE-lacZ mice significantly attenuates both β-galactosidase and NPY expression in this part of the hypothalamus. We also noted attenuation of VGF induction in the medial arcuate nucleus in leptin-treated fasted mice and observed elevated levels of VGF in the arcuate nuclei of ob/ob anddb/db mice compared with ad libitum fed wild-type mice (Fig. 2). Apparently then, the Vgf and Npygenes are similarly regulated by CREB and by leptin in the medial arcuate, and VGF, NPY, and AGRP are coexpressed in these neurons, perhaps coreleased through a common secretory pathway, and may subserve complementary or related downstream functions.
Fig. 7.

Schematic model indicating the sites where VGF may regulate energy balance, based on genetic, histochemical, and lesion data. Arcuate orexigenic and anorexigenic projections are shown asgray and white, respectively. Regions of the nervous system in which VGF mRNA or protein is abundantly expressed and/or regulated by feeding or GI manipulation are indicated by aspeckled pattern. The dashed linesidentify areas susceptible to GTG- or MSG-induced injury.Dark-gray projections outlined inblack, downstream of agouti effects on the MC4-R, represent possible candidate circuits that might be regulated by locally synthesized VGF (speckled areas).ARC, Arcuate nucleus; DRG, dorsal root ganglia; LH, lateral hypothalamus; VMN, ventromedial nucleus.
Where do AGRP-, α-MSH-, NPY-, and VGF-expressing arcuate neurons project? Previous studies indicate that many NPY- and AGRP-containing arcuate neurons, a large number of which likely coexpress VGF (Fig. 1), project to the PVN (Legradi and Lechan, 1999; Palkovits, 1999). In the PVN, these processes terminate on thyrotropin-releasing hormone neurons (Legradi and Lechan, 1999) and on neurons in the dorsal and ventral parvocellular subdivisions, an area rich in MC4-R mRNA (Mountjoy et al., 1994), that project to parasympathetic and sympathetic centers in the brainstem and spinal cord (Sawchenko and Swanson, 1982). Use of transneuronal pseudorabies virus tract tracers and analysis of sympathetic nerve activity indicate that the PVN is the major forebrain source and that the raphe pallidus is the major brainstem source of innervation to preganglionic sympathetic neurons that project to BAT (Bamshad et al., 1999; Morrison, 2001a,b). Thus VGF release from PVN-projecting arcuate neurons may have direct effects in the PVN, or the absence of VGF may indirectly alter release of NPY, AGRP, and/or α-MSH from these neurons.
The genetic data presented above argue that the predominant site(s) of VGF action is likely to be downstream of hypothalamic, brainstem, or spinal cord MC4-Rs (Mountjoy et al., 1994; van der Kraan et al., 1999), and these data do not support a critical role for VGF in NPY and/or AGRP secretion. Obesity in Ay/amice results from blockade of the MC4-R by the agouti protein, preventing α-MSH binding and leading to inhibition of satiety-inducing pathways (Barsh et al., 2000). Because the agouti protein is ubiquitously expressed and secreted inAy/a mice (Miller et al., 1993), it seems unlikely that VGF deficiency would inhibit agouti release or binding to melanocortin receptors. The inbreeding of VGF and MC4-R mutant mice that is underway should indirectly clarify this issue. Regions of the brain in which melanocortin receptor blockade could lead to alterations in energy balance include the hypothalamus and brainstem (Fig. 7). In the brainstem, feeding and body weight are controlled by the DMV (Grill et al., 1998), a region of high MC4-R density that receives afferent projections from the gastrointestinal (GI) tract and is located adjacent to the NTS, a site of POMC synthesis. Consistent with a role for VGF in the brainstem, VGF mRNA levels are regulated in the NTS and DMV in response to gastroduodenal irritation (Kanemasa et al., 1995a,b).
Failure of Vgf−/Vgf−,ob/ob mice to phenocopy Npy−/Npy−,ob/ob mice (Erickson et al., 1996) is an indication that NPY synthesis or release is unlikely to be dramatically decreased by targeted deletion of VGF. That double-mutant ob/ob,Vgf−/Vgf− andAy/a,Vgf−/Vgf− mice are not hyperphagic despite robust increases in hypothalamic NPY and AGRP mRNA levels suggests that feeding pathways in VGF-deficient mice are abnormal. Consistent with this possibility, VGF mutant mice have increased hypothalamic NPY and AGRP mRNA levels; however, despite decreased energy stores, these mice are hypermetabolic and consume amounts of food similar to what is consumed by as control mice (Hahm et al., 1999). However, comparable feeding responses of control and VGF mutant mice after fasting (Hahm et al., 1999) and to intracerebroventricular injection of NPY or AGRP (S. Hahm et al., unpublished data) do not indicate gross feeding deficiencies in the latter.
Both GTG and MSG treatments have been shown previously to lead to obesity in NPY-deficient mice (Hollopeter et al., 1998) and to decreased hypothalamic NPY levels in normal mice (Bergen et al., 1998), indicating that obesity in these lesioned animals is not attributable to NPY overexpression. Lower levels of hypothalamic POMC mRNA (Bergen et al., 1998) in GTG-treated mice suggest rather that a decrease in satiety signaling, caused by reduced activity and α-MSH release from injured glucose-responsive neurons in the lateral arcuate and ventromedial hypothalamus leads to GTG-induced obesity. VGF mutant mice fail to develop hyperphagia and obesity after GTG treatment, similar to the phenotype ofAy/a,Vgf−/Vgf− double-mutant mice, indicating that VGF may be a required component in either parallel or downstream pathways that are active when satiety signaling is decreased. Neonatal MSG treatment leads to an extensive degenerative lesion in the arcuate nucleus, reduced hypothalamic POMC mRNA, and virtual elimination of hypothalamic NPY mRNA and AGRP- and NPY-immunoreactive neurons and nerve fibers (Bergen et al., 1998;Legradi and Lechan, 1998; Legradi et al., 1998). Unlike our results with GTG, we noted that MSG treatment led to obesity in VGF mutant mice. This could have resulted from the additional damage to sympathetic postganglionic fibers that innervate BAT (Fig. 7), as suggested by previous studies that have demonstrated impaired UCP induction in response to cold exposure (Tsukahara et al., 1998) and decreased GLUT4 (Morris et al., 1998) transporter levels in BAT from MSG-treated obese mice.
VGF deficiency may therefore lead to increased sympathetic activity to peripheral metabolic tissues including BAT, WAT, and skeletal muscle, and damage to these pathways by MSG might be responsible for failure of the hypermetabolic, lean phenotype to develop.Vgf−/Vgf− mice are normothermic (Hahm et al., 1999), double-mutant ob/ob,Vgf−/Vgf− mice are equally as hypothermic as ob/ob mice, and preliminary studies suggest that UCP1 mRNA levels are not elevated inVgf−/Vgf− BAT (E. Watson, T. Mizuno, Hahm, S. Salton, unpublished data). VGF might consequently function in leptin-independent, melanocortin-regulated sympathetic projections to WAT that control lipolysis. Consistent with this hypothesis, lipoprotein lipase and α and β-3 adrenergic receptor mRNA levels are significantly elevated in Vgf−/Vgf− WAT (Watson, J. Windsor, Salton, unpublished data). Additional studies of sympathetic nervous system activity, adrenergic receptor signaling, and UCP expression in lean VGF mutant mice are warranted, and these should result in a better understanding of the mechanisms responsible for the selective resistance of VGF mutant mice to lesion-induced, diet-induced, and genetically induced obesity.
Footnotes
This work was supported by grants from the National Institutes of Health, the Culpeper Foundation, and Amgen Inc. and by a Career Scientist Award from the Irma T. Hirschl and Monique Weill-Caulier Trusts.
Correspondence should be addressed to Stephen R. J. Salton, Fishberg Research Center for Neurobiology, Box 1065, Mount Sinai School of Medicine, One Gustave L. Levy Place, New York, NY 10029. E-mail:stephen.salton@mssm.edu.
S. Hahm's present address: Tularik Inc., Two Corporate Drive, South San Francisco, CA 94080.
REFERENCES
- 1.Ahima RS, Saper CB, Flier JS, Elmquist JK. Leptin regulation of neuroendocrine systems. Front Neuroendocrinol. 2000;21:263–307. doi: 10.1006/frne.2000.0197. [DOI] [PubMed] [Google Scholar]
- 2.Bamshad M, Song CK, Bartness TJ. CNS origins of the sympathetic nervous system outflow to brown adipose tissue. Am J Physiol. 1999;276:R1569–R1578. doi: 10.1152/ajpregu.1999.276.6.R1569. [DOI] [PubMed] [Google Scholar]
- 3.Barsh G. From Agouti to Pomc: 100 years of fat blonde mice. Nat Med. 1999;5:984–985. doi: 10.1038/12415. [DOI] [PubMed] [Google Scholar]
- 4.Barsh GS, Farooqi IS, O'Rahilly S. Genetics of body-weight regulation. Nature. 2000;404:644–651. doi: 10.1038/35007519. [DOI] [PubMed] [Google Scholar]
- 5.Bergen HT, Mizuno TM, Taylor J, Mobbs CV. Hyperphagia and weight gain after gold-thioglucose: relation to hypothalamic neuropeptide Y and proopiomelanocortin. Endocrinology. 1998;139:4483–4488. doi: 10.1210/endo.139.11.6324. [DOI] [PubMed] [Google Scholar]
- 6.Bonni A, Ginty DD, Dudek H, Greenberg ME. Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci. 1995;6:168–183. doi: 10.1006/mcne.1995.1015. [DOI] [PubMed] [Google Scholar]
- 7.Chung WK, Belfi K, Chua M, Wiley J, Mackintosh R, Nicolson M, Boozer CN, Leibel RL. Heterozygosity for Lep(ob) or Lep(rdb) affects body composition and leptin homeostasis in adult mice. Am J Physiol. 1998;274:R985–R990. doi: 10.1152/ajpregu.1998.274.4.R985. [DOI] [PubMed] [Google Scholar]
- 8.D'Arcangelo G, Habas R, Wang S, Halegoua S, Salton SR. Activation of codependent transcription factors is required for transcriptional induction of the vgf gene by nerve growth factor and Ras. Mol Cell Biol. 1996;16:4621–4631. doi: 10.1128/mcb.16.9.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Rocco G, Pennuto M, Illi B, Canu N, Filocamo G, Trani E, Rinaldi AM, Possenti R, Mandolesi G, Sirinian MI, Jucker R, Levi A, Nasi S. Interplay of the E box, the cyclic AMP response element, and HTF4/HEB in transcriptional regulation of the neurospecific, neurotrophin-inducible vgf gene. Mol Cell Biol. 1997;17:1244–1253. doi: 10.1128/mcb.17.3.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol. 1998;402:442–459. [PubMed] [Google Scholar]
- 11.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 12.Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 13.Erickson JC, Hollopeter G, Palmiter RD. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science. 1996;274:1704–1707. doi: 10.1126/science.274.5293.1704. [DOI] [PubMed] [Google Scholar]
- 14.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 15.Fekete C, Legradi G, Mihaly E, Huang QH, Tatro JB, Rand WM, Emerson CH, Lechan RM. α-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J Neurosci. 2000a;20:1550–1558. doi: 10.1523/JNEUROSCI.20-04-01550.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fekete C, Mihaly E, Luo LG, Kelly J, Clausen JT, Mao Q, Rand WM, Moss LG, Kuhar M, Emerson CH, Jackson IM, Lechan RM. Association of cocaine- and amphetamine-regulated transcript-immunoreactive elements with thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and its role in the regulation of the hypothalamic-pituitary-thyroid axis during fasting. J Neurosci. 2000b;20:9224–9234. doi: 10.1523/JNEUROSCI.20-24-09224.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 18.Grill HJ, Ginsberg AB, Seeley RJ, Kaplan JM. Brainstem application of melanocortin receptor ligands produces long-lasting effects on feeding and body weight. J Neurosci. 1998;18:10128–10135. doi: 10.1523/JNEUROSCI.18-23-10128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunn TM, Miller KA, He L, Hyman RW, Davis RW, Azarani A, Schlossman SF, Duke-Cohan JS, Barsh GS. The mouse mahogany locus encodes a transmembrane form of human attractin. Nature. 1999;398:152–156. doi: 10.1038/18217. [DOI] [PubMed] [Google Scholar]
- 20.Hahm S, Mizuno TM, Wu TJ, Wisor JP, Priest CA, Kozak CA, Boozer CN, Peng B, McEvoy RC, Good P, Kelley KA, Takahashi JS, Pintar JE, Roberts JL, Mobbs CV, Salton SR. Targeted deletion of the Vgf gene indicates that the encoded secretory peptide precursor plays a novel role in the regulation of energy balance. Neuron. 1999;23:537–548. doi: 10.1016/s0896-6273(00)80806-5. [DOI] [PubMed] [Google Scholar]
- 21.Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. 1998;1:271–272. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- 22.Hawley RJ, Scheibe RJ, Wagner JA. NGF induces the expression of the VGF gene through a cAMP response element. J Neurosci. 1992;12:2573–2581. doi: 10.1523/JNEUROSCI.12-07-02573.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hollopeter G, Erickson JC, Palmiter RD. Role of neuropeptide Y in diet-, chemical-, and genetic-induced obesity of mice. Int J Obes Relat Metab Disord. 1998;22:506–512. doi: 10.1038/sj.ijo.0800615. [DOI] [PubMed] [Google Scholar]
- 24.Kanemasa K, Okamura H, Kodama T, Ibata Y. Induction of VGF mRNA in neurons of the rat nucleus tractus solitarius and the dorsal motor nucleus of vagus in duodenal ulceration by cysteamine. Brain Res Mol Brain Res. 1995a;32:55–62. doi: 10.1016/0169-328x(95)00059-2. [DOI] [PubMed] [Google Scholar]
- 25.Kanemasa K, Okamura H, Kodama T, Kashima K, Ibata Y. Time course of the induction of VGF mRNA in the dorsal vagal complex in rats with cysteamine-induced peptic ulcers. Brain Res Mol Brain Res. 1995b;34:309–314. doi: 10.1016/0169-328x(95)00183-s. [DOI] [PubMed] [Google Scholar]
- 26.Legradi G, Lechan RM. The arcuate nucleus is the major source for neuropeptide Y-innervation of thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 1998;139:3262–3270. doi: 10.1210/endo.139.7.6113. [DOI] [PubMed] [Google Scholar]
- 27.Legradi G, Lechan RM. Agouti-related protein containing nerve terminals innervate thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 1999;140:3643–3652. doi: 10.1210/endo.140.8.6935. [DOI] [PubMed] [Google Scholar]
- 28.Legradi G, Emerson CH, Ahima RS, Rand WM, Flier JS, Lechan RM. Arcuate nucleus ablation prevents fasting-induced suppression of ProTRH mRNA in the hypothalamic paraventricular nucleus. Neuroendocrinology. 1998;68:89–97. doi: 10.1159/000054354. [DOI] [PubMed] [Google Scholar]
- 29.Luc PV, Wagner JA. Regulation of the neural-specific gene VGF in PC12 cells. Identification of transcription factors interacting with NGF-responsive elements. J Mol Neurosci. 1997;8:223–241. doi: 10.1007/BF02736835. [DOI] [PubMed] [Google Scholar]
- 30.Miller MW, Duhl DM, Vrieling H, Cordes SP, Ollmann MM, Winkes BM, Barsh GS. Cloning of the mouse agouti gene predicts a secreted protein ubiquitously expressed in mice carrying the lethal yellow mutation. Genes Dev. 1993;7:454–467. doi: 10.1101/gad.7.3.454. [DOI] [PubMed] [Google Scholar]
- 31.Mizuno TM, Mobbs CV. Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology. 1999;140:814–817. doi: 10.1210/endo.140.2.6491. [DOI] [PubMed] [Google Scholar]
- 32.Mizuno TM, Bergen H, Kleopoulos S, Bauman WA, Mobbs CV. Effects of nutritional status and aging on leptin gene expression in mice: importance of glucose. Horm Metab Res. 1996a;28:679–684. doi: 10.1055/s-2007-979877. [DOI] [PubMed] [Google Scholar]
- 33.Mizuno TM, Bergen H, Funabashi T, Kleopoulos SP, Zhong YG, Bauman WA, Mobbs CV. Obese gene expression: reduction by fasting and stimulation by insulin and glucose in lean mice, and persistent elevation in acquired (diet-induced) and genetic (yellow agouti) obesity. Proc Natl Acad Sci USA. 1996b;93:3434–3438. doi: 10.1073/pnas.93.8.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and [corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47:294–297. doi: 10.2337/diab.47.2.294. [DOI] [PubMed] [Google Scholar]
- 35.Mizuno TM, Makimura H, Silverstein J, Roberts JL, Lopingco T, Mobbs CV. Fasting regulates hypothalamic neuropeptide Y, agouti-related peptide, and proopiomelanocortin in diabetic mice independent of changes in leptin or insulin. Endocrinology. 1999;140:4551–4557. doi: 10.1210/endo.140.10.6966. [DOI] [PubMed] [Google Scholar]
- 36.Morris MJ, Tortelli CF, Filippis A, Proietto J. Reduced BAT function as a mechanism for obesity in the hypophagic, neuropeptide Y-deficient monosodium glutamate-treated rat. Regul Pept. 1998;75–76:441–447. doi: 10.1016/s0167-0115(98)00100-1. [DOI] [PubMed] [Google Scholar]
- 37.Morrison SF. Differential control of sympathetic outflow. Am J Physiol Regul Integr Comp Physiol. 2001a;281:R683–R698. doi: 10.1152/ajpregu.2001.281.3.R683. [DOI] [PubMed] [Google Scholar]
- 38.Morrison SF. Differential regulation of sympathetic outflows to vasoconstrictor and thermoregulatory effectors. Ann NY Acad Sci. 2001b;940:286–298. doi: 10.1111/j.1749-6632.2001.tb03684.x. [DOI] [PubMed] [Google Scholar]
- 39.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 40.Nagle DL, McGrail SH, Vitale J, Woolf EA, Dussault BJ, Jr, DiRocco L, Holmgren L, Montagno J, Bork P, Huszar D, Fairchild-Huntress V, Ge P, Keilty J, Ebeling C, Baldini L, Gilchrist J, Burn P, Carlson GA, Moore KJ. The mahogany protein is a receptor involved in suppression of obesity. Nature. 1999;398:148–152. doi: 10.1038/18210. [DOI] [PubMed] [Google Scholar]
- 41.Palkovits M. Interconnections between the neuroendocrine hypothalamus and the central autonomic system. Geoffrey Harris Memorial Lecture, Kitakyushu, Japan, October 1998. Front Neuroendocrinol. 1999;20:270–295. doi: 10.1006/frne.1999.0186. [DOI] [PubMed] [Google Scholar]
- 42.Pizzi WJ, Barnhart JE. Effects of monosodium glutamate on somatic development, obesity, and activity in the mouse. Pharmacol Biochem Behav. 1976;5:551–557. doi: 10.1016/0091-3057(76)90268-9. [DOI] [PubMed] [Google Scholar]
- 43.Salton SR, Volonte C, D'Arcangelo G. Stimulation of vgf gene expression by NGF is mediated through multiple signal transduction pathways involving protein phosphorylation. FEBS Lett. 1995;360:106–110. doi: 10.1016/0014-5793(95)00086-o. [DOI] [PubMed] [Google Scholar]
- 44.Salton SR, Hahm S, Mizuno TM. Of mice and MEN: what transgenic models tell us about hypothalamic control of energy balance. Neuron. 2000a;25:265–268. doi: 10.1016/s0896-6273(00)80892-2. [DOI] [PubMed] [Google Scholar]
- 45.Salton SR, Ferri GL, Hahm S, Snyder SE, Wilson AJ, Possenti R, Levi A. VGF: a novel role for this neuronal and neuroendocrine polypeptide in the regulation of energy balance. Front Neuroendocrinol. 2000b;21:199–219. doi: 10.1006/frne.2000.0199. [DOI] [PubMed] [Google Scholar]
- 46.Sawchenko PE, Swanson LW. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J Comp Neurol. 1982;205:260–272. doi: 10.1002/cne.902050306. [DOI] [PubMed] [Google Scholar]
- 47.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 48.Shimizu-Albergine M, Ippolito DL, Beavo JA. Downregulation of fasting-induced cAMP response element-mediated gene induction by leptin in neuropeptide Y neurons of the arcuate nucleus. J Neurosci. 2001;21:1238–1246. doi: 10.1523/JNEUROSCI.21-04-01238.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsukahara F, Uchida Y, Ohba K, Ogawa A, Yoshioka T, Muraki T. The effect of acute cold exposure and norepinephrine on uncoupling protein gene expression in brown adipose tissue of monosodium glutamate-obese mice. Jpn J Pharmacol. 1998;77:247–249. doi: 10.1254/jjp.77.247. [DOI] [PubMed] [Google Scholar]
- 50.van der Kraan M, Tatro JB, Entwistle ML, Brakkee JH, Burbach JP, Adan RA, Gispen WH. Expression of melanocortin receptors and pro-opiomelanocortin in the rat spinal cord in relation to neurotrophic effects of melanocortins. Brain Res Mol Brain Res. 1999;63:276–286. doi: 10.1016/s0169-328x(98)00291-5. [DOI] [PubMed] [Google Scholar]