

Abstract
Activation of group I metabotropic glutamate receptors (mGluRs) can induce acute depression of excitatory synaptic transmission and long-term depression (LTD) in area CA1 of the rat hippocampus. The underlying mechanisms for both forms of depression are unknown. By measuring presynaptic calcium transients, we show that a reduction in the stimulation-induced presynaptic calcium rise that triggers vesicular release causes the acute depression of transmission by group I mGluRs. In contrast, the mechanism underlying mGluR-induced LTD does not involve a persistent change in stimulation-induced calcium influx. However, analysis of paired-pulse facilitation experiments suggests a presynaptic location for expression of this form of LTD. Furthermore, we show that mGluR-induced LTD can be completely blocked by a specific mGluR5 antagonist, whereas mGluR1 antagonists strongly attenuate the acute depression of transmission. These results support the hypothesis that the acute depression of transmission caused by activation of group I mGluRs involves regulation of stimulation-induced presynaptic calcium transients, whereas mGluR-induced LTD involves a distinct presynaptic modulation downstream of calcium influx.
Keywords: mGluR, DHPG, LTD, presynaptic, calcium, hippocampus
Metabotropic glutamate receptors (mGluRs) play many important roles in regulating neuronal excitability and synaptic transmission (Conn and Pin, 1997). In hippocampal area CA1, activation of mGluRs can reduce both excitatory and inhibitory transmission (Baskys and Malenka, 1991; Desai et al., 1992, 1994;Gereau and Conn, 1995; Manzoni and Bockaert, 1995). In addition, activation of mGluRs is inducing a certain form of long-term depression (LTD) of excitatory synaptic transmission at the Schaffer collateral–CA1 (SCC) synapse (Stanton et al., 1991; Bashir et al., 1993; Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Kemp and Bashir, 1999; Huber et al., 2000; Fitzjohn et al., 2001). This form of LTD (mGluR-LTD) is NMDA receptor independent and can be induced simultaneously with NMDA receptor-dependent LTD (Oliet et al., 1997).
mGluR-LTD can be selectively induced by (RS)-3,5-dihydroxyphenylglycine (DHPG), an agonist that specifically activates group I mGluRs, consisting of mGluR1 and mGluR5 (Palmer et al., 1997; Fitzjohn et al., 1999; Schnabel et al., 1999;Huber et al., 2000, 2001; Fitzjohn et al., 2001; Schnabel et al., 2001;Snyder et al., 2001) (for review, see Kemp and Bashir, 2001). The mechanisms underlying the expression of mGluR-LTD are still unclear. Although some studies show that stimulation-induced LTD is expressed presynaptically (Oliet et al., 1997), others suggest a postsynaptic expression of mGluR-LTD (Snyder et al., 2001) or that the expression is at least dependent on postsynaptic protein synthesis (Huber et al., 2000, 2001). Based on paired-pulse facilitation (PPF) data, it has been suggested recently that chemically induced mGluR-LTD is expressed presynaptically (Fitzjohn et al., 2001).
Several neurotransmitters, including adenosine, acetylcholine, GABA, and neuropeptide Y, reduce stimulation-induced presynaptic calcium influx at the CA3–CA1 synapse and, in this way, strongly attenuate synaptic transmission (Wu and Saggau, 1994a, 1995, 1997; Qian and Saggau, 1997; Qian et al., 1997). Preliminary findings in adult animals (Faas et al., 2000) and previous studies in neonatal rats showed that activation of mGluRs can similarly reduce synaptic transmission at this synapse by modulating presynaptic calcium influx, although the mGluR subtypes involved in this modulation of calcium influx were not known (Yoshino and Kamiya, 1995). These investigations were limited to acute depression by mGluR activation and did not address mGluR-LTD. In the present study, we sought to test the hypothesis that both acute and long-term depression of transmission by DHPG at the CA3–CA1 synapse involves modulation of stimulation-induced presynaptic calcium influx.
MATERIALS AND METHODS
Sprague Dawley rats (4–6 weeks old) were anesthetized with halothane and quickly decapitated in accordance with the guidelines of the National Institutes of Health, as approved by the animal care and use committee of Baylor College of Medicine. Transverse hippocampal slices of 400 μm were prepared on a tissue cutter (Vibratome 1000 plus; St. Louis, MO). For the dissection and the cutting of brain slices, an ice-cold solution with no sodium and high magnesium concentration was used. This solution contained the following (in mm): 2.5 KCl, 1.25 NaH2PO4, 1 CaCl2, 7 MgCl2, 7 dextrose, 190 sucrose, 1 ascorbic acid, 3 pyruvic acid, and 28 NaHCO3. Brain slices were stored in artificial CSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2 MgCl2, 25 dextrose, 1 ascorbic acid, 3 pyruvic acid, and 25 NaHCO3. Both solutions were constantly aerated with 95% CO2–5% O2 to maintain a pH of 7.4 and O2 saturation. Measurements were done in storing solution without the ascorbic acid and pyruvic acid. Recordings were made in small, constantly perfused chambers (1–2 ml) at 30–32°C.
Calcium measurements in presynaptic structures. Presynaptic structures were filled with the fluorescent calcium indicator fura-2 as described previously (Wu and Saggau, 1994a). In short, small amounts of the membrane-permeant form of fura-2 (50 μg of fura-2 AM, 5–10 μl of DMSO with 25% pluronic acid, and 50 μl of extracellular solution buffered to pH 7.3 with 10 mm HEPES) were pressure injected into the SCC using a Picospritzer II (General Valve, Fairfield, NJ) with pipettes of 2–3 μm tip diameter (Fig. 1a). After being taken up by axons, intracellular esterases cleaved the AM form to the membrane-impermeant indicator, which filled the presynaptic terminals. One to 2 hr after injection, brain slices were illuminated at 380 nm, in a small spot (∼150 μm in diameter) in area CA1, 300–500 μm away from the injection site to avoid any contamination of the optical recordings by accidental postsynaptic indicator loading. Fluorescence was detected using a single photodiode connected to a low-noise current-to-voltage converter and amplifier, and fractional changes (ΔF/F) were calculated.
Fig. 1.

Field potentials and corresponding calcium signals. a, Membrane-permeant fura-2 AM was pressure injected into the SCC tract and taken up by axons, in which intracellular esterases cleaved the AM form to the membrane-impermeant calcium indicator, which filled the presynaptic terminals. Fluorescence was recorded from a small spot as shown. CA3 axons were stimulated with a bipolar electrode placed in the SCC tract, and field recordings were made in area CA1. b, Bath application of ionotropic GluR antagonists abolished fEPSPs, although presynaptic volleys persisted. Simultaneously measured Ca2+ signals remained unchanged, indicating their presynaptic origin.
CA3 axons were stimulated with bipolar tungsten electrodes placed in the SCC tract, and extracellular field recordings were made using glass microelectrodes filled with extracellular solution that were placed in the middle of the optical recording spot (Fig. 1a).
Synaptic activity was evoked by current pulses (100–600 μA for 200 μsec) applied through the stimulation electrode. In every experiment, two pulses with a 20 msec interval were given. The amplitude of ΔF/F induced by the second stimulus always exceeded that of the first stimulus, indicating that the calcium indicator was not saturated during the first stimulus.
To verify the presynaptic origin of the observed calcium transients, ionotropic glutamate receptor antagonists (20 μm CNQX, 25 μmd-APV, or 50 μmdl-APV) were added to the bath at the end of each experiment. The antagonists always abolished synaptic transmission without affecting the presynaptic fiber volley or the corresponding Ca2+ rise, indicating that the Ca2+ signals were of presynaptic origin (Fig. 1b).
Field recordings were made using a model 5A amplifier (Getting Instruments, San Diego, CA). Data were acquired using a 16 bit analog-to-digital converter and processed with custom-made software. The same software was used together with Microsoft (Seattle, WA) Excel to analyze the data.
Some of the experiments testing the effects of the mGluR5 antagonists on synaptic transmission were performed on a separate setup at room temperature in which we only measured the field EPSPs (fEPSPs). All drugs used to determine receptor specificity were added 5 min before and throughout the period of DHPG application. Stimulation and recording electrodes were placed in hippocampal slice as described above. Recordings were made using a A310 Accupulser (World Precision Instruments, Sarasota, FL), a low-pass Bessel Filter 4 pole amplifier (Warner Instruments, Hamden, CT), an Axoclamp 2B, and pClamp 8.0 software (Axon Instruments, Foster City, CA). The same program was used together with Microsoft Excel to analyze the data.
Relative values reported throughout the text are with respect to control situations. Statistical comparisons were made using the Student's t test, and differences are considered significant when p < 0.05.
All drugs were purchased from Sigma (St. Louis, MO), except for APV, DHPG, DNQX, 2-methyl-6-(phenylethynyl) pyridine (MPEP), 7-(hydroxyimino)cyclopropa(β)chromen-1a-carboxylate ethyl ester (CPCCOEt), and LY367385, which were obtained from Tocris Cookson (Ballwin, MO).
RESULTS
The role of mGluR5 and mGluR1 in DHPG-induced LTD and acute depression of transmission in area CA1
Consistent with previous reports, we found that application of DHPG (100 μm, 20 min) produced a partially reversible depression of evoked fEPSPs (Fig.2a). During application of DHPG, the slope of the fEPSP was reduced to 25 ± 4% of its control value (n = 12; p < 0.001). This effect only partially recovered during wash out, resulting in mGluR-LTD that was sustained for the entire observation period (up to 1 hr). Forty minutes after washout of DHPG, the fEPSP had recovered to 65 ± 6% (p < 0.001) (Fig.1b). When the specific NMDA antagonist APV (50 μm) was present during the whole procedure, the fEPSP recovered to 72 ± 9% (n = 5; data not shown) after washout of DHPG. This confirmed previous findings that mGluR-LTD is independent of NMDA receptor activation (Palmer et al., 1997; Huber et al., 2001). The acute DHPG-induced depression consists of two components, the mGluR-LTD and a reversible component. The latter was calculated as the difference between the slope of the fEPSP during the acute depression and the slope of the fEPSP during mGluR-LTD. The reversible component by itself would reduce the fEPSP to 60 ± 6% of the pre-DHPG control value (Fig. 2b).
Fig. 2.
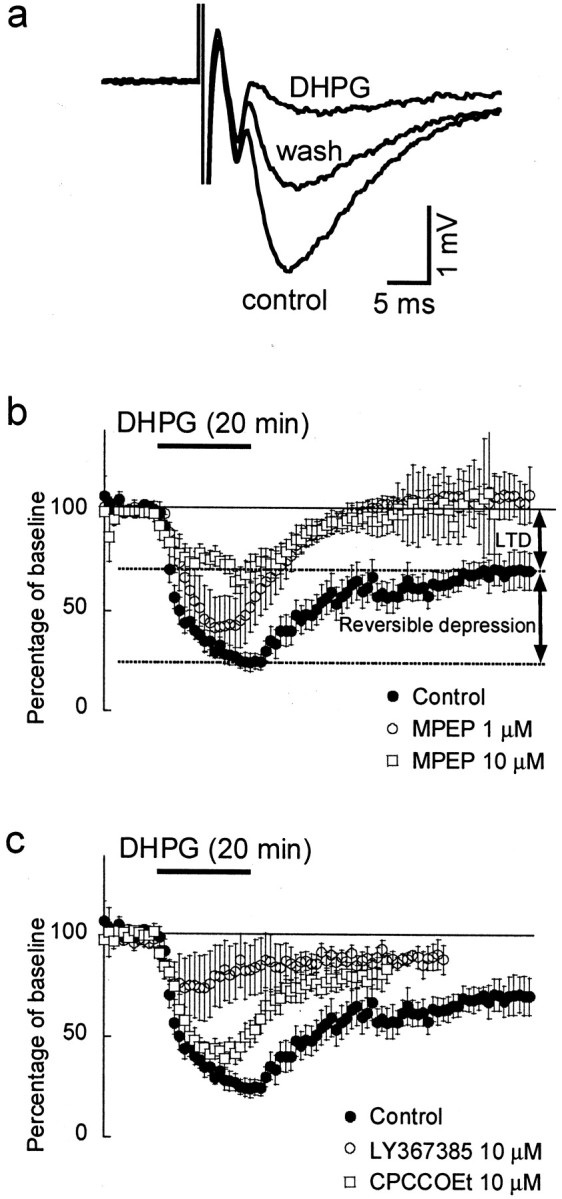
mGluR5 activation is required for mGluR-LTD.a, Evoked fEPSPs measured in stratum radiatum; stimulation artifacts are truncated. The amplitude and slope of the fEPSPs were reduced during application of DHPG (100 μm) and recovered partially during washout, revealing a DHPG-induced LTD. b, Slope of fEPSPs evoked at 1 min interstimulus interval, as percentage of baseline values. In control experiments (filled circles), DHPG caused a strong acute depression of the fEPSPs followed by LTD. The total response can be separated into two parts, a reversible suppression by DHPG and the LTD. Application of the mGluR5 antagonist MPEP at 1 μm (open circles) or 10 μm(open squares) did not abolish the acute depression but prevented the induction of LTD. c, Application of the mGluR1 antagonists LY367385 (10 μm; open circles) or 10 μm CPCCOEt (open squares) strongly attenuated the acute depression but not LTD.
mGluR-LTD has been shown to be absent in mGluR5 knock-out mice (Huber et al., 2001). Although these studies strongly implicate mGluR5 in mGluR-LTD, studies using standard knock-out mice can be confounded by developmental compensatory changes in the mice. We therefore investigated the role of mGluR5 in the acute DHPG-induced depression of synaptic transmission and mGluR-LTD using the selective mGluR5 antagonist MPEP. We found that MPEP (1 μm) did not significantly reduce the acute DHPG effect (n = 5;p > 0.1) (Fig. 1b); however, this concentration of MPEP completely abolished mGluR-LTD. The fEPSP recovered to 107 ± 7% of control after washout of DHPG (p < 0.005). A higher, but less specific, concentration of 10 μm MPEP (n= 8) (Fig. 1b) significantly reduced, but did not abolish, the acute DHPG effect (p < 0.05) to 67 ± 7% of pre-DHPG control.
To test the role of mGluR1 in the DHPG-induced depression of synaptic transmission and mGluR-LTD, the selective mGluR1 antagonists CPCCOEt and LY367385 were used. In the presence of CPCCOEt (10 μm), DHPG still induced both an acute and sustained depression of the fEPSP. During application of DHPG, the slope of the fEPSP was reduced to 43 ± 10% of its control value (n = 4; p < 0.001) (Fig.2c). After washout of DHPG, the fEPSP returned to 80 ± 5% of its control value (p < 0.01) (Fig.2c). These reductions in fEPSP were not significantly different compared with the reduction with DHPG alone. We also used the more potent mGluR1 antagonist LY367385 (10 μm). During DHPG, the fEPSP decreased to 75 ± 4% of its control value (n = 6; p < 0.001) (Fig.2c) and returned to 87 ± 3% after washout (p < 0.005) (Fig. 2c). Compared with the depression caused by DHPG alone, under LY367385, both the acute (p < 0.001) and long-term depression (p < 0.05) were significantly attenuated. Specifically, LY367385 seems to almost abolish the reversible component: with DHPG alone, the fEPSP would be reduced to 60 ± 6% by the reversible component, whereas with LY367385 present, it would be only reduced to 89 ± 6% (p < 0.01).
These experiments suggest that, in hippocampal area CA1, mGluR5 activation is essential for mGluR-LTD induction. Furthermore, we show that the acute depression caused by DHPG is probably dependent on mGluR1 activation.
Modulation of presynaptic calcium transients by DHPG
Our previous studies suggest that DHPG reduces synaptic transmission at the CA3–CA1 synapse via a presynaptic mechanism (Gereau and Conn, 1995). To test the hypothesis that depression by DHPG involves modulation of presynaptic calcium influx, we tested whether DHPG could reduce the stimulation-induced increase in presynaptic calcium in Schaffer collateral terminals. We selectively loaded presynaptic axons at the CA3–CA1 synapse with the calcium indicator fura-2. We then performed simultaneous measurements of presynaptic calcium transients and fEPSPs and examined the relationship between changes in calcium and synaptic transmission (n = 8).
Application of DHPG significantly reduced the stimulation-induced ΔF/F to 87 ± 2% (n = 8;p < 0.005) (Fig.3a,b), indicating that activation of group I mGluRs diminished the stimulation-induced presynaptic calcium influx. However, during washout of DHPG, the stimulation-induced calcium rise returned to baseline amplitude (99 ± 1%; p > 0.1) (Fig.3a,b), whereas the fEPSP only partially recovered to 67 ± 7% (Fig. 3b).
Fig. 3.
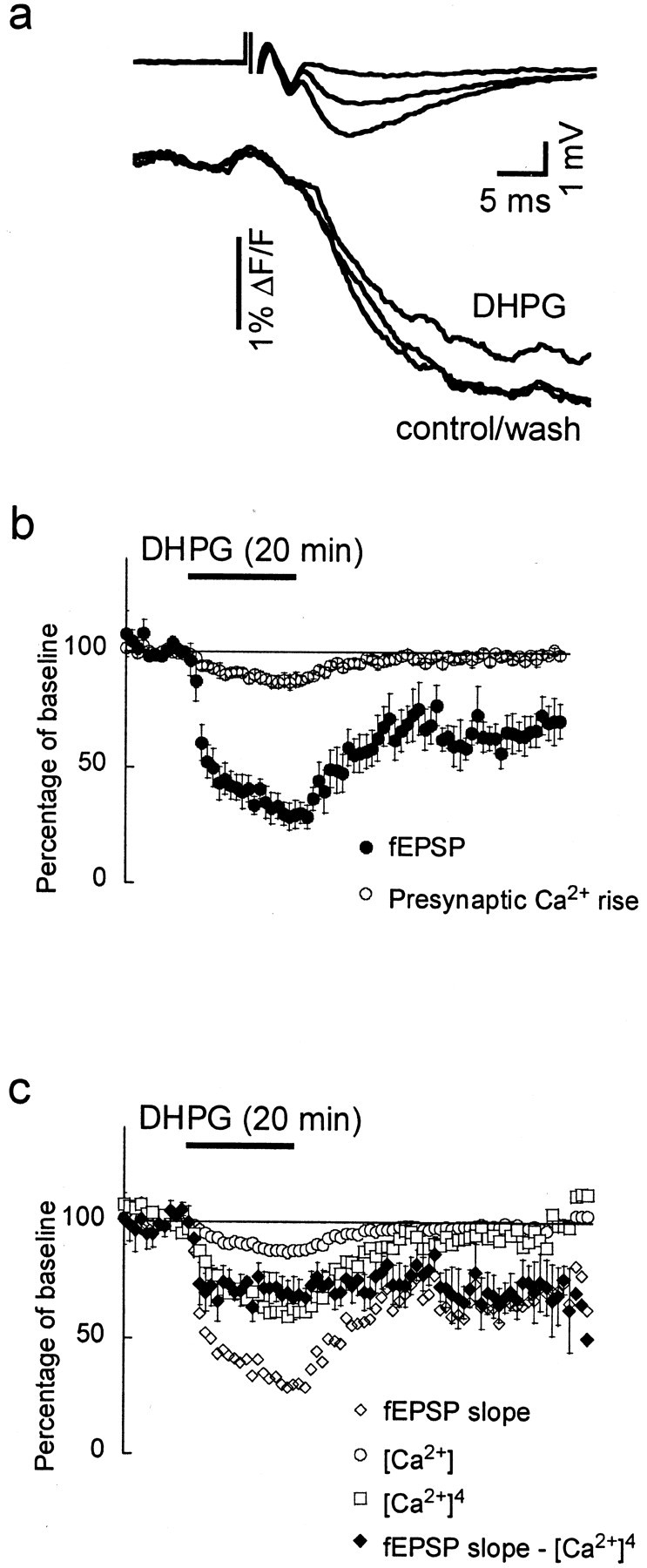
Acute depression of transmission by DHPG but not mGluR-LTD involves a reduction in stimulation-induced calcium influx.a, Simultaneously recorded fEPSPs and presynaptic calcium signals. Fura-2 was excited at 380 nm; thus, a decrease in ΔF/F indicates an increase in calcium concentration. In the presence of DHPG (100 μm), the calcium signal was attenuated but returned to control values after washout. b, The slope of the fEPSPs (dots) and the ΔF/Fchanges (triangles), measured every minute, are shown before, during, and after DHPG application, revealing both acute depression and mGluR-LTD of transmission. c, The mGluR-LTD (filled diamonds) was isolated by subtracting the predicted reduction of the fEPSP, calculated as [Ca2+]4 (open squares), from the measured fEPSP (open diamonds). For clarity, error bars are only shown for the calculated mGluR-LTD.
The dependence of neurotransmitter release on presynaptic calcium concentration is nonlinear and can be expressed by the a power relation: Release ∝ [Ca2+]m, wherem = 4, although some exceptions are known.
During DHPG application, the calculated power relationship between the fEPSP and the presynaptic calcium rise was m = 12.3 ± 3.6. Thus, the observed reduction in calcium rise alone cannot account for the total reduction in the fEPSP. However, the power for the reversible component alone was m = 4.0 ± 0.9, suggesting that the fEPSP reduction during DHPG has two components: first, a reversible component that is mediated by a reduction of the stimulation-induced presynaptic calcium rise, and second, a long-lasting component that is independent of the change in stimulation-induced calcium influx (i.e., mGluR-LTD).
In an attempt to isolate the calcium-independent component of the DHPG-induced depression of transmission, we calculated the calcium-dependent component as [Ca2+]4 and subtracted this result from the measured fEPSP (Fig. 3c). This revealed that the calcium-independent reduction of transmission was established within 2 min after application of DHPG and remained stable throughout the experiment. The calculated fEPSP was 70 ± 8% of control 2 min after the DHPG application and 70 ± 9% of control 40 min after washout of DHPG (Fig. 3c). This calcium-independent component did not change after washout of DHPG and appears to be the mGluR-LTD. Thus, changes underlying the mGluR-LTD must be downstream of the presynaptic calcium rise that triggers vesicular neurotransmitter release. This mGluR-LTD could be presynaptic (e.g., affecting the release machinery), postsynaptic (e.g., decreasing the glutamate sensitivity), or both.
mGluR-dependent LTD can also be induced using a more physiological synaptic stimulation. It has been shown previously that paired-pulse low-frequency stimulation (PP-LFS) induces both NMDA-dependent and NMDA-independent LTD (Kemp and Bashir, 1997; Kemp and Bashir, 1999;Huber et al., 2000). In the presence of the NMDA antagonist APV (50 μm), delivery of PP-LFS (50 msec interstimulus interval) at 1 Hz for 15 min produces a stable form of LTD. This form of LTD (PP-LFS-LTD) is completely blocked by application of the nonspecific mGluR antagonist LY341495 (Huber et al., 2000). We used the same protocol to study the modulation of presynaptic calcium transients in synaptically induced mGluR-LTD. PP-LFS stimulation induced a stable form of NMDA-independent LTD, in which the fEPSP was reduced to 78 ± 3% of control (n = 4; p < 0.05). The induction of PP-LFS-LTD was completely blocked when MPEP (10 μm) was present. Under MPEP, the fEPSP returned to 99 ± 3% of control (n = 5) (Fig.4a), indicating that PP-LFS-LTD, like mGluR-LTD, is dependent on mGluR5 activation.
Fig. 4.

Paired-pulse low-frequency stimulation induces NMDA-independent LTD. Long-lasting (15 min) PP-LFS (50 to 1 Hz) during application of the NMDA antagonist d-APV (25 μm) induces NMDA-independent LTD of synaptic transmission (filled circles in a andb). a, PP-LFS-LTD was completely blocked by 10 μm MPEP (open circles), indicating an essential role for mGluR5 activation. b, Stimulus-induced presynaptic calcium (open circles) rise was not changed during PP-LFS-LTD.
Just as with mGluR-LTD, the stimulation-induced presynaptic calcium rise returned to baseline during the stable component of PP-LFS-LTD (99 ± 1%) (Fig. 4b).
Modulation of paired-pulse facilitation in mGluR-LTD and PP-LFS-LTD
To elucidate the possible presynaptic or postsynaptic expression of mGluR-LTD, we tested for changes in PPF (n = 11). A recent study demonstrated that PPF with a 50 msec interstimulus interval increases during mGluR-LTD (Fitzjohn et al., 2001). This finding was interpreted as support for the hypothesis that mGluR-LTD is mediated presynaptically. We repeated this experiment using a 20 msec interstimulation interval, which should minimize possible polysynaptic contributions to modulation of PPF, allowing a more clear analysis of the presynaptic versus postsynaptic locus of depression. With a 20 msec interstimulus interval, we observed a control PPF of 1.5 ± 0.1. This ratio significantly increased to 141 ± 12% during DHPG-induced acute depression (p < 0.01) and 120 ± 5% (p < 0.01) when DHPG was washed out (Fig. 5a,b). When inducing LTD with the PP-LFS protocol, the ratio also increased similarly to 122 ± 4% (n = 3; p< 0.05) (Fig. 3b). Different interstimulus intervals will have different disadvantages. Whereas longer interstimulus intervals used could allow polysynaptic modulation of the presynaptic or postsynaptic activation that may confound this interpretation, shorter interstimulus intervals increase the likelihood of AMPA desensitization. Therefore, we repeated these experiments for different intervals varying from 20 to 150 msec (n = 3). We found that, for every tested interval in this range, the PPF ratio was increased during and after the application of DHPG (data not shown).
Fig. 5.
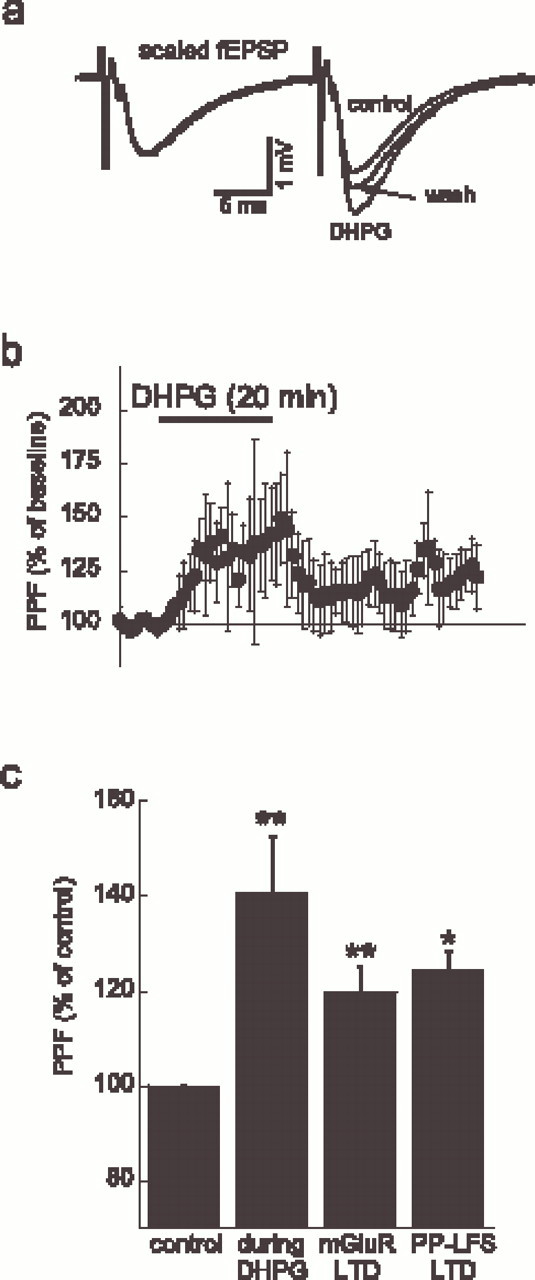
mGluR-LTD and PP-LFS-LTD both modulate paired-pulse facilitation. a, fEPSPs at control, during DHPG, and after washout. The traces are normalized to the first fEPSP at control (calibration applies to control), revealing the change in PPF during DHPG and after washout. b, DHPG increased PPF, which partially returned to control after washout.c, Histogram summarizing the PPF increase during DHPG (n = 8) and after induction of mGluR-LTD, as well as in PP-LFS-LTD (n = 4). *p < 0.05; **p < 0.01.
These results support and extend the previous findings (Fitzjohn et al., 2001) and strongly suggest that both mGluR-LTD and PP-LFS-LTD are, at least partially, maintained by presynaptic mechanisms.
DISCUSSION
Group I metabotropic glutamate receptor activation reduces excitatory synaptic transmission in hippocampal area CA1 (Gereau and Conn, 1995; Huber et al., 2001; Mannaioni et al., 2001). We showed previously that the modulation underlying this reduction is at least partially located presynaptically, because activation of group I mGluRs alters paired-pulse facilitation (Gereau and Conn, 1995). Our current findings suggest that group I mGluR activation reduces stimulation-induced presynaptic calcium transients, which likely accounts for the reversible reduction of synaptic transmission. We also found a component of the reduction of synaptic transmission that is downstream from the presynaptic calcium rise that triggers vesicular release. This component likely underlies the mGluR-LTD observed after washout of DHPG.
Previous studies have provided conflicting evidence for the identity of the group I mGluR that underlies the depression of transmission induced by DHPG in area CA1. For example, one group has shown that the mGluR5 antagonist MPEP has no effect on the depression of EPSCs induced by DHPG in area CA1 (Mannaioni et al., 2001). In contrast, two other studies showed that the depression of transmission induced by 1S,3R-ACPD and DHPG is absent in mGluR5 knock-out mice, suggesting that mGluR5 mediates this effect (Lu et al., 1997;Huber et al., 2001). Our studies support differential roles for mGluR5 in acute and long-term depression of transmission in area CA1. We found that the mGluR5 antagonist MPEP (1 μm) had no effect on the acute depression of transmission induced by DHPG but completely blocked DHPG-induced LTD. In contrast, the specific mGluR1 antagonist LY367385 strongly suppressed acute depression of transmission induced by DHPG but only partially blocked DHPG-induced LTD. These results suggest that, in hippocampal area CA1, mGluR5 activation is essential for mGluR-LTD, whereas the acute depression caused by DHPG is mainly dependent on mGluR1 activation. The fact that higher concentration of MPEP (10 μm) also partially blocked the reversible component and the mGluR-LTD is partially blocked by LY367385 seems to be caused by unspecific antagonistic effects of these drugs.
The reason for the discrepancy in the ability of 10 μm MPEP to reduce DHPG-induced depression of transmission between the present study and that of Mannaioni et al. (2001) is not clear but may be accounted for by differences in the age of the animals used (we used 30- to 40-d-old animals, whereas Mannaioni et al. used 2- to 3-week-old animals) or differences in technique (we used field recordings in intact slices, whereas Mannaioni et al. used whole-cell recordings in the presence of bicuculline with area CA3 removed from the hippocampal slice).
Several studies have addressed the question of whether LTD at the CA3–CA1 synapse is expressed presynaptically or postsynaptically. There are reports that LTD expression is associated with a decrease in miniature EPSC (mEPSC) frequency but not amplitude (Oliet et al., 1997; Fitzjohn et al., 2001), an increase in coefficient of variation of EPSC amplitude (Bolshakov and Siegelbaum, 1994), and an increase in PPF (Fitzjohn et al., 2001), all suggesting a presynaptic mechanism. We confirmed the findings of Fitzjohn et al. (2001) that PPF is increased in mGluR-LTD, indicating a presynaptic location for mGluR-LTD. Furthermore, we found comparable results for a stimulation-induced form of mGluR-dependent LTD that it is considered comparable with mGluR-LTD (Kemp and Bashir, 1999; Huber et al., 2000).
Our results show that, in DHPG-induced depression, the attenuation of stimulation-induced presynaptic calcium transients is only observed during the DHPG application. After washout of DHPG, LTD was established, but the presynaptic calcium rise returned to baseline levels. This finding suggests that the expression of mGluR-LTD is downstream of the presynaptic calcium rise that triggers transmitter release. We cannot rule out, however, that for the induction of LTD a transiently reduced presynaptic calcium influx is needed. On the other hand, mGluR-LTD can also be induced by DHPG without any stimulation during the presence the drug, which makes a dependence on a phase of reduced presynaptic calcium rise unlikely.
Other publications suggest a strong postsynaptic role in the expression of mGluR-LTD. It has been shown that the induction of mGluR-LTD is dependent on a postsynaptic protein synthesis (Huber et al., 2000,2001), which is expressed as internalization of AMPA receptors (Snyder et al., 2001), and that it is dependent on postsynaptic activation of voltage-dependent calcium channels (VDCCs) (Oliet et al., 1997). As discussed extensively by Fitzjohn et al. (2001), internalization of AMPA receptors can cause a change in PPF when it involves preferential silencing of high release probability synapses. Silencing of synapses could also explain the previously reported reduction in mEPSC frequency and an increase in coefficient of variation of EPSC amplitude. Our data cannot rule out a postsynaptic role in the induction and maintenance of LTD. We show that mGluR-LTD is independent of a change in the presynaptic calcium rise and is probably induced within 2 min after application of DHPG. Taking this fast induction into account, we propose that the initial phase of mGluR-LTD is not dependent on protein synthesis.
The mechanism by which group I mGluR activation reduces presynaptic calcium transients is not clear. Three possible mechanisms can directly attenuate the presynaptic calcium rise: (1) a direct inhibition of VDCCs, which could be mediated by decreased conductance, open time or probability, or number of active channels, (2) modulation of the ionic currents involved in the shaping of the presynaptic action potential, leading to decreased activation of VDCCs, or (3) reduced calcium release from intracellular stores, which may contribute to the observed calcium transients. Future experiments should attempt to elucidate the mechanisms underlying the modulation of calcium transients by DHPG. Nonetheless, our results clearly demonstrate that the acute and long-term depression of synaptic transmission induced by activation of group I mGluRs are mediated by at least two distinct presynaptic mechanisms, one involving modulation of stimulation-induced calcium transients, and the other mediated downstream of calcium influx.
We examined previously the mechanism underlying paired-pulse facilitation at short interstimulus intervals. Increased residual calcium levels correlate strongly with the amplitude of PPF, and therefore the residual calcium is thought to underlie PPF (Wu and Saggau, 1994b). However, our findings here show that presynaptic calcium influx is not decreased in mGluR-LTD. One possibility is that the extrusion and/or the buffering speed are altered, leading to different presynaptic calcium concentrations at the beginning of the second stimulus. Unfortunately, we could not accurately obtain changes in the speed of calcium extrusion–buffering because we used a high-affinity indicator, which unbinds too slowly from calcium to determine extrusion and buffering properties. If the total affinity of the endogenous calcium buffers were lower in mGluR-LTD, this would result in slower buffering of the calcium. It would also lead to a higher resting level of calcium. Preliminary experiments performed in our laboratory (data not shown) suggest that levels of resting presynaptic calcium are slightly increased during DHPG and after washout. Such varying levels of calcium might influence multiple secondary processes, possibly underlying changes in transmitter release. A change in the affinity of the presynaptic calcium buffers could thus underlie the mGluR-LTD.
We conclude that there are likely two presynaptic components in DHPG-induced depression. One component is reversible and is based on a reduction in stimulation-induced calcium transients. Furthermore, this component involves mGluR1 activation. The second component, which mediates mGluR-LTD, is expressed downstream from the presynaptic calcium influx and is dependent on mGluR5 activation. This latter component may involve buffering or extrusion of calcium and/or mechanisms directly related to vesicular release. Finally, we showed that PP-LFS-induced LTD, the stimulation-induced counterpart of this mGluR-LTD, occurs trough a similar mechanism.
Footnotes
This work was supported by National Institutes of Health Grants NS33147 (P.S.) and MH60230 (R.W.G.). We thank Dr. D. Johnston for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Peter Saggau, Baylor College of Medicine, Neuroscience, 1 Baylor Plaza, Room S603, Houston, TX 77030. E-mail: psaggau@bcm.tmc.edu.
REFERENCES
- 1.Bashir ZI, Jane DE, Sunter DC, Watkins JC, Collingridge GL. Metabotropic glutamate receptors contribute to the induction of long-term depression in the CA1 region of the hippocampus. Eur J Pharmacol. 1993;239:265–266. doi: 10.1016/0014-2999(93)91009-c. [DOI] [PubMed] [Google Scholar]
- 2.Baskys A, Malenka RC. Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol (Lond) 1991;444:687–701. doi: 10.1113/jphysiol.1991.sp018901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- 4.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 5.Desai MA, Smith TS, Conn PJ. Multiple metabotropic glutamate receptors regulate hippocampal function. Synapse. 1992;12:206–213. doi: 10.1002/syn.890120305. [DOI] [PubMed] [Google Scholar]
- 6.Desai MA, McBain CJ, Kauer JA, Conn PJ. Metabotropic glutamate receptor-induced disinhibition is mediated by reduced transmission at excitatory synapses onto interneurons and inhibitory synapses onto pyramidal cells. Neurosci Lett. 1994;181:78–82. doi: 10.1016/0304-3940(94)90564-9. [DOI] [PubMed] [Google Scholar]
- 7.Faas GC, Adwanikar H, Gereau RW, Saggau P. Activation of metabotropic glutamate receptors affects presynaptic calcium influx. Soc Neurosci Abstr. 2000;26:332.8. doi: 10.1523/JNEUROSCI.22-16-06885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzjohn SM, Kingston AE, Lodge D, Collingridge GL. DHPG-induced LTD in area CA1 of juvenile rat hippocampus; characterisation and sensitivity to novel mGlu receptor antagonists. Neuropharmacology. 1999;38:1577–1583. doi: 10.1016/s0028-3908(99)00123-9. [DOI] [PubMed] [Google Scholar]
- 9.Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL. A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro. J Physiol (Lond) 2001;537:421–430. doi: 10.1111/j.1469-7793.2001.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gereau RW, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1256. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 12.Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- 13.Kemp N, Bashir ZI. NMDA receptor-dependent and -independent long-term depression in the CA1 region of the adult rat hippocampus in vitro. Neuropharmacology. 1997;36:397–399. doi: 10.1016/s0028-3908(96)90015-5. [DOI] [PubMed] [Google Scholar]
- 14.Kemp N, Bashir ZI. Induction of LTD in the adult hippocampus by the synaptic activation of AMPA/kainate and metabotropic glutamate receptors. Neuropharmacology. 1999;38:495–504. doi: 10.1016/s0028-3908(98)00222-6. [DOI] [PubMed] [Google Scholar]
- 15.Kemp N, Bashir ZI. Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol. 2001;65:339–365. doi: 10.1016/s0301-0082(01)00013-2. [DOI] [PubMed] [Google Scholar]
- 16.Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci. 2001;21:5925–5934. doi: 10.1523/JNEUROSCI.21-16-05925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manzoni O, Bockaert J. Metabotropic glutamate receptors inhibiting excitatory synapses in the CA1 area of rat hippocampus. Eur J Neurosci. 1995;7:2518–2523. doi: 10.1111/j.1460-9568.1995.tb01051.x. [DOI] [PubMed] [Google Scholar]
- 19.Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 20.Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- 21.Qian J, Saggau P. Presynaptic inhibition of synaptic transmission in the rat hippocampus by activation of muscarinic receptors: involvement of presynaptic calcium influx. Br J Pharmacol. 1997;122:511–519. doi: 10.1038/sj.bjp.0701400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian J, Colmers WF, Saggau P. Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: modulation of presynaptic Ca2+ entry. J Neurosci. 1997;17:8169–8177. doi: 10.1523/JNEUROSCI.17-21-08169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnabel R, Kilpatrick IC, Collingridge GL. An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology. 1999;38:1585–1596. doi: 10.1016/s0028-3908(99)00062-3. [DOI] [PubMed] [Google Scholar]
- 24.Schnabel R, Kilpatrick IC, Collingridge GL. Protein phosphatase inhibitors facilitate DHPG-induced LTD in the CA1 region of the hippocampus. Br J Pharmacol. 2001;132:1095–1101. doi: 10.1038/sj.bjp.0703905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- 26.Stanton PK, Chattarji S, Sejnowski TJ. 2-Amino-3-phosphonopropionic acid, an inhibitor of glutamate-stimulated phosphoinositide turnover, blocks induction of homosynaptic long-term depression, but not potentiation, in rat hippocampus. Neurosci Lett. 1991;127:61–66. doi: 10.1016/0304-3940(91)90895-z. [DOI] [PubMed] [Google Scholar]
- 27.Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 1994a;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- 28.Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci. 1994b;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu LG, Saggau P. GABA(b) receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J Physiol (Lond) 1995;485:649–657. doi: 10.1113/jphysiol.1995.sp020759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 31.Yoshino M, Kamiya H. Suppression of presynaptic calcium influx by metabotropic glutamate receptor agonists in neonatal rat hippocampus. Brain Res. 1995;695:179–185. doi: 10.1016/0006-8993(95)00743-a. [DOI] [PubMed] [Google Scholar]