

Abstract
Amyotrophic lateral sclerosis (ALS) is a relentlessly progressive lethal disease that involves selective annihilation of motoneurons. Glial cell line-derived neurotrophic factor (GDNF) is proposed to be a promising therapeutic agent for ALS and other motor neuron diseases. Because adeno-associated virus (AAV) has been developed as an attractive gene delivery system with proven safety, we explored the therapeutic efficacy of intramuscular delivery of the GDNF gene mediated by an AAV vector (AAV-GDNF) in the G93A mouse model of ALS. We show here that AAV-GDNF leads to substantial and long-lasting expression of transgenic GDNF in a large number of myofibers with its accumulation at the sites of neuromuscular junctions. Detection of GDNF labeled with FLAG in the anterior horn neurons, but not β-galactosidase expressed as a control, indicates that most of the transgenic GDNF observed there is retrogradely transported GDNF protein from the transduced muscles. This transgenic GDNF prevents motoneurons from their degeneration, preserves their axons innervating the muscle, and inhibits the treated-muscle atrophy. Furthermore, four-limb injection of AAV-GDNF postpones the disease onset, delays the progression of the motor dysfunction, and prolongs the life span in the treated ALS mice. Our finding thus indicates that AAV-mediated GDNF delivery to the muscle is a promising means of gene therapy for ALS.
Keywords: amyotrophic lateral sclerosis, motoneuron, adeno-associated virus vector, glial cell line-derived neurotrophic factor, gene therapy, retrograde transport
Amyotrophic lateral sclerosis (ALS) is one of the most tragic neurodegenerative diseases affecting motoneurons. Because the mechanism leading to motoneuron degeneration in ALS is not understood, currently there is no therapy available to prevent or cure ALS. Approximately 20% of familial ALS is linked to mutations in the Cu/Zn superoxide dismutase (SOD1) gene (Julien, 2001), transgenic mice overexpressing this mutant (mSOD1G93A) gene found to develop a dominantly inherited adult-onset paralytic disorder that has many of the clinical and pathological features of familial ALS (Gurney et al., 1994).
Glial cell line-derived neurotrophic factor (GDNF), which has been demonstrated to be the most potent neurotrophic factor for the proliferation, differentiation, and survival of spinal motoneurons, exhibits very good therapeutic potential for ALS (Henderson et al., 1994; Oppenheim et al., 1995; Yan et al., 1995; Sagot et al., 1996;Bohn, 1999; Mohajeri et al., 1999). Systemic administration of GDNF as a recombinant protein to ALS patients, however, is not beneficial, because of its short plasma half-life and poor access to motoneurons, on the one hand, and, on the other hand, because of its severe side effects that prevent its administration at an adequate dose (Haase et al., 1997; Alisky and Davidson, 2000). If ways that make continuous and motoneuron-confined delivery of GDNF possible are established, the disadvantages of its systemic administration will be overcome. Gene therapy involving the injection of a vector encoding a gene for GDNF into skeletal muscles will be a good candidate for such a method.
Three different viral vectors have been examined to transfer different genes to different tissues for ALS gene therapy (Alisky and Davidson, 2000). An adeno-associated virus (AAV) vector is one of the most attractive gene delivery vehicles and one that might be more practical with respect to safety. Skeletal muscle is a good platform for gene delivery (Xiao et al., 1996), and only at neuromuscular junctions (NMJs) are the nerve terminals in contact with myofibers, in which a barrier against various substances is absent, allowing them to reach the CNS. Furthermore, intramuscular injection is much safer and easier compared with intraspinal injection. Indeed, muscle-directed gene therapy mediated by an AAV vector has led to tremendous success in numerous animal models of human diseases (Li et al., 1999; Kay et al., 2000; Wang et al., 2000).
Given the existence of endogenous GDNF in muscles and the spinal cord and its upregulation in ALS patients (Yamamoto et al., 1996; Golden et al., 1998; Suzuki et al., 1998; Grundstrom et al., 1999), we constructed the AAV-GDNF vector expressing a GDNF-FLAG fusion protein that can be readily distinguished from the endogenous one with FLAG. We showed that the intramuscular administration of an AAV vector harboring the GDNF gene to ALS mouse models can significantly delay the onset of disease, lengthen the life span, abate the behavioral impairment, and promote motoneuron survival. Moreover, we obtained direct evidence that the product of GDNF boosted by gene delivery in the muscle is retrogradely transported to the motoneurons of the spinal cord.
MATERIALS AND METHODS
Administration of an AAV vector. Male transgenic mice with the G93A human SOD1 mutation (SOD1G93A) were obtained from The Jackson Laboratory (Bar Harbor, ME). AAV vector plasmid pAAV-GDNFflag contains mouse GDNF cDNA tagged with a FLAG sequence (DYKDDDDK) at the C terminus under the human cytomegalovirus immediate-early promoter, with the human growth hormone first intron and simian virus 40 polyadenylation signal sequence between the inverted terminal repeats of the AAV type 2 genome (Wang et al., 2002). AAV vector plasmid pAAV-LacZ, auxiliary plasmid pHLP19, and pladenol were described previously (Matsushita et al., 1998). AAV vectors were produced in human embryonic kidney 293 (HEK293) cells by triple transfection of vector plasmid and helper plasmids listed above as described previously (Wang et al., 2002). In brief, subconfluent HEK293 cells were transiently transfected by calcium phosphate method. Seventy-two hours after transfection, the cells were collected and subjected to three cycles of freeze–thaw lysis (alternating between dry-ice–ethanol and 37°C baths). AAV vectors were purified by two sequential continuous cesium chloride density gradients and estimated for final particle titer by quantitative DNA dot-blot hybridization. Before administration, AAV vectors were diluted in PBS to 1 × 1011 genome copies/100 μl. At 9 weeks of age, ALS mice were randomly assigned to one treatment group that was injected with AAV-GDNF vector (n = 12) or one of two control groups that were injected with AAV-LacZ vector (n = 6) and the vehicle (n = 5), respectively, into four limbs (gastrocnemius and triceps brachii muscles). The dosage was 30 μl for gastrocnemius and 20 μl for triceps brachii muscles. Because mice injected with AAV-LacZ vector and the vehicle were indistinguishable with regard to all variables tested during the experimental period, the two groups were considered as one control group for analysis. In another subgroup (n = 7), all of the mice had AAV-GDNF vector injected into the muscles of the left forelimbs and hindlimbs and AAV-LacZ vector into those of the right ones.
Behavioral testing and mortality. Mice were first given 3 d to become acquainted with the rotarod apparatus (Rota-Rod/7650; Ugo Basile, Comerio, Italy) before the test. For detection, mice were placed on the rotating rod at the speeds of 5, 10, and 20 rpm, and the time each mouse remained on the rod was registered automatically. The onset of disease was defined as the time when the mouse could not remain on the rotarod for 7 min at a speed of 20 rpm, as described previously (Li et al., 2000). If the mouse remained on the rod for >7 min, the test was completed and scored as 7 min. Mice were tested every 2 d until they could no longer perform the task. Mortality was scored as the age of death when the mouse was unable to right itself within 30 sec when placed on its back in a supine position (Li et al., 2000).
Tissue preparation. One week before being killed, mice were bilaterally injected with neural tracer cholera toxin subunit B (CTB) (0.1% in distilled H2O, 3 μl; List Biologic, Campbell, CA) into gastrocnemius muscles to selectively label motoneurons that retained axons innervating the treated muscles. At the indicated times, gastrocnemius muscles were dissected out, weighed, rapidly frozen in liquid nitrogen-cooled isopentane, and then stored at − 80°C for immunohistochemistry or GDNF ELISA analysis. After dissecting out the muscles, the mice were perfused with ice-cold PBS, followed by 4% paraformaldehyde (PFA). The spinal cord was dissected out, postfixed for 4 hr in 4% PFA, and then cryoprotected sequentially in sucrose.
GDNF ELISA. To determine muscle GDNF levels, tissues were homogenized at a w/v ratio of 100 mg/ml in lysis buffer (137 × 10−3 mol/l NaCl, 20 × 10−3 mol/l Tris, pH 8.0, 1% NP-40, and 10% glycerol) containing protease and phosphatase inhibitors, ultrasonicated, and then centrifuged at 12,000 × g. The supernatants were acidified and neutralized to pH 7.4 before assaying. The tissue levels of GDNF were measured with an ELISA kit (GDNF Emax ImmunoAssay System; Promega, Madison, WI), according to the protocol of the supplier. The levels of GDNF were expressed as picograms per milligram of protein. The assay sensitivity ranged from 16 to 1000 pg/ml.
Immunohistochemistry. Muscle sections (10 μm) were fixed in cold acetone, followed by incubation with rabbit anti-FLAG polyclonal antibodies (1:1000; Sigma, St. Louis, MO) as primary antibodies and biotinylated anti-rabbit antibodies as secondary ones (1:400; Santa Cruz Biotechnology, Santa Cruz, CA). Sections were visualized by the avidin–biotin–peroxidase complex procedure (Vectastain ABC kits; Vector Laboratories, Burlingame, CA) using 3,3-diaminobenzidine as a chromogen.
For double-immunofluorescence staining of muscles, sections were sequentially incubated with blocking solution, polyclonal rabbit anti-FLAG antibodies (1:500; Sigma), FITC-conjugated goat anti-rabbit IgG (1:200; Santa Cruz Biotechnology), and tetramethylrhodamine-conjugated α-bungarotoxin (Molecular Probes, Eugene, OR). Sections were examined and photographed under a confocal laser scanning microscope (TCS NT; Leica, Heidelberg, Germany).
For morphological analysis of the spinal cord, serial transverse sections (30 μm) were obtained for Nissl, SMI-32, or CTB immunostaining. Free-floating sections were immunohistochemically stained for SMI-32 with a Mouse-on-Mouse kit (M.O.M kit) (Vector Laboratories, Burlingame, CA), according to the protocol of the manufacturer. Sections processed for CTB immunoreactivity were blocked with 5% rabbit serum, followed by incubation with anti-CTB antibodies (1:10000, goat antiserum to CTB; List Biologic). Sections were visualized by standard ABC methods.
For double immunostaining of the spinal cord, sections were blocked with 10% normal goat serum and the blocking solution supplied with the M.O.M kit for 1 hr, respectively, and then sequentially incubated with polyclonal rabbit anti-FLAG antibodies (1:250; Sigma) and monoclonal mouse anti-SMI-32 antibodies (1:500) overnight at 4°C. After incubation with FITC-conjugated goat anti-rabbit IgG (mouse absorbed, 1:200; Santa Cruz Biotechnology) and rhodamine-conjugated goat anti-mouse IgG (1:200; Santa Cruz Biotechnology) for 2 hr at room temperature, the sections were examined and photographed under confocal laser scanning microscope.
Morphometric analysis and cell counting. Morphometric analysis was performed on images captured with a CCD camera using KS 400 image analysis software (Zeiss, Oberkochen, Germany). The mean area of muscle fibers was calculated from counts of >1000 fibers in randomly selected areas. To compare the number of motoneurons in the spinal cord, we counted neurons in Nissl-stained and SMI-32- and CTB-immunostained sections spanning the cervical and lumbrosacral enlargements in each group, as described previously (Lewis et al., 2000). For each mouse, at least 20 sections in each sixth serial section were subjected to counting. Only large cell profiles meeting the following criteria were included: location in the ventral horn below a lateral line from the central canal, containing a distinct nucleus with a nucleolus, and possession of at least one thick process.
Statistical analyses. The data were statistically analyzed using repeated-measures ANOVA, followed by a Tukey's honestly significance difference test for multiple comparisons between groups (StatView 5.0 software; SAS, Cary, NC).
RESULTS
GDNF transgene expression in muscles of ALS mice
We determined the amount of GDNF in gastrocnemius muscles by ELISA. At 110 d of age (7 weeks after injection), the GDNF levels in AAV-GDNF vector-treated mice were 7985.0 ± 874.0 pg/mg protein, which is >120-fold higher than that in the control ALS group (62.2 ± 20.5 pg/mg protein; p < 0.01;n = 4). At the time of death, AAV-GDNF vector-treated ALS mice tended to show a decrease in intramuscular GDNF expression (3281.7 ± 667.0 pg/mg protein; n = 4). We assumed that the reduction of GDNF was attributable to the severe atrophy of the transduced muscle fibers in ALS mice, because stable GDNF expression can last for at least 8 months in age-matched wild-type mice (our unpublished data). These data suggested that AAV-GDNF vector could drive substantial transgenic GDNF expression in ALS mice until the end stage of the disease.
We next investigated the pattern of distribution of transgenic GDNF in muscles by means of immunodetection. Here, FLAG was used as a tag to distinguish transgenic GDNF from its endogenous counterpart. In AAV-GDNF vector-injected mice, strong FLAG immunoreactivity was detected in a large number of myofibers, both at 110 d of age and at the end stage of the disease. Punctured and reticular staining was observed in transverse sections of muscles, with intense immunoreactivity mainly localized in the vicinity of the sarcolemma, indicating that transgene-derived GDNF was efficiently secreted into the surrounding regions (Fig.1b,c). Substantial FLAG signals could still be detected in atrophied myofibers at the end stage of the disease.
Fig. 1.

Characterization and muscle expression of AAV vector-transduced genes after injection into gastrocnemius muscles. 5-Bromo-4-chloro-3-indolyl-β-d-galactopyranoside-stained cross sections from control ALS mice after an injection with AAV-LacZ vector at 110 d of age (a). FLAG immunoreactivity is observed around the injection sites in AAV-GDNF vector-treated ALS mice of the same age (b; low magnification). At higher magnification (c), intense immunoreactivity can be seen to be mainly localized in the vicinity of the sarcolemma as well as surrounding regions, suggesting secreted expression of transgene-derived GDNF after intramuscular AAV-GDNF vector injection. More intense immunoreactivity for FLAG (d) was localized to postsynaptic AChR-rich regions, as confirmed by double staining with rhodamine-labeled α-bungarotoxin (e), indicating the accumulation of transgene-derived GDNF at neuromuscular junctions. f, Merging of c and d. Scale bars: (inc) a, 100 μm; b, 200 μm; c, 50 μm; (in f)d–f, 50 μm.
Furthermore, we performed double-immunofluorescence staining with anti-FLAG antibodies and α-bungarotoxin. α-Bungarotoxin is a molecular probe that specifically binds to the acetylcholine receptor (AChR) with high affinity on the postsynaptic membranes of NMJs. The results showed that more intense immunoreactivity for FLAG was colocalized with α-bungarotoxin signals, indicating that transgenic GDNF was concentrated primarily in the regions of NMJs (Fig. 1d–f). As expected, the muscles treated with AAV-LacZ vector or the vehicle exhibited no immunostaining for anti-FLAG at any time point.
Preservation of vector-treated muscles
At 110 d of age, the gastrocnemius muscles in the control ALS mice weighed only approximately half those in the age-matched wild-type mice (95.8 ± 19.4 vs 183.0 ± 22.2 mg; n = 5). However, the gastrocnemius muscles of AAV-GDNF vector-treated ALS mice were ∼1.68 times (160.1 ± 32.9 mg; p < 0.01; n = 5) heavier than those of control ALS mice at the same age.
Histological analysis of muscles in control ALS mice at 110 d of age revealed widespread groups of small, acutely angulated fibers, consistent with severe neurogenic atrophy (Fig.2b). The mean myofiber area was greatly decreased (1053.8 ± 581.0 μm2; n = 4), being ∼30% of that in age-matched wild-type mice (3517.6 ± 613.5 μm2; n = 5). In contrast, the muscles treated with AAV-GDNF vector showed little evidence of neurogenic atrophy with a more consistent fiber size (Fig.2c), the mean myofiber area (2252.8 ± 1035.2 μm2; n = 5) reaching ∼71% of that in wild-type mice and more than two times that in the control ALS group. Additionally, the notable shift of myofibers toward a smaller diameter observed in control ALS mice was evidently moderated in the AAV-GDNF vector-treated group (Fig. 2d), and the percentage of atrophied myofibers of <20 μm was significantly decreased (24% in control ALS group vs 9% in AAV-GDNF-treated group).
Fig. 2.

Photomicrographs of sections of gastrocnemius muscles from ALS mice at 110 d of age. a, Wild-type mouse. Note that large groups of angulated and atrophic muscle fibers were observed in control ALS mice (b), suggesting severe neurogenic atrophy in these mice. However, AAV-GDNF vector treatment markedly attenuated this denervation atrophy (c), consistent with a greater fiber area and a decreased shift in fiber size toward smaller ones (d) compared with in control ALS mice. Scale bar, 100 μm.
Retrograde transport of transgenic GDNF into spinal motoneurons
Retrograde axonal transport of GDNF into spinal lumbar motoneurons has been demonstrated in adult rats (Leitner et al., 1999). Here we examined whether or not transgenic GDNF could also be retrogradely transported to spinal motoneurons in ALS mice. For this purpose, we took advantage of the FLAG tag in transgenic GDNF to avoid interference of the results by endogenous GDNF. SMI-32 is a well characterized antibody that specifically recognizes nonphosphorylated neurofilaments (NP-NFs) and therefore serves as a reliable marker for motoneurons (Carriedo et al., 1996). Thus, we performed double immunostaining with SMI-32 and FLAG antibodies on spinal cord sections from ALS mice. At 110 d of age, FLAG immunosignals could be detected in SMI-32-positive cells in the corresponding ventral horn in ALS mice at 7 weeks after intramuscular AAV-GDNF vector injection, whereas no FLAG signal was detected in the spinal cords of the control group ALS mice. This was further demonstrated in the subgroup of unilaterally treated ALS mice; FLAG signals could only be detected in motoneurons of the ventral horn ipsilateral to the AAV-GDNF vector-injected side (Fig.3a–c) and none in those on the contralateral AAV-LacZ vector-injected side. Although β-galactosidase signals were widely detected in AAV-LacZ vector-injected muscles, they were not observed at all in the corresponding ventral horn of the spinal cord.
Fig. 3.

The retrograde transport of transgene-derived GDNF in motoneurons of the spinal cord. At 110 d of age (7 weeks after vector administration), transgene-derived GDNF was detected in the ventral horn of the corresponding spinal cord ipsilateral to the AAV-GDNF vector-injected side on double-immunofluorescence staining with anti-FLAG (a) and SMI-32 (b) antibodies. Merging of a and b showed colocalization of FLAG and SMI-positive neurons in the anterior horns, suggesting that the retrograde transport of transgene-derived GDNF occurred in motoneurons (c). d–f, The contralateral side injected with AAV-LacZ vector. Scale bar, 20 μm.
Effect of transgenic GDNF on spinal motoneuron survival
To assess the neuroprotective effect of GDNF on the survival of motoneurons, we compared the numbers of spinal motoneurons in the different groups at 110 d of age. Nissl staining of the spinal cord showed a severe loss of motoneurons in the ventral horns of the control ALS mice (Fig.4b,g). In contrast, in AAV-GDNF vector-treated mice, a significantly larger number of motoneurons remained in both cervical and lumbar segments (Fig.4c,g), suggesting a markedly protective effect of the transgenic GDNF on motoneurons.
Fig. 4.
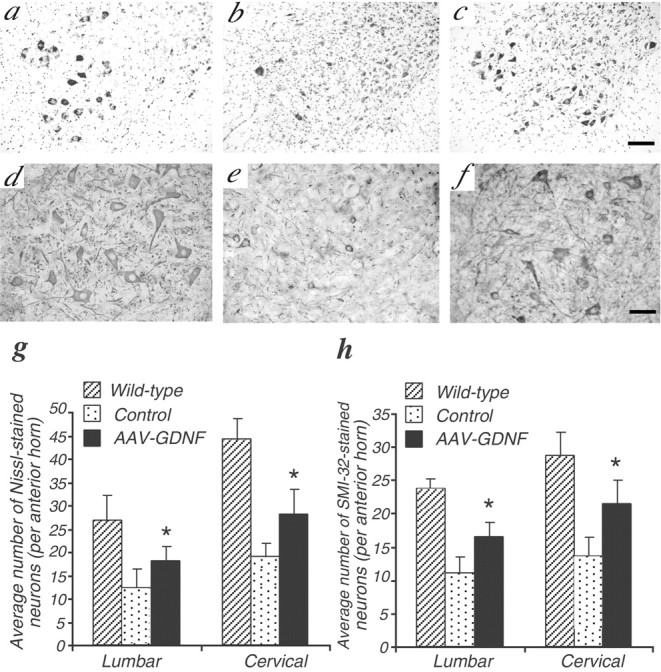
AAV-GDNF treatment significantly inhibits the loss of spinal motoneurons in ALS mice. Nissl staining (a–c) and SMI-32 immunochemistry (d–f) in wild-type (a, d), control (b,e), and AAV-GDNF vector-treated (c,f) ALS mice at 110 d of age after intramuscular injection. Scale bars: a–c, 100 μm;d–f, 50 μm. g and hshow the significant presence of both Nissl-stained and SMI-32-positive motoneurons in the ventral horns of the spinal cord, respectively (n = 4; *p < 0.01). The data represent the average numbers of neurons per anterior horn.
Staining for NP-NF is a reliable means of assessing the extent of motoneuron loss in ALS, in which it has been shown that motoneuron degeneration induces dephosphorylation of NP-NF, resulting in SMI-32 staining resistance (Tsang et al., 2000). We performed this staining on serial sections to evaluate motoneurons with NP-NF. Consistent with the Nissl-staining results, AAV-GDNF vector-treated ALS mice had significantly greater numbers of SMI-32-positive motoneurons compared with in the control ALS group (Fig. 4d–f,h). Thus, the motoneuron degeneration, as well as the aberrant NF dephosphorylation in the spinal cord ventral horn of ALS mice, is also significantly inhibited after AAV-GDNF vector administration.
In the unilaterally treated subgroup of ALS mice killed at 110 d of age, many more motoneurons survived in the lumbar spinal cord ventral horns ipsilateral to the AAV-GDNF vector-injected side than on the contralateral side treated with AAV-LacZ vector (Nissl staining, 17.1 ± 3.2 vs 10.3 ± 1.1; SMI-32-positive neurons, 15.6 ± 1.8 vs 8.8 ± 3.2; p < 0.01;n = 5). Thus, these findings further suggested that the therapeutic effect on motoneurons resulted from retrograde transport of transgenic GDNF on the same side rather than from systemic delivery.
Effect on the maintenance of motoneuron axonal projections to muscles
To further quantitatively assess surviving motoneurons that retained functioning neuromuscular projections to the injected muscles, we selectively labeled such motoneurons by injection of a neural tracer CTB into the bilateral gastrocnemius muscles of mice 1 week before being killed. At 110 d of age, there were much fewer CTB-labeled motoneurons in control ALS mice than those in wild-type mice. However, with AAV-GDNF vector treatment, more CTB-labeled motoneurons were maintained than in the control ALS group (20.7 ± 4.9 vs 11.0 ± 2.5%; n = 4; p < 0.01) (Fig.5d). Transgenic GDNF delivery to muscle thus played an important role in the maintenance of the axonal projections of corresponding motoneurons.
Fig. 5.

Effect of GDNF on motoneurons that retained axonal projections. CTB-labeled motoneurons in the ventral horns at 110 d of age in wild-type (a), control (b), and AAV-GDNF vector-treated (c) ALS mice after intramuscular injection. Scale bar, 50 μm. Note that AAV-GDNF vector-treated mice had significantly more large CTB-labeled motoneurons than control ALS mice (d). The value represents the CTB/Nissl ratio (average number of neurons per anterior horn). The shift in motoneuron size toward a smaller diameter was markedly retarded in AAV-GDNF vector-treated mice compared with in control ALS mice (e).
For more accurate morphometric evaluation of surviving motoneurons labeled with CTB, we next determined the size and distribution of such neurons in the lumbar spinal cord. The control group of ALS mice killed at 110 d of age exhibited a significantly smaller mean area of CTB-labeled neurons than that in age-matched wild-type mice (353.1 ± 173.8 vs 733.7 ± 252.0 μm2;n = 4; p < 0.01), the size distribution being shifted toward smaller ones, indicating significant atrophy of CTB-positive motoneurons (Fig. 5e). In contrast, AAV-GDNF vector treatment of ALS mice markedly decreased the motoneuron atrophy (605.8 ± 248.2 μm2;n = 4; p < 0.01 vs control group), and the size distribution shifted toward smaller ones. Together, these results may directly show that GDNF gene delivery to muscles can promote the survival and inhibit the atrophy of motoneurons with axonal projections to target muscles in ALS mice.
GDNF delays the onset of disease, improves motor performance, and prolongs survival in transgenic ALS mice
Any group of ALS mice that had AAV-GDNF vector, AAV-LacZ vector, or the vehicle injected in the four limbs at 9 weeks of age showed similar motor performance, as quantified with a rotarod, until 12 weeks of age. Thereafter, it deteriorated quickly in control ALS mice, whereas the performance deterioration was significantly delayed in AAV-GDNF vector-treated mice (p < 0.05) (Fig.6a–c), indicating significantly prolonged maintenance of their motor strength.
Fig. 6.
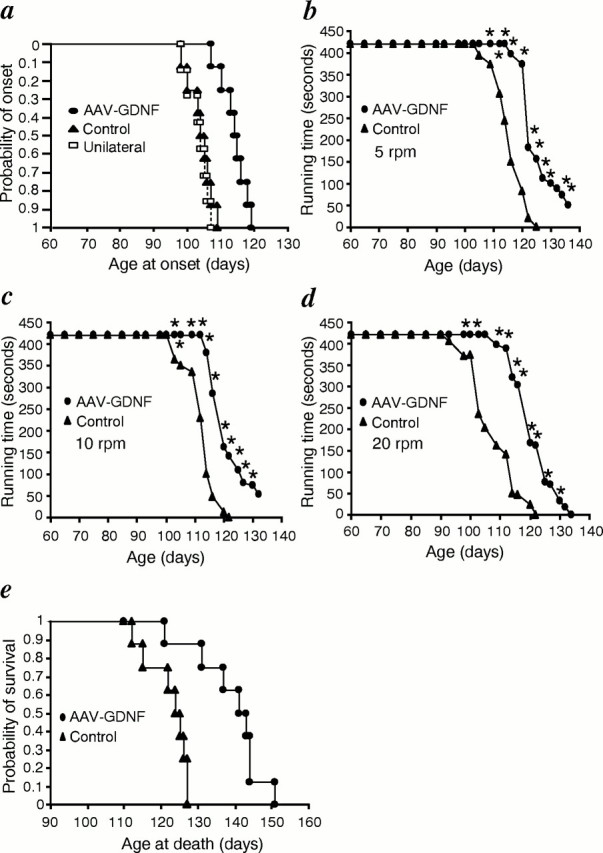
a, General behavior test for ALS mice. Cumulative probability of onset of rotarod deficits in ALS mice. The AAV-GDNF vector-treated mice had an age of onset of 114.0 ± 4.0 d (n = 12) compared with 101.3 ± 5.4 d (n = 11) for control ALS mice and 102.7 ± 3.1 d (n = 7) for unilaterally AAV-GDNF vector-treated mice. AAV-GDNF vector treatment significantly delayed disease onset by ∼13 d compared with in control ALS mice (p < 0.01), whereas the onset in unilateral AAV-GDNF vector-treated mice did not show a significant difference from that in control ALS mice. Performance of ALS mice in the rotarod test at 5 rpm (b), 10 rpm (c), and 20 rpm (d). AAV-GDNF vector-treated ALS mice performed significantly better than control ALS mice (n = 8; *p < 0.05).e, Cumulative probability of survival. Survival was significantly prolonged by ∼17 d in AAV-GDNF vector-treated ALS mice when compared with in control transgenic ALS littermates treated with the vehicle or AAV-LacZ vector (n = 8;p < 0.01).
The average age of motor deficit onset in AAV-GDNF vector-treated ALS mice was 114.0 ± 4.0 d (n = 12), whereas it was 101.3 ± 5.4 d (n = 11) in control ALS mice, the difference being significant (p < 0.01) (Fig. 6a). AAV-GDNF vector treatment prolonged the mean survival by 16.6 ± 4.1 d compared with in the control ALS mice (138.9 ± 9.2 d in AAV-GDNF vector-treated mice vs 122.3 ± 5.7 d in control ALS mice; n = 8;p < 0.01) (Fig. 6e). These results mean that bilateral intramuscular injection of AAV-GDNF vector delayed the onset of disease by ∼13% and prolonged the survival of transgenic ALS mice by ∼14%.
However, weakness and atrophy of the skeletal muscles, especially in the hindlimbs, ultimately developed in all mice of all groups once motor symptoms had appeared. The duration of the disease, as evaluated as the number of days that elapsed from the onset to the end stage, did not differ between the AAV-GDNF vector-treated and control ALS mice (24.0 ± 3.5 vs 21.0 ± 3.5 d; p > 0.05).
Because GDNF is a secreted protein, we assessed whether the therapeutic benefit of transgene-derived GDNF also resulted from systemic circulation after AAV-GDNF vector administration or not. In a subgroup of unilaterally treated ALS mice that had AAV-GDNF vector injected into their left limbs and AAV-LacZ vector injected into their right ones, each mouse moved the AAV-GDNF vector-injected limbs almost normally until 110 d of age. However, the contralateral limbs developed muscle weakness at as early as 93 d of age, there being a waddling gait. Despite the better motor functions of AAV-GDNF vector-treated limbs, the mice showed no significant difference in the running time on a rotarod at any speed tested compared with the control ALS mice (data not shown). The average onset time of motor deficit in this subgroup also showed no significant difference compared with in the control ALS group (102.7 ± 3.1 d; n = 7;p > 0.05) (Fig. 6d).
DISCUSSION
We report here that intramuscular injection of AAV-GDNF vector into the transgenic ALS mice model results in sustained substantial biosynthesis of GDNF in the muscles, with findings demonstrating its retrograde transport to the corresponding spinal motoneurons. Furthermore, this transgene expression not only significantly prevents the loss of motoneurons but also leads to marked attenuation of the manifestation of the disease and prolongs survival of the transgenic ALS mice.
Here, we demonstrated substantial expression of the GDNF transgene after an intramuscular AAV-GDNF vector injection, which is sustained until the terminal stage in ALS mice, thus probably guaranteeing a continual supply of biologically synthesized GDNF to the motoneuronal axon terminals in the muscles. This will well meet the demands for long-term availability of therapeutic factors that is necessary because of the chronicity and progression of ALS. Although we failed to detect transgenic GDNF in the spinal cord at the death stage in ALS mice, this is probably attributable to severe loss of motoneurons or/and the impaired capacity of axonal transport at the end stage (Warita et al., 1999; Williamson and Cleveland, 1999).
It has been assumed that the neuroprotective effect of GDNF on motoneurons is based on its retrograde axonal transport from a target tissue to neuronal cell bodies, but no direct evidence for this hypothesis in gene therapy has been presented yet (Mohajeri et al., 1999; Alisky and Davidson, 2000). We successfully detected transgenic GDNF in spinal neurons of the ventral horn ipsilateral to the AAV-GDNF-injected side. These cells are confirmed to be motoneurons by means of double immunofluorescence. Because the antibody we used recognizes the FLAG epitope tagging transgenic GDNF, the interference with this result by endogenous GDNF in the spinal cord is excluded.
The transgenic GDNF that appeared in the motoneurons may have been derived through three possible ways: systemic delivery, retrograde transport of AAV vectors, or retrograde transport of GDNF fusion protein itself. The restricted distribution and ipsilateral presentation of transgenic GDNF in motoneurons, as well as its known inability to pass through the blood–brain barrier, exclude the possibility of its systematic delivery to the spinal cord. To date, most reports show that AAV vectors are not retrogradely transported or are transported in only a very limited manner (Chamberlin et al., 1998;Klein et al., 1998; Alisky et al., 2000). One recent report, however, has revealed retrograde transport of an AAV vector itself in the CNS, a reporter green fluorescent protein being used as a tracer (Kaspar et al., 2002). Thus, in our study we cannot completely role out the possibility that AAV particles may also have been transported to the corresponding motoneurons. The vectors carried to the motoneurons, however, are assumed to be very limited because no β-galactosidase was detected in the corresponding spinal motoneurons, despite its wide distribution in the transduced muscles.
In contrast, the transgenic GDNF is abundantly detected in both transduced muscles and the corresponding motoneurons after AAV-GDNF injection. This finding, combined with the previous reports as well as our observation for β-galactosidase activity, indicates that the transgenic GDNF in the motoneurons is mainly derived through retrograde axonal transport of the GDNF protein. This is consistent with a previous study (Kordower et al., 2000) and our recently published data for the nigrostriatal system (Wang et al., 2002), showing that transgenic GDNF is retrogradely transported. The finding that transgenic GDNF in muscle fibers was predominantly accumulated to the regions of NMJs is also compatible with its retrograde transport hypothesized because it is in the axon terminals in which substances secreted from muscle fibers are taken up to be retrogradely transported.
Both SMI-32 staining and Nissl staining have confirmed significant rescue of motoneurons by AAV-GDNF vector delivery. Because CTB can be axonally transported to neuronal cell bodies in a retrograde direction and be detected throughout the neuronal cytoplasm (Llewellyn-Smith et al., 2000), the detection of CTB-positive motoneurons means that they maintain intact axonal connection with the AAV-GDNF vector-injected muscles. Thus, CTB labeling makes it possible to assess the effect of the transgenic GDNF on the spinal motoneurons more accurately than with Nissl or NP-NF staining alone. This method reveals greater numbers of larger spinal motoneurons labeled with CTB in AAV-GDNF vector-treated ALS mice. These findings together indicate that intramuscular injection of AAV-GDNF vector can delay the degeneration of motoneurons, thereby allowing prolonged functioning axons in ALS mice.
AAV-GDNF vector-treated ALS mice with four-limb injections, in contrast to the control ALS group, show much better behavioral performance, with delayed onset of disease and a prolonged life span, which is in agreement with the attenuation of the motoneuron pathology. In the subgroup with unilateral AAV-GDNF treatment, the therapeutic effects of GDNF on behavioral and pathological features are limited to the same treated side, with obvious deterioration of motor performance on the AAV-LacZ vector-treated side. However, the motor performance on a rotarod and the onset of disease remain similar to those in the control group. Thus, it is assumed that the therapeutic benefit mostly resulted from direct action of transgenic GDNF on motoneurons after its retrograde transport rather than from the systemic delivery.
Although bilateral administration of AAV-GDNF vector markedly delays the onset of disease and improves the survival of ALS mice, it fails to prolong the length of time from disease onset to death. What is more, despite the substantial expression of GDNF, the AAV-GDNF vector-treated mice ultimately reach the end stage, when morphological assessment demonstrates such severe atrophy of myofibers and massive loss of spinal motoneurons as in the control ALS mice (data not shown). It has been reported that pathological changes occur at asymptomatic stages in ALS mice, and massive motoneuron death occurs at the end stage (Dal Canto and Gurney, 1995; Wong et al., 1995; Mourelatos et al., 1996; Tu et al., 1996; Bruijn et al., 1998; Shibata et al., 1998). Thus, it is assumed that the transgenic GDNF only exhibits its protective function for motoneurons in ALS mice at asymptomatic stages when the ventral horns may have a mild pathology. Once the disease develops, however, GDNF gene therapy cannot effectively inhibit the massive motoneuron death or interfere with the rapidly inevitable progression of the disease. In this study, we began the treatment at the age of 9 weeks. Administration of GDNF at earlier times and/or together with other neurotrophic factors (Bilak et al., 2001) may lead to better results.
In summary, we showed that intramuscular injection of AAV-GDNF vector in ALS mice results in long-term expression of GDNF in muscles, bringing about obvious benefits in behavioral, functional, and pathological features. The transgenic GDNF protein observed in the spinal motoneurons suggests its retrograde transport from the nerve terminals to motoneuronal cell bodies. Together, these data imply that AAV-mediated GDNF delivery to muscle will be a promising means of gene therapy for ALS.
Footnotes
This study was supported in part by the following: a Grant-in-Aid for Scientific Research on Priority Areas and Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science and Technology, The Japanese Government; by Health Sciences Research Grants from the Ministry of Health Labour and Welfare of Japan; by Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation; by Research on Neurodegenerative Diseases, Health Science Research Grants from the Ministry of Health, Labor and Welfare; and by a Grant-in-Aid for Research on Specific Diseases from the Ministry of Health, Labour and Welfare of Japan. We thank Yaeko Nagatsuka and Yoshie Sato for their excellent technical assistance. We thank Masashi Urabe and Dongsheng Fan for the helpful advice. We also thank Avigen Inc. for providing the AAV vector production system.
Correspondence should be addressed to either Dr. Imaharu Nakano or Dr. Shin-ichi Muramatsu, Division of Neurology, Department of Medicine, Jichi Medical School, 3311-1 Yakushiji, Minamikawachi-machi, Tochigi 329-0498, Japan. E-mails: inakano@ms.jichi.ac.jp andmuramats@ms.jichi.ac.jp.
Y.-Y. Lu's present address: Stem Cell Research Center, Health Science Center, Peking University, Beijing 100083, China.
REFERENCES
- 1.Alisky JM, Davidson BL. Gene therapy for amyotrophic lateral sclerosis and other motor neuron diseases. Hum Gene Ther. 2000;11:2315–2329. doi: 10.1089/104303400750038435. [DOI] [PubMed] [Google Scholar]
- 2.Alisky JM, Hughes SM, Sauter SL, Jolly D, Dubensky TW, Jr, Staber PD, Chiorini JA, Davidson BL. Transduction of murine cerebellar neurons with recombinant FIV and AAV5 vectors. NeuroReport. 2000;11:2669–2673. doi: 10.1097/00001756-200008210-00013. [DOI] [PubMed] [Google Scholar]
- 3.Bilak MM, Corse AM, Kuncl RW. Additivity and potentiation of IGF-I and GDNF in the complete rescue of postnatal motor neurons. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2:83–91. doi: 10.1080/146608201316949523. [DOI] [PubMed] [Google Scholar]
- 4.Bohn MC. A commentary on glial cell line-derived neurotrophic factor (GDNF). From a glial secreted molecule to gene therapy. Biochem Pharmacol. 1999;57:135–142. doi: 10.1016/s0006-2952(98)00280-9. [DOI] [PubMed] [Google Scholar]
- 5.Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motoneuron toxicity of an ALS-linked SOD1 mutant independent from wild type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 6.Carriedo SG, Yin HZ, Weiss JH. Motoneurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlin NL, Du B, de Lacalle S, Saper CB. Recombinant adeno-associated virus vector: use for transgene expression and anterograde tract tracing in the CNS. Brain Res. 1998;793:169–175. doi: 10.1016/s0006-8993(98)00169-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu, Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 1995;676:25–40. doi: 10.1016/0006-8993(95)00063-v. [DOI] [PubMed] [Google Scholar]
- 9.Golden JP, Baloh RH, Kotzbauer PT, Lampe PA, Osborne PA, Milbrandt J, Johnson EM., Jr Expression of neurturin, GDNF, and their receptors in the adult mouse CNS. J Comp Neurol. 1998;398:139–150. doi: 10.1002/(sici)1096-9861(19980817)398:1<139::aid-cne9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Grundstrom E, Askmark H, Lindeberg J, Nygren I, Ebendal T, Aquilonius SM. Increased expression of glial cell line-derived neurotrophic factor mRNA in muscle biopsies from patients with amyotrophic lateral sclerosis. J Neurol Sci. 1999;162:169–173. doi: 10.1016/s0022-510x(98)00333-5. [DOI] [PubMed] [Google Scholar]
- 11.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motoneuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 12.Haase G, Kennel P, Pettmann B, Vigne E, Akli S, Revah F, Schmalbruch H, Kahn A. Gene therapy of murine motor neuron disease using adenoviral vectors for neurotrophic factors. Nat Med. 1997;3:429–436. doi: 10.1038/nm0497-429. [DOI] [PubMed] [Google Scholar]
- 13.Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simmons L, Moffet B, Vandlen RA, Simpson LC. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- 14.Julien JP. Amyotrophic lateral sclerosis: unfolding the toxicity of the misfolded. Cell. 2001;104:581–591. doi: 10.1016/s0092-8674(01)00244-6. [DOI] [PubMed] [Google Scholar]
- 15.Kaspar BK, Erickson D, Schaffer D, Hinh L, Gage FH, Peterson DA. Targeted retrograde gene delivery for neuronal protection. Mol Ther. 2002;5:50–56. doi: 10.1006/mthe.2001.0520. [DOI] [PubMed] [Google Scholar]
- 16.Kay MA, Manno CS, Ragni MV, Larson PJ, Couto LB, McClelland A, Glader B, Chew AJ, Tai SJ, Herzog RW, Arruda V, Johnson F, Scallan C, Skarsgard E, Flake AW, High KA. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet. 2000;24:257–261. doi: 10.1038/73464. [DOI] [PubMed] [Google Scholar]
- 17.Klein RL, Meyer EM, Peel AL, Zolotukhin S, Meyers C, Muzyczka N, King MA. Neuron-specific transduction in the rat septohippocampal or nigrostriatal pathway by recombinant adeno-associated virus vectors. Exp Neurol. 1998;150:183–194. doi: 10.1006/exnr.1997.6736. [DOI] [PubMed] [Google Scholar]
- 18.Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, Brown WD, Holden JE, Pyzalski R, Taylor MD, Carvey P, Ling Z, Trono D, Hantraye P, Deglon N, Aebischer P. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science. 2000;290:767–773. doi: 10.1126/science.290.5492.767. [DOI] [PubMed] [Google Scholar]
- 19.Leitner ML, Molliver DC, Osborne PA, Vejsada R, Golden JP, Lampe PA, Kato AC, Milbrandt J, Johnson EM., Jr Analysis of the retrograde transport of glial cell line-derived neurotrophic factor (GDNF), neurturin, and persephin suggests that in vivo signaling for the GDNF family is GFRalpha coreceptor-specific. J Neurosci. 1999;19:9322–9331. doi: 10.1523/JNEUROSCI.19-21-09322.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Dressman D, Tsao YP, Sakamoto A, Hoffman EP, Xiao X. rAAV vector-mediated sarcogylcan gene transfer in a hamster model for limb girdle muscular dystrophy. Gene Ther. 1999;6:74–82. doi: 10.1038/sj.gt.3300830. [DOI] [PubMed] [Google Scholar]
- 22.Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- 23.Llewellyn-Smith IJ, Martin CL, Arnolda LF, Minson JB. Tracer-toxins: cholera toxin B-saporin as a model. J Neurosci Methods. 2000;103:83–90. doi: 10.1016/s0165-0270(00)00298-3. [DOI] [PubMed] [Google Scholar]
- 24.Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman GJ, Iwaki Y, Colosi P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 25.Mohajeri MH, Figlewicz DA, Bohn MC. Intramuscular grafts of myoblasts genetically modified to secrete glial cell line-derived neurotrophic factor prevent motoneuron loss and disease progression in a mouse model of familial amyotrophic lateral sclerosis. Hum Gene Ther. 1999;10:1853–1866. doi: 10.1089/10430349950017536. [DOI] [PubMed] [Google Scholar]
- 26.Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motoneurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oppenheim RW, Houenou LJ, Johnson JE, Lin LF, Li L, Lo AC, Newsome AL, Prevette DM, Wang S. Developing motoneurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;373:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- 28.Sagot Y, Tan SA, Hammang JP, Aebischer P, Kato AC. GDNF slows loss of motoneurons but not axonal degeneration or premature death of pmn/pmn mice. J Neurosci. 1996;16:2335–2341. doi: 10.1523/JNEUROSCI.16-07-02335.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shibata N, Hirano A, Kobayashi M, Dal Canto MC, Gurney ME, Komori T, Umahara T, Asayama K. Presence of Cu/Zn superoxide dismutase (SOD) immunoreactivity in neuronal hyaline inclusions in spinal cords from mice carrying a transgene for Gly93Ala mutant human Cu/Zn SOD. Acta Neuropathol. 1998;95:136–142. doi: 10.1007/s004010050777. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki H, Hase A, Miyata Y, Arahata K, Akazawa C. Prominent expression of glial cell line-derived neurotrophic factor in human skeletal muscle. J Comp Neurol. 1998;402:303–312. [PubMed] [Google Scholar]
- 31.Tsang YM, Chiong F, Kuznetsov D, Kasarskis E, Geula C. Motoneurons are rich in non-phosphorylated neurofilaments: cross-species comparison and alterations in ALS. Brain Res. 2000;861:45–58. doi: 10.1016/s0006-8993(00)01954-5. [DOI] [PubMed] [Google Scholar]
- 32.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Muramatsu S, Lu Y, Ikeguchi K, Fujimoto K, Okada T, Mizukami H, Hanazono Y, Kume A, Urano F, Ichinose H, Nagatsu T, Nakano I, Ozawa K. Delayed delivery of AAV-GDNF prevents nigral neurodegeneration and promotes functional recovery in a rat model of Parkinson's disease. Gene Ther. 2002;9:381–389. doi: 10.1038/sj.gt.3301682. [DOI] [PubMed] [Google Scholar]
- 35.Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999;819:120–131. doi: 10.1016/s0006-8993(98)01351-1. [DOI] [PubMed] [Google Scholar]
- 36.Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motoneurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 37.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motoneuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 38.Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto M, Sobue G, Yamamoto K, Terao S, Mitsuma T. Expression of glial cell line-derived growth factor mRNA in the spinal cord and muscle in amyotrophic lateral sclerosis. Neurosci Lett. 1996;204:117–120. doi: 10.1016/0304-3940(96)12342-9. [DOI] [PubMed] [Google Scholar]
- 40.Yan Q, Matheson C, Lopez OT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motoneurons. Nature. 1995;373:341–344. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]